A new version of this title is available
See the updated version of this chapter
Lysosomes are intracellular membrane-bound organelles that turn over and degrade many types of macromolecules, via the action of lysosomal enzymes (also called acid hydrolases because of the low-pH characteristic of lysosomes). These enzymes are synthesized in the endoplasmic reticulum (ER) on membrane-bound ribosomes and traverse the ER–Golgi pathway along with other newly synthesized proteins. At the terminal Golgi compartment, they are segregated from other glycoproteins and selectively delivered to lysosomes. In most “higher” animal cells, this specialized trafficking is achieved primarily through the recognition of N-glycans containing mannose 6-phosphate (M6P) by “P-type” lectins. As the first clear-cut example of a biological role for glycans on mammalian glycoproteins and the first shown link between glycoprotein biosynthesis and human disease, the interesting history of its discovery is described in some detail. More recent data on other proteins with “P-type” lectin domains are also mentioned.
HISTORICAL BACKGROUND
I-Cell Disease and the Common Recognition Marker of Lysosomal Enzymes
Early studies of human genetic storage disorders by Elizabeth Neufeld indicated a failure to degrade cellular components, which therefore accumulated in lysosomes (Chapter 44). Soluble corrective factors from normal cells could reverse these defects when added to the culture media. These factors were later identified as lysosomal enzymes that were found to be deficient in patients with storage disorder diseases. These enzymes, secreted in small quantities by normal cells in culture, or by cells from patients with a different “complementary” defect (Figure 33.1), exist in two forms: a high-uptake form, recognized by saturable, high-affinity receptors that could correct the defect in deficient cells, and an inactive low-uptake form that could not correct the defect.

FIGURE 33.1.
Historical background regarding cross-correction of lysosomal enzyme deficiencies in cultured cells. Small amounts of high-uptake lysosomal enzymes secreted by normal fibroblasts (thin arrows) were found to be taken up by fibroblasts from a patient with (more...)
Fibroblasts from patients with a rare genetic disease showing prominent inclusion bodies in cultured cells (therefore termed I-cell disease) were found to be deficient in almost all lysosomal enzymes. Interestingly, all of these enzymes are actually synthesized by I-cells, but are mostly secreted into the medium, instead of being retained in lysosomes. Neufeld showed that although I-cells incorporate the high-uptake enzymes secreted by normal cells, enzymes secreted by I-cells are not taken up by other cells (Figure 33.1). She therefore proposed that I-cell disease results from a failure to add a common recognition marker to lysosomal enzymes. This marker was assumed to be responsible for the proper trafficking of newly synthesized enzymes to lysosomes in normal cells. Because the high-uptake property was destroyed by strong periodate treatment, which oxidizes vicinal hydroxyl groups in sugars, it was predicted that the recognition marker was a glycan.
Discovery of the M6P Recognition Marker
High uptake of lysosomal enzymes was next found by William Sly to be blocked by M6P and its stereoisomer fructose 1-phosphate, but not by other sugar phosphates. Treatment of lysosomal enzymes with alkaline phosphatase also abolished high-uptake activity. By this time, the general pathway for N-glycan processing had been defined (see Chapter 9). Because oligomannosyl N-glycans are rich in mannose residues, these were predicted to be phosphorylated on lysosomal enzymes. Indeed, the M6P moiety detected on oligomannosyl N-glycans was released by endo-β-N-acetylglucosaminidase H (endo H) from high-uptake forms of lysosomal enzymes, confirming the hypothesis. Surprisingly, the groups of Stuart Kornfeld and Kurt von Figura then found that a portion of the M6P moieties were “blocked” by α-linked N-acetylglucosamine residues attached to the phosphate residue, creating a phosphodiester.
Enzymatic Mechanism for Generation of the M6P Recognition Marker
Comparison of glycans with phosphodiesters and phosphomonoesters predicted that the metabolic precursor of the M6P determinant was a phosphodiester and that phosphorylation was mediated not by an ATP-dependent kinase but by a UDP-GlcNAc-dependent GlcNAc-1-phosphotransferase. This was proven using a double-labeled donor and Golgi extracts:
Another Golgi enzyme was shown to remove the outer N-acetylglucosamine residue and uncover the M6P moiety. Pulse-chase studies confirmed the order of events, documenting that multiple glycans on a given lysosomal enzyme could acquire one or two phosphate residues and that removal of some mannose residues on the N-glycan by processing Golgi mannosidases was also required (Figure 33.2). In most cell types, the phosphate is eventually lost from the M6P, presumably after exposure to acid phosphatase in lysosomes. Thus, the overall biochemical pathway is as follows:

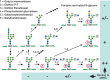
FIGURE 33.2.
Pathways for biosynthesis of N-glycans bearing the mannose 6-phosphate (M6P) recognition marker. Following early N-glycan processing (Chapter 9), a single GlcNAc phosphodiester is added to the N-glycans of lysosomal enzymes on one of three mannose (Man) (more...)
The first two enzymes that mediate these reactions were then purified and characterized and the genes encoding these proteins were cloned. UDP-GlcNAc:lysosomal enzyme GlcNAc-1-phosphotransferase (GlcNAc-P-T) is an α2β2γ2 complex encoded by two genes. The α and β subunits are encoded by the GNPTAB gene, whose 1256 amino acid type-III transmembrane product undergoes proteolysis in the Golgi to give rise to the two subunits, whereas the 305 amino acid γ-subunit is encoded by the GNPTG gene (Figure 33.3). The α/β subunits contain the catalytic function of the enzyme (Stealth domains) as well as elements (Notch1, Notch2, and DMAP [DNA methylase-associated protein]) that mediate binding to protein determinants present on the lysosomal enzymes. The γ-subunit facilitates the phosphorylation of a subset of the lysosomal enzymes. It contains an M6P receptor homology (MRH) domain that is thought to bind oligomannosyl N-glycans on the lysosomal enzymes and present them to the catalytic site for phosphorylation, and a DMAP recognition domain. The second enzyme, α-N-acetylglucosamine-1-phosphodiester glycosidase encoded by the NAGPA gene, is a 272-kDa complex of four identical 68-kDa subunits, arranged as two disulfide-linked homodimers. Unlike other Golgi enzymes, this is a type-I membrane-spanning glycoprotein with its amino terminus in the lumen of the Golgi.

FIGURE 33.3.
GlcNAc-P-T is an α2β2γ2 hexamer encoded by two genes. The GNPTAB gene encodes a catalytically inactive type 3 transmembrane precursor that undergoes a proteolytic cleavage between Lys-928 and Asp-929 in the Golgi to generate the (more...)
The oligomannosyl N-glycans of lysosomal enzymes are identical to those of many other glycoproteins passing through the ER–Golgi pathway. Thus, specific recognition by GlcNAc-P-T is crucial to achieve selective trafficking. This recognition is not explained by any similarities in the primary polypeptide sequences of lysosomal enzymes. Indeed, denatured lysosomal enzymes lose their specialized GlcNAc-P-T acceptor activity, indicating that features of secondary or tertiary structure are critical for recognition. Two complementary approaches have been used to define elements of this recognition marker. In loss-of-function studies, various amino acids of the lysosomal enzyme have been replaced with alanine, with the effect on phosphorylation determined. In gain-of-function experiments, residues of the lysosomal protease cathepsin D have been substituted into the homologous secretory protease glycopepsinogen. These studies revealed that selected lysine residues have a critical role in the interaction with GlcNAc-P-T. In fact, as few as two lysines in the correct orientation to each other and to an N-glycan can serve as minimal elements of the recognition domain. However, additional amino acid residues function to enhance the interaction with GlcNAc-P-T. In some instances (e.g., cathepsin D), the enzyme may contain a very extended determinant, or perhaps, more than one recognition domain.
The GlcNAc phosphodiester glycosidase that catalyzes the exposure of the M6P recognition marker is found primarily in the trans-Golgi network (TGN), and it cycles between this compartment and the plasma membrane. Thus, uncovering the recognition marker is a late event in the Golgi apparatus, occurring just before loading of the enzymes onto the M6P receptors (MPRs).
Enzymatic Basis for I-Cell Disease and Pseudo-Hurler Polydystrophy
Analysis of fibroblasts from patients with I-cell disease (also called mucolipidosis-II; ML-II) revealed a deficiency in GlcNAc-P-T enzyme activity. A milder variant called pseudo-Hurler polydystrophy (mucolipidosis-III; ML-III) showed a less severe deficiency of enzyme activity. Metabolic radiolabeling of fibroblasts corroborated the failure to phosphorylate mannose residues in these diseases, and asymptomatic obligate heterozygotes showed a partial deficiency, with slightly elevated levels of serum lysosomal enzymes. Mutations of various types in the two GlcNAc-P-T genes have since been detected in all examined patients with ML-II and -III, indicating that deficiency of this enzyme is the primary genetic cause of the disorder.
COMMON FEATURES OF P-TYPE LECTINS (M6P RECEPTORS)
The first candidate (∼275-kDa) receptor for the M6P recognition marker was isolated by affinity chromatography and was found to bind M6P in the absence of cations (CI-MPR [cation-independent mannose 6-phoshate receptor]). Certain cells deficient in this receptor still showed M6P-inhibitable binding of lysosomal enzymes, leading to the discovery of a second MPR of ∼45 kDa, which required divalent cations for optimal binding (CD-MPR [cation-dependent MPR]). Both receptors bind with highest affinity to glycans carrying two M6P residues (Figure 33.2, structure C) and poorly to molecules bearing GlcNAc-P-Man phosphodiesters (Figure 33.2, structures A and B). Binding to molecules carrying one M6P (Figure 33.2, structure D) is intermediate in affinity. Removal of outer mannose residues by processing Golgi mannosidases enhances binding.
Genes encoding both MPRs have been cloned and extensively characterized. Both are type I transmembrane glycoproteins with large extracytoplasmic domains, transmembrane regions and carboxy-terminal cytoplasmic domains. The CI-MPR has 15 contiguous repetitive units of ∼145 amino acids with partial identity to one another. The CD-MPR has a single extracellular domain, showing homology with the repeating domains of the CI-MPR. Together with conservation of certain intron–exon boundaries, this homology suggests that the two genes evolved from a common ancestor. On the basis of their sequence relationships and unique binding properties to M6P, the two MPRs have been formally classified as P-type lectins. Structural homologs of the MPRs are present in yeast and Drosophila, but these proteins lack M6P-binding ability.
The CD-MPR exists mainly as a dimer, with each monomer binding one M6P residue. However, monomeric and tetrameric forms of the CD-MPR exist, and the equilibrium between forms is affected by temperature, pH, and the presence of ligands. The CI-MPR also seems to be a dimer in the membrane, although it readily dissociates on solubilization. Two of its repeating units (3 and 9) bind M6P phosphomonoesters with high affinity, a third (5) binds M6P phosphodiesters with low affinity, and a fourth (15) binds both M6P phosphomonoesters and diesters with equal affinity. Mutagenesis studies have identified the specific residues of these receptors involved in M6P binding, and crystal structures of several M6P-binding domains have been obtained in a complex with M6P. The CD-MPR has been crystallized as a dimer, with each monomer folded into a nine-stranded flattened β-barrel that has a striking resemblance to the protein folds in avidin (Figure 33.4). The distance between the two ligand-binding sites of the dimer provides a good explanation for the differences in binding affinity shown by the CD-MPR toward various lysosomal enzymes. The crystal structure of the amino-terminal 432 residues of the CI-MPR, encompassing domains 1–3 (domain 3 is one of the M6P-binding domains), has also been solved. Each domain shows a topology similar to that of the CD-MPR, and the three domains assemble into a compact structure that provides insight into the arrangement of the entire extracellular region of the CI-MPR. The proposed model does not position the two M6P-binding domains (3 and 9) sufficiently close to bind a single, diphosphorylated N-glycan. This suggests that the high-affinity binding of this N-glycan is due to the spanning of binding sites located on different monomers of the CI-MPR. An alternative possibility is that the receptor is dynamic, with the spacing between the two M6P-binding sites being flexible, to enhance interactions with lysosomal enzymes containing phosphorylated glycans at various positions on their protein backbones.

FIGURE 33.4.
Ribbon diagram of the bovine cation-dependent M6P receptor (CD-MPR). Shown are the two monomers (magenta and cyan ribbons) of the dimer as well as the ligand M6P (gold ball-and-stick model). (Modified, with permission, from Roberts et al. 1998. Cell (more...)
INDUCED GENETIC DEFECTS IN THE MPRs
Targeted disruption of the CD-MPR gene in mice is associated with normal or only slightly elevated levels of lysosomal enzymes in the circulation and an otherwise grossly normal phenotype. However, thymocytes or primary cultured fibroblasts from such mice show an increase in the amount of phosphorylated lysosomal enzymes secreted into the medium. This indicates that there are mechanisms that compensate for the deficiency in vivo. Intravenous injection of inhibitors of other glycan-specific receptors capable of mediating endocytosis (e.g., the mannose receptor of macrophages and the asialoglycoprotein receptor of hepatocytes; see Chapter 34) give rise to a marked increase in lysosomal enzymes in the serum of the deficient mice. Thus, such receptors are likely part of the compensatory mechanisms in vivo.
Like fibroblasts that lack only CD-MPR, fibroblasts that lack only CI-MPR have a partial impairment in sorting. Fibroblasts from embryos that lack both receptors show a massive missorting of multiple lysosomal enzymes. Thus, both receptors are required for efficient intracellular targeting of lysosomal enzymes. Comparison of lysosomal enzymes secreted by the different cell types indicates that the two receptors may interact preferentially with different subgroups of enzymes. Thus, the structural heterogeneity of the M6P recognition marker within a single lysosomal enzyme and between different enzymes is one explanation for the evolution of two MPRs with complementary binding properties: that is, to provide an efficient but varied targeting of lysosomal proteins in different cell types or tissues. In the final analysis, what initially appeared to be a precise lock-and-key mechanism turns out to be a far more complex and flexible system, with functionally useful biological flexibility.
SUBCELLULAR TRAFFICKING OF THE MPRs
At steady state, MPRs are concentrated in the TGN and late endosomes, but they cycle constitutively between these organelles, early (sorting) endosomes, recycling endosomes, and the plasma membrane (Figure 33.5). MPRs avoid delivery to lysosomes, in which they would be degraded. This trafficking is governed by a number of short amino acid sorting signals in the cytoplasmic tails of the receptors. The TGN is the site where newly synthesized lysosomal enzymes bind to MPRs that are then collected into clathrin-coated pits and packaged into clathrin-coated vesicles for delivery to the early endosome. This process involves interaction of the MPRs with two types of coat proteins: the GGAs (Golgi-localized, γ-ear–containing, ADP-ribosylation factor binding) and AP1 (adapter protein 1). In addition to binding MPRs, the coat proteins recruit clathrin for the assembly of clathrin-coated vesicles. Following delivery to early endosomes, lysosomal enzymes are released from MPRs as the endosomes mature to late endosomes and the pH decreases. Late endosomes then undergo dynamic fusion/fission with lysosomes, allowing selective transfer of lysosomal enzymes to the lysosomes and leaving the MPRs behind in subdomains of the late endosomes. These MPRs may then either return to the TGN or move to the plasma membrane, in which internalization via clathrin-coated pits occurs, mediated by the coat protein AP2. There are several pathways for the MPRs to be returned to the TGN from the various endosomal compartments, although the relative importance of the different pathways is unclear.
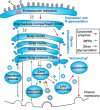
FIGURE 33.5.
Subcellular trafficking pathways of glycoproteins, lysosomal enzymes, and M6P receptors (MPRs). Newly synthesized glycoproteins originating from the rough endoplasmic reticulum (ER) pass through the Golgi stacks and are then sorted to various destinations. (more...)
The CI-MPR Mediates Uptake of Phosphorylated Lysosomal Enzymes—Implications for Enzyme Replacement Therapy
The original experiments of Neufeld showed that a portion of newly synthesized lysosomal enzyme molecules are secreted into the medium, but may be recaptured by the same cell or by adjacent cells expressing cell-surface CI-MPRs (Figure 33.1). Enzyme molecules that bind to such cell-surface MPRs are endocytosed via clathrin-coated pits and vesicles, eventually reaching the same late endosomal compartments in which newly synthesized molecules arrive from the Golgi. This secretion–recapture pathway is a minor one in most cells, but plays a critical role in enzyme replacement therapy.
As described in Chapter 44, there are many genetic disorders in glycan degradation that result from decreased activity of a given lysosomal enzyme. Some of these enzymes that are targeted to lysosomes via the M6P pathway have been prepared in large quantities as recombinant soluble proteins and used in enzyme replacement therapy. To date, the benefits have been variable but less than optimal. There are a number of potential reasons for this. First, some of the preparations may not contain the physiologic complement of the phosphomannosyl recognition marker. It is reasonable to suggest that a greater M6P content may improve the efficacy of enzyme replacement in these patients. This has been shown to be the case in mouse and dog model systems. However, even with fully phosphorylated enzymes there may be obstacles that are difficult to overcome. For example, some cell types in the body may not express adequate levels of the CI-MPR on their surfaces to endocytose sufficient enzyme to restore normal lysosomal function. Also, the organ that is most seriously affected in many of these diseases (the brain) is inaccessible because of the blood–brain barrier. This is an area where further studies are needed.
EVOLUTIONARY ORIGINS OF THE M6P RECOGNITION SYSTEM
Although the MPR pathway has a major role in vertebrate lysosomal enzyme trafficking, its contribution in invertebrate systems is not prominent. Lysosomal enzymes are targeted in organisms such as Saccharomyces, Trypanosoma, and Dictyostelium, without the aid of identifiable MPRs. The slime mold Dictyostelium discoideum produces a novel methylphosphomannose structure on some of its lysosomal enzymes that can be recognized in vitro by the mammalian CI-MPR (not the CD-MPR). However, despite the presence of a GlcNAc-P-T that recognizes α1-2-linked mannose residues, the enzyme does not specifically recognize lysosomal enzymes, and no receptor for M6P has been found in this organism. The protozoan Acanthamoeba produces a GlcNAc-P-T that specifically recognizes lysosomal enzymes. However, this organism lacks a gene that could encode an “uncovering” enzyme, and so would not be expected to form M6P available to an MPR. Although some additional “lower” organisms do show evidence for an uncovering enzyme, no MPR activity has yet been found. The evolutionary divergence point at which the complete MPR system became established remains to be defined.
THE CI-MPR BINDS MANY OTHER LIGANDS
Although originally discovered as a receptor for lysosomal enzyme trafficking, the CI-MPR turns out to be a remarkably multifunctional molecule. It binds IGF-II with high affinity even though this polypeptide lacks M6P residues. Many studies have explored potential interactions between these two disparate ligands. Although these two ligands bind to distinct sites on the CI-MPR receptor, there are conflicting reports regarding synergistic or antagonistic interactions between the two activities. Regardless, it appears that the CI-MPR acts primarily as a general sink for excess IGF-II in the extracellular fluid, carrying it to the lysosome for degradation, and reducing the amount available to bind to the IGF-I receptor. Several reports indicate that binding of IGF-II to the CI-MPR regulates motility and growth in some cell types. It has also been found that the CI-MPR binds retinoic acid with high affinity at a site that is distinct from those for M6P and IGF-II. This binding of retinoic acid seems to enhance the primary functions of the CI-MPR receptor, and the biological consequence appears to be the suppression of cell proliferation and/or induction of apoptosis. The significance of this unexpected observation is still being explored. Other ligands include urokinase-type plasminogen activator receptor (uPAR) and plasminogen.
There are also some unexplained changes in CI-MPR expression in relation to malignancy. Loss of heterozygosity at the CI-MPR locus occurs in dysplastic liver lesions and in hepatocellular carcinomas associated with the high-risk factors of hepatitis virus infection and liver cirrhosis. Mutations in the remaining allele were detected in ∼50% of these tumors, which also seem to frequently develop from clonal expansions of phenotypically normal, CI-MPR-mutated hepatocytes. Thus, the CI-MPR fulfills many of the classic criteria to be classified as a liver “tumor-suppressor” gene.
SIGNIFICANCE OF M6P ON NONLYSOSOMAL PROTEINS
Interestingly, M6P-containing N-glycans have been found on a variety of nonlysosomal proteins. Some are hydrolytic enzymes that seem to have taken on a predominantly secretory route, for example, uteroferrin and DNase I. In the first case, the failure of removal of the blocking N-acetylglucosamine residues may be the cause for secretion. With DNase I, the native level of phosphorylation simply appears to be low. M6P has been found on the transforming growth factor β (TGF-β) precursor and the phosphate is lost from the mature form. It appears that M6P may serve to target the precursor to CI-MPR for activation. Other nonlysosomal proteins reported to carry M6P include proliferin, CREG (cellular repressor of E1A-stimulated genes), LIF (leukemia inhibitory factor), and thyroglobulin. In the last case, M6P-containing N-glycans are suggested as a mechanism to target the protein for degradation and release of thyroid hormones. But one should not necessarily assume that M6P-containing N-glycans on all of these proteins are involved in intracellular trafficking. Just as phosphorylation of serine residues has diverse biological roles, M6P might be used for more than one purpose in a complex multicellular organism.
Herpes simplex virus (HSV) and Varicella zoster virus (VZV) glycoproteins have also been shown to carry N-glycans with M6P. In these cases, M6P is on complex N-glycans, suggesting that it originates from a distinct biosynthetic pathway. Regardless of its mode of synthesis, interaction of cell-free VZV with CI-MPR at the cell surface is required for viral entry into endosomes. Interestingly, intracellular CI-MPR can also divert newly synthesized enveloped VZV to late endosomes in which the virions are inactivated before exocytosis. This is suggested as the mechanism by which this successful parasite limits immediate excessive spread and avoids killing the host or possibly, a form of host defense. Biopsies of VZV-infected human skin showed that CI-MPR expression is lost in maturing superficial epidermal cells, preventing diversion of VZV to endosomes and allowing constitutive secretion of infectious VZV. These data implicate CI-MPR in the complex biology of VZV infection.
OTHER PATHWAYS FOR TRAFFICKING LYSOSOMAL ENZYMES
Although the M6P recognition marker has a crucial role in trafficking newly synthesized lysosomal enzymes to vertebrate lysosomes, alternate mechanisms exist in some cell types. In patients with I-cell disease some cells and tissues, such as the liver and circulating granulocytes, have essentially normal levels of enzymes. B-lymphoblast lines derived from these patients also do not show the complete phenotype of enzyme deficiency seen in fibroblasts. One possible explanation is that the M6P pathway for trafficking of lysosomal enzymes is a specialized form of targeting, superimposed on some other basic mechanisms that more primitive organisms use, some of which remain undefined. In this regard, two lysosomal enzymes, acid phosphatase and β-glucocerebrosidase, are not at all affected in their distribution in I-cell disease fibroblasts. With acid phosphatase, the enzyme is synthesized initially as a membrane-bound protein, and once in the lysosome, it is proteolytically cleaved to generate the mature soluble form. Glucocerebrosidase is targeted to lysosomes independently of the MPR pathway by forming a complex with LIMP-2, a lysosomal membrane protein that contains a targeting signal in its cytoplasmic tail. Likewise, other integral membrane proteins of the lysosome use similar targeting motifs in their cytoplasmic tails to traffic to the lysosome.
Mannose 6-Phosphate Receptor Homology Domain-Containing Proteins Function in the Secretory Pathway
A number of proteins have now been identified as P-type lectins because of the presence of MRH domains. These MRH domains contain the residues that bind mannose, but lack the residues that bind phosphate. In several instances, the proteins have been shown to bind to oligomannosyl structures. Most of these proteins reside in the ER where they function as lectins in the ER quality control pathway. Interestingly, the γ-subunit of GlcNAc-P-T has an MRH domain that plays a critical role in the phosphorylation of a subset of lysosomal enzymes. The precise biologic role of these MRH domains remains to be elucidated.
ACKNOWLEDGMENTS
The authors appreciate helpful comments and suggestions from Sally Riewe Boyd, Sumit Rai, and Patience Sanderson.
FURTHER READING
- Fratantoni JC, Hall CW, Neufeld EF. 1968. Hurler and Hunter syndromes: Mutual correction of the defect in cultured fibroblasts. Science 162: 570–572. [PubMed: 4236721]
- Hickman S, Neufeld EF. 1972. A hypothesis for I-cell disease: Defective hydrolases that do not enter lysosomes. Biochem Biophys Res Commun 49: 992–999. [PubMed: 4345092]
- Kaplan A, Achord DT, Sly WS. 1977. Phosphohexosyl components of a lysosomal enzyme are recognized by pinocytosis receptors on human fibroblasts. Proc Natl Acad Sci 74: 2026–2030. [PMC free article: PMC431066] [PubMed: 266721]
- Hasilik A, Klein U, Waheed A, Strecker G, von Figura K. 1980. Phosphorylated oligosaccharides in lysosomal enzymes: Identification of α-N-acetylglucosamine(1)phospho(6)mannose diester groups. Proc Natl Acad Sci 77: 7074–7078. [PMC free article: PMC350443] [PubMed: 6938953]
- Tabas I, Kornfeld S. 1980. Biosynthetic intermediates of β-glucuronidase contain high mannose oligosaccharides with blocked phosphate residues. J Biol Chem 255: 6633–6639. [PubMed: 7391040]
- Reitman ML, Varki A, Kornfeld S. 1981. Fibroblasts from patients with I-cell disease and pseudo-Hurler polydystrophy are deficient in uridine 5′-diphosphate-N-acetylglucosamine: Glycoprotein N-acetylglucosaminylphosphotransferase activity. J Clin Invest 67: 1574–1579. [PMC free article: PMC370727] [PubMed: 6262380]
- Varki A, Kornfeld S. 1983. The spectrum of anionic oligosaccharides released by endo-β-N-acetylglucosaminidase H from glycoproteins. Structural studies and interactions with the phosphomannosyl receptor. J Biol Chem 258: 2808–2818. [PubMed: 6298207]
- Kornfeld S. 1986. Trafficking of lysosomal enzymes in normal and disease states. J Clin Invest 77: 1–6. [PMC free article: PMC423299] [PubMed: 3003148]
- von Figura K, Hasilik A. 1986. Lysosomal enzymes and their receptors. Annu Rev Biochem 55: 167–193. [PubMed: 2943218]
- Kornfeld S, Mellman I. 1989. The biogenesis of lysosomes. Annu Rev Cell Biol 5: 483–525. [PubMed: 2557062]
- Munier-Lehmann H, Mauxion F, Hoflack B. 1996. Function of the two mannose 6-phosphate receptors in lysosomal enzyme transport. Biochem Soc Trans 24: 133–136. [PubMed: 8674621]
- Bonifacino JS, Traub LM. 2003. Signals for sorting of transmembrane proteins to endosomes and lysosomes. Annu Rev Biochem 72: 395–447. [PubMed: 12651740]
- Ghosh P, Dahms NM, Kornfeld S. 2003. Mannose 6-phosphate receptors: New twists in the tale. Nat Rev Mol Cell Biol 4: 202–212. [PubMed: 12612639]
- Braulke T, Pohl S, Storch S. 2008. Molecular analysis of the GlcNAc-1-phosphotransferase. J Inherit Metab Dis 31: 253–257. [PubMed: 18425436]
- Dahms NM, Olson LJ, Kim JJ. 2008. Strategies for carbohydrate recognition by the mannose 6-phosphate receptors. Glycobiology 18: 664–678. [PMC free article: PMC2733771] [PubMed: 18621992]
- Vogel P, Payne BJ, Read R, Lee WS, Gelfman CM, Kornfeld S. 2009. Comparative pathology of murine mucolipidosis types II and IIIC. Vet Pathol 46: 313–324. [PMC free article: PMC2705191] [PubMed: 19261645]
- Castonguay AC, Olson LJ, Dahms NM. 2011. Mannose 6-phosphate receptor homology (MRH) domain-containing lectins in the secretory pathway. Biochim Biophys Acta 1810: 815–826. [PMC free article: PMC3150748] [PubMed: 21723917]
- van Meel E, Lee WS, Liu L, Qian Y, Doray B, Kornfeld S. 2017. Multiple domains of GlcNAc-1-phosphotransferase mediate recognition of lysosomal enzymes. J Biol Chem 291: 8295–8307. [PMC free article: PMC4825028] [PubMed: 26833567]
Publication Details
Author Information and Affiliations
Authors
Ajit Varki and Stuart Kornfeld.Publication History
Published online: 2017.
Copyright
PDF files are not available for download.
Publisher
Cold Spring Harbor Laboratory Press, Cold Spring Harbor (NY)
NLM Citation
Varki A, Kornfeld S. P-Type Lectins. 2017. In: Varki A, Cummings RD, Esko JD, et al., editors. Essentials of Glycobiology [Internet]. 3rd edition. Cold Spring Harbor (NY): Cold Spring Harbor Laboratory Press; 2015-2017. Chapter 33. doi: 10.1101/glycobiology.3e.033




