
 1
1 {kind=link}
Copyright Notice
Diabetes in America is in the public domain of the United States. You may use the work without restriction in the United States.
Bookshelf ID: NBK597411PMID: 38117927

An official website of the United States government

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Lawrence JM, Casagrande SS, Herman WH, et al., editors. Diabetes in America [Internet]. Bethesda (MD): National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK); 2023-.
Maria J. Redondo, MD, PhD, MPH, Suna Onengut-Gumuscu, PhD, and Kyle J. Gaulton, PhD.
Author Information and AffiliationsInitial Posting: December 20, 2023.
Type 1 diabetes is a complex disease that has both genetic and environmental determinants. Based on twin and family studies from largely European-ancestry populations, the estimated contribution of genetic factors to type 1 diabetes risk is ~50%. Genes and their variants within the human major histocompatibility complex (MHC), the human leukocyte antigen (HLA) loci, including class I (HLA-A, -B, and -C) and class II (HLA-DR, -DQ, and -DP), account for ~50% of the genetic risk of type 1 diabetes. In addition to the MHC region, type 1 diabetes risk loci were initially identified through candidate gene and linkage studies, including variants in or near the INS, CTLA4, IL2RA, and PTPN22 genes. Genome-wide association approaches have revealed additional loci containing common variants with relatively small individual effects on type 1 diabetes risk. International efforts led by the Type 1 Diabetes Genetics Consortium and others have identified more than 90 non-MHC loci and narrowed the likely candidate genes and variants substantially through fine-mapping and functional studies. Ancestry-specific genetic associations of type 1 diabetes have emerged. The majority of non-MHC variants are in noncoding sequences and most likely affect gene regulation rather than directly altering protein structure. In addition to genetic factors, epigenetic changes in the genome are implicated in type 1 diabetes. Research on large cohorts of non-European ancestry, longitudinal follow-up of individuals in the preclinical stages of type 1 diabetes, and identification of the genetic and environmental underpinnings of distinct phenotypes that are emerging within the wide spectrum of type 1 diabetes are needed. Analytic and molecular studies to assess the functional significance of type 1 diabetes susceptibility gene variants would help identify critical biologic pathways that could lead to novel interventions and therapeutics.
Type 1 diabetes begins with precipitating events in an individual with genetic susceptibility. Twin and family studies provide evidence for genetic factors contributing to type 1 diabetes risk and estimates of the familial aggregation of the disease based on risk in relatives of an affected individual. In monozygotic twins, who share 100% of their genes, when a member of the pair has type 1 diabetes (proband twin), the risk to the co-twin is approximately 50%, suggesting that both genetic and nongenetic factors contribute to risk (1,2). This concordance for type 1 diabetes increases to 65% by age 60 years and 89% for autoantibody-positive pairs (2). High-risk genotypes of the human leukocyte antigen (HLA) loci (HLA-DR3, -DR4) in the major histocompatibility complex (MHC) (3) and the insulin (INS) gene (4) are found in higher frequency in concordant pairs, suggesting a major impact of genetic factors and heterogeneity in concordance rates. Dizygotic twins and non-twin siblings share only 50% of their genes, and accordingly, their concordance rate for type 1 diabetes is lower—under 8% (5,6). In contrast, the estimated prevalence of type 1 diabetes in the general population of the United States is approximately 4 per 1,000. Highlighting the role of the environment, siblings, who have lesser extent of common exposures than twins, have lower risk of development of islet autoimmunity and clinical type 1 diabetes than dizygotic twins (6).
Type 1 diabetes is most common in populations of European ancestry, but it also occurs in those of African, Hispanic, and Asian ancestry. The incidence and prevalence of type 1 diabetes in youth have been increasing in the United States over the past two decades, particularly among Hispanic and non-Hispanic Black youth (7,8). From 2002–2003 to 2017–2018, the adjusted annual incidence increase (95% confidence interval) of type 1 diabetes among African American, Hispanic American, and Asian American and Pacific Islander U.S. youth age <20 years was 2.93 (1.91–3.96), 4.14 (3.19–5.10), and 4.84 (2.57–7.15), respectively, compared to 0.60 (0.04–1.16) in non-Hispanic White youth. Despite extensive epidemiologic data on the incidence and prevalence of type 1 diabetes in various ethnic groups, there is a critical absence of data on risk to siblings in diverse populations, preventing estimation of the genetic impact on type 1 diabetes risk in non-White populations. Nearly one-half of type 1 diabetes cases in non-Hispanic White populations are diagnosed after age 20 years, and the incidence of type 1 diabetes seems to steadily increase throughout the adult years (9,10). However, the absence of information on the risk to siblings of individuals with adult-onset type 1 diabetes prevents estimation of the impact of genetic factors on risk of type 1 diabetes in adults of any population ancestry.
The genetic risk ratio in siblings (lambda-s), which is defined as the ratio of the risk in siblings of individuals with the disease to the population prevalence, is ~16 in type 1 diabetes of European ancestry (11). Yet, this figure provides no insight into either the number of genes contributing to type 1 diabetes risk or the sizes of their effects. About 50% of the risk of type 1 diabetes can be attributed to genetic (familial) factors; however, the contribution of specific genes and their variants to the overall genetic risk is variable. For example, in families with two children with type 1 diabetes, the expectation under the hypothesis that a variant in a gene has no effect on type 1 diabetes risk is that 25% of sibling pairs would share both copies of the variant (same genotype), 50% would share one copy of the variant in the genotype, and 25% would share no copies of the variant (completely different genotypes). In fact, the risk to siblings who share two HLA haplotypes is 55%, much greater than 25%, indicating that a significant proportion of the total genetic risk can be attributed to the HLA region (11).
The recognition that approximately half of risk for type 1 diabetes is genetic and half of that genetic risk is due to factors in the MHC provided the stimulus for two subsequent research paths. The first was to delineate the specific genes and mechanisms in the MHC (the HLA genes and other genes and variants) that account for the majority of type 1 diabetes genetic risk. The second was to identify the non-MHC genes that account for the remaining genetic risk for type 1 diabetes. Two primary study designs were used for these efforts: family-based (typically, families with two parents and two type 1 diabetes-affected children, i.e., affected sib pair [ASP] families) and case-control (a series of type 1 diabetes cases and a series of unaffected controls). The family-based designs were used for linkage analyses (i.e., to look for evidence of increased sharing of alleles or chromosomal segments in the ASP) or for association in the presence of linkage analyses (i.e., to evaluate whether the frequency of transmission of an allele from heterozygous parents to their affected child deviates from the expected 50% rate when there is no linkage). As collection of large numbers of ASP families (for robust statistical power) was difficult, much of the genetic evaluation of genes contributing to type 1 diabetes employed a case-control design.
Genome-wide association studies (GWAS) of cases of type 1 diabetes and controls with very large sample sizes, on the order of thousands, allowed the identification of multiple additional single nucleotide polymorphisms (SNPs) that are associated with type 1 diabetes, although each of them confers small risk (i.e., low odds ratio [OR]). Replication of findings to confirm the associations was followed by fine-mapping and functional studies to identify the causal genes and pathophysiological mechanisms that underlie the associations. Since the initial studies included participants mostly from European ancestry, there is great interest in cohorts from other ancestries to advance our understanding of type 1 diabetes in diverse populations.
Genetics improves the ability to predict type 1 diabetes in individuals at risk (12,13,14,15,16), is shedding light on the heterogeneity of the disease (17,18), and may provide evidence of underlying pathophysiological mechanisms (19) that could be targeted for prevention and treatment. The following sections expand on the evolution of genetics technology and findings related to the genetic basis of type 1 diabetes, including progress made since publication of Diabetes in America, 3rd edition (20).
Between the 1970s and 2000, research efforts focused on the localization and identification of genes that contribute to the occurrence of type 1 diabetes, as well as other autoimmune diseases, particularly genes involved in the immune response. Clear candidates in this regard were genes encoding the HLA molecules, which are highly polymorphic and play critical roles in the immunologic distinction between self and non-self, as well as in the presentation of peptide antigens to the T lymphocytes. In humans, the MHC is a gene-rich region on chromosome 6p21.3 that includes genes encoding the HLA class I (HLA-A, -B, and -C) and class II (HLA-DR, -DQ, and -DP) molecules (Figure 1) (21). The importance of HLA in type 1 diabetes risk is likely through its role in antigen presentation; genetic variation in this system could interfere with central tolerance during T cell development in the thymus or with peripheral tolerance by altering the repertoire of antigens presented (Figure 2) (22). The cellular immune response is centered on antigen presentation, in which foreign antigens are recognized by antigen-presenting cells (APCs), processed into peptides, complexed to HLA class I or II (depending on the source of those antigens), and presented on the cell surface, where they can be recognized by T cells. Since the T cell does not recognize unbound foreign peptides, antigen presentation requires antigen processing, binding to the HLA molecule, and display of the HLA-bound peptide on the surface of the APC. Thus, a critical aspect of immunological protection is the ability of the HLA system to provide a broad array of molecules to bind and present the foreign peptide, which is determined by genetic variation.


The HLA genes contain many alternative forms (i.e., many alleles) and thereby provide extensive variation in humans, resulting in the most variable region of the human genome. This extraordinary variability is critical to the breadth of immune responses. The high allelic polymorphism of HLA class I and II genes makes it extremely unlikely that two unrelated individuals should share the same combination of HLA alleles. All normal cells in the body, except for erythrocytes, express HLA class I and constantly present self peptides within those HLA class I molecules to CD8 T cells. In addition, professional APCs, such as dendritic cells or macrophages, express HLA class II and present foreign peptides within either their HLA class I or class II molecules to CD8 or CD4 T cells, respectively. Recognition of the HLA-peptide by the T cell receptor (TCR) initiates an intracellular signal cascade that leads to T cell activation.
The initial report of a strong association of HLA with type 1 diabetes occurred in 1973 with alleles of the class I HLA-A (or then, HL-A) locus (23). Many reports confirmed and extended the association of type 1 diabetes with antigens/alleles of the class I (HLA-A and -B) and class II (HLA-DR, -DQ, and -DP) loci. Studies on the effect of HLA-A, -B, and -DR were conducted in small samples of cases and controls, which were robust due to the large effect of the HLA alleles on risk/protection. This approach had two consequences—a focus on the immune system in a number of candidate gene studies and the simultaneous publication of underpowered candidate gene studies (as the expected effect sizes were far too large, as evidenced by the Wellcome Trust Case-Control Consortium [WTCCC], powered to detect OR ~1.5 with SNPs at MAF [minor allele frequency] >0.05). Of course, a limitation of early studies was the absence of cost-effective, genome-wide coverage of reliable markers, with “candidate gene” evaluations primarily of a few coding, sometimes promoter, variants.
The MHC region on chromosome 6p21.3 spans ~4 Mb, is gene rich (Figure 1) (21), and lacks informative recombination events, resulting in strong linkage disequilibrium (LD). LD is the occurrence of combinations of alleles at adjacent loci more often than expected from the frequencies of the individual alleles; groups of alleles on a chromosomal segment can be inherited as a unit or haplotype. The human MHC displays strong LD, and specific HLA haplotypes are associated with type 1 diabetes risk, providing a “fingerprint” of the transmission of disease-associated variation across populations.
Statistical methods can help to construct HLA haplotypes from unrelated individuals; however, the most precise method for observation and estimation of haplotypic association comes from family studies. As noted, multiple family studies have been conducted in non-Hispanic White populations, typically restricted to probands diagnosed with type 1 diabetes before age 16 years but up to age 35 years in siblings. A common measure of association is the odds ratio, which measures the relationship between an exposure (i.e., genotype or haplotype) and an outcome (i.e., type 1 diabetes). As the odds ratio is used to compare the relative odds of the outcome given the exposure, it can be interpreted as “risk” (OR >1), “protection” (OR <1), or neutral (OR 1). A family study of type 1 diabetes (24) demonstrated that the strongest class I associations with type 1 diabetes occurred with the HLA-B8 (OR 3.20) and HLA-B15 (OR 3.69) alleles and the class II associations with the HLA-DR3 (OR 3.54) and HLA-DR4 (OR 6.81) alleles (risk), as well as the HLA-DR2 (OR 0.21), HLA-DR5 (OR 0.30), and HLA-DR7 (OR 0.24) alleles (protective). Presence of the HLA-B8, -B15, -DR3, and -DR4 alleles increased type 1 diabetes risk, while the presence of HLA-DR2, -DR5, and -DR7 decreased type 1 diabetes risk. However, the haplotypes formed by HLA-B7-DR2 (OR 0.10) and HLA-B15-DR4 (OR 7.55) exhibited stronger association than the individual alleles, highlighting the genetic complexity within the MHC association with type 1 diabetes.
Of note, type 1 diabetes-associated alleles and haplotypes were obtained from studies that primarily recruited individuals of European ancestry. The association of HLA alleles and haplotypes with type 1 diabetes remains strong across populations of non-European ancestry, but the specific alleles and their strength vary. Studies of the HLA region have identified ancestry-specific HLA alleles that harbor risk for type 1 diabetes and demonstrate the necessity of studying diverse populations (25,26) (reviewed in Redondo et al. (27)). Figure 3 shows the distribution of five HLA-DR-DQ haplotypes and their effect on risk or protection in different world regions (27). For example, in Asians, a high risk is seen for DRB1*09:01, an allele found at low frequency in Europeans and, therefore, not associated with type 1 diabetes risk in Europeans. There are also ethnic differences in HLA allele frequencies that differ from European populations. For instance, African-specific HLA-DR3 and HLA-DR7 haplotypes have opposite effects on type 1 diabetes susceptibility than their European counterparts. Assembling large cohorts of non-European type 1 diabetes cases or families is difficult, given differences in disease risk among races/ethnicities. The study of type 1 diabetes in non-European populations, from both epidemiologic and genetic perspectives, is an important area of research.
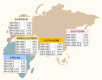
Regional Frequencies of Five HLA-DR-DQ Haplotypes. This map depicts the prevalence of five well-known HLA-DR-DQ haplotype groups that confer susceptibility or resistance to type 1 diabetes in five different world regions.
Subsequent studies have investigated the association of class I and class II alleles with type 1 diabetes, adjusting for LD within the MHC in numerous populations. The most comprehensive evaluation of these associations occurred at the HLA and Immunogenetics Workshop (28). The class I alleles most significantly associated with type 1 diabetes are HLA-B*57:01 (protective) and HLA-B*39:06 (susceptible). The HLA-B*39:06 allele increases the risk of HLA-DR4 susceptibility haplotypes in Scandinavian populations, as well as other risk haplotypes (DR3, DR1, and DR8) in larger cohorts. Other significantly associated class I alleles include: HLA-A*24:02, HLA-A*02:01, HLA-B*18:01, and HLA-C*05:01 (susceptibility); and HLA-A*11:01, HLA-A*32:01, HLA-A*66:01, HLA-B*07:02, HLA-B*44;03, HLA-B*35:02, HLA-C*16:01, and HLA-C*04:01 (protective) (28,29).
HLA allele and haplotype frequencies differ among populations, perhaps contributing to the differences in type 1 diabetes prevalence across geographic areas. There is a North-South gradient in type 1 diabetes prevalence, with Scandinavians having the highest type 1 diabetes risk. An exception to this rule is Sardinia, an island in the Mediterranean Sea, in the South of Europe. The high prevalence of type 1 diabetes in Sardinia has been attributed to the founder effect that established particularly high frequencies of the type 1 diabetes-susceptible HLA-DR3 and HLA-DR4 alleles and haplotypes and low frequency of the type 1 diabetes-protective HLA-DR2 allele and haplotype (30). In addition, most of the Sardinian HLA-DR2 haplotypes carry a DQB1*05:02 allele that is not protective (31).
The influence of the MHC region on type 1 diabetes risk is highly complex. Variation in the genes encoding the HLA-DR and HLA-DQ molecules is associated with risk of type 1 diabetes with odds ratios in excess of 10 for susceptible genotypes (e.g., DR3/DR4 or DRB1*03:01-DQB1*02:01/DRB1*04-DQB1*03:02) or under 0.1 for protective genotypes (e.g., DR2/DRX, where “X” is neither DR3 nor DR4 [or DR2]; the dominantly protective haplotype is DRB1*15:01-DQB1*06:02). The class II molecules encoded by these genes combine to form heterodimeric (αβ) protein receptors that are expressed on the surface of APCs. HLA-DQ is determined by a polymorphic α-chain (encoded by the HLA-DQA1 locus) and a polymorphic β-chain (encoded by the HLA-DQB1 locus). On the other hand, HLA-DR is determined by a monomorphic α-chain (encoded by the HLA-DRA locus) paired with a highly polymorphic β-chain. Most DR haplotypes in the population contain two loci: DRB1 and a second DRB locus (either DRB3, DRB4, or DRB5). A few DR haplotypes (e.g., DRB1*01, DRB1*08, and DRB1*10) do not have a second DRB locus. A heterozygous individual could encode up to four different DRB molecules (32). Thus, type 1 diabetes risk is determined by specific genotype combinations. The genotype HLA-DRB1*03-DQB1*02/HLA-DRB1*04-DQB1*03:02 confers the highest risk and has the highest frequency in type 1 diabetes cases with youngest onset.
The allelic variations in the critical HLA class II genes (HLA-DRB1, HLA-DQA1, and HLA-DQB1) are thought to alter specific amino acid residues that affect binding of peptides leading to initiation of the autoimmune disease process. Independent amino acid positions and large-scale genetic studies allowed examination of the contribution of HLA class II alleles and their interactions in type 1 diabetes (33). A large series of European cases, controls, and families allowed the study of more than 7,000 SNPs and amino acid residues at 399 positions for eight HLA genes. As expected, the single most associated risk variant was HLA-DQB1*03:02 that encodes an alanine at HLA-DQB1 position 57, while the aspartic acid at this position is most protective (Figure 4) (33). The second independent association is at HLA-DRB1 position 13, where a histidine residue (tagging HLA-DR4) confers risk (arginine is protective), as does a serine residue that tags HLA-DR3 (tyrosine is protective). The HLA-DRB1 residue at position 71 is a third independent association, with lysine conferring strong risk (tagging HLA-DR3 and HLA-DR4), while an alanine residue at this site is protective.

The HLA-DQB1 position 57, HLA-DRB1 position 13, and HLA-DRB1 position 71 are each located in the peptide-binding groove of the HLA molecule (Figure 5) (33). HLA-DRB1 positions 13 and 71 line the P4 pocket of HLA-DR. The HLA-DQB1 position 57 alone accounts for ~15% of the total risk of type 1 diabetes, and HLA-DRB1 position 13 and HLA-DRB1 position 71 account for an additional 12% of risk (33). Therefore, together, these three amino acid positions capture ~27% of type 1 diabetes risk or ~80% of the MHC-associated risk. The total type 1 diabetes risk explained by independent (additive) effects in the classical HLA genes is ~34% (Figure 6) (33). Structurally, the known association of HLA-DQB1 position 57 would alter the HLA-DQ P9 pocket; however, the associations with the HLA-DRB1 position 13 and HLA-DRB1 position 71 sites would alter the HLA-DR P4 pocket. This structural site may be crucial in binding specific autoantigens that are associated with type 1 diabetes risk.
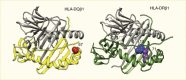

Although the influence of HLA class II genes on type 1 diabetes risk remains unquestioned, there is significant residual evidence of association with type 1 diabetes in the MHC region after controlling for the effects of HLA class II loci (33). Analysis of HLA haplotypes indicates that nonadditive effects are common within the MHC. HLA-DQB1 position 57 and HLA-DRB1 position 13 are the strongest factors in both additive and interactive effects on type 1 diabetes risk in the MHC (34). These two strongest interacting positions are in separate HLA molecules (HLA-DQ for position 57 and HLA-DR for position 13). In a detailed interaction analysis of the MHC, the effect of interaction within a site (dominance) was shown to have a small (~1%), but significant, impact on the contribution to risk of type 1 diabetes (34). Including interactions at the individual haplotypes, rather than alleles at a given site, resulted in an additional 3% of risk of type 1 diabetes. Jointly, the additive and interaction effects of the classical HLA sites and amino acid residues account for more than 90% of the MHC-type 1 diabetes association.
The two common methods by which genetic variation in association with type 1 diabetes risk is evaluated rely on assessing DNA sequence variation and studying either co-segregation with type 1 diabetes in families or differences between type 1 diabetes cases and controls. One such DNA variant occurs when a single nucleotide in the genome differs in a population with greater than 5% frequency; this variant is then termed a SNP. Millions of SNPs exist throughout the human genome—roughly one SNP every 2,000 bases.
The major goal of genome-wide linkage scans in type 1 diabetes was to identify regions of the genome that exhibited a significant deviation of “sharing” (identity-by-descent, IBD) by sibling pairs from that expected for Mendelian segregation of chromosomes (e.g., 25% sharing two alleles IBD, 50% sharing one allele IBD, 25% sharing no alleles IBD). In type 1 diabetes, this process was typically performed on families with multiple affected siblings, with a large number of families required to achieve strong statistical power given the small expected size of risk associated to non-MHC genes (35,36). This strategy effectively detected genes with variant alleles that have a large effect on risk of type 1 diabetes.
Unlike the MHC, for which a sample of 100 ASP families could detect significant evidence of linkage, the lower risk anticipated for other genes suggested a sample size of as many as 4,000 ASP families. The Type 1 Diabetes Genetics Consortium (T1DGC) was created to assemble a collection of ASP families to conduct genome-wide linkage scans for type 1 diabetes (37). Although the resolution of the linkage approach was limited to ~1 Mb regions, the first major analysis of 2,658 ASP families confirmed the contribution of the HLA region, INS, and CTLA4 (35). With additional collection of ASP families, analysis of 4,422 ASP families continued to identify the contribution of the MHC and the INS locus, but no compelling evidence for other susceptibility genes with similar large effects was found (36). These results suggested that additional loci contributing as much as 50% of the genetic risk have smaller effects and may be common, with allele frequency greater than 5% in the population, or private (unique) to individual families, and thus, they require a different experimental approach for detection.
With the improved genomic coverage through SNP discovery from the International HapMap Project (38) and decreasing costs of genotyping, the use of GWAS with robust statistical power became feasible. The primary source of participants for GWAS is unrelated type 1 diabetes cases and controls, as the statistical method is association and not linkage. Association studies take advantage of LD. Generally, the lead variant identified by GWAS is most likely not the “true” type 1 diabetes causal variant but in high LD with it. However, family-based association analyses provide important complementary results because of their resistance to stratification bias, which occurs, for example, when genetic differences in cases and controls occur due to sampling from populations of different ancestry and are unrelated to disease.
The first GWAS publications in type 1 diabetes focused on nonsynonymous SNPs (i.e., those that modify the amino acid sequence of protein), with the assumption that variants resulting in an amino acid variation would increase the likelihood of detecting a functional change associated with type 1 diabetes risk (39). This design led to the discovery of a novel gene, IFIH1, as having multiple, rare, protein-coding variants associated with type 1 diabetes. The function of IFIH1 was not in the obvious immune pathway of cytokines or beta cell proteins—IFIH1 encodes a cytoplasmic sensor of viral RNA that is part of the innate immune system and the response to viral infection. Notably, IFIH1 is involved in the response to picornaviruses, a family that includes viruses previously implicated in type 1 diabetes risk (40). Rare, inactivating alleles of IFIH1 are underrepresented among those with type 1 diabetes, suggesting that loss-of-function is protective from type 1 diabetes. In this manner, GWAS results can lead to discovery of genes that identify novel biologic pathways and avenues to novel drug targets to prevent or reduce the burden of type 1 diabetes.
The WTCCC conducted the first extensive GWAS using the Affymetrix GeneChip 500K Mapping Array Set in participants from the United Kingdom (41). Approximately 2,000 subjects for each of seven diseases and a shared set of approximately 3,000 controls were genotyped. Case-control analyses identified seven significant, independent associations with type 1 diabetes. Six genes/regions had strong preexisting statistical support for a role in type 1 diabetes susceptibility: namely, MHC, INS, CTLA4, PTPN22, IL2RA/CD25, and IFIH1. The WTCCC study demonstrated the power of the GWAS approach to identify novel loci associated with type 1 diabetes in an unbiased manner and suggested that larger samples with greater density of SNP genotyping could uncover additional novel genes and pathways related to type 1 diabetes risk. Following the WTCCC study, the 16p13 region containing DEXI and KIAA0350 was identified in a second GWAS (42) and as a novel locus of susceptibility for other autoimmune diseases, consistent with pervasive sharing of genetic risk across immune-mediated disorders (43).
The T1DGC expanded the results from the WTCCC by starting a new GWAS of 3,983 type 1 diabetes cases and 3,999 controls, followed by a combined meta-analysis with previously published studies that added 3,715 additional cases and 5,069 controls with information from more than 841,622 SNPs. From the meta-analysis of 7,514 cases and 9,045 controls with confirmation in 4,267 cases, 4,670 controls, and 2,319 ASP families, 28 novel regions were shown to be significantly associated with type 1 diabetes risk (44). Eighteen novel regions were strongly replicated from the GWAS meta-analysis.
In total, the number of loci contributing significantly to type 1 diabetes risk reached 42 in 2009, although each locus varied in the size of the interval, number of candidate genes in the locus, size of the effect on type 1 diabetes risk for the most associated SNP, and strength of the evidence supporting the most likely candidate. The genes (loci) of largest impact on type 1 diabetes risk were identified in the 30-year period from the early 1970s (HLA) to 2000 (INS) as the result of small case-control and family studies, reflecting the fact that variants with larger effects can be identified with smaller sample sizes. In 2001–2006, with the advent of large-scale genotyping arrays and assembly of larger case-control series, investigators identified several additional loci (including PTPN22, CTLA4, and IFIH1) that slightly increased the amount of genetic risk identified. In 2007–2008, the first well-powered GWAS by the WTCCC greatly increased the number of loci of modest to small contribution to risk (e.g., IL2RA). In 2009, the T1DGC GWAS meta-analysis (44) with almost 8,000 cases and 9,000 controls plus replication, nearly doubled the number of loci, with each having small effect. Of interest, this meta-analysis in the T1DGC ASP families confirmed the directions of the risk allele effect. However, the size of the effect was lower in the families than in the case-control study, and statistical evidence for replication in families was not reached for 11 of the 18 loci. This finding may indicate the greater role of the MHC in type 1 diabetes risk when multiple family members express disease.
The T1DGC further examined the 18 novel loci (45) by expanding the analyses in 2,322 additional families, which combined with the original 2,319 families, provided improved protection from population stratification bias and increased power for further replication support. With this larger sample of families, only one (C14orf181/14q24.1) of the 18 novel loci did not attain statistical significance. Furthermore, all of the novel type 1 diabetes risk loci had consistent direction of effects (e.g., risk or protective for type 1 diabetes risk) with the original GWAS meta-analysis (44,45) without evidence of variability in the disease associations across family collections, despite significant SNP genotype frequency differences. After clear replication of type 1 diabetes risk loci, the next steps involved dense SNP mapping to better describe causal genes and variants. Identification of genes with both positional and functional evidence for a role in type 1 diabetes pathogenesis could shed light on the pathways underlying type 1 diabetes.
In 2021, Chiou et al. (46) increased the power of type 1 diabetes GWAS by combining datasets to achieve a larger sample size. The compiled dataset included 18,942 individuals with type 1 diabetes and 501,638 control individuals (total of 520,580 European ancestry samples). Through meta-analysis, Chiou et al. identified 81 loci that reached genome-wide significance (P<5x10-8), including 33 loci that were previously unreported (Table 1).
Type 1 Diabetes-Associated Genomic Regions From Fine-Mapping Efforts
Most common variants identified through a GWAS are located between genes (intergenic), in regions that contain regulatory DNA, or if located in a gene, in a region that is not protein coding (intronic). Relatively few robustly type 1 diabetes-associated variants with likely deleterious effects have been located in coding regions (exons). Thus, the variants that are associated with type 1 diabetes from GWAS typically do not have obvious functional consequences. This result may suggest that many of the disease-associated variants affect the regulation of gene expression. However, initial GWAS results do not allow exclusion of the possibility that these variants are associated with one or more causative variants within a coding region. The information provided by GWAS on a likely causal gene is limited because of the sparse coverage of a given region of the genome even with high-density SNP genotyping arrays. Thus, prior to detailed examination of genes and regulation, dense SNP mapping of a type 1 diabetes-associated locus is required to firmly establish the most likely associated SNPs and causal genes.
For example, the T1DGC GWAS meta-analysis (44) identified more than 40 loci that are significantly associated with type 1 diabetes risk. However, each type 1 diabetes locus contained an average of seven genes, with a range of 0 to 27 (44,47). Some regions harbored potential candidate genes, for example, involved in the immune response, but in other cases, only a single gene without obvious functional relevance to type 1 diabetes or autoimmunity was found. Indeed, for a few regions, noncoding genes were present. The novel loci identified in the GWAS meta-analysis typically had modest or small effects on type 1 diabetes risk, with C10orf159 (RNLS, renalase) having the largest effect (OR ~1.3) and others with smaller effect sizes, consistent with the dependence of their detection on large sample sizes. A large number of previously detected loci had small effects, including those with strong or confirmed candidate causal genes (e.g., CTLA4, IFIH1).
In most GWAS regions, the locus associated with disease is defined by few (perhaps 100) SNPs. The size of the average type 1 diabetes susceptibility locus from the T1DGC meta-analysis (44) was defined by an ad hoc algorithm based on the most associated SNP at the center with boundaries for that region defined by sites of high recombination. Since there are many candidate genes in each type 1 diabetes region, functional studies are needed to define a role for a particular gene and/or allelic variant in type 1 diabetes pathogenesis. Such studies require significant time and resources, and unless at least the disease-relevant gene within a region is known, those efforts could well target the incorrect gene or variant. To address this concern and refine GWAS regions of susceptibility for type 1 diabetes, dense genotyping (fine-mapping) was required that could comprehensively evaluate the influence of thousands of SNPs in a region.
A GWAS seeks to capture genomic architecture with high information content using a standard set of SNPs. In contrast, fine-mapping requires saturation of specific regions of the genome, with the predetermined boundaries of each region defining the set of SNPs to be genotyped. The ImmunoChip Consortium was established with the purpose of meeting the needs of type 1 diabetes and other autoimmune diseases with genome-wide significant regions to be interrogated. The Consortium consisted of investigators who provided data (including unpublished) to design a genotyping array (the ImmunoChip) for fine-mapping autoimmune disease susceptibility genes. The ImmunoChip is a custom Illumina Infinium high-density genotyping array comprising 196,524 SNPs over 186 regions of the human genome.
For type 1 diabetes, the initial analyses of ImmunoChip data focused on European ancestry subjects and included 6,670 type 1 diabetes cases, 9,416 controls, 2,601 T1DGC ASP families, and 69 T1DGC trio (affected child and both parents) families (48). A large proportion of these samples (98% of cases, 76% of controls, and 57% of family samples) were part of the previous GWAS meta-analysis that initially identified many of the type 1 diabetes genetic regions (44). Fifty-five non-MHC loci were evaluated, including loci that were significantly associated with another autoimmune disease but only nominally with type 1 diabetes. Of the non-MHC type 1 diabetes-associated regions, 36 were significantly associated with type 1 diabetes risk from the ImmunoChip data (including 33 of 45 that had been reported by the T1DGC or others previously). In addition, three loci were nominally associated with type 1 diabetes that were also robustly associated with other autoimmune diseases (49) and were, therefore, included as new type 1 diabetes loci: 1q32.1, also associated with ulcerative colitis, Crohn’s disease, and celiac disease; 4q32.3, also implicated in celiac disease; and 5p13.2, also associated with multiple sclerosis (50,51,52,53,54,55).
While the majority of the large-scale GWAS in type 1 diabetes focused on European ancestry populations, the second phase of T1DGC ImmunoChip fine-mapping analyses focused on ancestrally diverse type 1 diabetes cases, controls, and affected families. Robertson et al. (56) assigned each study subject (n=61,427) to one of five ancestry groups by utilizing principal-component analysis: European (n=47,319), Finnish (n=6,991), East Asian (n=588), African admixed (n=4,290), and other admixed (n=2,239). To improve discovery and fine-mapping resolution, genotypes within ImmunoChip regions were imputed using the Trans-Omics for Precision Medicine (TOPMed) multiancestry reference panel (57). A meta-analysis of the case-control and family results identified 78 chromosome regions associated with type 1 diabetes (P<5×10−8), including 36 new regions (Table 1). Fine-mapping efforts both by Robertson et al. (56) and Chiou et al. (46) revealed that more than one-third of the type 1 diabetes risk loci contain more than one independent association. For example, both studies identified three independent association signals in the PTPN2 locus. This observation suggests that much work remains to be accomplished to identify the causal genes, variants within the genes that modify risk, and functional impact of those variants.
Until the late 2010s, studies of the genetics of type 1 diabetes focused largely on European populations where the ascertainment of large numbers of individual cases or multiple-case families is easier. The lack of knowledge on the genetic architecture of type 1 diabetes and other common diseases in non-European ancestry populations is widely recognized as a major problem in the era where we aim to utilize genetics to apply precision medicine with the ultimate goal of improving health outcomes in all individuals (58,59). The Major Histocompatibility Complex and Type 1 Diabetes Risk section covers information on ancestry-specific HLA alleles that confer risk for type 1 diabetes. As of 2023, the number of studies with sufficient sample size to discover genetic risk variants outside the HLA region in populations of non-European ancestry is limited.
As yet, the largest GWAS focused on type 1 diabetes in individuals of African ancestry included 1,021 participants with type 1 diabetes and 2,928 control individuals (60). Association studies in the African-ancestry cohort provided concordant findings for some type 1 diabetes loci identified in European-ancestry populations, i.e., 2q33.2 (CTLA4), 6p32 (MHC), 6q22.32 (CENPW), 11p15.5 (INS), 12q13.2 (IKZF4, RPS26, and ERBB3), and 17q12 (IKZF3, ORMDL3, and GSDMB). In addition, it revealed alternative risk alleles at 10q23.31 (RNLS).
In East Asian populations, two GWAS have been conducted. The first study was carried out in 2,596 type 1 diabetes cases and 5,082 control participants with Chinese Han ancestry (61). Zhu et al. identified four loci with genome-wide significance (P<5×10−8), including two known type 1 diabetes risk regions—6p32 (MHC) and 12q13.2 (IKZF4, RPS26, and ERBB3)—and two novel regions—one in 6q22.2 (BTN3A1) and one in 10p14 (GATA3). The 10p14 region harbors type 1 diabetes risk variants also found in European-ancestry individuals (46). A second study analyzed 638 type 1 diabetes cases from Biobank Japan but, likely due to the small sample size, only identified type 1 diabetes risk variants in 6p32 (MHC) (62).
Data on the genetic architecture of type 1 diabetes risk in populations from other regions of the world, such as Ethiopia (63), the Philippines (64), and Indonesia (65), or in the United States, Hispanic individuals (66) and American Indian, Alaska Native, Native Hawaiian, or Pacific Islander populations, are still limited.
At a small number of non-MHC loci, a fine-mapped risk variant alters the protein coding sequence of a gene. Genes with protein-altering type 1 diabetes risk variants include PTPN22 (R620W, rs2476601), TYK2 (P1104A, rs34536443; I684S, rs12720356), IFIH1 (I923V, rs35667974; N160D, rs74162074; A946T, rs1990760), SH2B3 (W262R, rs3184504), C1QTNF6 (G21V, rs229527), SIRPG (V263A, rs6043409), CTSH, AIRE (R471C, rs74203920), and PRF1 (A91V, rs35947132). Most protein-altering variants affecting type 1 diabetes are missense and, thus, change an amino acid. An exception is FUT2, where the fine-mapped type 1 diabetes-associated variant W143X (rs601338) introduces an early stop codon, leading to a truncated, nonfunctional protein. Protein-altering risk variants encourage functional studies of the allelic forms of specific proteins to determine whether they impact cellular or organismal phenotypes that could plausibly contribute to type 1 diabetes pathogenesis. In addition, variants can be evaluated for association to other traits and endophenotypes related to type 1 diabetes, such as in phenome-wide association studies (PheWAS) (67), which can also provide insight into variant function.
Functional studies of genes with protein-altering risk variants, including of different protein-coding alleles, support a prominent role for T cells in type 1 diabetes pathophysiology, as well as APCs, beta cells, thymus, and other cell types that modulate immune responses. Variants affecting adaptive immune cell function include PTPN22 R620W, which impacts TCR signaling and T cell activation (68,69,70,71,72), as well as B cell signaling (73,74,75); TYK2 P1104A and I684S, which regulate cellular responses to cytokine signaling (76,77,78,79,80,81,82); PRF1 A91V (83,84); IFIH1 I923V and A946T (85,86); and SIRPG V263A (87,88). Protein-altering variants in other genes instead impact innate immune cell function, including SH2B3 W262R (89) and C1QTNF6 G21V (90,91). Protein-coding variants that affect the beta cell include IFIH1 (92), which modulates intracellular responses to viral RNA, as well as CTSH (93) and TYK2 (76). Finally, protein-altering variants at AIRE R471C and FUT2 W143X implicate such processes as thymic selection and response to bacterial or viral infection, respectively (94,95,96,97).
For most type 1 diabetes-associated loci identified and fine-mapped in GWAS studies, risk variants map to noncoding sequences and likely affect gene regulation (48). Annotating noncoding variants requires detailed maps of cis-regulatory elements in the human genome. High-throughput epigenomic assays that measure hallmarks of gene regulatory activity, such as accessible chromatin, histone modifications, transcription factor binding, and DNA methylation, have been extensively applied to human cell lines, tissues, and primary cells as part of such projects as ENCODE (98,99) and the NIH Epigenome Roadmap (100). These projects have defined tissue and cell-type maps of regulatory elements, including regions of enhancer and promoter activity (98,99,100,101). Technological developments have further enabled profiling the epigenome of single cells, where regulatory elements can be defined in each specific cell type within a heterogeneous tissue (102).
As shown by the T1DGC ImmunoChip study (48) and others (46,56,103), type 1 diabetes-associated variants preferentially map in enhancer regions active in CD4 and CD8 T cells, as well as other immune cell types, such as B cells and macrophages. While type 1 diabetes-associated variants are not localized in pancreatic beta cell enhancers in the basal state, they preferentially map in the subset of beta cell enhancers activated upon proinflammatory cytokine exposure (104,105). Type 1 diabetes-associated variants also map in enhancers specifically active in other cell types, such as acinar and ductal cells of the exocrine pancreas (46).
Risk variants overlapping regulatory elements in type 1 diabetes-relevant cells may not necessarily be functional, and therefore, mapping variant effects on regulatory activity is needed to distinguish functional from benign variants. Approaches to derive risk variant effects on regulatory activity in T cells and other relevant cell types include chromatin accessibility or histone quantitative trait locus (caQTL or hQTL) mapping (106,107,108,109,110,111), allelic imbalance (AI) mapping (112), massively parallel gene reporter assays (MPRAs) (113,114), and machine learning predictions (115,116,117). For example, caQTL mapping in CD4 T cells identified type 1 diabetes-associated variants at multiple loci affecting T cell chromatin (56,118), and an hQTL mapping study determined that type 1 diabetes-associated variants affected enhancer-related histone marks in T cells more often than in other tested cell types (108).
Type 1 diabetes-associated variants that affect enhancer activity can regulate the expression of one or more genes, including over large distances (119,120). Techniques to link enhancers to the genes they affect include 3D physical interactions with Hi-C (a genomic method that captures chromatin conformation) and its derivations (121), co-activity (118,122), the activity-by-contact (ABC) method (123,124), and CRISPR deletion or interference (CRISPRi) (125,126). Enhancer interactions with gene promoters mapped using promoter capture Hi-C (pcHi-C) in 17 primary immune cell types (127) demonstrated that type 1 diabetes-associated variants interacted with genes involved in TCR signaling and other immune signaling pathways in CD4 and CD8 T cells. Another approach for determining genes affected by noncoding variants is QTL mapping of gene expression levels (eQTL) (128). Variants at multiple type 1 diabetes loci are eQTLs in immune cell types, such as monocytes and T cells (108,129,130).
Type 1 diabetes risk loci that affect T cell regulatory activity include IL2RA (131,132,133,134), CTLA4 (135,136,137,138,139), BACH2 (56,108,114,127,140,141), and UBASH3A (142,143). For example, at BACH2, type 1 diabetes-associated variants affect CD4 T cell chromatin accessibility, transcription factor binding, and BACH2 expression, and reduced BACH2 activity affects T cell activation and differentiation (114,144). Other loci likely affect beta cell regulatory activity, such as GLIS3 (145,146,147,148), 16p13 for DEXI and SOCS1 (104,105), and TNFSF18 (104,149). A subset of type 1 diabetes loci also affect pancreatic exocrine cell regulatory activity and function, for example, CFTR and CTRB1/2 (46,150,151,152). At the INS locus, the INS promoter VNTR (variable number of tandem repeats region) has been argued to affect expression of insulin in the thymus, as well as in beta cells (153,154,155,156,157), suggesting that it may have dual mechanisms.
Many additional candidate genes at type 1 diabetes loci affect relevant beta cell or immune cell phenotypes in cell or animal models, e.g., PTPN2 (158), RNLS (159), ERBB3 (160), TNFAIP3 (161,162), STAT6 (163), BCL2L11 (158), ALOX15 (164), CAMK4 (165,166), IL10 (167,168,169), IL2 (170,171), IL27 (172), and IL7R (173), but have not necessarily been linked to type 1 diabetes risk variant activity directly. For IL10, IL2, and IL7R, these are almost certainly the genes through which type 1 diabetes risk is mediated in humans given their ability in mouse models to directly cause or prevent type 1 diabetes, but risk variant effects on their activities remain unknown.
As additional studies profile the activity of protein-coding and noncoding risk variants, the interpretation of type 1 diabetes variant function will reveal novel mechanisms of disease risk that can be targeted for prevention and treatment.
Diagnosis of type 1 diabetes follows a preclinical phase of variable duration during which autoimmunity attacks beta cells in the pancreatic islets. As a consequence, beta cell function is reduced below the threshold required to maintain normal glucose concentration in blood. The discovery of biomarkers, namely detectable serum islet autoantibodies, enabled the exploration of this process and its contributors, including genetics. The first GWAS of islet autoantibody positivity in type 1 diabetes was conducted in T1DGC cases with measurement of two islet autoantibodies (glutamate decarboxylase [GAD] and insulinoma-associated antigen 2 [IA-2A]), antibodies to the autoimmune thyroid (Graves’) disease autoantigen thyroid peroxidase, and antibodies against gastric parietal cells (PCA) that are associated with autoimmune gastritis (174). Genome-wide, variants in FCRL3/1q23 were associated with IA-2A positivity, and variants in ABO/9q34 were associated with the presence of PCA (174). The T1DGC samples were obtained from individuals with type 1 diabetes (i.e., prevalent cases), which is a limitation because the influential genetic (and environmental factors) may vary along this process, and islet autoantibodies can become undetectable in individuals with long-duration type 1 diabetes. Therefore, the evaluation of genetic factors that initiate type 1 diabetes requires a different approach: namely, identifying and following initially nondiabetic individuals who will develop type 1 diabetes.
Since the incidence of type 1 diabetes in the general population is relatively low, studies aiming to identify individuals who will develop type 1 diabetes require extremely large sample sizes, which limits the feasibility of this research. To lower the estimated sample size, studies increase the expected incidence of the disease by focusing on individuals with higher risk. For example, The Environmental Determinants of Diabetes in the Young (TEDDY) study (175) longitudinally follows children with HLA genotypes known to increase the risk of type 1 diabetes. The Diabetes and Autoimmunity Study in the Young (DAISY) included a cohort with siblings or parents of children with type 1 diabetes and another cohort of children from the general population who carried high-risk HLA genotypes (13,176). The observational arm of the Type 1 Diabetes TrialNet study Pathway to Prevention follows autoantibody-positive relatives of individuals with type 1 diabetes (177). The Finnish Type 1 Diabetes Prediction and Prevention (DIPP) study longitudinally followed newborns carrying type 1 diabetes-associated HLA genotypes (178). These and other studies have provided invaluable insights on the genetic differences in the initiation and progression of type 1 diabetes.
In a study of almost 8,000 genetically at-risk infants participating in TEDDY, HLA-DR3-DQ2/DR4-DQ8 was the best predictor for the initiation of islet autoimmunity (179). Another analysis of TEDDY participants found additional genetic influences for autoimmunity initiation from non-HLA SNPs, such as rs2476601 in PTPN22, rs2292239 in erb-b2 receptor tyrosine kinase 3 (ERBB3), rs3184504 in SH2B adaptor protein 3 (SH2B3), and rs1004446 in INS (180). In the TEDDY cohort, PTPN22 rs2476601 was the best predictor for insulin autoantibodies appearing first and SNPs rs12708716 in CLEC16A and rs2292239 in ERBB3 for GAD autoantibodies as the first-appearing autoantibody (179). In DAISY, among non-HLA genes, the appearance of islet autoantibodies was associated with SNPs in PTPN22 (rs2476601) and UBASH3A (rs11203203 and rs9976767) (181,182).
The influence of HLA-DR3-DQ2/DR4-DQ8 was demonstrated to be stronger on the initiation of islet autoimmunity than on the progression to clinical diabetes (183,184). In approximately 3,400 participants in the TrialNet Pathway to Prevention study, HLA-DRB1*15:01-DQA1*01:02-DQB1*06:02 was shown to offer protection from islet autoimmunity initiation, as well as progression from single to multiple autoantibody positivity and then to type 1 diabetes, which occurs in very few individuals with this haplotype (185). Progression from single to multiple autoantibody positivity was influenced by HLA genotypes and rs3087243 in CTLA4, while diabetes progression was affected by HLA genotypes, rs6476839 in GLIS family zinc finger 3 (GLIS3), and rs3184504 in SH2B3 (186); in this study, age-related differential genetic influences were seen. DAISY also found that INS, UBASH3A, and IFIH1 were associated with progression from islet autoimmunity to diabetes (181,182). An analysis in the TrialNet Pathway to Prevention study demonstrated that type 2 diabetes-associated variants in the TCF7L2 locus influenced progression from single to multiple positivity with different effects depending on the autoantibody specificity and body mass index (BMI) (187), suggesting complex interactions among different diabetogenic mechanisms that may be present in a given individual. Critically, genome-wide (rather than candidate gene) approaches are necessary to interrogate and discover those variants contributing to initiation, as well as progression, of the autoimmune process.
The age at onset of type 1 diabetes varies widely from infancy to adulthood. There have been conflicting reports of possible effects of type 1 diabetes risk variants on age at onset, largely from studies of modest sample size. However, the role of HLA alleles, particularly HLA-DR and HLA-DQ, on age at onset has been reported by multiple studies (188). Children who are diagnosed under age 6 years are more likely to have the HLA-DR3 allele and the HLA-DR3/DR4 genotype. For example, children who develop type 1 diabetes before age 7 years have increased insulitis, beta cell stress, and beta cell loss compared with older children and adolescents (189,190). TEDDY longitudinally follows newborns with high genetic risk for type 1 diabetes and has reported differences associated with the specific sequence of islet autoantibody seroconversion before the clinical onset of diabetes. In this study, insulin autoantibodies appeared first in very young children who carried HLA-DR4-DQ8 haplotypes and progressed rapidly to clinical disease, while GAD65 autoantibodies appeared first in older children who carried HLA-DR3-DQ2 (191,192). Among children with newly diagnosed type 1 diabetes, insulin autoantibody positivity and higher titers were associated with younger age at onset of type 1 diabetes, while GADA65 positivity was more common in females and those with thyroid peroxidase antibodies (193).
Well-powered studies allowed testing of the association of age of onset with type 1 diabetes-associated SNPs identified in GWAS and fine-mapping studies (194). Two loci, RNLS at 10q23.31 and IL2 at 4q27, displayed significant associations with age at onset. For IL2/4q27, the G allele for the rs2069763 variant is “protective”: that is, the age at diagnosis increases with the number of protective alleles (mean age at diagnosis for those with the TT genotype is 8.7 years, the GT genotype 9.0 years, and the GG genotype 9.3 years). This effect is also seen for the RNLS/10q23.3 rs10500540 variant, in that the mean age at diagnosis increases with the number of protective C alleles. The influence of HLA-DQB1*03:02 (tagged by rs9273363 SNP) on younger age of onset was confirmed by a GWAS that also demonstrated the effect on the same direction of the 6q22.33 region that contains the genes encoding protein tyrosine phosphatase receptor kappa (PTPRK) and thymocyte-expressed molecule involved in selection (THEMIS) (195). Furthermore, children with type 1 diabetes diagnosed under the age of 7 years had increased frequency of the HLA-DR3-DQ2/DR4-DQ8 genotype, A*2402 and B39*06 alleles, as well as non-HLA regions involved in the immune system (e.g., ILSRA, IL10, THEMIS, Ikaros family zinc finger 3 [IKZF3]) or the beta cell (e.g., GLIS3) (196).
Adults who present with type 1 diabetes can develop rapid insulin insufficiency, which is important for clinicians to bear in mind (197). However, most studies to date have shown that compared with childhood type 1 diabetes, adult-onset type 1 diabetes has weaker associations with HLA genotypes and stronger associations with INS and some loci associated with type 2 diabetes (198). Similarly, adult-onset type 1 diabetes seems to progress more slowly overall, and at the extreme of the spectrum, some individuals, although diagnosed with diabetes, do not become insulin dependent for months or years after clinical onset; this subtype of autoimmune type 1 diabetes is often referred to as latent autoimmune diabetes in adults (LADA). Genetically, LADA is closer to type 1 diabetes but has commonalities with type 2 diabetes (199). HLA genotypes typically associated with type 1 diabetes are less common in individuals who develop type 1 diabetes at an older age, with higher BMI, and lower number of islet autoantibodies. In these individuals, their relatively conserved C-peptide production suggests insulin resistance as a contributing factor to their diabetes (200). Furthermore, compared with individuals with multiple positive islet autoantibodies, those with single autoantibody positivity are more likely to carry a TCF7L2 genetic variant that confers strong risk for type 2 diabetes (18).
Overall, although clusters of characteristics suggest common pathogenic mechanisms, clear endotypes (defined as pathophysiologically distinct subtypes of disease that may respond differently to treatment (201)) have not emerged yet. Uncovering the function of genetic factors associated with type 1 diabetes may help improve the current understanding of the heterogeneity of diabetes with the ultimate goal of applying precision medicine, that is, applying “the right treatment, for each patient, at the right time” (202).
One of the goals of increasing the understanding of type 1 diabetes pathogenesis is to recognize the risk factors and biomarkers that can be used for prediction of disease. Individuals identified as having high risk for type 1 diabetes are less likely to present with diabetic ketoacidosis (203) and can participate in clinical trials to prevent diabetes (204), or if meeting the approved indications, they can receive a recently approved treatment with teplizumab to delay the onset of clinical type 1 diabetes (205).
The first attempts to predict type 1 diabetes began more than 30 years ago (206,207) after the discovery of assays to measure islet autoantibodies in serum and the knowledge of HLA genotypes that confer risk. Since then, multiple islet autoantibodies have been discovered, and high-throughput methods for standardized detection of islet autoantibodies in serum have been developed. Furthermore, it has become clear that other factors, in particular age, have a profound influence on risk and rate of progression and the characteristics of the disease. The trajectory of metabolic measures, e.g., C-peptide and glucose, before clinical onset of diabetes has also been described and used for prediction (208).
Improved understanding of the sequence of events in the natural history of type 1 diabetes allowed investigators to break down the preclinical course of type 1 diabetes into stages beginning with the expression of multiple islet autoantibodies, when the risk of developing clinical disease within 10 years is 70% and virtually 100% within 20 years (209). Therefore, stage 1 type 1 diabetes is marked by positivity for multiple islet autoantibodies and normal glucose metabolism; stage 2, by multiple autoantibody positivity and dysglycemia (i.e., subclinical abnormalities that do not meet criteria for diabetes); and stage 3, by glucose in the diabetic range (i.e., clinical type 1 diabetes) (Figure 7) (210). Although refinements to this system are likely to be developed, the concept of stages provides a framework for prediction and prevention of type 1 diabetes.
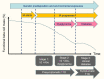
Phases in the Natural History of Type 1 Diabetes. Abs, islet autoantibodies; IA, islet autoimmunity; T1D, type 1 diabetes.
Aiding in type 1 diabetes prediction, more than 90 genetic regions within and outside the HLA region that are associated with type 1 diabetes risk have been identified. As reviewed in the Genes and Initiation of Type 1 Diabetes section, genetic influences on the initiation of islet autoimmunity may differ from those that affect further progression of islet autoimmunity and, finally, onset of type 1 diabetes (179,181,182,185,186,187). There are also differences by age at seroconversion (191,192,194,195,196,198,199). This information has been integrated to predict the development of type 1 diabetes in individuals at various preclinical stages. Survival analysis-based estimation of risks of first autoantibody seroconversion, spread to multiple autoantibody positivity, and then progression to clinical type 1 diabetes demonstrated interactions, with multiplicative effects, of combinations of high-risk HLA genotypes (e.g., HLA-DR3-DQ2/DR4-DQ8) and non-HLA loci (e.g., INS, PTPN22) (211).
While metabolic milestones, such as the development of dysglycemia, are the strongest markers of short-term risk, the value of genetics is with long-term horizons before metabolic deterioration occurs (15). Similarly, genetics may have less predictive power in the presence of multiple islet autoantibodies or autoantibodies that reflect advanced progression (e.g., IA-2A autoantibodies). On the other hand, in earlier stages, genetics has the advantage of being permanent and, thus, not needing repeated assessment, as opposed to other indicators of risk, such as autoantibodies (212).
The higher prevalence of additional autoimmune conditions in individuals with type 1 diabetes may be explained, at least in part, by common genetic factors (213). For example, HLA-DR3-DQ2 and/or, to a lesser degree, HLA-DR4-DQ8 are found in most patients with celiac disease (214,215); HLA-DR4-DQ8 and non-HLA genes, such as PTPN22 and CTLA4, are associated with rheumatoid arthritis (216); and while autoimmune thyroid disease has specific genetic influences (e.g., thyroglobulin, thyrotropin receptor, and CD40), HLA-DR3-DQ2, HLA-DR4-DQ8, PTPN22, and CTLA4 are shared with type 1 diabetes (217). These commonalities highlight pathways that underlie autoimmunity in general and might offer the potential pragmatic use of predicting additional autoimmune diseases in individuals with type 1 diabetes.
Additional gaps in knowledge must be covered to optimize the use of genetics for type 1 diabetes prediction. As reviewed in previous sections, the specific genes that facilitate triggering of islet autoimmunity differ from the genes that regulate its progression and the loss of beta cell function (218), but the latter are less well known in the setting of type 1 diabetes. In addition, resolving discrepancies in results among studies with different populations and designs is still necessary. The genetic underpinnings of the differences marked by age are only beginning to be understood (195,196), and data remain scarce in adult-onset type 1 diabetes. Research in this area may reveal the differential pathogenic mechanisms that underlie the development of type 1 diabetes across the lifespan (219). Although most knowledge on the natural history of type 1 diabetes before clinical onset stems from observational studies conducted in participants with high genetic risk or a relative with type 1 diabetes, prediction models and screening strategies for type 1 diabetes risk must be tested in the general population (212,220). This endeavor has become particularly pressing since the demonstration of effective and safe interventions to delay the onset of type 1 diabetes in individuals with stage 2 type 1 diabetes (205).
An additional barrier to using genetics in type 1 diabetes for prediction and selection of candidates for clinical trials or standard treatment was the pragmatic problem of how to integrate the elevated number of genetic loci that contribute to the development of type 1 diabetes, with variable effect sizes, in the predictive models and algorithms. This need has been addressed by the development of type 1 diabetes genetic risk scores.
Incorporating the influences of a very large number of genes for pragmatic uses was difficult. For a long time, the two highest risk HLA haplotypes, i.e., HLA-DR4-DQ8 and HLA-DR3, and the strongest protective haplotype, i.e., HLA-DR2-DQB1*06:02, were the only genetic factors commonly used in type 1 diabetes prediction. However, the development of polygenic scores that combine multiple genes known to affect type 1 diabetes risk into a single number (reviewed in Redondo et al. (221)) has facilitated the use of genetics in classification of diabetes and type 1 diabetes prediction.
Distinguishing between diabetes types can be a challenge because of the absence of pathognomonic features for type 1 or type 2 diabetes, which are the most frequent forms. Type 1 diabetes polygenic scores can help discriminate type 1 and type 2 diabetes (222), even in young adults (223) and children (224), and predict clinical course (225). These scores can also help distinguish between type 1 diabetes and monogenic diabetes (226). Since the constellation of characteristics used to diagnose type 1 diabetes was established in study cohorts of primarily European-ancestry individuals with pediatric-onset, clinically-diagnosed type 1 diabetes, misclassification is more frequent in individuals of non-European ancestry, adults, and those with atypical features. The addition of a type 1 diabetes polygenic score improves phenotypic algorithms to identify individuals with type 1 diabetes (227).
Type 1 diabetes polygenic scores have been successfully incorporated in the predictive model for type 1 diabetes in individuals at risk. A type 1 diabetes genetic risk score improved prediction over only HLA genotypes and other markers in children followed since birth (15,228) and in relatives of patients across the lifespan (14). As the genetics of type 1 diabetes in non-European ancestries are uncovered, investigators are developing ancestry-specific genetic risk scores, such as Japanese (229), African (60), and Chinese Han (61) specific scores (reviewed in Redondo et al. (27)). However, given the increasing frequency of individuals who have multiple ancestries, developing scores that can be used in populations of any ancestry would be beneficial, and thus, more studies in global populations are warranted.
In addition to genetic factors, epigenetic changes in the genome have been implicated in type 1 diabetes. Epigenetics refers to modifications that affect gene regulation without altering DNA sequence directly, such as methylation of cytosine residues or post-translational modification of histone tails (230). Cytosine methylation is found most prominently at CpG dinucleotides, which are preferentially located at gene promoters and are associated with gene repression (231). Histone tail residues can be modified in several ways, such as methylation or acetylation, and depending on the residue and modification, can lead to activated or repressed transcription (232). Importantly, epigenetic marks, including DNA methylation and histone modifications, reflect both genetic and external influences (233) and, therefore, can help capture disease processes driven by environmental factors.
A common strategy is to profile monozygotic twin pairs, which have nearly identical genome sequence, discordant for type 1 diabetes to determine whether differences in epigenetic profiles contribute to disease risk (234,235,236,237). For example, an epigenome-wide association study (EWAS) compared CpG site methylation in discordant twin pairs in T cells, B cells, and monocytes and found thousands of CpG sites in each cell type associated with type 1 diabetes (238). The variability in CpG site methylation did not appear to be due to genetic factors; therefore, environmental factors are a likely contributor to these changes.
Further studies have shown differences in immune cell-type CpG methylation in pre-stages of disease, such as in islet autoantibody-positive individuals, suggesting that methylation changes precede disease onset and may potentially be useful biomarkers (234,239,240). Methylation profiles of beta cell-specific genes detected in circulating blood have also been evaluated as biomarkers of beta cell death in type 1 diabetes (241,242,243).
In addition to DNA methylation, studies have profiled changes in histone modification in type 1 diabetes. For example, researchers have measured levels of histone modifications, such as dimethylation and acetylation of H3 lysine 9 (H3K9me2, H3K9ac), in lymphocytes and monocytes and found altered levels in type 1 diabetes compared to controls, including in type 1 diabetes risk loci, e.g., CTLA4 and class II MHC (244,245). Changes in histone modification levels, as well as histone acetyltransferase localization, have been observed in immune cells in disease-relevant environmental conditions, such as hyperglycemia (246,247).
A caveat to many of these studies is that epigenetic changes observed in type 1 diabetes may reflect consequences of the disease process that do not contribute to disease pathogenesis directly. Mapping epigenetic changes mediated by genetic risk variants can help to define which epigenetic features may play a causal role in disease. For example, CpG sites at the CTSH locus are affected by both environmental stimuli and genetic risk variants, supporting the hypothesis that methylation of these CpG sites may causally affect disease (248). Compared to genetic studies, the sample sizes of epigenetic studies have been relatively small, and consequently, results across different studies have had limited consistency. Therefore, larger studies are needed to enable well-powered analyses.
Type 1 diabetes is a complex disorder that results from the individual and, possibly, the combined effects of multiple genetic and environmental risk factors. The advent of high-throughput genotyping methodologies enabling GWAS combined with large collections of biospecimens from type 1 diabetes cases and unaffected control individuals has allowed the identification of a large number of genetic loci that contribute to type 1 diabetes risk. Fine-mapping studies building on GWAS results have substantially lowered the number of possible causal genes. Some genes involved in type 1 diabetes risk have been identified and have consistent and strong effects (e.g., HLA class I and class II loci or INS). Other genes have small individual contributions to risk; nevertheless, these genes may represent components in common biologic pathways with rate-limiting reactions or products that may be amenable to manipulation for therapy. While a subset of genes involved in type 1 diabetes risk are associated with changes to the protein product (e.g., PTPN22, TYK2, IFIH1, FUT2), most genes are affected by noncoding variants that modulate expression in disease-relevant cell types. Research on the function of protein-coding and noncoding variants associated with type 1 diabetes is shedding light on the pathophysiological mechanisms that underlie type 1 diabetes risk, as well as helping to dissect disease heterogeneity.
Future studies of the genetics of type 1 diabetes focusing on the relationship of genotype and phenotype would help elucidate the molecular, cellular, and organismal effects of individual risk variants on gene activity. As most variants affecting type 1 diabetes risk are located in noncoding regions of the genome, more detailed maps of regulatory elements and genes regulated by these elements would enable researchers to link risk variants to their gene targets. These studies, building upon unbiased searches for risk genes and variants, hold the promise of providing novel insights into the pathogenesis of type 1 diabetes, targets for potential preventive therapies, and markers of response that can be used to predict type 1 diabetes and select candidates who may respond to specific preventive agents. In addition, genetic studies in global populations, as well as the incorporation of ancestry in prediction models, would fill a gap in identifying individuals of any race or ethnicity who are at high risk of developing type 1 diabetes. Learning to use genetics, along with environmental factors, to screen for type 1 diabetes risk in the general population offers a key opportunity for finding those who are most likely to benefit from preventive therapies. The development of genetic risk scores and the lower cost of genotyping methods will facilitate the incorporation of genetics in clinical practice to help in diagnosis of diabetes type, prognosis of clinical course (e.g., C-peptide decline, development of additional autoimmune diseases, etc.), and potentially, selection of best clinical management approaches.
antigen-presenting cell
affected sib (sibling) pair
body mass index
chromatin accessibility QTL
Diabetes and Autoimmunity Study in the Young
deoxyribonucleic acid
expression QTL
glutamate decarboxylase
genome-wide association study
human leukocyte antigen
histone QTL
insulinoma-associated antigen 2
identity-by-descent
insulin gene
latant autoimmune diabetes in adults
linkage disequilibrium
major histocompatibility complex
odds ratio
parietal cell antibodies
quantitative trait locus
single nucleotide polymorphism
Type 1 Diabetes Genetics Consortium
T cell receptor
The Environmental Determinants of Diabetes in the Young study
Wellcome Trust Case-Control Consortium
Support was provided by the National Institute of Diabetes and Digestive and Kidney Diseases grants DK124395 (MJR, SOG), DK121843 (MJR), DK114650 (KJG), DK122607 (KJG), and DK120429 (KJG).
This is an update of: Rich SS, Erlich H, Concannon P: Genetics of Type 1 Diabetes. Chapter 12 in Diabetes in America, 3rd ed. Cowie CC, Casagrande SS, Menke A, Cissell MA, Eberhardt MS, Meigs JB, Gregg EW, Knowler WC, Barrett-Connor E, Becker DJ, Brancati FL, Boyko EJ, Herman WH, Howard BV, Narayan KMV, Rewers M, Fradkin JE, Eds. Bethesda, MD, National Institutes of Health, NIH Pub No. 17-1468, 2018, p. 12.1–12.16
Received in final form on June 16, 2023.
Drs. Redondo and Onengut-Gumuscu reported no conflicts of interest. Dr. Gaulton consults for Genentech and holds stock in Neurocrine Biosciences.
Diabetes in America is in the public domain of the United States. You may use the work without restriction in the United States.
Your browsing activity is empty.
Activity recording is turned off.
See more...