Summary
Monogenic diabetes is a term used to describe a group of single-gene causes of diabetes. Traditional nomenclature has focused on the types of diabetes known as MODY, defined as maturity-onset diabetes of the young; infantile (or perinatal) onset diabetes, including permanent and transient neonatal diabetes mellitus; and syndromic types. These syndromes include genes associated with insulin resistance, such as the insulin receptor itself. A growing field is also focused on single gene variants that are responsible for multi-autoimmune syndromes that include type 1 diabetes. Altogether, monogenic diabetes may account for up to 3% of all diabetes diagnosed before age 35 years, amounting to an estimated 300,000 cases in the United States. However, a systematic study of incidence and prevalence in the United States has not been done.
Introduction
The term “monogenic diabetes” includes “maturity-onset diabetes of the young (MODY),” a term coined by Fajans and Tattersall only a few decades ago to describe a presentation of non-insulin-dependent diabetes that occurs in young people with a strong autosomal dominant inheritance (1,2). The initial case reports and, then, meticulous studies of several families by Fajans and colleagues (1) led to the identification of the first MODY genes by international teams, including those of Bell, Hattersley, Froguel, Njølstad, and others. Fajans suggested identification of these forms could be aided by using simple criteria: appearance of diabetes in adolescents and young adults age <30 years; a strong family history affecting two to three linear generations in an autosomal dominant pattern; usually not obese; and in some cases, high sensitivity to sulfonylureas (2).
To date, over 20 genes have been labeled as “monogenic diabetes genes.” Previously, many of these genes (particularly those linked to MODY) were numbered based on their order of discovery and frequently described using uninformative numbers rather than gene names. As the list of MODY genes grew and the designation of some MODY genes was refuted (3), it became more appropriate to refer to these entities by the gene name, such as GCK-MODY (rather than MODY 2) or HNF1B-MODY (rather than MODY 5), to avoid confusion. Accordingly, these gene names are used throughout the text, but both gene names and the older MODY number designations are utilized in the tables. This approach is particularly important as some of the more recent putative “MODY” genes were given the same numerical designation by different authors, and several “MODY” genes listed in the tables are either not causal for diabetes at all or are very rare. Establishing an accessible nomenclature is an important part of efforts to make recognition and diagnosis of these entities commonplace.
Monogenic diabetes-related genes constitute about 1.2%–2.5% of all new-onset childhood diabetes (4), amounting to an estimated 300,000 cases in the United States (5). The glucokinase gene, GCK, is the most common, followed by HNF1A (at least in several reports). Individuals with monogenic diabetes may come to attention when they have a urinalysis showing glucosuria (see the HNF1A-MODY section) but still have normal blood glucose and glycated hemoglobin (A1c) tests.
This article focuses on common issues in the presentation and treatment of monogenic diabetes and updates Chapter 7: Monogenic Forms of Diabetes, published in Diabetes in America, 3rd edition (6). For forms that appear to be most common, the incidence, prevalence, clinical features, and approaches to treatment are reviewed. Very uncommon monogenic diabetes forms are listed in the tables, and the reader is referred to focused reviews for more information.
Monogenic Diabetes
Epidemiology
Overall, monogenic diabetes accounts for approximately 2%–3% of all patients diagnosed with diabetes before age 35 years, based on data from the University of Exeter group; in the United Kingdom, the minimum prevalence of MODY was estimated to be 108 cases per million (4,7). Monogenic diabetes has been studied in registries across Europe and to a lesser extent in Asian, African, Latino, and Middle Eastern populations, with variations in prevalence and relative prevalence of subtypes (8). In European countries (United Kingdom, Germany, The Netherlands, Norway, Poland), the most common subtype is HNF1A-MODY, followed by GCK-, HNF4A-, and HNF1B-MODY (8,9,10). In 2020, the first MODY cases were reported in patients from Africa, where the predominant subtype, HNF1A-MODY, represented 5.9% of the study population (11).
The Exeter group showed that mutations in recessive genes are major contributors to childhood-onset monogenic diabetes in a population with a high rate of consanguinity (12). In addition to true differences in prevalence, geographic variations in prevalence also likely result from differences in ascertainment (e.g., more GCK-MODY cases where asymptomatic glucose screening is standard of care), genotyping/sequencing methodology, and reduced ability to classify genetic variants in non-White populations due to limited available data (8).
In the U.S. SEARCH for Diabetes in Youth Study (5), of approximately 5,000 newly diagnosed children with diabetes (diagnosed age <20 years), 730 youth (14.5%) were autoantibody-negative and fasting C-peptide-positive (also termed A-B+). Of these children, 586 underwent sequencing of GCK, HNF1A, and HNF4A, and 48 (8.2%) were positive for a likely pathogenic mutation in one of these three genes. Thus, about 1.2% of the original cohort of children had genetically confirmed monogenic diabetes. Non-Hispanic White (35%), African American (24%), Hispanic (26%), and Asian and Pacific Islander (13%) individuals were all found in the monogenic diabetes group. Almost all were previously considered to have type 1 diabetes or type 2 diabetes and treated with insulin or metformin and other oral hypoglycemic medications, respectively.
The U.S. Treatment Options for Type 2 Diabetes in Adolescents and Youth (TODAY) Study enrolled 699 racially and ethnically diverse individuals who were diagnosed with type 2 diabetes, overweight or obese (body mass index [BMI] ≥85th percentile), and A-B+ in a metformin +/- rosiglitazone or lifestyle intervention clinical trial. From this cohort, 488 individuals with available DNA samples underwent targeted sequencing for 40 genes implicated in monogenic diabetes or related disorders (13). Of these individuals, 22 (4.5%) were found to have variants classified as pathogenic or likely pathogenic for monogenic diabetes (seven HNF4A, seven GCK, five HNF1A, two insulin gene [INS], and one KLF11) based on the 2015 American College of Medical Genetics and Genomics/Association of Molecular Pathology (ACMG/AMP) guidelines. Only one of these cases was in a gene (KLF11) now refuted as a monogenic diabetes gene by the Clinical Genomic Resource (ClinGen) Monogenic Diabetes Expert Panel Gene Curation Expert Panel (MDEP GCEP), bringing the total to 4.3%. There may have been more individuals with monogenic diabetes in the cohort given that 30 variants of uncertain significance (VUS) were identified in the common and rare MODY genes described in Table 1 (14). These data provided evidence that monogenic diabetes should be considered in the differential diagnosis for diabetes in children and adolescents, regardless of adiposity. Importantly, the adverse impact of a missed monogenic diabetes diagnosis on treatment response was demonstrated in the TODAY study. The seven individuals with HNF4A-MODY were significantly more likely to fail to respond to the insulin-sensitizing treatments used in the study as measured by the primary outcome of reaching an A1c level >8% (>64 mmol/mol) in 6 months or the inability to wean the participant off insulin within 3 months after treatment initiation (hazard ratio [HR] 5.03, 95% confidence interval 2.18–11.58, p=0.0002) (13). This is expected given that HNF4A-MODY represents an insulin secretory rather than sensitivity defect. Conversely, none of the seven individuals with GCK-MODY failed treatment (HR undefined due to no events), which is expected given that A1c in individuals with GCK-MODY is rarely, if ever, >7.6% (>60 mmol/mol) regardless of treatment (15).
TABLE 1.
Classification of Maturity-Onset Diabetes of the Young, U.S., 2022
A larger, overlapping sample comprising 492 SEARCH participants with clinician diagnosed-diabetes before age 20 years, a slightly larger sample from TODAY (n=511), and a cross-sectional sample of 2,330 additional U.S. pediatric type 2 diabetes cases (diagnosed before age 18 years with BMI ≥85th percentile) underwent exome sequencing as part of the Progress in Diabetes Genetics in Youth (ProDiGY) collaboration and were evaluated for variants in the rare and common MODY genes (16). Of the individuals sequenced, 93 (2.8%) had a disease-causing variant (44 in HNF1A, 23 in GCK, 16 in HNF4A, 5 in PDX1, 4 in INS, and 1 in CEL), with 83 (89%) variants having management implications. While metabolic traits differed between those with and without a known variant, no trait or set of traits could be used reliably to make the distinction without testing. These results underscored in a larger sample of overweight or obese individuals with youth-onset diabetes without autoantibodies the considerable likelihood (nearly 3%) of a true diagnosis of monogenic diabetes underlying apparent pediatric type 2 diabetes (16). Moreover, this outcome may be an underestimate given that over 300 VUS were identified. The findings also underscore the importance of utilizing biomarker information (presence of C-peptide and absence of diabetes autoantibodies) to identify individuals who should have genetic testing for monogenic diabetes.
In 2021, the University of Chicago Monogenic Diabetes Registry reported the relative frequency of monogenic diabetes and neonatal diabetes in the registry (14). Among genes that cause a MODY phenotype, GCK mutations were the most common (59%), followed by HNF1A mutations (28%). Mutations in KCNJ11 were the most common cause among genes associated with neonatal diabetes (35%), followed by INS mutations (16%) (14).
Presentation of Monogenic Forms of Diabetes
Monogenic diabetes includes neonatal, childhood, and young adult onset of diabetes (also known as MODY types) with diagnosis typically occurring at age <35 years. The initial clinical diagnosis may be erroneously reported as either type 1 diabetes or type 2 diabetes. Anti-islet antibody tests are almost always negative. In the case of a mildly positive anti-GAD (glutamic acid decarboxylase) test only, one must consider that positive anti-GAD does not always correlate with autoimmune diabetes, as there is a false positive rate in the general population. Anti-islet antibodies can also fade and become undetectable over time. Testing multiple autoantibodies at diabetes presentation, including anti-GAD, anti-IA2 (tyrosine phosphatase-related islet antigen-2), anti-ZnT8 (zinc transporter 8), and anti-insulin, can be helpful, but negative antibodies are not definitive for the absence of autoimmune type 1 diabetes. Other autoimmune diseases in the patient or their close family members may be suggestive of type 1 diabetes. For research evaluation, the type 1 diabetes genetic risk score (T1DGRS) developed by Patel, Oram, and colleagues (17) can be suggestive and has been validated in several populations. Measuring C-peptide is generally not helpful until 3–5 years after diagnosis. Detectable levels of C-peptide can persist for many years after the diagnosis of type 1 diabetes, but by 3–5 years after diagnosis, stimulated or fasting C-peptide levels collected with a simultaneous glucose level are usually reduced in those with type 1 diabetes (18). Interest is growing in evaluating urinary C-peptide levels (also developed by the Exeter group), since this assay can give a time-averaged estimate of beta cell function and can be more stable than a blood draw (19). This test is not yet generally available in the United States on a clinical basis.
Figure 1 provides an algorithm for the diagnosis of monogenic diabetes (9). A search for possible syndromic features is extremely helpful when considering monogenic diabetes testing. These features are variable in their presentation and might include developmental delay, deafness, ocular muscle defects, retinal changes, liver adenomas, renal or other organ cysts, genitourinary abnormalities, alkalosis, and hypomagnesemia. Birthweight (high or low) and any episodes of neonatal hypoglycemia are also important to review. A detailed family history can help with a diagnosis. Important findings include any first-degree relative with diabetes of any kind, as monogenic diabetes family members are also likely to be assigned an incorrect diabetes type. A family history of syndromic features or autoimmune conditions may be helpful.
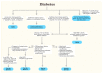
While most monogenic diabetes syndromes are dominantly inherited, recessive and maternal inheritance forms are known. Pedigrees with GCK-, HNF1A-, HNF4A-, or HNF1B-MODY may show three or more generations with dominantly inherited diabetes, with extended family members also possibly affected (Figure 2). Once a variant causing monogenic diabetes is confirmed, a referral should be made for genetic counseling to further discuss the findings with the patient and their family and to provide them with the necessary information to share with additional family members about the potential benefits of testing (Figure 3). Most variants are dominant, so there is a 50% chance that each child of an affected individual will have the same form of diabetes.
Common Forms of Monogenic Diabetes
GCK-MODY
GCK was the first actual MODY gene to be identified, although it was previously referred to as MODY 2 because it was the second gene to be localized to a chromosomal region through family-based linkage studies (20,21). GCK-MODY results from decreased beta cell sensing of glucose due to loss-of-function mutations in the GCK gene on chromosome 7p15-p13, encoding the enzyme glucokinase. The estimated prevalence rate of GCK-MODY is 0.2% in the general population and 1.3% in patients with diabetes (22). These patients have stable fasting hyperglycemia (99–144 mg/dL [5.49–7.99 mmol/L]) present at birth. Hyperglycemia may be discovered as an incidental finding in a child or adult during a routine blood chemistry check performed for an unrelated illness or pregnancy screening; islet cell antibodies are negative and glucose tolerance testing shows mild hyperglycemia, rarely exceeding 200 mg/dL (11.10 mmol/L) at 2 hours with delayed return to modestly elevated basal glucose concentrations in the range of 90–130 mg/dL (5.00–7.22 mmol/L) (Figure 4). GCK-MODY patients have a uniform phenotype despite considerable functional differences in mutation severity, possibly due to compensation by overexpression of the normal allele (23,24).
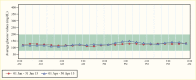
Insulin levels, if measured, are appropriate but occur at higher-than-normal glucose concentrations. A1c values are generally in the 5.6%–7.6% (38–60 mmol/mol) range (24,25). In the absence of a family history, such patients may be considered to have type 1 diabetes early in their clinical course and may be inappropriately treated with insulin. Alternatively, they may be considered to have type 2 diabetes and be treated with oral hypoglycemic agents or insulin sensitizers. Patients with GCK-MODY lack a glycemic response to oral hypoglycemic agents or low-dose insulin. Beta cell function shows minimal deterioration with increasing age, consistent with the general population. Microvascular complications occur very infrequently. Nonproliferative retinopathy is the only complication that has been reported in GCK-MODY, and its occurrence is only slightly increased when compared to healthy controls (26).
All pharmacological treatments used for GCK-MODY are minimally effective due to decreased glucokinase activity in the beta cells, liver, and brain, which also affects counterregulation. One study showed a significant drop in fasting plasma glucose after one tablet of dapagliflozin; additional studies are needed to evaluate this effect (27). Long-term clinical studies of patients with GCK-MODY treated with sodium-glucose cotransporter 2 (SGLT2) inhibitors are required to further assess this treatment due to reported risk of euglycemic diabetic ketoacidosis (DKA) (26,27). However, published data support the idea that treatment with SGLT2 inhibitors is unnecessary, based on the lack of long-term complications. In any case, precisely due to the lack of long-term complications in most people with GCK mutations, specific glucose-lowering treatment is not needed and may be harmful. Diabetes practitioners in many cases remain skeptical of this recommendation based on reasonably large-scale studies. It may be that since most people with GCK mutations are not obese and have normal lipid levels and normal blood pressure, the usual combination of features in type 2 diabetes that lead to complications is not present. If a patient’s A1c level is high, the diagnosis should be reconsidered, or the presence of additional factors, including obesity, steroid use, and even type 1 diabetes, should be considered. Rare cases have been described of patients with GCK-MODY who have developed co-occurring autoimmune (antibody-positive) type 1 diabetes; hence, an evaluation for other diabetes types, including type 1 diabetes, is recommended if glycemic levels deteriorate beyond the typical range for GCK-MODY. In these rare cases, insulin treatment would be required.
Pregnancy is an exception when treatment may be recommended for individuals with GCK mutations causing hyperglycemia based on known or suspected fetal genotype. If the fetus inherits the GCK mutation, the fetus will have the same elevated glucose set point as their mother and will regulate their blood glucose levels to that higher set point; thus, treatment of the mother is not recommended. If the fetus does not inherit the GCK mutation, the fetus will be exposed to maternal hyperglycemia in utero. In this situation, insulin treatment is recommended, aimed at lowering maternal blood glucose levels to normal pregnancy levels and avoiding possible complications. Supraphysiological insulin doses (>1 unit/kg/day) are usually required to overcome maternal counterregulatory mechanisms and can result in severe hypoglycemia in pregnant women (28). For further information on fetal testing and management of GCK-MODY during pregnancy, the reader is encouraged to refer to more detailed discussions (25,29).
HNF1A-MODY
HNF1A-MODY is due to heterozygous dominant variants in the gene encoding the transcription factor hepatocyte nuclear factor 1-alpha (HNF1α), which is expressed primarily in liver, pancreatic beta cells, and kidney cells and can be detected in many other cell types. HNF1A was the third gene chromosomally localized by linkage mapping in MODY families, hence the historical designation of HNF1A-MODY as MODY 3 (30,31). HNF1A gene expression is partially regulated by HNF4A, providing a link between these two forms of monogenic diabetes. HNF1A and HNF1B are homeobox transcription factors that function as dimers, interacting with DNA at specific target sequences and with other specific proteins. Point mutations are predominantly located in the DNA-binding domain but can occur anywhere, along with insertions and deletions. Diabetes primarily results from haploinsufficiency, meaning the functional absence of one allele is the key. The correct temporal and cell/organ-specific expression of the genes is critical for proper developmental formation of specific organs, including the islets/beta cells, and later for the maintenance of the mature cells where the factor is expressed. No specific genotype-phenotype correlation with respect to the presentation of diabetes has been found, although a relationship between the type and position of the variant with age of diagnosis has been observed (32). Birth weight is normal in patients with heterozygous HNF1A variants. Diabetes usually appears towards the end of the first decade into the third decade of life.
Presentations of HNF1A-MODY can be extremely mild with minimal symptoms, and the diagnosis may be made serendipitously, following a urinalysis for a sports physical, for example. Others present with overt diabetes and possible ketosis, although DKA is rare, which can lead to temporary or intermittent treatment with insulin. Most patients with HNF1A-MODY are initially quite responsive to sulfonylureas, even to the point of hypoglycemia with low doses. Any patient who has severe hypoglycemia in response to the usual starting doses of a sulfonylurea should be carefully considered for a HNF1A variant. Some patients with HNF1A-MODY may have progressive failure of sulfonylurea therapy, requiring insulin for treatment; excessive weight gain seems to be a risk factor for sulfonylurea failure (24). However, newer agents, including glucagon-like peptide-1 receptor agonist (GLP-1 RA) and dipeptidyl peptidase 4 (DPP4) inhibitors, combined with sulfonylureas may prove useful for good glycemic control in the absence of exogenous insulin. Variability in the age of onset of HNF1A-MODY can occur even within the same pedigree (Figure 2). The degree of sulfonylurea responsiveness can also vary. These features do not seem to be related to the specific variant (33).
Another feature of HNF1A-MODY that may occur is liver adenomas that might be large but benign, although bleeding has been reported. Subtle changes in lipid levels may occur that should lead to early consideration of lipid-lowering treatment, particularly given increased risk for adverse cardiovascular outcomes in people with HNF1A-MODY compared to their unaffected relatives. Interestingly, the kidney protein SGLT2, which is responsible for re-uptake of filtered glucose in the renal tubule, is regulated by HNF1A. In the case of HNF1A-MODY, reduction in the HNF1α protein leads to a decrease in the expression of SGLT2. This, in turn, can result in appearance of glucosuria despite normal blood glucose levels, the equivalent of reducing the renal tubular maximum for glucose from 220 mg/dL (12.21 mmol/L). This observation explains the diagnosis of HNF1A-MODY in young people with glucosuria, but normal blood glucose and A1c levels, and has resulted in the misdiagnosis of type 1 diabetes or type 2 diabetes in these individuals. Interestingly, expression of C-reactive protein (CRP), which is produced in the liver, may be partially driven by HNF1A, so that patients with HNF1A variants also have low high-sensitivity CRP (hsCRP) levels compared to all other forms of diabetes, including other forms of monogenic diabetes; however, this finding has not been confirmed in all studies (34).
Complications of diabetes due to HNF1A-MODY can be just as severe as in uncontrolled type 2 diabetes, if inadequately treated. While sulfonylurea treatment may be preferable to insulin treatment for some patients, others may not continue to show the same high degree of responsiveness with time. Several anecdotal or small studies have suggested that GLP-1 RAs may be a useful approach, possibly restoring sulfonylurea responsiveness (35). A large systematic study has not yet been reported. Other studies have suggested that SGLT2 inhibitors might be useful. A concern with that approach is that given the decrease in SGLT2 expression already seen in these individuals, further reduction in SGLT2 function may lead to a normalization of blood glucose with insufficient insulin secretion to suppress ketones. This phenomenon of euglycemic DKA has not been reported in patients with HNF1A-MODY but certainly has been seen in individuals with type 1 diabetes and type 2 diabetes with low insulin secretion. Caution in pursing this therapy outside of a clinical trial is recommended, as the lower expression of SGLT2 in these patients may increase their risk for this complication.
Genome-wide association studies have linked variants in HNF1A to the more common forms of type 2 diabetes, suggesting the existence of a spectrum of mutations with variable penetrance. Indeed, the Framingham Heart Study (36) showed that incomplete penetrance of HNF1A-MODY variants does occur. This result further suggests that other genes may be able to modulate or overcome the loss of HNF1A function in some instances.
HNF4A-MODY
HNF4A-MODY occurs due to variants in the gene encoding HNF4α. It was the first gene chromosomally localized in families for MODY (37), hence the historical designation as MODY 1, although it was the third actual gene to be identified as having variants causing MODY (38). HNF4A is expressed in the liver, kidney, intestine, and islets and at lower levels in many cell types. HNF4α is a known key regulator of hepatic gene expression, and HNF4A-MODY, like HNF1A-MODY, is characterized by progressive loss of insulin secretion with increasing age. Likewise, the clinical features of HNF4A-MODY are similar to those of patients with HNF1A-MODY. Clinical onset of symptoms and recognition of hyperglycemia can occur from the early childhood years to the third decade of life. Many affected individuals are sensitive to sulfonylurea drugs, which restore insulin secretion and lower glucose. Other individuals may develop progressive loss of insulin secretion and require insulin for treatment.
Two interesting features of HNF4A-MODY reported in some individuals are that: (1.) affected participants have been found to weigh about 800 grams more at birth than their unaffected siblings and (2.) hyperinsulinemia at birth with symptomatic neonatal hypoglycemia that resolves spontaneously may occur (39). Diazoxide has been used to reduce hypoglycemia in these babies. These findings suggest that at least some HNF4A variants at first cause excessive secretion of insulin, directly or indirectly, and only later result in impaired beta cell function with hypoinsulinemia and diabetes. Whether all babies with macrosomia and neonatal hypoglycemia go on to develop HNF4A-related diabetes is not yet known. These children should be followed with the expectation that diabetes will develop, possibly near the time of puberty.
Patients with HNF4A-MODY have lower levels of lipoprotein A2 and high-density lipoprotein (HDL) cholesterol, features commonly seen in type 2 diabetes (40). Reports of markedly lower concentrations of CRP in patients with HNF1A variants have not been replicated in HNF4A-MODY patients. Hence, hsCRP cannot be considered as a screening test to identify patients suspected of having HNF4A-MODY before embarking on molecular diagnosis (1,2,40,41,42).
HNF1B-MODY
HNF1B-MODY has historically been called MODY 5 because HNF1B was the fifth MODY gene both localized and identified (43,44). Heterozygous single nucleotide variants and large deletions in the HNF1B gene each account for about 50% of a variable, multi-organ phenotype with HNF1B-MODY, either in isolation or with other affected organs (45). Haploinsufficiency is thought to underlie the disease mechanism, and notably, no clear genotype-phenotype distinction between single nucleotide variants and large deletions of the HNF1B gene in regards to diabetes has been identified. HNF1B regulates HNF4A and, hence, may provide a link between the three main transcription factor gene causes of MODY.
Variants in HNF1B most frequently result in abnormally developed kidneys, and a common presentation is the Renal Cysts and Diabetes (RCAD) syndrome (43). Other associated anomalies include morphological and functional anomalies of the exocrine pancreas, with reduced pancreatic size and pancreatic exocrine deficiency. These anomalies may initially be subclinical but can progress with time, making surveillance with fecal elastase testing appropriate. Additionally, reproductive tract abnormalities with fertility implications, abnormal liver tests, hyperuricemia, hypomagnesemia due to renal magnesium wasting, alkalosis, and early appearance of proteinuria that is not due to diabetic renal disease are observed. End-stage kidney disease may occur, requiring hemodialysis and kidney transplantation. HNF1B variants may come to light because a patient with diabetes has siblings or parents with renal cysts and early renal failure.
The pathophysiology of HNF1B-MODY is due to a combination of beta cell dysfunction and insulin resistance. Unlike patients with HNF1A-MODY or HNF4A-MODY, most individuals with HNF1B-MODY do not respond to sulfonylurea drugs, and a large percentage require insulin for management, although alternative diabetes drugs have not been rigorously studied (1,2,41,42,46). Ray et al. published a report of three cases of patients with diabetes and refractory hypomagnesemia (47). Two patients had confirmed HNF1B mutation, and hnf1b deletions could not be ruled out in the third patient. The study demonstrated that SGLT2 inhibitors can ameliorate refractory hypomagnesemia in patients with urinary magnesium wasting, potentially by enhancing renal tubular reabsorption of magnesium. The authors did not report serum glucose levels and/or glomerular filtration rate after SGLT2 inhibitor treatment (47).
In addition to single nucleotide variants and large deletions in HNF1B (which may, in fact, be due to chromosome microdeletion), chromosome 17q12 contiguous gene microdeletions are also a cause of HNF1B-MODY. These deletions encompass 14 genes in addition to HNF1B and are associated with neurodevelopmental disorders, such as autism spectrum disorder, attention-deficit hyperactivity disorder (ADHD), and cognitive impairments. There is not yet complete agreement whether a neurocognitive phenotype is seen in HNF1B intragenic variants (48,49). Dubois-Laforgue et al. found an increase in patients with intragenic mutations, albeit at lower rates than in the patients with 17q12 deletion (49). Clissold et al. identified several genes that are differentially methylated in HNF1B-associated disease, some of which are specific to individuals with 17q12 deletion (50). They documented organized changes in DNA methylation across a large deletion region suggestive of a compensational role of this epigenetic modification. However, the relationship between the DNA methylation status of the 17q12 deletion and the increased prevalence of psychiatric symptoms was not studied due to lack of psychiatric data. Interestingly, more recent publications have suggested a genotype-phenotype correlation for renal disease in HNF1B-associated disease with 17q12 deletion, resulting in better renal function than HNF1B intragenic mutations that cause loss of function (49,50,51,52).
Care for people and families with HNF1B-associated disease is complex. The combined guidance of experts in diabetes, kidney function, gynecology/urology, gastroenterology, and neurology may be required.
Rare Forms of Monogenic Diabetes
More than 20 genes associated with monogenic diabetes with onset beyond the first few years of life have been reported. Some of these genetic variants remain unconfirmed as causes of diabetes. Some of the genes are of considerable interest as they can also cause neonatal diabetes, or syndromic forms, or cause acinar pancreatic dysfunction that includes loss of beta cell function. Rather than describe these extremely rare forms of dominantly inherited diabetes, the reader is referred to a 2021 review (53) and to websites listed in the Resources section for information on manifestations and approaches to treatment.
Maternally Inherited Diabetes and Deafness and Mitochondrial Encephalopathy With Lactic Acidosis and Stroke-Like Episodes
Maternally inherited diabetes and deafness (MIDD) and mitochondrial encephalopathy with lactic acidosis and stroke-like episodes (MELAS) are complex forms of maternally inherited diabetes that have extremely varied syndromic presentations. These forms are caused by both mitochondrial DNA point mutations and deletions that may affect <1% of patients with diabetes (54); studies using comprehensive monogenic testing have found that about 5% of cases are due to mitochondrial mutations (55). About 80% of MELAS patients harbor the maternally inherited variant m.3243A>G mapping in the MT-TL1 gene encoding the mitochondrial transfer RNA, tRNALeu(UUR) (54). Other variants include, for example, an m.5541C>T mutation in the tRNATrp gene found in a large MELAS family (56). These gene variants are typically inherited from the mother, since mitochondrial DNA is present in oocytes and only to a very limited extent in mature spermatozoa. Therefore, relatives on the maternal side frequently have manifestations of mitochondrial gene defects. Mutations in nuclear genes that encode mitochondrial structures are also rare and have somewhat different complex phenotypes.
For the mitochondrial tRNA gene variants, diabetes is common. Other systemic manifestations vary according to heteroplasmy, meaning that the distribution of damaged mitochondria to tissues varies within the same pedigree and can include cardiac defects (conduction and heart failure), short stature, retinopathy, lactic acidosis, glomerulopathy with focal segmental glomerulosclerosis (FSGS), and strokes or stroke-like episodes with cerebellar and cerebral atrophy. The variable appearance of defective mitochondria can make diagnosis from blood or saliva samples difficult, as well as limit the understanding of the overall burden of disease. One approach is to collect urine and sequence mitochondrial genes in the urinary sediment. This strategy allows an estimation of the ratio of damaged to normal mitochondrial genomes.
Because mitochondrial variants result in diminished adenosine triphosphate (ATP) production, tissues with high energy turnover, such as pancreatic islets and key auditory cells, are highly affected, explaining the presence of diabetes and deafness alone in mild cases. Diabetes may be variable in its severity, even presenting as DKA in young patients, and may be misdiagnosed as type 1 diabetes. However, anti-islet antibodies will be absent. Alternatively, milder forms may present later in life and be (mis)diagnosed as type 2 diabetes. With advancing myopathy, insulin resistance can be extreme. Renal failure is also a feature of the advancing FSGS. The prognosis for MIDD is determined by the associated systemic manifestations, including cardiac failure and central nervous system dysfunction, and diabetes requires insulin for effective management. The associated retina changes are also diagnostic for mitochondrial disease.
The use of metformin in mitochondrial diabetes is debated. Given the possibility of metformin-induced lactic acidosis resulting from mitochondrial toxicity, the recommendation is to avoid the use of metformin and indeed any agent that might be a mitochondrial toxin. Insulin remains the cornerstone of therapy. Several groups have reported some improvement using high-dose taurine or arginine therapy (57,58), but randomized controlled trials are lacking. Genetic counseling is important in clarifying the maternal inheritance of this entity to affected family members; all offspring of an affected mother are expected to have some copies of the variant.
Wolfram Syndrome
Wolfram syndrome, also known as DIDMOAD (diabetes insipidus, insulin-deficient diabetes mellitus, optic nerve atrophy, and deafness), is a rare neurodegenerative disorder of autosomal recessive inheritance (59). Over 200 pathogenic variants have been reported in association with this disease (60). Suspicion of the diagnosis is usually based on history and clinical manifestations, and genetic testing is useful to confirm the diagnosis. Diabetes mellitus and diabetes insipidus are the most common clinical manifestations of Wolfram syndrome, and follow-up and management should be done in a standard way. As Wolfram syndrome affects different organs and systems in the body, multidisciplinary care by physicians and healthcare professionals from a range of disciplines is required. Wolfram syndrome was initially categorized as a mitochondrial disease due to its symptoms and several reports of mitochondrial mutations. However, Wolfram syndrome is now established as a prototype endoplasmic reticulum (ER) disease. Currently, novel therapeutics for Wolfram syndrome use small molecules to target mediators of ER calcium homeostasis (60).
Genetic Lipodystrophies
Genetic lipodystrophies are autosomal dominant and recessive disorders. Autosomal dominant forms include familial partial lipodystrophies (FPL; FPLD 1, 2, 3, 4 and AKT2), atypical progeroid syndrome (LMNA), and SHORT syndrome (PIK3R1). Autosomal recessive disorders include the congenital generalized lipodystrophies (CGL1, 2, 3, and 4), mandibuloacral dysplasia (MAD) types A and B, and familial partial lipodystrophy (FPLD5). The reader is referred elsewhere for more complete discussion (61).
Neonatal Diabetes Mellitus
Neonatal diabetes mellitus (NDM) is the term used to define a special, rare category of diabetes defined by its extremely early onset, whereby those diagnosed with diabetes at age <6 months are very likely to be due to underlying monogenic causes. Although these monogenic etiologies can very rarely cause diabetes that occurs between ages 6 and 9 months or even later, the incidence of autoimmune diabetes (type 1 diabetes) approaches ≥95% of cases. Therefore, sequencing the various genes known to be associated with NDM should be undertaken in all individuals with diabetes diagnosed at age <6 months, as well as those diagnosed at age 6–12 months (or potentially even older), if testing for islet antibodies is negative or there are other features that suggest the possibility of an underlying monogenic etiology. A classification of NDM is shown in Table 2. NDM can be permanent (PNDM) or transient (TNDM), with some overlap of genes associated with later-onset MODY-type diabetes and with type 2 diabetes.
TABLE 2.
Classification of Neonatal Diabetes Mellitus, 2022
Epidemiology of Neonatal Diabetes
The incidence of NDM is approximately 1:100,000 live births; although in regions with high rates of consanguinity, the incidence has been reported to be as high as 1:21,000 live births (62,63,64). Infants with NDM are characterized by insulin deficiency and generally are small at birth, reflecting the important role of insulin as a fetal growth factor. In the majority of cases, the diabetes will be permanent, whereas for some causes or certain mild variants, individuals will experience spontaneous remission of the diabetes, with near-normal glucose homeostasis in the absence of any treatment. The majority of TNDM cases are due to overexpression of genes at the maternally imprinted 6q24 locus (6q24-TNDM) (Table 2) (65). Most remaining cases of TNDM are due to mildly activating mutations in ABCC8 and KCNJ11, the two genes encoding the subunits of the pancreatic ATP-regulated potassium (KATP) channel (reviewed in the next section), whereas most KATP-NDM cases will have PNDM. Individuals with 6q24-TNDM will often have relapse of the diabetes later in life (especially around puberty or older), while the recurrence of diabetes in KATP-NDM is less predictable and glucose levels may be elevated episodically, such as when ill (63,65,66,67,68,69,70). Heterozygous mutations in INS are the second most common cause of PNDM.
KCNJ11, INS, and ABCC8 are now well recognized to also be rarer causes of MODY, with diabetes being diagnosed for the first time in adolescence or young adulthood. Any infant diagnosed with diabetes at age <12 months (or even older, if antibody testing is negative or other features are present) should undergo comprehensive genetic testing that preferably includes all known causes or, at the very least, KCNJ11, ABCC8, and INS, along with determination of 6q24 imprinting defects (which requires more than just sequencing for complete testing).
KATP Channel Diabetes: KCNJ11 and ABCC8
The most common cause of NDM is activating mutations in either of the two genes encoding the KATP channel; these mutations comprise nearly half of cases of neonatal diabetes. The KATP channel is a key ion channel that plays the major role in regulating insulin secretion by maintaining a resting membrane potential when the channels are open. Insulin secretion occurs when the membrane is depolarized due to closure of channels in response to higher levels of ATP that are generated in the presence of elevated glucose levels. Activating mutations in either gene (usually heterozygous but may also be biallelic) keep KATP channels inappropriately open even in the presence of high glucose levels. Around 95% of KATP channel mutations will respond to treatment with high doses of oral sulfonylurea drugs (most often glyburide in doses ranging from 0.5 to 2 mg/kg/day or more) that cause channel closure and restore insulin secretion. Importantly, these channels are also expressed widely in the brain, and consequently, individuals carrying these activating mutations can exhibit a spectrum of neurodevelopmental dysfunction where the severity of central nervous system features (from mild learning difficulties and ADHD to severe global developmental delay and unremitting seizures) correlates with the degree of mutation effect on channel function (71,72,73,74).
Most children with KATP channel diabetes have de novo spontaneous mutations that were not inherited, although a family history of NDM (in either parent and possibly other family members) is present in 10%–15% of cases. Sequencing will confirm presence of the causal variant in affected family members, who should receive a trial of high-dose sulfonylurea therapy (up to 2 mg/kg/day or more of glyburide), as it will often still be effective even in adults who have been treated with insulin only for decades. Parents of children with what appears to be a de novo variant must be counseled with the possibility of a second child being affected, because of the potential for germline mosaicism, in which the variant is present in the gonadal tissue (ovaries or testes) but not necessarily in the peripheral blood.
High-dose oral sulfonylurea therapy restores near-normal insulin secretion via non-KATP channel pathways induced by such factors as the incretin response to oral feeding (75). Although severe hypoglycemia is very rare, mild to moderate hypoglycemia can occur following meals lacking carbohydrates, since insulin secretion is prompted even in response to protein/fat and is no longer regulated by glucose-stimulated KATP channel closure (71,72,76,77,78,79,80). Clinical experience supports the notion that some improvement in neurologic sequelae follows the initiation of sulfonylureas (72,80). The degree of improvement likely depends on treatment starting as early as possible in brain development, but because dysfunction may occur early in utero, the potential for improvement may be limited (81).
Transient Neonatal Diabetes Mellitus Due to Overexpression of Imprinted Genes on Chromosome 6q24 (6q24-TNDM)
Children born with 6q24-TNDM generally weigh approximately 2,000 grams at birth, which is significantly underweight for a near-term neonate. Diabetes is typically diagnosed in the first week or so of life but is rarely detected within a few weeks later. Initial significant hyperglycemia requires treatment that is most often insulin, but some cases may respond to sulfonylurea therapy (82,83). Unlike with other forms of NDM characterized by insulin deficiency, DKA rarely occurs (84). This observation suggests a low level of beta cell function that gradually improves over several months, during which treatment can be slowly weaned off based on glucose measurements. Remission occurs in all cases, with a median of about 4 months of life, and very rare cases have been described as requiring treatment up to about one year. Some patients with this form of TNDM have macroglossia and/or umbilical hernia and rarely may have other features, such as neurodevelopmental delay, especially when the cause is related to a global defect of maternal methylation (sometimes due to recessive mutations in ZFP57, but often idiopathic). During remission, insulin production by beta cells may appear normal, and individuals have near-normal glucose metabolism. An uncertain, but large, fraction of cases will experience relapse of the diabetes around the time of puberty or sometimes as young adults. This relapse can sometimes be treated with oral hypoglycemic agents and may not necessarily require insulin (65,70).
The pathophysiology of 6q24-TNDM is poorly understood but results from at least three different mechanisms that all cause overexpression of the maternally imprinted genes at the chromosome 6q24 locus. Two of the more common mechanisms are spontaneous nonrecurrent paternal uniparental disomy or paternal duplications involving 6q24 that may be inherited. The remainder of cases are due to defects in maternal imprinting that are most often idiopathic but may rarely be due to biallelic mutations in ZFP57 and can involve other maternally imprinted loci in addition to 6q24.
Insulin Gene Mutations
Heterozygous dominant mutations in INS are the next most common cause of NDM after KATP channel and 6q24 defects. These mutations tend to occur in critical regions of the preproinsulin molecule (such as near disulfide bridges) and generally cause misfolding of the molecule and impair normal insulin processing and secretion, associated with ER stress and beta cell apoptosis (85,86). Individuals with INS mutations do not usually have other features, such as neurological dysfunction, but the diabetes is most often permanent and characterized by progressive beta cell deficiency requiring replacement doses of insulin, clinically managed similarly to type 1 diabetes. Diagnosis of diabetes may be delayed to age 6–12 months or later in some cases, while certain mutations causing less severe insulin deficiency can present later in life with a MODY-like phenotype (41,42,86,87,88).
Recessive mutations in INS causing NDM also have been reported (85). Compared to dominant mutations, homozygous recessive defects can cause a more severe form of insulin deficiency characterized by lower birth weight (-3.2 standard deviation [SD] score vs. -2.0 SD score) and earlier diagnosis (median diagnosis at age 1 week vs. 10 weeks) (85,89). Notably, some patients with certain recessively inherited INS mutations have a TNDM phenotype, and others may not manifest clinical diabetes until the teen years or later. These mutations may affect mRNA splicing or promoter function or alter the dibasic cleavage sites in proinsulin. Some heterozygous carriers of the recessive mutations may have MODY and are diagnosed later in life, suggesting less severe insulin deficiency (85). See Table 1, where INS is listed as a rare cause of a MODY phenotype.
Monogenic Autoimmune Neonatal Diabetes: IPEX and IPEX-Like Syndromes
Mutations in the X-linked FOXP3 gene can cause a syndrome termed IPEX, which stands for immune dysregulation, with polyendocrinopathy and enteropathy on the X chromosome. IPEX is characterized by intractable diarrhea, skin rashes, and immune dysregulation, and it often requires stem cell transplant; however, in some cases, the clinical manifestations are less severe. A similar phenotypic spectrum can result from defects in other genes that are essential for immune regulation, such as STAT3, LRBA, IL2RA, and others (90).
Other Causes of Neonatal Diabetes Mellitus
Table 2 lists the causes of NDM. In addition to the causes described above, defects in several genes (GATA6, GATA4, PDX1, PTF1A) can cause pancreatic agenesis or hypoplasia. The majority of the remaining causes are exceedingly rare recessive syndromes. EIF2AK3 mutations are the most common. These syndromes provide insight into the regulation of pancreas formation and insulin secretion in humans, and several reviews are available (41,42,53,88,91). No monogenic cause has yet been identified in a fraction of cases; several cases of unknown cause have Down syndrome (91).
Genetic Defects in Insulin Action: Insulin Receptor and Postreceptor Defects
The insulin receptor gene, INSR, codes for a heterotetramer protein complex consisting of two extracellular α subunits that bind insulin and two membrane-spanning β subunits with an intracellular tyrosine kinase domain that initiates a signaling cascade. Both subunits are encoded by the single gene, and variants in this gene may cause distinct syndromes, depending on the severity of disrupted insulin signaling. As many as 0.1%–1% of the population are suspected of having some variants that interfere with insulin action, but most can be overcome by increasing insulin secretion (92). Among the INSR variants that impair insulin action and cause diabetes are three syndromes: insulin resistance syndrome type A, Donohue syndrome, and Rabson-Mendenhall syndrome. All are rare, and no data on the precise incidence of these syndromes are available.
The insulin resistance syndrome type A is a relatively mild and more common form of insulin receptor abnormality defined by the presence of hyperandrogenism, acanthosis nigricans, and marked insulin resistance with glucose intolerance and hyperinsulinemia (93). Donohue syndrome is the most severe and rare form of insulin resistance and is due to near total absence of insulin receptor function. Affected individuals are characterized by severe intrauterine growth retardation, dysmorphic features, hyperinsulinemia, and abnormal glucose homeostasis. The abnormal glucose homeostasis is characterized by hyperglycemia during and after feeding, due to virtual absence of insulin action, which is essential to enable peripheral glucose uptake and utilization, as well as hypoglycemia during fasting due to the absence of insulin action, which is necessary to enable storage of glucose as glycogen (94). Patients with Rabson-Mendenhall syndrome have some residual insulin action and, therefore, somewhat different dysmorphic features, primarily involving abnormalities of teeth and nails, which are dysplastic and differ from those with Donohue syndrome. Pineal hyperplasia has been reported as well in Rabson-Mendenhall syndrome. Because the variant is not as severe, these patients are not at risk of dying early and, later in life, manifest the classic signs of insulin resistance, including acanthosis nigricans and hirsutism. Like patients with Donohue syndrome, those with Rabson-Mendenhall syndrome also manifest postprandial hyperglycemia and fasting hypoglycemia (94).
Gaps in Monogenic Diabetes Diagnosis
Multiple barriers arise on the journey to a diabetes diagnosis. A 2020 study on patient perspectives identified barriers at different levels: (a) patient-related (nature of MODY symptoms, perceived test utility, individual personality); (b) provider-related (provider awareness and knowledge, provider communication); and (c) healthcare system-related (cost of testing, access to knowledgeable providers, patient education, and support resources) (95).
Clinical Genome Resource (ClinGen) Monogenic Diabetes Expert Panels (MDEP) for Gene and Variant Curation
Next-generation sequencing has enabled high-throughput genetic testing for monogenic diabetes at a dramatically reduced cost, thereby increasing the number of patients who can benefit from a diagnosis of monogenic diabetes (along with other single gene disorders). However, this growth in testing has also increased the number of VUS identified and highlights the complexity of determining whether a variant in a relevant gene causes disease or is merely one of the millions of harmless variants in the genome.
To respond to growing awareness that many genetic variants were being interpreted differently with respect to disease causation by different labs, the ACMG and AMP developed and published a consensus statement on recommendations for “standards and guidelines for the interpretation of sequence variants.” This consensus statement provided guidance for using available clinical, family, population, and functional data on rare variants for classifying pathogenicity into five categories ranging from pathogenic to benign (96).
Subsequently, the National Institutes of Health-funded ClinGen (97) and ClinVar (98) resources have worked with members of the scientific and medical genetics and genomics communities to curate and publicize gene-disease relationship and gene-variant relationship, to continue refining the guidelines, and to develop disease- and gene-specific versions of the guidelines. This latter activity was accomplished by convening disease- and gene-specific expert panels (EPs) comprised of experts in the clinical and molecular diagnosis, management, genetics, and molecular biology of specific disease areas (99). Formation of these EPs enables members to inform curations not only through sharing of diverse expertise but also through the sharing of unpublished case data. Sharing of case data enables confident assertations to be made of the pathogenicity of a variant that may be only observed once in a single laboratory but several times in laboratories across the world with a consistent phenotype. A ClinGen EP for monogenic diabetes was convened in 2017 and approved for gene curation in 2019 (100) and variant curation in 2021 (101). ClinVar contains variant curation by both individual laboratories and by EPs; those curations completed by MDEPs, like all EP curations, are distinguished by a 3-star confidence rating and by indication of the U.S. Food and Drug Administration oversight of the EP process. MDEP specification development, variant curation, and deposit of curations into ClinVar are ongoing and expected to reduce some of the ambiguity involved in monogenic diabetes diagnosis and, thus, improve access to an accurate diagnosis for individuals with monogenic diabetes.
Concluding Remarks
Monogenic forms of diabetes are uncommon, but combined, they result in ~2%–3% of new cases of diabetes under age 35 years. Rates are 1:100,000 births for neonatal diabetes and rarer still for the syndromic forms of diabetes. Any clinician commonly seeing persons with diabetes will see people with monogenic diabetes. Mitochondrial causes of diabetes may be more common than appreciated because they are rarely considered. Defining the genetic bases for monogenic diabetes has shed light on the complexity of human pancreas formation and function, the regulation of insulin production and secretion, and important aspects of insulin action. This research has enabled a clearer understanding of the nature of dominantly and recessively inherited forms of early-onset diabetes. The use of rational treatments based on the pharmacology of the KATP channel has led to a spectacular improvement in the treatment of this rare form of KATP channel-mediated diabetes. This line of research also has led to an understanding that some of the same genes that cause monogenic diabetes are involved in the more common forms of what clinically appears to be type 1 diabetes and type 2 diabetes. Variable penetrance may affect gene variants thought previously to be highly penetrant, if not always disease-causing. Variations in onset or severity of diabetes occur within and between pedigrees and suggest the presence of modifying genes or at least compensation.
Correct diagnosis requires molecular confirmation, which enables precision treatment, avoids unnecessary use of insulin or other inappropriate agents, may lead to cascade testing of family members, and permits genetic counseling. In any autosomal dominant disease, the diagnosis in a proband should prompt the identification of affected members; in the case of MODY, any family member should be clinically assessed to decide if testing is needed. Next-generation sequencing has reduced the cost of genetic testing to the level of many routine tests, yet large barriers remain for insurance coverage, as well as diagnostic inertia, due to often convoluted approaches to ordering genetic testing. These discoveries are likely to have wide impact on clinical practice.
Resources
The following websites provide access to expert advice and assistance with possible mutation analysis in patients suspected of having monogenic forms of diabetes:
- https://monogenicdiabetes.uchicago.edu/, a monogenic diabetes registry located in the United States at the University of Chicago
- www.diabetesgenes.org, an information resource on genetic types of diabetes located in the United Kingdom at the University of Exeter
- https://www.ncbi.nlm.nih.gov/gtr/, Genetic Testing Registry, a compendium of sites and locations of mutational analyses performed for research or via CLIA-certified laboratories
- Individuals with 17q recurrent deletion syndrome may benefit from contact with the 17q12 Foundation (www.chromo17q12.org).
List of Abbreviations
- A-B+
autoantibody-negative and fasting C-peptide-positive
- A1c
glycated/glycosylated hemoglobin
- ACMG
American College of Medical Genetics and Genomics
- ADHD
attention-deficit hyperactivity disorder
- AMP
Association for Molecular Pathology
- ATP
adenosine triphosphate
- BMI
body mass index
- ClinGen
Clinical Genomic Resource
- CRP
C-reactive protein
- DKA
diabetic ketoacidosis
- EP
expert panel
- ER
endoplasmic reticulum
- FSGS
focal segmental glomerulosclerosis
- GAD
glutamic acid decarboxylase
- GCK
glucokinase gene
- GLP-1 RA
glucagon-like peptide-1 receptor agonist
- HNF
hepatocyte nuclear factor
- HR
hazard ratio
- hsCRP
high-sensitivity C-reactive protein
- INS
insulin gene
- INSR
insulin receptor gene
- IPEX
immune dysregulation, with polyendocrinopathy and enteropathy on the X chromosome
- KATP
ATP-regulated potassium (channel)
- MDEP
Monogenic Diabetes Expert Panel
- MELAS
Mitochondrial Encephalopathy With Lactic Acidosis and Stroke-Like Episodes
- MIDD
Maternally Inherited Diabetes and Deafness
- MODY
maturity-onset diabetes of the young
- NDM
neonatal diabetes mellitus
- PNDM
permanent neonatal diabetes mellitus
- SEARCH
SEARCH for Diabetes in Youth Study
- SGLT2
sodium-glucose cotransporter 2
- TNDM
transient neonatal diabetes mellitus
- TODAY
Treatment Options for Type 2 Diabetes in Adolescents and Youth Study
- tRNA
transfer ribonucleic acid
- VUS
variants of uncertain significance
Conversions
A1c: (% x 10.93) - 23.50 = mmol/mol
C-peptide: ng/mL x 0.333 = nmol/L
Glucose: mg/dL x 0.0555 = mmol/L
Funding
Maria V. Salguero’s training is funded by the Clinical Therapeutics Training Grant (T32GM00719). This work was supported by R01DK104942 (Philipson/Greeley/Naylor), U54DK118612 (Philipson/Naylor), DRTC P30DK020595 (Graeme Bell, PhD, University of Chicago), and ADA grant 7-22-ICTSPM-17 (Naylor).
Acknowledgment
This is an update of: Sperling MA, Garg A: Monogenic Forms of Diabetes. Chapter 7 in Diabetes in America, 3rd ed. Cowie CC, Casagrande SS, Menke A, Cissell MA, Eberhardt MS, Meigs JB, Gregg EW, Knowler WC, Barrett-Connor E, Becker DJ, Brancati FL, Boyko EJ, Herman WH, Howard BV, Narayan KMV, Rewers M, Fradkin JE, Eds. Bethesda, MD, National Institutes of Health, NIH Pub No, 17-1468, 2018, p 7.1–7.27
Article History
Received in final form on April 17, 2023.
References
- 1.
- Fajans SS, Bell GI, Polonsky KS: Molecular mechanisms and clinical pathophysiology of maturity-onset diabetes of the young. N Engl J Med 345:971–980, 2001 [PubMed: 11575290]
- 2.
- Fajans SS, Bell GI: MODY: history, genetics, pathophysiology, and clinical decision making. Diabetes Care 34:1878–1884, 2011 [PMC free article: PMC3142024] [PubMed: 21788644]
- 3.
- ClinGen: Clinical Genome Resource. Gene-Disease Validity: Curations by Monogenic Diabetes Expert Panel [article online], 2021. Available from https://search
.clinicalgenome .org/kb/affiliate/10016. Accessed 2 Feb 2022 - 4.
- Shepherd M, Shields B, Hammersley S, Hudson M, McDonald TJ, Colclough K, Oram RA, Knight B, Hyde C, Cox J, Mallam K, Moudiotis C, Smith R, Fraser B, Robertson S, Greene S, Ellard S, Pearson ER, Hattersley AT: Systematic population screening, using biomarkers and genetic testing, identifies 2.5% of the U.K. pediatric diabetes population with monogenic diabetes. Diabetes Care 39:1879–1888, 2016 [PMC free article: PMC5018394] [PubMed: 27271189]
- 5.
- Pihoker C, Gilliam LK, Ellard S, Dabelea D, Davis C, Dolan LM, Greenbaum CJ, Imperatore G, Lawrence JM, Marcovina SM, Mayer-Davis E, Rodriguez BL, Steck AK, Williams DE, Hattersley AT: Prevalence, characteristics and clinical diagnosis of maturity onset diabetes of the young due to mutations in HNF1A, HNF4A, and glucokinase: results from the SEARCH for Diabetes in Youth. J Clin Endocrinol Metab 98:4055–4062, 2013 [PMC free article: PMC3790621] [PubMed: 23771925]
- 6.
- Sperling MA, Garg A: Monogenic Forms of Diabetes. Chapter 7 in Diabetes in America, 3rd ed. Cowie CC, Casagrande SS, Menke A, Cissell MA, Eberhardt MS, Meigs JB, Gregg EW, Knowler WC, Barrett-Connor E, Becker DJ, Brancati FL, Boyko EJ, Herman WH, Howard BV, Narayan KMV, Rewers M, Fradkin JE, Eds. Bethesda, MD, National Institutes of Health, NIH Pub No. 17-1468, 2018, p. 7.1–7.27. Available from https://www
.ncbi.nlm .nih.gov/books/NBK567994/. Accessed 8 Dec 2023 - 7.
- Shields BM, Hicks S, Shepherd MH, Colclough K, Hattersley AT, Ellard S: Maturity-onset diabetes of the young (MODY): how many cases are we missing? Diabetologia 53:2504–2508, 2010 [PubMed: 20499044]
- 8.
- Kleinberger JW, Pollin TI: Undiagnosed MODY: time for action. Curr Diab Rep. 2015 Oct 12 [Epub] doi: 10.1007/s11892-015-0681-7 [PMC free article: PMC4785020] [PubMed: 26458381] [CrossRef]
- 9.
- Nkonge KM, Nkonge DK, Nkonge TN: The epidemiology, molecular pathogenesis, diagnosis, and treatment of maturity-onset diabetes of the young (MODY). Clin Diabetes Endocrinol. 2020 Nov 4 [Epub] doi: 10.1186/s40842-020-00112-5 [PMC free article: PMC7640483] [PubMed: 33292863] [CrossRef]
- 10.
- Shepherd MH, Shields BM, Hudson M, Pearson ER, Hyde C, Ellard S, Hattersley AT, Patel KA: A UK nationwide prospective study of treatment change in MODY: genetic subtype and clinical characteristics predict optimal glycaemic control after discontinuing insulin and metformin. Diabetologia 61:2520–2527, 2018 [PMC free article: PMC6223847] [PubMed: 30229274]
- 11.
- Matsha TE, Raghubeer S, Tshivhase AM, Davids SF, Hon GM, Bjorkhaug L, Erasmus RT: Incidence of HNF1A and GCK MODY variants in a South African population. Appl Clin Genet 13:209–219, 2020 [PMC free article: PMC7754620] [PubMed: 33363396]
- 12.
- Patel KA, Ozbek MN, Yildiz M, Guran T, Kocyigit C, Acar S, Siklar Z, Atar M, Colclough K, Houghton J, Johnson MB, Ellard S, Flanagan SE, Cizmecioglu F, Berberoglu M, Demir K, Catli G, Bas S, Akcay T, Demirbilek H, Weedon MN, Hattersley AT: Systematic genetic testing for recessively inherited monogenic diabetes: a cross-sectional study in paediatric diabetes clinics. Diabetologia 65:336–342, 2022 [PMC free article: PMC8741690] [PubMed: 34686905]
- 13.
- Kleinberger JW, Copeland KC, Gandica RG, Haymond MW, Levitsky LL, Linder B, Shuldiner AR, Tollefsen S, White NH, Pollin TI: Monogenic diabetes in overweight and obese youth diagnosed with type 2 diabetes: the TODAY clinical trial. Genet Med 20:583–590, 2018 [PMC free article: PMC5955780] [PubMed: 29758564]
- 14.
- Bowden T, Letourneau-Freiberg L, Kandasamy B, Sanyoura M, Tian P, Harris A, Bell G, Philipson L, Naylor R, Greeley SA: Insight on diagnosis and treatment from over a decade of research through the University of Chicago Monogenic Diabetes Registry. Front Clin Diabetes Healthc. 2021 Nov 5 [Epub] doi: 10.3389/fcdhc.2021.735548 [PMC free article: PMC9629510] [PubMed: 36330312] [CrossRef]
- 15.
- Steele AM, Wensley KJ, Ellard S, Murphy R, Shepherd M, Colclough K, Hattersley AT, Shields BM: Use of HbA1c in the identification of patients with hyperglycaemia caused by a glucokinase mutation: observational case control studies. PLoS One. 2013 Jun 14 [Epub] doi: 10.1371/journal.pone.0065326 [PMC free article: PMC3683003] [PubMed: 23799006] [CrossRef]
- 16.
- Todd JN, Kleinberger JW, Zhang H, Srinivasan S, Tollefsen SE, Levitsky LL, Levitt Katz LE, Tryggestad JB, Bacha F, Imperatore G, Lawrence JM, Pihoker C, Divers J, Flannick J, Dabelea D, Florez JC, Pollin TI: Monogenic diabetes in youth with presumed type 2 diabetes: results from the Progress in Diabetes Genetics in Youth (ProDiGY) Collaboration. Diabetes Care 44:2312–2319, 2021 [PMC free article: PMC8929184] [PubMed: 34362814]
- 17.
- Patel KA, Oram RA, Flanagan SE, De Franco E, Colclough K, Shepherd M, Ellard S, Weedon MN, Hattersley AT: Type 1 diabetes genetic risk score: a novel tool to discriminate monogenic and type 1 diabetes. Diabetes 65:2094–2099, 2016 [PMC free article: PMC4920219] [PubMed: 27207547]
- 18.
- Jones AG, Hattersley AT: The clinical utility of C-peptide measurement in the care of patients with diabetes. Diabet Med 30:803–817, 2013 [PMC free article: PMC3748788] [PubMed: 23413806]
- 19.
- Bowman P, McDonald TJ, Shields BM, Knight BA, Hattersley AT: Validation of a single-sample urinary C-peptide creatinine ratio as a reproducible alternative to serum C-peptide in patients with type 2 diabetes. Diabet Med 29:90–93, 2012 [PubMed: 21883437]
- 20.
- Vionnet N, Stoffel M, Takeda J, Yasuda K, Bell GI, Zouali H, Lesage S, Velho G, Iris F, Passa P, Froguel P, Cohen D: Nonsense mutation in the glucokinase gene causes early-onset non-insulin-dependent diabetes mellitus. Nature 356:721–722, 1992 [PubMed: 1570017]
- 21.
- Froguel P, Vaxillaire M, Sun F, Velho G, Zouali H, Butel MO, Lesage S, Vionnet N, Clement K, Fougerousse F, Tanizama Y, Weissenbach J, Beckmann JS, Lathrop GM, Passa P, Permutt MA, Cohen D: Close linkage of glucokinase locus on chromosome 7p to early-onset non-insulin-dependent diabetes mellitus. Nature 356:162–164, 1992 [PubMed: 1545870]
- 22.
- Sanyoura M, Letourneau L, Knight Johnson AE, Del Gaudio D, Greeley SAW, Philipson LH, Naylor RN: GCK-MODY in the US Monogenic Diabetes Registry: description of 27 unpublished variants. Diabetes Res Clin Pract 151:231–236, 2019 [PMC free article: PMC6544496] [PubMed: 31063852]
- 23.
- Hattersley AT, Patel KA: Precision diabetes: learning from monogenic diabetes. Diabetologia 60:769–777, 2017 [PMC free article: PMC5907633] [PubMed: 28314945]
- 24.
- Naylor R, Knight Johnson A, del Gaudio D: Maturity-Onset Diabetes of the Young Overview. In GeneReviews(®) [Internet]. Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Gripp KW, Mirzaa GM Amemiya A, Eds. Seattle, WA, University of Washington, 2018. Available from https://www
.ncbi.nlm .nih.gov/books/NBK500456/ [PubMed: 29792621] - 25.
- Chakera AJ, Steele AM, Gloyn AL, Shepherd MH, Shields B, Ellard S, Hattersley AT: Recognition and management of individuals with hyperglycemia because of a heterozygous glucokinase mutation. Diabetes Care 38:1383–1392, 2015 [PubMed: 26106223]
- 26.
- Broome DT, Pantalone KM, Kashyap SR, Philipson LH: Approach to the patient with MODY-monogenic diabetes. J Clin Endocrinol Metab 106:237–250, 2021 [PMC free article: PMC7765647] [PubMed: 33034350]
- 27.
- Hohendorff J, Szopa M, Skupien J, Kapusta M, Zapala B, Platek T, Mrozinska S, Parpan T, Glodzik W, Ludwig-Galezowska A, Kiec-Wilk B, Klupa T, Malecki MT: A single dose of dapagliflozin, an SGLT-2 inhibitor, induces higher glycosuria in GCK- and HNF1A-MODY than in type 2 diabetes mellitus. Endocrine 57:272–279, 2017 [PMC free article: PMC5511327] [PubMed: 28593615]
- 28.
- Rudland VL: Diagnosis and management of glucokinase monogenic diabetes in pregnancy: current perspectives. Diabetes Metab Syndr Obes 12:1081–1089, 2019 [PMC free article: PMC6628087] [PubMed: 31372018]
- 29.
- Chakera AJ, Carleton VL, Ellard S, Wong J, Yue DK, Pinner J, Hattersley AT, Ross GP: Antenatal diagnosis of fetal genotype determines if maternal hyperglycemia due to a glucokinase mutation requires treatment. Diabetes Care 35:1832–1834, 2012 [PMC free article: PMC3425005] [PubMed: 22773699]
- 30.
- Vaxillaire M, Boccio V, Philippi A, Vigouroux C, Terwilliger J, Passa P, Beckmann JS, Velho G, Lathrop GM, Froguel P: A gene for maturity onset diabetes of the young (MODY) maps to chromosome 12q. Nat Genet 9:418–423, 1995 [PubMed: 7795649]
- 31.
- Yamagata K, Oda N, Kaisaki PJ, Menzel S, Furuta H, Vaxillaire M, Southam L, Cox RD, Lathrop GM, Boriraj VV, Chen X, Cox NJ, Oda Y, Yano H, Le Beau MM, Yamada S, Nishigori H, Takeda J, Fajans SS, Hattersley AT, Iwasaki N, Hansen T, Pedersen O, Polonsky KS, Turner RC, Velho G, Chevre J-C, Froguel P, Bell GI: Mutations in the hepatocyte nuclear factor-1alpha gene in maturity-onset diabetes of the young (MODY3). Nature 384:455–458, 1996 [PubMed: 8945470]
- 32.
- Bellanné-Chantelot C, Carette C, Riveline JP, Valéro R, Gautier JF, Larger E, Reznik Y, Ducluzeau PH, Sola A, Hartemann-Heurtier A, Lecomte P, Chaillous L, Laloi-Michelin M, Wilhem JM, Cuny P, Duron F, Guerci B, Jeandidier N, Mosnier-Pudar H, Assayag M, Dubois-Laforgue D, Velho G, Timsit J: The type and the position of HNF1A mutation modulate age at diagnosis of diabetes in patients with maturity-onset diabetes of the young (MODY)-3. Diabetes 57:503–508, 2008 [PubMed: 18003757]
- 33.
- Hoffman LS, Fox TJ, Anastasopoulou C, Jialal I: Maturity Onset Diabetes in the Young. In StatPearls [Internet]. Treasure Island, FL, StatPearls Publishing, 2022. Available from https://www
.ncbi.nlm .nih.gov/books/NBK532900/ [PubMed: 30422495] - 34.
- McDonald TJ, Shields BM, Lawry J, Owen KR, Gloyn AL, Ellard S, Hattersley AT: High-sensitivity CRP discriminates HNF1A-MODY from other subtypes of diabetes. Diabetes Care 34:1860–1862, 2011 [PMC free article: PMC3142017] [PubMed: 21700917]
- 35.
- Ostoft SH, Bagger JI, Hansen T, Pedersen O, Faber J, Holst JJ, Knop FK, Vilsboll T: Glucose-lowering effects and low risk of hypoglycemia in patients with maturity-onset diabetes of the young when treated with a GLP-1 receptor agonist: a double-blind, randomized, crossover trial. Diabetes Care 37:1797–1805, 2014 [PubMed: 24929431]
- 36.
- Flannick J, Beer NL, Bick AG, Agarwala V, Molnes J, Gupta N, Burtt NP, Florez JC, Meigs JB, Taylor H, Lyssenko V, Irgens H, Fox E, Burslem F, Johansson S, Brosnan MJ, Trimmer JK, Newton-Cheh C, Tuomi T, Molven A, Wilson JG, O’Donnell CJ, Kathiresan S, Hirschhorn JN, Njølstad PR, Rolph T, Seidman JG, Gabriel S, Cox DR, Seidman CE, Groop L, Altshuler D: Assessing the phenotypic effects in the general population of rare variants in genes for a dominant Mendelian form of diabetes. Nat Genet 45:1380–1385, 2013 [PMC free article: PMC4051627] [PubMed: 24097065]
- 37.
- Bell GI, Xiang KS, Newman MV, Wu SH, Wright LG, Fajans SS, Spielman RS, Cox NJ: Gene for non-insulin-dependent diabetes mellitus (maturity-onset diabetes of the young subtype) is linked to DNA polymorphism on human chromosome 20q. Proc Natl Acad Sci U S A 88:1484–1488, 1991 [PMC free article: PMC51043] [PubMed: 1899928]
- 38.
- Yamagata K, Furuta H, Oda N, Kaisaki PJ, Menzel S, Cox NJ, Fajans SS, Signorini S, Stoffel M, Bell GI: Mutations in the hepatocyte nuclear factor-4alpha gene in maturity-onset diabetes of the young (MODY1). Nature 384:458–460, 1996 [PubMed: 8945471]
- 39.
- Pearson ER, Boj SF, Steele AM, Barrett T, Stals K, Shield JP, Ellard S, Ferrer J, Hattersley AT: Macrosomia and hyperinsulinaemic hypoglycaemia in patients with heterozygous mutations in the HNF4A gene. PLoS Med. 2007 Apr 4 [Epub] doi: 10.1371/journal.pmed.0040118 [PMC free article: PMC1845156] [PubMed: 17407387] [CrossRef]
- 40.
- Pearson ER, Pruhova S, Tack CJ, Johansen A, Castleden HA, Lumb PJ, Wierzbicki AS, Clark PM, Lebl J, Pedersen O, Ellard S, Hansen T, Hattersley AT: Molecular genetics and phenotypic characteristics of MODY caused by hepatocyte nuclear factor 4alpha mutations in a large European collection. Diabetologia 48:878–885, 2005 [PubMed: 15830177]
- 41.
- Murphy R, Ellard S, Hattersley AT: Clinical implications of a molecular genetic classification of monogenic beta-cell diabetes. Nat Clin Pract Endocrinol Metab 4:200–213, 2008 [PubMed: 18301398]
- 42.
- Steck AK, Winter WE: Review on monogenic diabetes. Curr Opin Endocrinol Diabetes Obes 18:252–258, 2011 [PubMed: 21844708]
- 43.
- Horikawa Y, Iwasaki N, Hara M, Furuta H, Hinokio Y, Cockburn BN, Lindner T, Yamagata K, Ogata M, Tomonaga O, Kuroki H, Kasahara T, Iwamoto Y, Bell GI: Mutation in hepatocyte nuclear factor-1 beta gene (TCF2) associated with MODY. Nat Genet 17:384–385, 1997 [PubMed: 9398836]
- 44.
- Pearson ER, Badman MK, Lockwood CR, Clark PM, Ellard S, Bingham C, Hattersley AT: Contrasting diabetes phenotypes associated with hepatocyte nuclear factor-1alpha and -1beta mutations. Diabetes Care 27:1102–1107, 2004 [PubMed: 15111528]
- 45.
- Edghill EL, Stals K, Oram RA, Shepherd MH, Hattersley AT, Ellard S: HNF1B deletions in patients with young-onset diabetes but no known renal disease. Diabet Med 30:114–117, 2013 [PubMed: 22587559]
- 46.
- Lindner TH, Njølstad PR, Horikawa Y, Bostad L, Bell GI, Sovik O: A novel syndrome of diabetes mellitus, renal dysfunction and genital malformation associated with a partial deletion of the pseudo-POU domain of hepatocyte nuclear factor-1β. Hum Mol Genet 8:2001–2008, 1999 [PubMed: 10484768]
- 47.
- Ray EC, Boyd-Shiwarski CR, Liu P, Novacic D, Cassiman D: SGLT2 inhibitors for treatment of refractory hypomagnesemia: a case report of 3 patients. Kidney Med 2:359–364, 2020 [PMC free article: PMC7380441] [PubMed: 32734255]
- 48.
- Clissold RL, Shaw-Smith C, Turnpenny P, Bunce B, Bockenhauer D, Kerecuk L, Waller S, Bowman P, Ford T, Ellard S, Hattersley AT, Bingham C: Chromosome 17q12 microdeletions but not intragenic HNF1B mutations link developmental kidney disease and psychiatric disorder. Kidney Int 90:203–211, 2016 [PMC free article: PMC4915913] [PubMed: 27234567]
- 49.
- Dubois-Laforgue D, Bellanné-Chantelot C, Charles P, Jacquette A, Larger E, Ciangura C, Saint-Martin C, Rastel C, Keren B, Timsit J: Intellectual disability in patients with MODY due to hepatocyte nuclear factor 1B (HNF1B) molecular defects. Diabetes Metab 43:89–92, 2017 [PubMed: 27838256]
- 50.
- Clissold RL, Ashfield B, Burrage J, Hannon E, Bingham C, Mill J, Hattersley A, Dempster EL: Genome-wide methylomic analysis in individuals with HNF1B intragenic mutation and 17q12 microdeletion. Clinical Epigenetics. 2018 Jul 18 [Epub] doi: 10.1186/s13148-018-0530-z [PMC free article: PMC6052548] [PubMed: 30021660] [CrossRef]
- 51.
- Heidet L, Decramer S, Pawtowski A, Morinière V, Bandin F, Knebelmann B, Lebre AS, Faguer S, Guigonis V, Antignac C, Salomon R: Spectrum of HNF1B mutations in a large cohort of patients who harbor renal diseases. Clin J Am Soc Nephrol 5:1079–1090, 2010 [PMC free article: PMC2879303] [PubMed: 20378641]
- 52.
- Moreno-De-Luca D; SGENE Consortium; Mulle JG; Simons Simplex Collection Genetics Consortium; Kaminsky EB, Sanders SJ; GeneSTAR; Myers SM, Adam MP, Pakula AT, Eisenhauer NJ, Uhas K, Weik L, Guy L, Care ME, Morel CF, Boni C, Salbert BA, Chandrareddy A, Demmer LA, Chow EWC, Surti U, Aradhya S, Pickering DL, Golden DM, Sanger WG, Aston E, Brothman AR, Gliem TJ, Thorland EC, Ackley T, Iyer R, Huang S, Barber JC, Crolla JA, Warren ST, Martin CL, Ledbetter DH: Deletion 17q12 is a recurrent copy number variant that confers high risk of autism and schizophrenia. Am J Hum Genet 87:618–630, 2010 [PMC free article: PMC2978962] [PubMed: 21055719]
- 53.
- Zhang H, Colclough K, Gloyn AL, Pollin TI: Monogenic diabetes: a gateway to precision medicine in diabetes. J Clin Invest. 2021 Feb 1 [Epub] doi: 10.1172/jci142244 [PMC free article: PMC7843214] [PubMed: 33529164] [CrossRef]
- 54.
- Murphy R, Turnbull DM, Walker M, Hattersley AT: Clinical features, diagnosis and management of maternally inherited diabetes and deafness (MIDD) associated with the 3243A>G mitochondrial point mutation. Diabet Med 25:383–399, 2008 [PubMed: 18294221]
- 55.
- Pang L, Colclough KC, Shepherd MH, McLean J, Pearson ER, Ellard S, Hattersley AT, Shields BM: Improvements in awareness and testing have led to a threefold increase over 10 years in the identification of monogenic diabetes in the U.K. Diabetes Care 45:642–649, 2022 [PMC free article: PMC7612472] [PubMed: 35061023]
- 56.
- Ji K, Lin Y, Xu X, Wang W, Wang D, Zhang C, Li W, Zhao Y, Yan C: MELAS-associated m.5541C>T mutation caused instability of mitochondrial tRNA(Trp) and remarkable mitochondrial dysfunction. J Med Genet 59:79–87, 2022 [PubMed: 33208382]
- 57.
- Guerrero-Molina MP, Morales-Conejo M, Delmiro A, Morán M, Domínguez-González C, Arranz-Canales E, Ramos-González A, Arenas J, Martín MA, González de la Aleja J: High-dose oral glutamine supplementation reduces elevated glutamate levels in cerebrospinal fluid in patients with mitochondrial encephalomyopathy, lactic acidosis and stroke-like episodes syndrome. Eur J Neurol 30:538–547, 2023 [PubMed: 36334048]
- 58.
- Barcelos I, Shadiack E, Ganetzky RD, Falk MJ: Mitochondrial medicine therapies: rationale, evidence, and dosing guidelines. Curr Opin Pediatr 32:707–718, 2020 [PMC free article: PMC7774245] [PubMed: 33105273]
- 59.
- Urano F: Wolfram syndrome: diagnosis, management, and treatment. Curr Diab Rep. 2016 Jan 7 [Epub] doi: 10.1007/s11892-015-0702-6 [PMC free article: PMC4705145] [PubMed: 26742931] [CrossRef]
- 60.
- Abreu D, Urano F: Current landscape of treatments for Wolfram syndrome. Trends Pharmacol Sci 40:711–714, 2019 [PMC free article: PMC7547529] [PubMed: 31420094]
- 61.
- Garg A: Clinical review#: Lipodystrophies: genetic and acquired body fat disorders. J Clin Endocrinol Metab 96:3313–3325, 2011 [PMC free article: PMC7673254] [PubMed: 21865368]
- 62.
- Kanakatti Shankar R, Pihoker C, Dolan LM, Standiford D, Badaru A, Dabelea D, Rodriguez B, Black MH, Imperatore G, Hattersley A, Ellard S, Gilliam LK: Permanent neonatal diabetes mellitus: prevalence and genetic diagnosis in the SEARCH for Diabetes in Youth Study. Pediatr Diabetes 14:174–180, 2013 [PMC free article: PMC4101463] [PubMed: 23050777]
- 63.
- Habeb AM, Al-Magamsi MS, Eid IM, Ali MI, Hattersley AT, Hussain K, Ellard S: Incidence, genetics, and clinical phenotype of permanent neonatal diabetes mellitus in northwest Saudi Arabia. Pediatr Diabetes 13:499–505, 2012 [PubMed: 22060631]
- 64.
- De Franco E, Flanagan SE, Houghton JA, Lango Allen H, Mackay DJ, Temple IK, Ellard S, Hattersley AT: The effect of early, comprehensive genomic testing on clinical care in neonatal diabetes: an international cohort study. Lancet 386:957–963, 2015 [PMC free article: PMC4772451] [PubMed: 26231457]
- 65.
- Mackay DJ, Temple IK: Transient neonatal diabetes mellitus type 1. Am J Med Genet C Semin Med Genet 154C:335–342, 2010 [PubMed: 20803656]
- 66.
- Sperling MA: Neonatal diabetes mellitus: from understudy to center stage. Curr Opin Pediatr 17:512–518, 2005 [PubMed: 16012265]
- 67.
- Russo L, Iafusco D, Brescianini S, Nocerino V, Bizzarri C, Toni S, Cerutti F, Monciotti C, Pesavento R, Iughetti L, Bernardini L, Bonfanti R, Gargantini L, Vanelli M, Aguilar-Bryan L, Stazi MA, Grasso V, Colombo C, Barbetti F: Permanent diabetes during the first year of life: multiple gene screening in 54 patients. Diabetologia 54:1693–1701, 2011 [PMC free article: PMC3110270] [PubMed: 21544516]
- 68.
- Ma D, Shield JP, Dean W, Leclerc I, Knauf C, Burcelin RR, Rutter GA, Kelsey G: Impaired glucose homeostasis in transgenic mice expressing the human transient neonatal diabetes mellitus locus, TNDM. J Clin Invest 114:339–348, 2004 [PMC free article: PMC484972] [PubMed: 15286800]
- 69.
- Hattersley AT: Unlocking the secrets of the pancreatic beta cell: man and mouse provide the key. J Clin Invest 114:314–316, 2004 [PMC free article: PMC484987] [PubMed: 15286795]
- 70.
- Mackay DJ, Callaway JL, Marks SM, White HE, Acerini CL, Boonen SE, Dayanikli P, Firth HV, Goodship JA, Haemers AP, Hahnemann JM, Kordonouri O, Masoud AF, Oestergaard E, Storr J, Ellard S, Hattersley AT, Robinson DO, Temple IK: Hypomethylation of multiple imprinted loci in individuals with transient neonatal diabetes is associated with mutations in ZFP57. Nat Genet 40:949–951, 2008 [PubMed: 18622393]
- 71.
- Clark RH, McTaggart JS, Webster R, Mannikko R, Iberl M, Sim XL, Rorsman P, Glitsch M, Beeson D, Ashcroft FM: Muscle dysfunction caused by a KATP channel mutation in neonatal diabetes is neuronal in origin. Science 329:458–461, 2010 [PMC free article: PMC5890903] [PubMed: 20595581]
- 72.
- Busiah K, Drunat S, Vaivre-Douret L, Bonnefond A, Simon A, Flechtner I, Gérard B, Pouvreau N, Elie C, Nimri R, De Vries L, Tubiana-Rufi N, Metz C, Bertrand AM, Nivot-Adamiak S, de Kerdanet M, Stuckens C, Jennane F, Souchon PF, Le Tallec C, Désirée C, Pereira S, Dechaume A, Robert JJ, Phillip M, Scharfmann R, Czernichow P, Froguel P, Vaxillaire M, Polak M, Cavé H; French NDM Study Group: Neuropsychological dysfunction and developmental defects associated with genetic changes in infants with neonatal diabetes mellitus: a prospective cohort study [corrected]. Lancet Diabetes Endocrinol 1:199–207, 2013 [PubMed: 24622368]
- 73.
- Bowman P, Hattersley AT, Knight BA, Broadbridge E, Pettit L, Reville M, Flanagan SE, Shepherd MH, Ford TJ, Tonks J: Neuropsychological impairments in children with KCNJ11 neonatal diabetes. Diabet Med 34:1171–1173, 2017 [PubMed: 28477417]
- 74.
- Carmody D, Pastore AN, Landmeier KA, Letourneau LR, Martin R, Hwang JL, Naylor RN, Hunter SJ, Msall ME, Philipson LH, Scott MN, Greeley SA: Patients with KCNJ11-related diabetes frequently have neuropsychological impairments compared with sibling controls. Diabet Med 33:1380–1386, 2016 [PMC free article: PMC5654490] [PubMed: 27223594]
- 75.
- Pearson ER, Flechtner I, Njølstad PR, Malecki MT, Flanagan SE, Larkin B, Ashcroft FM, Klimes I, Codner E, Iotova V, Slingerland AS, Shield J, Robert JJ, Holst JJ, Clark PM, Ellard S, Søvik O, Polak M, Hattersley AT; Neonatal Diabetes International Collaborative Group: Switching from insulin to oral sulfonylureas in patients with diabetes due to Kir6.2 mutations. N Engl J Med 355:467–477, 2006 [PubMed: 16885550]
- 76.
- Lanning MS, Carmody D, Szczerbinski L, Letourneau LR, Naylor RN, Greeley SAW: Hypoglycemia in sulfonylurea-treated KCNJ11-neonatal diabetes: mild-moderate symptomatic episodes occur infrequently but none involving unconsciousness or seizures. Pediatr Diabetes 19:393–397, 2018 [PMC free article: PMC5918230] [PubMed: 29205704]
- 77.
- Bowman P, McDonald TJ, Knight BA, Flanagan SE, Leveridge M, Spaull SR, Shields BM, Hammersley S, Shepherd MH, Andrews RC, Patel KA, Hattersley AT: Patterns of postmeal insulin secretion in individuals with sulfonylurea-treated KCNJ11 neonatal diabetes show predominance of non-K(ATP)-channel pathways. BMJ Open Diabetes Res Care. 2019 Dec 18 [Epub] doi: 10.1136/bmjdrc-2019-000721 [PMC free article: PMC6936449] [PubMed: 31908791] [CrossRef]
- 78.
- Kooptiwut S, Plengvidhya N, Chukijrungroat T, Sujjitjoon J, Semprasert N, Furuta H, Yenchitsomanus PT: Defective PAX4 R192H transcriptional repressor activities associated with maturity onset diabetes of the young and early onset-age of type 2 diabetes. J Diabetes Complications 26:343–347, 2012 [PubMed: 22521316]
- 79.
- Babenko AP, Polak M, Cavé H, Busiah K, Czernichow P, Scharfmann R, Bryan J, Aguilar-Bryan L, Vaxillaire M, Froguel P: Activating mutations in the ABCC8 gene in neonatal diabetes mellitus. N Engl J Med 355:456–466, 2006 [PubMed: 16885549]
- 80.
- Carmody D, Philipson L: Neonatal diabetes: the brain comes into focus. Lancet Diabetes Endocrinol 1:167–168, 2013 [PubMed: 24622356]
- 81.
- Dalgin G, Tryba AK, Cohen AP, Park SY, Philipson LH, Greeley SAW, Garcia AJ, 3rd: Developmental defects and impaired network excitability in a cerebral organoid model of KCNJ11 p.V59M-related neonatal diabetes. Sci Rep. 2021 Nov 3 [Epub] doi: 10.1038/s41598-021-00939-7 [PMC free article: PMC8566525] [PubMed: 34732776] [CrossRef]
- 82.
- Carmody D, Bell CD, Hwang JL, Dickens JT, Sima DI, Felipe DL, Zimmer CA, Davis AO, Kotlyarevska K, Naylor RN, Philipson LH, Greeley SA: Sulfonylurea treatment before genetic testing in neonatal diabetes: pros and cons. J Clin Endocrinol Metab 99:E2709–E2714, 2014 [PMC free article: PMC4255121] [PubMed: 25238204]
- 83.
- Garcin L, Kariyawasam D, Busiah K, Fauret-Amsellem AL, Le Bourgeois F, Vaivre-Douret L, Cavé H, Polak M, Beltrand J: Successful off-label sulfonylurea treatment of neonatal diabetes mellitus due to chromosome 6 abnormalities. Pediatr Diabetes 19:663–669, 2018 [PubMed: 29504184]
- 84.
- Letourneau LR, Carmody D, Wroblewski K, Denson AM, Sanyoura M, Naylor RN, Philipson LH, Greeley SAW: Diabetes presentation in infancy: high risk of diabetic ketoacidosis. Diabetes Care 40:e147–e148, 2017 [PMC free article: PMC5606305] [PubMed: 28779000]
- 85.
- Garin I, Edghill EL, Akerman I, Rubio-Cabezas O, Rica I, Locke JM, Maestro MA, Alshaikh A, Bundak R, del Castillo G, Deeb A, Deiss D, Fernandez JM, Godbole K, Hussain K, O’Connell M, Klupa T, Kolouskova S, Mohsin F, Perlman K, Sumnik Z, Rial JM, Ugarte E, Vasanthi T, Johnstone K, Flanagan SE, Martínez R, Castano C, Patch AM, Fernandez-Rebollo E, Raile K, Morgan N, Harries LW, Castano L, Ellard S, Ferrer J, Perez de Nanclares G, Hattersley AT: Recessive mutations in the INS gene result in neonatal diabetes through reduced insulin biosynthesis. Proc Natl Acad Sci U S A 107:3105–3110, 2010 [PMC free article: PMC2840338] [PubMed: 20133622]
- 86.
- Stoy J, Edghill EL, Flanagan SE, Ye H, Paz VP, Pluzhnikov A, Below JE, Hayes MG, Cox NJ, Lipkind GM, Lipton RB, Greeley SA, Patch AM, Ellard S, Steiner DF, Hattersley AT, Philipson LH, Bell GI: Insulin gene mutations as a cause of permanent neonatal diabetes. Proc Natl Acad Sci U S A 104:15040–15044, 2007 [PMC free article: PMC1986609] [PubMed: 17855560]
- 87.
- Stoy J, De Franco E, Ye H, Park SY, Bell GI, Hattersley AT: In celebration of a century with insulin—update of insulin gene mutations in diabetes. Mol Metab. 2021 Jun 24 [Epub] doi: 10.1016/j.molmet.2021.101280 [PMC free article: PMC8513141] [PubMed: 34174481] [CrossRef]
- 88.
- Rubio-Cabezas O, Hattersley AT, Njølstad PR, Mlynarski W, Ellard S, White N, Chi DV, Craig ME; International Society for Pediatric and Adolescent Diabetes: ISPAD Clinical Practice Consensus Guidelines 2014. The diagnosis and management of monogenic diabetes in children and adolescents. Pediatr Diabetes 15(Suppl 20):47–64, 2014 [PubMed: 25182307]
- 89.
- Carmody D, Park SY, Ye H, Perrone ME, Alkorta-Aranburu G, Highland HM, Hanis CL, Philipson LH, Bell GI, Greeley SA: Continued lessons from the INS gene: an intronic mutation causing diabetes through a novel mechanism. J Med Genet 52:612–616, 2015 [PMC free article: PMC4744477] [PubMed: 26101329]
- 90.
- Strakova V, Elblova L, Johnson MB, Dusatkova P, Obermannova B, Petruzelkova L, Kolouskova S, Snajderova M, Fronkova E, Svaton M, Lebl J, Hattersley AT, Sumnik Z, Pruhova S: Screening of monogenic autoimmune diabetes among children with type 1 diabetes and multiple autoimmune diseases: is it worth doing? J Pediatr Endocrinol Metab 32:1147–1153, 2019 [PubMed: 31483759]
- 91.
- Johnson MB, De Franco E, Greeley SAW, Letourneau LR, Gillespie KM, Wakeling MN, Ellard S, Flanagan SE, Patel KA, Hattersley AT: Trisomy 21 is a cause of permanent neonatal diabetes that is autoimmune but not HLA associated. Diabetes 68:1528–1535, 2019 [PMC free article: PMC6609990] [PubMed: 30962220]
- 92.
- Taylor SI: Lilly Lecture: molecular mechanisms of insulin resistance. Lessons from patients with mutations in the insulin-receptor gene. Diabetes 41:1473–1490, 1992 [PubMed: 1327927]
- 93.
- Kahn CR, Flier JS, Bar RS, Archer JA, Gorden P, Martin MM, Roth J: The syndromes of insulin resistance and acanthosis nigricans. Insulin-receptor disorders in man. N Engl J Med 294:739–745, 1976 [PubMed: 176581]
- 94.
- Longo N, Wang Y, Smith SA, Langley SD, DiMeglio LA, Giannella-Neto D: Genotype-phenotype correlation in inherited severe insulin resistance. Hum Mol Genet 11:1465–1475, 2002 [PubMed: 12023989]
- 95.
- Guan Y, Maloney KA, Pollin TI: Patient perspectives on the diagnostic journey to a monogenic diabetes diagnosis: barriers and facilitators. J Genet Couns 29:1106–1113, 2020 [PMC free article: PMC7486254] [PubMed: 32162750]
- 96.
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL; ACMG Laboratory Quality Assurance Committee: Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 17:405–424, 2015 [PMC free article: PMC4544753] [PubMed: 25741868]
- 97.
- Rehm HL, Berg JS, Brooks LD, Bustamante CD, Evans JP, Landrum MJ, Ledbetter DH, Maglott DR, Martin CL, Nussbaum RL, Plon SE, Ramos EM, Sherry ST, Watson MS: ClinGen—the Clinical Genome Resource. N Engl J Med 372:2235–2242, 2015 [PMC free article: PMC4474187] [PubMed: 26014595]
- 98.
- Landrum MJ, Chitipiralla S, Brown GR, Chen C, Gu B, Hart J, Hoffman D, Jang W, Kaur K, Liu C, Lyoshin V, Maddipatla Z, Maiti R, Mitchell J, O’Leary N, Riley GR, Shi W, Zhou G, Schneider V, Maglott D, Holmes JB, Kattman BL: ClinVar: improvements to accessing data. Nucleic Acids Res 48:D835–D844, 2020 [PMC free article: PMC6943040] [PubMed: 31777943]
- 99.
- Rivera-Munoz EA, Milko LV, Harrison SM, Azzariti DR, Kurtz CL, Lee K, Mester JL, Weaver MA, Currey E, Craigen W, Eng C, Funke B, Hegde M, Hershberger RE, Mao R, Steiner RD, Vincent LM, Martin CL, Plon SE, Ramos E, Rehm HL, Watson M, Berg JS: ClinGen Variant Curation Expert Panel experiences and standardized processes for disease and gene-level specification of the ACMG/AMP guidelines for sequence variant interpretation. Hum Mutat 39:1614–1622, 2018 [PMC free article: PMC6225902] [PubMed: 30311389]
- 100.
- ClinGen: Clinical Genome Resource. Monogenic Diabetes Gene Curation Expert Panel [article online], 2019. Available from https:
//clinicalgenome .org/affiliation/40016/. Accessed 14 Feb 2022 - 101.
- ClinGen: Clinical Genome Resource. Monogenic Diabetes Variant Curation Expert Panel [article online], 2021. Available from https:
//clinicalgenome .org/affiliation/50016/. Accessed 14 Feb 2022
All authors reported no conflicts of interest.
Publication Details
Author Information and Affiliations
Authors
Maria V. Salguero, MD, MSc, 1 Marilyn Arosemena, MD,2 Toni Pollin, PhD,3 Siri Atma W. Greeley, MD, PhD,4 Rochelle N. Naylor, MD,4 Lisa Letourneau-Freiberg, MPH, RD,5 Tiana L. Bowden,5 Diane Wei,6 and Louis H. Philipson, MD, PhD4.
1 Marilyn Arosemena, MD,2 Toni Pollin, PhD,3 Siri Atma W. Greeley, MD, PhD,4 Rochelle N. Naylor, MD,4 Lisa Letourneau-Freiberg, MPH, RD,5 Tiana L. Bowden,5 Diane Wei,6 and Louis H. Philipson, MD, PhD4.Affiliations
Department of Pediatrics
Section Pediatric Endocrinology
Oak Lawn, IL
Department of Medicine
San Antonio, TX
Baltimore, MD
Department of Pediatrics and Medicine
Section of Endocrinology, Diabetes and Metabolism
Chicago, IL
Kovler Diabetes Center
Chicago, IL
Toledo, OH
Corresponding author.Publication History
Initial Posting: December 20, 2023.
Copyright
Diabetes in America is in the public domain of the United States. You may use the work without restriction in the United States.
Publisher
National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK), Bethesda (MD)
NLM Citation
Salguero MV, Arosemena M, Pollin T, et al. Monogenic Forms of Diabetes. 2023 Dec 20. In: Lawrence JM, Casagrande SS, Herman WH, et al., editors. Diabetes in America [Internet]. Bethesda (MD): National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK); 2023-.