Summary
Since early 2007, the establishment of international consortia and biobanks has catalyzed the performance of large-scale genomic studies. These efforts have driven an explosion in the discovery of genetic variation associated with type 2 diabetes. Most studies have involved genetic data captured using genotyping arrays populated by common single nucleotide polymorphisms (SNPs), although a rapid drop in the cost of next-generation sequencing has facilitated a growing number of exome and genome sequencing studies, which can capture increasingly rare variation. Hundreds of independent SNPs have been associated with type 2 diabetes and glycemic traits using genome-wide association studies (GWAS), and their numbers continue to increase. Findings have pointed to both known and novel molecular pathways and increased the understanding of fundamental disease biology.
On the other hand, causal variants have been identified for only a small fraction of the loci identified by GWAS, and a substantial proportion of disease heritability remains unexplained. While combining genetic variation into polygenic scores improves prediction of type 2 diabetes risk substantially beyond that of single variants, such scores are not yet used in clinical practice due to inadequate predictive ability and implementation challenges. Despite including millions of individuals, genetic studies remain limited by the diversity of populations represented, are underpowered to fully capture rare variation of modest effect sizes, and have incomplete ascertainment of alternate (non-SNP) forms of genetic variation.
As the community continues to expand genetic discovery and pursues systematic fine-mapping, platforms that focus on functional variation, systems biology approaches, and expansion to non-European populations, the coming years will witness exponential growth in our understanding of the genetic architecture of metabolic phenotypes related to type 2 diabetes. Whether these findings prove useful in disease prediction or therapeutic decision-making must be tested in rigorously designed clinical trials.
Introduction
The explosive parallel growth in the prevalences of the related metabolic disorders of obesity and type 2 diabetes in much of the developed and developing world over the past few decades is almost certainly driven by environmental and behavioral factors, as genetic components do not change in an appreciable manner over such a short time. However, several lines of evidence suggest that variation in DNA sequence does contribute to type 2 diabetes risk. First, twin studies have shown that concordance for type 2 diabetes is greater for monozygotic twins (who share 100% of their DNA sequence) than for dizygotic twins (who, like siblings, share approximately 50% of their DNA sequence) (1,2,3,4,5). Second, the incidence of diabetes is much higher in certain racial/ethnic groups, despite environments that are relatively comparable to those of neighboring populations (6,7,8). Third, family history is an independent risk factor for the development of diabetes in population studies (9,10). And fourth, rare familial forms of diabetes, caused by mutations in single genes (hence, termed monogenic or Mendelian), prove that single base pair changes in the coding regions of key genes, which lead to alterations in protein sequence and function, are sufficient to cause hyperglycemia in the diabetic range (Figure 1) (11,12). Consistent with this notion, the estimated heritability of type 2 diabetes ranges from 25%–69% in a set of Scandinavian families (13) to 52% (95% confidence interval [CI] 26%–80%) in the Washington State Twin Registry (14) and as high as 72% in a large international meta-analysis of twin studies (15).
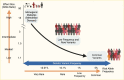
Taken together, these observations illustrate that rapid changes in the global epidemiology of type 2 diabetes are likely caused by environmental and behavioral factors overlaid on a background of genetic predisposition. This genetic predisposition may vary across populations, in some measure due to divergent genetic histories and unequal selection pressures in specific geographic regions; however, this is a topic of continued investigation (16).
Why is genetic exploration relevant? Regardless of whether genetic predictors become useful markers of disease onset or of subtypes in clinical practice, the identification of genetic variants associated with type 2 diabetes illustrates pathogenic mechanisms that may be targeted by future therapies. Because germline genetic variation always predates the onset of disease, the arrow of time establishes a causal relationship that is not evident with other biological associations. Thus, the genetic approach has a unique opportunity to shed light on the pathophysiology of diabetes in its various manifestations, helping to unravel its clinical heterogeneity and potentially refine therapeutic strategies. This article updates the information included in the Diabetes in America, 3rd edition chapter Genetics of Type 2 Diabetes (17).
Discovery of Genetic Risk Factors for Type 2 Diabetes
Before a draft sequence of the human genome was available, genetic mapping depended on the generation and anchoring of anonymous genetic markers to specific locations in the genome. This task was first achieved by identifying variation in DNA fragments with known cleavage sites (restriction fragment length polymorphisms). Subsequently other markers, such as DNA segments with characteristic repeats (microsatellites) or unique stretches of DNA (sequence tag sites), enabled the introduction of more sophisticated methods (whole-genome linkage analysis and positional cloning) for determining the position of disease-causing genetic variation within the genome. Linkage analysis depends on the co-segregation of a causal mutation and nearby anonymous markers with affected, but not unaffected, members across successive generations in pedigrees. This approach has been successful for gene discovery in diseases that are highly penetrant: where disease-causing alleles are almost always found in individuals with disease and are almost always absent in individuals without disease. For diabetes, linkage analysis facilitated the discovery of the genes that underlie various types of monogenic diabetes, such as maturity-onset diabetes of the young (MODY) (12,18) or neonatal diabetes (19,20,21) (Figure 1); these associations are described in detail in Diabetes in America: Monogenic Forms of Diabetes (22).
However, diabetes is a complex disease, for which liability to disease is determined by hundreds to thousands of genetic variants that are only slightly penetrant and may carry interactions with the environment. Thus, the use of linkage analysis to characterize the genetic basis of common, complex forms of diabetes was far less successful. For type 2 diabetes, no single genetic locus of substantial effect analogous to type 1 diabetes loci exerts a very strong effect in the general population or even in individual family pedigrees. Thus, the effect of genetic variation is probabilistic rather than deterministic; a substantial proportion of people with some risk variants may be disease-free, whereas others who carry protective alleles may instead have type 2 diabetes due to a constellation of other factors.
To demonstrate the effect of genetic variation on human phenotypes, an alternative approach was needed. Association testing quantifies whether a specific allele is significantly overrepresented in diabetes cases compared to controls without diabetes. With large sample size, this method has greater statistical power to detect a common variant of weak effect. The major limitation of this approach was that—prior to 2005—only a handful of variants could be tested at a time, which required prior knowledge of the existence of genetic variants and prior biological knowledge of a role of a given candidate gene in diabetes pathophysiology. Although multiple genetic associations were described before 2005, only two of these have stood the test of time. Both variants change the amino acid sequence in genes that encode antihyperglycemic drug targets: the p.Pro12Ala polymorphism in the peroxisome proliferator-activated receptor gamma 2 (encoded by PPARG) (23) and the p.Glu23Lys polymorphism in the islet ATP-dependent potassium channel Kir6.2 (encoded by KCNJ11) (23,24). A third locus, a noncoding variant in the transcription factor 7-like 2 gene (TCF7L2), was discovered by large-scale association testing in areas of suggestive linkage (25). The TCF7L2 common intronic rs7903146 polymorphism had the strongest statistical association (though with a modest odds ratio ~1.4) seen consistently across multiple studies (Figure 2) (26,27).
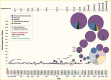
The panorama changed dramatically with the advent of genome-wide association studies (GWAS) (28). Several factors coalesced to enable GWAS: the discovery of millions of single nucleotide polymorphisms (SNPs) and their deposition in public databases; the manufacturing of genotyping arrays that could simultaneously query hundreds of thousands of SNPs with great precision and lower cost; the understanding of an underlying correlation structure between SNPs, driven by the finite number of recombination events in human history, which reduced the complexity of the variation to be interrogated; the recognition that the scientific imperative of reproducibility required the acceptance of strict statistical thresholds that accounted for the universe of possible hypotheses in the human genome; and the corollary of such awareness, that for these very small p-values to be achieved, very large sample sizes had to be assembled through international collaboration. Thus, for the first time, most common variants in the human genome (i.e., those with a minor allele frequency >5%) could be tested en masse (Figure 1).
Several independent GWAS (29,30,31,32,33) initially focused predominantly on European ancestry populations and, coupled with the growing scientific exchange that led to successive meta-analyses of ever-increasing size (34,35,36,37,38,39,40), produced a plethora of robust associations. This research changed the landscape of type 2 diabetes-associated variants, which grew from three prior to the GWAS era to several dozen leading up to 2018 and to more than 500 by 2022 (Figure 2). This list has been complemented by the implementation of similar approaches in the discovery of genetic determinants of quantitative glycemic traits (41,42,43,44,45,46,47,48,49,50,51) and other metabolic phenotypes, which share many signals with type 2 diabetes (Figure 3). Additionally, GWAS in non-European ancestry populations (52,53,54,55,56,57,58,59,60,61,62,63,64,65,66,67,68,69,70,71,72,73) and multi-ancestry meta-analyses of many of these studies (64,74,75,76,77,78,79) have identified hundreds of additional type 2 diabetes loci and demonstrated a large overlap in signals across populations. While a 2023 study suggested strong correlation in effect sizes among ancestries for type 2 diabetes (80), the largest GWAS for type 2 diabetes suggest some ancestry-correlated heterogeneity that may be explained by body mass index (BMI), which does vary across ancestries (81).
A particularly illustrative example of a combination of these approaches has been furnished by Moltke et al. (82). On studying the population isolate of Greenland, the study team selected a custom-made SNP array, the Metabochip (83), and focused on quantitative glycemic traits. The team followed up an original signal in TBC1D4 by sequencing the coding regions of this gene and identified a nonsense p.Arg684Ter variant of Inuit ancestry that is common in the Greenlandic population (frequency 17%) and associated with 2-hour glucose and insulin levels. Stop codon homozygotes harbor a tenfold increased risk of type 2 diabetes compared to wild-type allele carriers. Definitive identification of the implicated protein allowed for functional studies: the stop codon induces lower levels of the TBC1D4 protein in human skeletal muscle, causing reduced numbers of the glucose transporter GLUT4 and decreased insulin-stimulated glucose uptake, which lead, in turn, to postprandial hyperglycemia and impaired glucose tolerance.
The first generations of GWAS mainly tested genetic variation that is common across human populations (i.e., minor allele frequencies >5%). At first, only variants directly genotyped by the SNP arrays could be analyzed; subsequently, computational tools were developed to allow for statistical imputation that enabled accurate prediction of ungenotyped variants based on reference genome sequences (e.g., The 1000 Genomes Project Consortium (84)), which more readily allowed meta-analyses of variants tested in GWAS. As reference human genome sequence panels increased in sample size and diversity over time, three major developments occurred: first, high-accuracy imputation became possible for less common variants (i.e., allele frequencies <5%) (85,86); second, targeted genotyping arrays were designed that included less common but likely functional variation (e.g., coding variants) (87); and third, multi-ancestry GWAS analyses improved, with better capture of variants across ancestral groups (88). The combination of these techniques deployed from around 2017, coupled with the development of large population- and hospital-based biobanks, resulted in a dramatic increase in the number of discovered type 2 diabetes loci (Figure 2).
In addition to SNP arrays (which contain both protein-coding and noncoding variation), protein-coding arrays containing selected coding regions were developed with the hope of achieving a more direct link between associated signals and biological function. For example, using data from a genotyping array designed to focus on coding variation (i.e., the ExomeChip), Mahajan et al. identified 40 mostly common coding variant associations, including novel associations at POC5, PNPLA3, LPL, and ANKH (89). Interestingly, “fine-scale mapping” analyses (described further in the Determining the Function of Type 2 Diabetes Genes section) in this study indicated that often the associated coding variant was not the causal variant underlying the type 2 diabetes association signal. This finding suggested that a significant association between a protein-coding variant and disease was not enough to definitively implicate a gene in diabetes pathogenesis and may have identified the wrong gene altogether.
With the advent of new sequencing technologies, the cost of sequencing became dramatically reduced, enabling large-scale sequencing studies and discovery both within the protein-coding portion of the genome (the exome) (e.g., HNF1A p.Glu508Lys) (90,91,92,93), as well as from the full genome (94).
Gene burden analysis is an approach to implicate disease-causing genes by testing groups of variants within a gene together for their association with disease rather than testing single variants. This approach has become more feasible, including in type 2 diabetes, with the increase in the availability of sequencing data in large populations (Figure 4). Typically, the sets of variants within a gene aggregated for gene burden testing are those that are too rare to be tested individually (i.e., allele frequencies of ≤0.1%) and predicted to be functionally consequential (e.g., causing loss of gene function). For example, in a targeted gene burden analysis of SLC30A8, loss-of-function mutations were collectively associated with 65% reduction in risk of type 2 diabetes (95). Gene burden testing of all genes across the genome has also become increasingly possible with growing sequencing studies and development of high-throughput statistical tools. For example, Fuchsberger et al. performed a rare variant burden study in 12,940 individuals, although the study did not identify statistically compelling positive associations, which was likely due to a lack of statistical power (93). Subsequently, Flannick et al. performed gene burden testing using exome data from 20,791 type 2 diabetes cases and 24,440 controls and identified four genes (SLC30A8, MC4R, PAM, and UBE2NL) that were significantly associated with type 2 diabetes risk (91). More recently, large-scale exome sequencing in up to ~450,000 British subjects enrolled in the UK Biobank led to multiple additional gene-level associations with type 2 diabetes and glycemic traits, including some for genes known to cause monogenic diabetes (GCK, HNF1A, HNF4A, and PDX1), as well as novel diabetes genes (GIGYF1, MAP3K15, and FAM234A) (96,97,98,99).
Despite rapidly growing study sizes, exome and genome sequencing studies remain limited by the power to detect single, low frequency variant and gene burden associations. An additional challenge is inference of rare-variant burden contributed by noncoding variants, which is especially difficult owing to the lack of meaningful and well-powered approaches to aggregation, which is a subject of active investigation (100,101). As sequencing becomes more routine across large biobanks, data from millions of subjects will likely help to identify a plethora of such associations for type 2 diabetes.
Determining the Function of Type 2 Diabetes Genes
Once a region of the genome has been statistically associated with type 2 diabetes beyond reasonable doubt, new questions emerge, all of which are important to understanding genetic mechanisms and the viability of drug development. Such questions include: (i.) What causal variants underlie the signature of association? (ii.) What genes are causally implicated by these variants, and which tissues are etiologically relevant? (iii.) By what genetic and physiological mechanisms does variation in the region influence diabetes risk? (iv.) What direction of perturbation at this locus would be expected to produce a therapeutically beneficial effect on disease development and progression?
To identify where effort should be placed in the overwhelming space of functional experimentation, the research community has engaged in a variety of computational approaches to help prioritize candidate causal variants and genes for further work. For example, in focusing on the first question—identifying causal variants underlying an association signal identified by GWAS—the approach of statistical fine-mapping has been widely utilized (Figure 5) (102). This method can be conceptualized in two parts: first, locus fine-mapping, where the goal is to determine how many independent variants (or signals) contribute to the patterns of association observed in the region; second, signal fine-mapping, where the goal is to construct a “credible set” of variants statistically likely to harbor the true, causal variant for each identified signal. Fine-mapping methods can achieve these goals based on the statistical information alone; however, additional data can be used to further home in on the causal variant with that credible set. First, association data from multiple ancestries can narrow signals owing to different patterns of linkage disequilibrium (LD), meaning genomic variation that tracks together (77). In addition, annotations that label genomic sequences with a function due to their presence in either protein-coding sequence or noncoding sequence with presumed regulatory function (e.g., a promoter or enhancer) can be used to prioritize variants that are most likely to be driving the association signal (Figure 5) (38,77,103,104,105). These approaches are now routinely used in tandem. For example, in a 2022 multi-ancestry meta-analysis, the authors used both differences in LD across populations and functional annotation from diabetes-relevant tissues (e.g., DNA accessibility or chromatin state in islets, adipocytes, or hepatocytes) to determine the fewest number of variants that explained the genetic association at each signal (77).
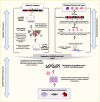
The second question—which genes are causally implicated by these variants—is challenging but must be addressed to enable appropriate mechanistic studies. It is worth remembering here that although a handful of signals identified for type 2 diabetes include coding variants in the credible set and thus provide a potential candidate gene, the majority of signals map exclusively noncoding variation without an obvious link to a candidate gene (89). As a result, different approaches have been utilized to link variants to genes to generate priorities for further study at the wet-lab bench. These approaches often combine empirical data derived from disease-relevant cell types (e.g., islets, adipocytes, hepatocytes, myocytes), including assessments of how the DNA is folded and which promoters interact with which enhancers (e.g., promoter capture Hi-C) or in silico methods that use known features, such as chromatin state, to predict which genes have altered expression due to a particular variant (Figure 5) (106,107,108). Combining such data in a probabilistic framework can provide estimates that a given gene is causal for type 2 diabetes (109,110). Initial functional studies focused on bulk tissue (i.e., human islets), but the advent of single-cell sequencing approaches allowed a shift to this higher-resolution approach (111,112). Most genes contain variants in their promoters that alter their expression, so it is very important to establish that signals for altered gene expression co-localize with those for diabetes risk (113,114). Advances in understanding the regulatory grammar of DNA have enabled machine learning approaches to predict variants that are most likely to alter gene expression in a given tissue (115,116). Understanding whether changes in gene expression result in a diabetes-relevant cellular phenotype is an important next step and is increasingly being assessed at scale through targeted and genome-wide cellular screening efforts (117,118).
Next, four examples of moving from “variant to function” are summarized, highlighting the challenges of this work, as well as the insights into disease biology that can be provided.
Coding variants provide a molecular “signpost” to the gene underlying the genetic association and have accelerated efforts to translate genetic discoveries into molecular mechanisms. The following examples showcase various approaches that can be taken to understand how genetic factors influence diabetes pathogenesis. For example, in 2007, a common coding variant (p.Arg375Trp) in SLC30A8 was identified in an early type 2 diabetes GWAS (29). SLC30A8 encodes a zinc transporter (ZnT8) that is located in pancreatic islet cells and is integral for zinc ion transport to stabilize insulin and trigger crystallization (119). Whether this diabetes-associated allele resulted in a loss or gain of function was not obvious. For years, there was uncertainty and debate as to how genetic variation in SLC30A8 influenced diabetes risk, since a common risk-associated allele from GWAS cannot inform on whether disease mechanism involves loss or gain of gene function, and mouse models did not have a consistent phenotype (120,121). Compelling evidence for the disease mechanism arrived in 2014 from a human gene burden analysis (Figure 4), described in the previous section, which showed that carriers of protein-truncating variants in SLC30A8 had a 65% reduced risk of type 2 diabetes compared to non-carriers, indicating that loss of function of the gene is protective for diabetes (95). In concordance, newer mouse models carrying the protective p.Trp138* allele had increased capacity to secrete insulin in high-glucose conditions, with secretion of 50% more insulin in response to hyperglycemia (122). Additional studies in humans supported these findings, demonstrating that particular loss-of-function missense mutations in SLC30A8 enhance proinsulin processing and insulin responsiveness to glucose, conferring protection against type 2 diabetes (123).
The SLC16A11 locus provides an illustrative second example both of discovery, this time in a non-European ancestry population, and of insight gained into disease biology from functional studies. A GWAS of populations in Mexico and East Asia first identified 17p13 as a type 2 diabetes genetic risk locus (67,69). The risk haplotype at this region is more common among individuals of Mexican or Latin American ancestry (minor allele frequencies ~30% compared to <2% in European populations) with each allele increasing diabetes risk by approximately 25% (67). Genetic fine-mapping at this locus revealed that some variants decreased SLC16A11 expression in liver, and other variants disrupted the interaction of the SLC16A11 protein (a monocarboxylate transporter) with basigin, a chaperone protein important for plasma membrane localization, leading to reduced cell-surface localization of the SLC16A11 protein (124). Homozygous carriers had up to 85% less SLC16A11 at the plasma membrane. SLC16 family members mediate transport of distinct substrates. Reduced SLC16A11 transport activity at the cell membrane results in alterations in fatty acid and lipid metabolism, driving cellular changes consistent with those seen with insulin resistance. As these findings implicated reduced SLC16A11 activity in risk for developing type 2 diabetes, increasing SLC16A11 function may offer a therapeutic target strategy.
The next example highlights the PAM locus, which was first identified in a glycemic trait exome-array study as harboring coding variants that alter beta cell function, as captured by the insulinogenic index (125). Initially, genetic data alone were insufficient to distinguish whether the association was due to the PAM missense allele or a second missense allele in near perfect LD in the neighboring PPIP5K2 gene. However, a second study in an Icelandic population identified a second missense variant in the PAM gene that was independently associated with altered type 2 diabetes risk, suggesting that PAM was more likely to be the effector gene at this locus (92). Subsequently, gene burden testing of missense variants predicted to be deleterious in PAM provided further support for the gene’s association with diabetes (91,98). Peptidylglycine alpha-amidating monooxygenase (PAM) is an enzyme in neuroendocrine cells that creates peptide amides, increasing peptide biological potency (126,127). PAM is widely expressed, including in both pancreatic beta and alpha cells, where it is involved in regulating insulin secretion (128). Type 2 diabetes-associated PAM alleles result in defective expression and reduced catalytic function, directly reducing pancreatic beta cell insulin content and impairing release in response to glucose (128).
A final example represents the more common signal that researchers need to dissect, as it is a regulatory variant. In an early study that combined fine-mapping with integration of genomic annotation from diabetes-relevant cell types, researchers established that the credible set of variants at the MTNR1B locus contained a signal variant. When they integrated the fine-mapping results with epigenomic data from diabetes-relevant cell types (e.g., pancreatic islets, hepatocytes), they showed that the variant was located in an enhancer and created a binding site for the transcription factor NEUROD1. Using in vitro cellular models, allelic-specific differences were found in NEUROD1 binding to the diabetes risk variant, and in human islets, the type 2 diabetes-associated allele was associated with increased MTNR1B expression (104). MTNR1B encodes the melatonin receptor, which is expressed in human beta cells and plays a role in inhibiting insulin secretion. The diabetes risk allele increases expression of the receptor and is predicted to reduce insulin secretion inappropriately.
Polygenic Scores for Prediction of Type 2 Diabetes
As a polygenic condition, type 2 diabetes is well appreciated to be genetically determined by thousands of variants. Most genetic discovery, as noted, has involved common variants identified via GWAS, which exert only modest penetrance (i.e., odds ratios typically <1.2) (Figure 2). In isolation, such risk variants have little effect on disease risk; however, combining risk variants together into polygenic scores can improve risk prediction.
Polygenic scores estimating disease risk have been considered in two categories: (i.) “restricted-to-significant” polygenic scores (rsPS), which include only genetic variants significantly associated with disease, and (ii.) “global extended” polygenic scores (gePS), which typically include hundreds of thousands of variants, taking into account redundancy of signals related to correlation (i.e., LD) of variants (129). The ability of a polygenic score to predict future or prevalent disease risk has commonly been quantified using the area under the receiver operator characteristic curve (AUROC) or C statistic. The AUROC measures the predictive efficacy of a test, with a value of 0.5 indicating no greater predictive ability than chance and a value of 1.0 indicating a perfectly predictive test.
The first polygenic scores for type 2 diabetes were rsPS and included between three to a few dozen genetic variants identified in early genetic studies (including pre-GWAS candidate gene studies). While such scores had better predictive ability for future diabetes onset than any single variant, they offered AUROCs on the order of 0.60, which were insufficient for clinical use and inferior to prediction models involving clinical data alone (130,131,132,133). One of the largest rsPS for type 2 diabetes, including 62 SNPs, was tested in the Framingham Offspring Study (FOS) and produced an AUROC of 0.726 for incident diabetes prediction when combined with age and sex, which represented a significant (p<0.001), but minimal, improvement from the model with age and sex alone (AUROC 0.698) (133). Studies have demonstrated that polygenic risk for type 2 diabetes can be offset by environmental factors. For example, the Diabetes Prevention Program showed that an intensive lifestyle intervention, consisting of dietary and physical activity components, is effective even in the quartile of participants with highest type 2 diabetes rsPS (134).
More recently, methods to develop gePS have allowed capture of substantially more variants than in rsPS, including hundreds of thousands of variants with sub-genome-wide significant associations with disease (129). Multiple derivations of gePS for type 2 diabetes have demonstrated some improvement in risk prediction beyond rsPS. Still, even the most recently developed gePS have not generated prediction levels suitable for clinical use, with maximum AUROC estimates of approximately 0.73 in models that include age and sex (Figure 6) (38,135,136). Like the rsPS, the gePS have contributed only incremental improvement when added to models with clinical variables. For example, a model with age, sex, and BMI demonstrated an AUROC of 0.818 for prediction of prevalent diabetes status in the FinnGen Biobank compared to an AUROC of 0.835 when the model also included the gePS (77).

The predictive ability of polygenic scores is poised to improve with forthcoming methods that can incorporate rarer genetic variation. For example, a combined rare and common variant polygenic score was developed for glycated hemoglobin (A1c) (137). This combined polygenic score for A1c identified 4.9 million misdiagnosed type 2 diabetes cases in the United States due to inaccuracies in A1c representation of glycemia given erythrocyte variation—nearly 1.5-fold more people than a common variant polygenic score alone.
Another related application of polygenic scores is identification of individuals with particularly high disease risk. Different score thresholds can be chosen to pinpoint individuals at the greatest genetic risk of diabetes. For example, multiple studies have demonstrated that those in the top 10% of the type 2 diabetes gePS have approximately fivefold increased risk of type 2 diabetes compared to the bottom 10% and around 2.75-fold increased risk compared to the remaining 90% of the population (76,77). For individuals with such high polygenic type 2 diabetes risk, this genetic information could have clinical utility, and studies are ongoing to formally assess this possibility. At the same time, placing such risk estimates in the context of genetic testing for monogenic diabetes, which is performed as part of standard clinical practice, can be helpful. Rare genetic variants causing monogenic diabetes have been estimated in the UK Biobank to confer more than eightfold increased diabetes risk (138,139). Furthermore, in the UK Biobank, people with the highest 1% of the type 2 diabetes gePS were at significantly lower risk of diabetes, with ~20% disease prevalence compared to ~56% prevalence in monogenic diabetes carriers (Figure 6) (138). Further research is necessary to determine whether identifying individuals with high type 2 diabetes polygenic scores, who have essentially moderate disease risk, will be useful clinically. Additionally, even if deemed potentially useful in a particular context (e.g., as part of a clinical risk tool), implementation of polygenic score generation in clinical labs has many challenges, including determining the appropriate risk estimates for individuals of different genetic ancestries.
In addition to predicting future diabetes risk, a potential application of the type 2 diabetes polygenic score is clinical discrimination between type 1 and type 2 diabetes to improve accuracy of subtype designation. A 69-SNP type 2 diabetes rsPS was assessed for this purpose in the Wellcome Trust Case Control Consortium and was unable to provide strong discrimination between type 1 and type 2 diabetes cases (AUROC 0.64). This performance was inferior to a 30-variant type 1 diabetes rsPS, which had an AUROC 0.88 and was only minimally improved (AUROC 0.89) by the addition of the type 2 diabetes score to it (140). Thus, as of 2023, the type 2 diabetes polygenic score is unable to effectively aid diabetes subtyping; however, as described in the next section, additional strategies are being investigated to leverage type 2 diabetes genetic discovery for this purpose.
Genetic Stratification of Type 2 Diabetes
Type 2 diabetes is a heterogenous disease, with patients displaying varying degrees of pancreatic beta cell dysfunction and insulin resistance (141). In theory, genetics could identify underlying disease pathways leading to type 2 diabetes and allow stratification of patients based on their inherited risk for such pathways contributing to their disease process. The research field has a great interest in grouping type 2 diabetes genetic variants into shared mechanistic categories or pathways.
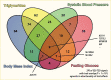
Prior to 2018, genetic clustering utilized approaches that restricted each locus to be a member of only one cluster, so-called “hard clustering” (36,89,142). These efforts identified some potential disease mechanisms, e.g., groups of loci broadly related to beta cell function and insulin resistance. However, loci with well-appreciated mechanisms were often not grouped in expected clusters, and some clusters were difficult to interpret. In 2018, two papers (89,143) suggested that applying “soft clustering,” where a locus could be involved with multiple pathways, could improve interpretability of genetic clusters. These two independent efforts also converged on a shared set of five clusters that could be readily linked to potential disease processes: two clusters related to distinct mechanisms of impaired beta cell function (termed beta cell and proinsulin by Udler et al. (143)) and three clusters related to different mechanisms of insulin response (termed lipodystrophy, obesity, and liver/lipid (143)) (Figure 7). A sixth, less well-defined cluster was identified by Mahajan et al. (89). Supporting a biological basis of the grouping of loci into clusters, the five clusters described in Udler et al. contained loci significantly enriched for active DNA regulatory elements that were specific to distinct tissues (e.g., the beta cell cluster loci were enriched in pancreatic islets).

A person’s aggregate genetic risk of loci in a given cluster is quantified by generating cluster-specific “partitioned” polygenic scores (pPS), which potentially capture risk for developing diabetes due to a particular disease process (143). In an analysis of ~17,000 individuals with type 2 diabetes, Udler et al. found that cluster pPS were significantly associated with distinct clinical features, suggesting that genetics could help disentangle the heterogeneity of type 2 diabetes. For example, an increased beta cell cluster pPS and proinsulin cluster pPS were each significantly associated with decreased C-peptide levels, supporting a shared disease mechanism of insulin deficiency. Additionally, the pPS could identify individuals with high risk specifically for a given genetic cluster. Thirty percent of individuals had a pPS uniquely in the top 10% of a single cluster, and these individuals also had distinct phenotypes compared to other individuals with type 2 diabetes. For example, those with extreme pPS for the lipodystrophy cluster had significantly decreased high-density lipoprotein (HDL), percent body fat, and BMI compared to all others with type 2 diabetes, mirroring the pattern of clinical phenotypes seen in patients with monogenic forms of lipodystrophy. Associations of the cluster pPS with distinct clinical phenotypes have been replicated in other datasets, and the pPS have differential associations with metabolic outcomes, including coronary artery disease, hypertension, and chronic kidney disease (144). For example, two clusters related to potential mechanisms of insulin resistance had opposite associations with coronary artery disease, with the lipodystrophy pPS increasing disease risk versus the liver/lipid cluster pPS decreasing risk. Subsequent expansion of the clustering work to include 323 type 2 diabetes variants and 64 trait associations identified 10 clusters, recapturing the original clusters along with additional clusters related to mechanisms of insulin deficiency and insulin resistance (145).
While genetic clusters have pointed to potential genetic mechanisms of type 2 diabetes, the currently derived pPS have small effect sizes and, as of 2023, are not useful for individual patient stratification in clinical practice. Nevertheless, analysis of cellular phenotypes in subcutaneous adipocytes derived from human biopsies has demonstrated differences in cellular features between individuals with high versus low lipodystrophy cluster pPS (146). Remarkably, cellular changes, such as the size of lipid droplets and intensity of mitochondrial staining, were visible on images, suggesting that while the effects conferred by the pPS on clinical biomarkers, such as serum lipid levels, may be small, there appear to be quite large effects on cellular features further upstream. As discussed in the Additional Insights Gained section, the specific cellular changes also point to convergence of polygenic and monogenic diabetes genetic pathways.
Genetic clustering has advanced simultaneously with clustering of individuals with diabetes based on clinical features (reviewed in Deutsch et al. (147)). Genetic analyses of some phenotypic diabetes clusters, including with use of pPS, have supported potential etiological differences in clusters (148,149). For example, Mansour Aly et al. demonstrated that in phenotypically derived clusters developed in Scandinavian populations (150), the two cluster pPS capturing forms of insulin deficiency (beta cell and proinsulin clusters) were both most strongly associated with their “Severe Insulin Deficient Diabetes” cluster (148).
Summary of Type 2 Diabetes Genetics
The tremendous success of GWAS and their follow-up for type 2 diabetes and other human phenotypes has resulted in several insights into the genetic architecture of disease:
- More than 500 independent loci have been associated with type 2 diabetes, with the vast majority of findings coming from GWAS.
- Most type 2 diabetes-associated loci are located near genes previously not suspected of playing a role in type 2 diabetes pathophysiology.
- The genes harboring the functional variants at most loci remain unknown.
- A handful of robust gene-level associations have been identified using gene burden analyses in sequencing data.
- The road from association to function is arduous and may involve fine-scale mapping, deployment of various -omics data, and wet-bench lab experiments. All of these techniques are important for characterizing the biological system to establish function. Techniques within the -omics field can probe the effect of genetic variation on RNA, protein, metabolites, and other biologically relevant structures, making these technologies key to this process.
- Type 2 diabetes polygenic scores have not yet demonstrated utility for standard clinical practice.
- Efforts to group type 2 diabetes genetic variants by shared physiology into clusters have enabled the generation of cluster-specific polygenic scores with distinct associations with clinical phenotypes, but such approaches are not yet clinically useful at the individual level.
Additional Insights Gained
Pleiotropy is the idea that a single genetic variant or gene can have multiple, unrelated biological effects. Pervasive pleiotropy exists for loci associated with type 2 diabetes—with considerable complexity—which can be exploited. One notable observation following the success of association studies is the extent to which specific signals for type 2 diabetes often demonstrate association with a wider spectrum of additional traits, including cardiometabolic risk factors. Pleiotropy—a term used to describe this general phenomenon—turns out to be pervasive across the human genome (151). This phenomenon is nicely exemplified for type 2 diabetes by the glucokinase regulatory protein gene, GCKR. Genetic variation in this gene has been associated with numerous traits, including type 2 diabetes, serum triglyceride levels, and fasting glucose (152), as well as dozens of others. Genomic regions containing type 2 diabetes-associated loci display considerable shared associations with other cardiometabolic traits, including fasting glucose, systolic blood pressure, triglycerides, and BMI (Figure 3). Looking exclusively at the lead variant associated with type 2 diabetes at each of the 520 established signals reported to date, 71% carry nominal association (at a less rigorous p<1x10-3) with at least one of these traits; 10% carry association with three or four traits. Rather than focus on associations that are nearby, work in the 2020s has taken this analysis a step further, utilizing computational analyses to map the set of loci where these associations implicate a singular genetic change with multiple predisposing risk factors and increased risk to type 2 diabetes though formal statistical colocalization (153).
Beyond quantifying subtypes within the spectrum of diabetes patients, as discussed in the Genetic Stratification of Type 2 Diabetes section, multi-trait association data may be quite useful to indicate the expected physiological effect broadly if the target in the region were perturbed, i.e., “on target” effects beyond the central type 2 diabetes indication. For example, taking this approach and focusing on coding variation at the glucagon-like peptide-1 (GLP-1) receptor gene, Scott et al. contrasted the effects expected from genetic perturbation with GLP-1 receptor agonists, showing consistency with (lower) fasting glucose, but also evidence of lower risk for type 2 diabetes and coronary heart disease (154). Data and examples like this one can help prioritize targets with ideal physiological drug-targeting properties to maximize success in downstream safety trials, as well as to deprioritize targets that may be predicted to cause negative physiological responses through on-target perturbations.
Type 2 diabetes and monogenic diabetes overlap genetically. While large-scale genomic studies for type 2 diabetes have implicated many genes not previously connected to diabetes, variation near or within monogenic diabetes genes has been consistently associated with risk of type 2 diabetes (90,155,156). For example, in HNF1A, a gene causing a subtype of MODY, the p.Glu508Lys variant, which is present in 0.3% of people of Latino ancestry, was identified in exome sequencing of Mexican people and found to confer approximately fourfold increased risk of type 2 diabetes. In experimental models capturing its impact on gene expression, this variant demonstrated reduced transactivation activity of its target promoter at an intermediate level between the wild-type HNF1A protein and those with mutations causing MODY. Subsequent large-scale sequencing studies, as noted, have demonstrated that individuals with type 2 diabetes carry more rare variants in monogenic diabetes genes than would be expected by chance (91,96,157). Additionally, type 2 diabetes risk variants generating the lipodystrophy cluster pPS (143), as described in the Genetic Stratification of Type 2 Diabetes section, define a cellular phenotype in human subcutaneous adipocytes (involving increased mitochondrial activity and decreased lipid accumulation) that mirrors cellular changes seen in monogenic lipodystrophy (146). Together, these findings suggest that patients with monogenic diabetes and type 2 diabetes exist along a continuum rather than in discrete binary categories. Further supporting the idea of a continuum, population-based studies of people with pathogenic genetic variants expected to cause monogenic diabetes have demonstrated less severe clinical phenotypes than classically described (138,139).
Genetic studies augment prior epidemiological observations. Several of the strongest genetic associations for BMI (e.g., the FTO-IRX3-IRX5 and MC4R loci) are also strongly associated with susceptibility to type 2 diabetes. Furthermore, the alleles that increase BMI also increase susceptibility to type 2 diabetes, which is broadly consistent with prior epidemiological studies. This result is fully expected because BMI is a causal risk factor for type 2 diabetes. One would expect that genetic variants that modify any type 2 diabetes causal risk factor should also modify type 2 diabetes susceptibility. This idea can, thereby, be used to generate novel discovery: that is, the observation that genetic variants associated with an exposure in aggregate also modify type 2 diabetes susceptibility is evidence that this exposure is causal.
Under specific assumptions, potential causal exposure/outcome hypotheses can be statistically evaluated by using genetic variants for the exposures in an instrumental variable analysis framework (158). This framework, dubbed Mendelian randomization, has seen dramatic expansion and utilization in the last decade, given the substantial growth of genetic association data, to search broadly for causal risk factors for disease. In the context of type 2 diabetes, this approach has led to some interesting (perhaps provocative) statistical observations (159). For example, genetically elevated testosterone levels increase susceptibility to type 2 diabetes in women, but decrease type 2 diabetes risk in men (160). In contrast to the well-established protective effects of lower low-density lipoprotein (LDL) on heart disease, reduced LDL cholesterol levels may increase susceptibility to type 2 diabetes (161,162). For example, there is a higher risk of type 2 diabetes in individuals who take statins, which lower cholesterol levels (163), and a lower prevalence of type 2 diabetes is observed in patients with familial hypercholesterolemia (164). Distinguishing between correlation and causality for these observations is certainly a key objective for future work.
Limitations of Current Approaches (and Their Solutions)
Despite the overwhelming success of large-scale human genetic studies in advancing knowledge of the genetic determinants of type 2 diabetes, a number of limitations must be recognized. These limitations have been identified by the research community and are being addressed to complement gaps in our understanding.
Persistent overrepresentation of individuals of European ancestry in GWAS. The past 15 years have seen a welcome and much-needed increase in the diversity of population groups where GWAS for type 2 diabetes have been performed. These groups include East Asian (56,57,58,59,60,61,62,63,69,71,165), South Asian (53,65,66,78), African American (54,70,76), Latin American and Hispanic (52,55,67,76), and Native American (68,75) populations (Figure 2). As consortia assembling these data perform large-scale, multi-ancestry meta-analyses (75,76,77,165), more novel findings are expected to come to light. Still, even if all existing data were combined, the proportion of samples for the genetics of type 2 diabetes would not reflect the national distribution of these groups across the United States, much less the distribution of these populations across the world. To maximize the power for discovery and the translational benefits of these discoveries for everyone, continued inclusion efforts at scale in GWAS and generally in multi-omics resources are essential. DNA Biobanks—like those of the Million Veteran Program (MVP) or the All of Us cohort, to name a couple—are poised to close some of the gaps, although additional efforts to expand collections and integrate with existing data are a clear direction of growth in years to come.
Polygenic scores derived from European populations do not translate well across other ancestral groups. As polygenic scores have been developed primarily in European populations, prediction has been most accurate in populations of matched ancestry, with poorer performance in other groups. For example, the performance of the 23andMe gePS score published in 2019 declined from an AUROC of 0.652 in a people of European ancestry to 0.588 in those of African American ancestry (136). Trans-ancestry polygenic scores are being developed using modeling to derive variant weights without the need for a priori population assignment (e.g., Ge et al. (166)). However, such scores still suffer from relatively low AUROC values, reflecting the need for further refinement before clinical applications are possible.
Most analyses have not considered interactions with the environment and sex. Type 2 diabetes results from complex interactions among multiple genetic and environmental factors, and the rapid rise of its prevalence cannot be attributed to genetics. Because most GWAS are designed to detect loci that have primary effects on type 2 diabetes risk, regardless of the environmental context, many variants whose impact varies according to an environmental parameter might be missed. This effect is due in part to the imprecision inherent to environmental measures and the noise introduced by a single cross-sectional environmental exposure. In contrast, genotyping methods are extremely accurate, and the genetic exposure is uniform across an individual’s lifetime. Nevertheless, analytical methods are being developed that allow for the joint inquiry of main gene effects and gene × environment interactions (167). These methods have already been deployed to identify loci for insulin sensitivity (43) and the regulation of BMI (168). In the future, genetic biobanks that are richly phenotyped, where environmental factors on diet, geography, or other factors are included, may offer the potential to further advance studies of gene × environment interactions. For example, a 2022 study reported numerous loci associated with a collection of physical activity traits (169), and identifying specific physical activity × type 2 diabetes interactions is a clear direction for future research. Finally, new statistical methods are available that can also identify effects of gene × environment even when the exact risk factor is not known (170). These methods are limited only by statistical power, and thus, the samples sizes provided by biobanks are uniquely powered for this type of discovery effort.
A second variable routinely included in genetic association studies of type 2 diabetes is biological sex. However, sex-stratified analyses have been performed for type 2 diabetes (39,165) and fasting glucose/insulin (49) to identify associations that may differ between sexes. While a scant handful of loci appear to have differences between sexes, this heterogeneity can be of epidemiological interest. For example, in a large-scale study in individuals of East Asian ancestry, Spracklen et al. identified a compelling type 2 diabetes association in males but not in females at the ALDH2 locus (165). ALDH2 is a well-known locus related to alcohol metabolism, suggesting that difference in type 2 diabetes susceptibility attributed at this locus may be explained in part by behavioral differences in alcohol consumption between sexes and the downstream effects on metabolic traits this may impart (165,171).
Progress on understanding the genetics of complications of diabetes. While large-scale genetic studies for type 2 diabetes have been performed, progress on understanding the basis of risk for diabetic complications has met with variable success depending on the trait. Certainly, environmental factors, particularly glycemic control, have a large effect on the risk of developing complications. Yet, these factors do not completely explain the risk for complications, which is further influenced by genetic background. Knowledge of genetic contributions to relatively common traits for which people with diabetes are at elevated risk has advanced significantly, including for coronary artery disease (172), peripheral artery disease (173), stroke (174), and chronic kidney disease (175). However, genetic loci that specifically elevate risk of these complications in type 2 diabetes patients have not been obviously detected despite relatively large sample sizes, for example in coronary artery disease (176). Thus, the excess risk for these conditions that is conferred by type 2 diabetes has not been explained. With complications specific to diabetes—diabetic retinopathy, diabetic kidney disease, or diabetic neuropathy—advances have been limited by the lack of sufficient well-phenotyped samples. Potentially, biobanks that are richly phenotyped may offer a pathway to address this challenge, e.g., predicting diabetic retinopathy using a type 2 diabetes polygenic risk score (76). However, electronic health record data, from which a diabetes complication phenotype could be defined, are quite noisy and require substantial effort to construct high-quality, accurate phenotypes where the proper order of disease progression to complication can be determined. Pooling efforts across multiple biobanks to make progress on understanding excess risk for complications, as well as predicting which type 2 diabetes patients are most liable to develop various comorbidities, offers a very promising direction for future work.
Most analyses are simple additive tests for association and do not explore more complex modes of inheritance. There are good statistical reasons for this approach, a model in which the presence of two copies of the risk allele in an individual essentially doubles the risk associated with a single allele. This assumption may not always be the case: in the two extreme examples, under a dominant model, two copies of the risk allele will not add any further risk to that conferred by a single copy, and under a recessive model, the risk will not be made manifest unless both copies are present. O’Connor et al. conducted a GWAS using a recessive model for type 2 diabetes and identified 51 loci associated with type 2 diabetes, including five variants undetected by prior additive analysis (177). One low-frequency variant, rs115018790, which colocalization analysis linked to reduced expression of PELO, had an odds ratio of 2.56 for type 2 diabetes in homozygous carriers. While most type 2 diabetes-associated SNPs exert their action via an additive model, this is expected as their discovery took place precisely under such a model. A comprehensive examination of alternative modes of inheritance is needed in existing GWAS datasets, which now are reaching adequate sizes to compensate for the smaller number of homozygous minor allele carriers and for the penalty incurred by additional statistical testing. Similarly, tests of gene × gene interactions (epistasis) and accounting for divergent effects depending on parental line of inheritance, where that information is available (178), are likely to yield additional loci.
The Future of Research on the Genetics of Type 2 Diabetes
Given the rapid progress achieved in genetic discovery in type 2 diabetes and the multipronged approach deployed to overcome experimental limitations, the research community holds great hope that the pace will be maintained and a substantial part of the genetic architecture of type 2 diabetes will be elucidated in the coming years. If this vision is realized, several conceptual advances can be expected:
The nosology of disease will be refined. Type 2 diabetes, diagnosed solely on the basis of the final common pathway of hyperglycemia, is likely a heterogeneous syndrome that can be caused by a variety of processes (179). Genetic etiologies have already helped classify the various forms of monogenic diabetes, and an analogous genetic subclassification of type 2 diabetes informed by physiology is beginning to emerge (e.g., Udler et al. (143)).
Additional disease pathways will be identified. Unsuspected biology has already been uncovered via genetic discovery. For example, human genomic studies followed by elegant functional studies of TM6SF2 and PNPLA3 have implicated liver lipid metabolism in the pathogenesis of both type 2 diabetes and fatty liver disease (180). The increasing number of genetic loci at hand and availability of -omics tools (e.g., transcriptomics, epigenomics, metabolomics) will facilitate discovery and refined understanding of disease pathways, some of which may be amenable to the development of new therapeutics.
Genetic discovery may identify drug targets. Among the initial type 2 diabetes genetic associations were coding variants in PPARG, the gene that encodes the target of thiazolidinediones (23), and KCNJ11/ABCC8, the genes that encode the targets for sulfonylureas (24,181). More recent studies have identified the target for GLP-1 receptor agonists as another type 2 diabetes-associated gene (182,183). Additionally, multiple companies have developed glucokinase activators, which piggyback on years of research of GCK’s role in regulating glucose homeostasis and causing MODY (41,184,185). While debate continues around whether glucokinase activators are a safe and effective treatment for diabetes (186,187,188,189), all of these examples represent proof of principle for the potential for genetic discovery to aid drug development. Aggregating -omics data in humans and model systems, as well as ancillary associations that might point to off-target effects, will be essential for the genomic revolution to catalyze new drug discovery.
Treatment paradigms will be modified. Pharmacotherapy in type 2 diabetes is often selected based on patient comorbidities, medication side effects, cost, and availability, without consideration of the underlying pathophysiology driving disease phenotype (190). Pharmacogenetics is a promising precision medicine approach to tailor treatment choices by a patient’s underlying genetic background (191,192). Specific loci have been implicated in the response to metformin (193,194,195), sulfonylureas (196), and GLP-1 receptor agonists (197,198,199,200). Using genetics to guide medication selection might orchestrate a greater improvement in A1c, decrease risk for comorbidities, or suggest lower medication doses, reducing side effects.
Stratification of patients may allow for better targeting of public health or clinical trial interventions. Some preventive or therapeutic measures may be too expensive to deploy in the population at large, or they may be futile in specific subgroups. Genetic characterization may help identify the groups of people more likely to benefit from particular public health strategies, for example, those at highest genetic, as well as socioeconomic, risk (201). Similarly, the efficiency of clinical trials may be enhanced by enrolling participants who are more likely to reach the desired endpoints or benefit from the agents being tested, as has been demonstrated already for an atherosclerotic disease gePS in a trial for evolocumab (202).
Genetics may facilitate the implementation of precision or personalized medicine. Though it is not yet clear that genetic information will be powerful enough to apply therapeutic decisions at the individual level, it may help do so for specific subgroups. For example, genetic approaches may identify groups of individuals who are more likely to develop a particular diabetes complication. For such an approach to be feasible, researchers envision that in the not-too-distant future any individual who joins a public or private health care system would be genotyped or sequenced for the full list of actionable genetic variants (e.g., those that modify risk of common diseases or response to available medications), such that his/her information is available in the electronic medical record. When the time comes to make specific screening or therapeutic decisions, genetic information filtered through appropriate decision support tools would automatically guide the practitioner into the course of action most appropriate to the person and situation at hand.
Large-scale consortia efforts are critical for advancement of precision medicine. The genetics community has demonstrated the value of large-scale collaborative science for the development and deployment of novel methods and the discovery of robust disease associations. As the field accelerates efforts to move from genetic discovery to molecular mechanism, this ethos needs to be extended to capture new communities and data types. The International Common Disease Alliance (ICDA) seeks to catalyze these efforts by bringing expertise together, thus reducing redundancy and increasing efficiency. For type 2 diabetes, the Accelerated Medicines Partnership for Common Metabolic Disorders (AMP-CMD) provides a focus for the diabetes community, initially bringing genetic and genomic data together, but increasingly extending this to epigenomic and cellular data. The American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD) support efforts to provide consensus guidelines on how genetics can be incorporated into precision medicine efforts for diabetes diagnosis, prognosis, and treatment. Moving forward, all efforts must be inclusive and aim to reduce health disparities.
Conclusion
In summary, the field of type 2 diabetes genetics has experienced a steep discovery curve, and efforts now focus on translation of findings to improve understanding of pathophysiology and augment disease risk prediction. Progress has been uneven, with most efforts focused on common variants and populations of European ancestry. However, concerted efforts are being made to improve the diversity of genetic studies, in part facilitated by the emergence and growth of biobanks. Larger and more diverse study populations with both clinical and genetic phenotyping, along with the improving affordability of sequencing technologies and the continued development of analytical tools, contribute to an optimistic outlook for the future. Whether this newfound knowledge will translate into improved patient care depends on the success of basic science to identify causal risk genes and their mechanisms, the development of polygenic scores with enhanced predictive ability, and the opportunity to design and execute genetically based and outcomes-driven clinical trials.
List of Abbreviations
- A1c
glycated/glycosylated hemoglobin
- AUROC
area under the receiver operator characteristic curve
- BMI
body mass index
- gePS
global extended polygenic score
- GLP-1
glucagon-like peptide-1
- GWAS
genome-wide association study
- LD
linkage disequilibrium
- MODY
maturity-onset diabetes of the young
- PAM
peptidylglycine alpha-amidating monooxygenase
- pPS
partitioned polygenic score
- rsPS
restricted-to-significant polygenic score
- SNP
single nucleotide polymorphism
Funding
Dr. Kreienkamp is supported by a training grant from the National Institute of General Medical Sciences (T32GM774844). Dr. Voight is supported by a grant from the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK; UM1DK126194). Dr. Gloyn is a Wellcome Senior Fellow in Basic Biomedical Science and is funded by Wellcome (200837), the NIDDK (UM1DK126185), and the Stanford Diabetes Research Center (P30DK116074). Dr. Udler is supported by the NIDDK (K23DK114551, R03DK131249), the Clinical Scientist Development Award from the Doris Duke Charitable Foundation, and the Massachusetts General Hospital Transformative Scholars Award.
Acknowledgments
The authors thank Jason Flannick for code and time provided to help generate Figure 2 and Kirk Smith for his assistance in developing Figure 7.
This is an update of: Florez JC, Udler MS, Hanson RL: Genetics of Type 2 Diabetes. Chapter 14 in Diabetes in America, 3rd ed. Cowie CC, Casagrande SS, Menke A, Cissell MA, Eberhardt MS, Meigs JB, Gregg EW, Knowler WC, Barrett-Connor E, Becker DJ, Brancati FL, Boyko EJ, Herman WH, Howard BV, Narayan KMV, Rewers M, Fradkin JE, Eds. Bethesda, MD, National Institutes of Health, NIH Pub No. 17-1468, 2018, p. 14.1–14.25
Article History
Received in final form on August 23, 2023.
References
- 1.
- Barnett AH, Eff C, Leslie RD, Pyke DA. Diabetes in identical twins. A study of 200 pairs. Diabetologia. 1981;20(2):87-93. doi:10.1007/BF00262007 [PubMed: 7193616] [CrossRef]
- 2.
- Newman B, Selby JV, King MC, Slemenda C, Fabsitz R, Friedman GD. Concordance for type 2 (non-insulin-dependent) diabetes mellitus in male twins. Diabetologia. 1987;30(10):763-768. doi:10.1007/BF00275741 [PubMed: 3428496] [CrossRef]
- 3.
- Kaprio J, Tuomilehto J, Koskenvuo M, et al. Concordance for type 1 (insulin-dependent) and type 2 (non-insulin-dependent) diabetes mellitus in a population-based cohort of twins in Finland. Diabetologia. 1992;35(11):1060-1067. doi:10.1007/BF02221682 [PubMed: 1473616] [CrossRef]
- 4.
- Meigs JB, Muller DC, Nathan DM, Blake DR, Andres R, Baltimore Longitudinal Study of Aging. The natural history of progression from normal glucose tolerance to type 2 diabetes in the Baltimore Longitudinal Study of Aging. Diabetes. 2003;52(6):1475-1484. doi:10.2337/diabetes.52.6.1475 [PubMed: 12765960] [CrossRef]
- 5.
- Poulsen P, Kyvik KO, Vaag A, Beck-Nielsen H. Heritability of type II (non-insulin-dependent) diabetes mellitus and abnormal glucose tolerance—a population-based twin study. Diabetologia. 1999;42(2):139-145. doi:10.1007/s001250051131 [PubMed: 10064092] [CrossRef]
- 6.
- Bennett PH, Burch TA, Miller M. Diabetes mellitus in American (Pima) Indians. Lancet. 1971;2(7716):125-128. doi:10.1016/s0140-6736(71)92303-8 [PubMed: 4104461] [CrossRef]
- 7.
- Ferrannini E, Nannipieri M, Williams K, Gonzales C, Haffner SM, Stern MP. Mode of onset of type 2 diabetes from normal or impaired glucose tolerance. Diabetes. 2004;53(1):160-165. doi:10.2337/diabetes.53.1.160 [PubMed: 14693710] [CrossRef]
- 8.
- Knowler WC, Pettitt DJ, Saad MF, Bennett PH. Diabetes mellitus in the Pima Indians: incidence, risk factors and pathogenesis. Diabetes Metab Rev. 1990;6(1):1-27. doi:10.1002/dmr.5610060101 [PubMed: 2192853] [CrossRef]
- 9.
- Lyssenko V, Almgren P, Anevski D, et al. Predictors of and longitudinal changes in insulin sensitivity and secretion preceding onset of type 2 diabetes. Diabetes. 2005;54(1):166-174. doi:10.2337/diabetes.54.1.166 [PubMed: 15616025] [CrossRef]
- 10.
- Wilson PW, Meigs JB, Sullivan L, Fox CS, Nathan DM, D’Agostino RB, Sr. Prediction of incident diabetes mellitus in middle-aged adults: the Framingham Offspring Study. Arch Intern Med. 2007;167(10):1068-1074. doi:10.1001/archinte.167.10.1068 [PubMed: 17533210] [CrossRef]
- 11.
- Velho G, Froguel P. Genetic, metabolic and clinical characteristics of maturity onset diabetes of the young. Eur J Endocrinol. 1998;138(3):233-239. doi:10.1530/eje.0.1380233 [PubMed: 9539292] [CrossRef]
- 12.
- Fajans SS, Bell GI, Polonsky KS. Molecular mechanisms and clinical pathophysiology of maturity-onset diabetes of the young. N Engl J Med. 2001;345(13):971-980. doi:10.1056/NEJMra002168 [PubMed: 11575290] [CrossRef]
- 13.
- Almgren P, Lehtovirta M, Isomaa B, et al. Heritability and familiality of type 2 diabetes and related quantitative traits in the Botnia Study. Diabetologia. 2011;54(11):2811-2819. doi:10.1007/s00125-011-2267-5 [PubMed: 21826484] [CrossRef]
- 14.
- Avery AR, Duncan GE. Heritability of type 2 diabetes in the Washington State Twin Registry. Twin Res Hum Genet. 2019;22(2):95-98. doi:10.1017/thg.2019.11 [PMC free article: PMC6557115] [PubMed: 31124778] [CrossRef]
- 15.
- Willemsen G, Ward KJ, Bell CG, et al. The concordance and heritability of type 2 diabetes in 34,166 twin pairs from international twin registers: the Discordant Twin (DISCOTWIN) Consortium. Twin Res Hum Genet. 2015;18(6):762-771. doi:10.1017/thg.2015.83 [PubMed: 26678054] [CrossRef]
- 16.
- Hanson RL, Van Hout CV, Hsueh WC, et al. Assessment of the potential role of natural selection in type 2 diabetes and related traits across human continental ancestry groups: comparison of phenotypic with genotypic divergence. Diabetologia. 2020;63(12):2616-2627. doi:10.1007/s00125-020-05272-8 [PMC free article: PMC7642101] [PubMed: 32886191] [CrossRef]
- 17.
- Florez JC, Udler MS, Hanson RL. Genetics of Type 2 Diabetes. In: Cowie CC, Casagrande SS, Menke A, et al, eds. Diabetes in America. 3rd ed. National Institutes of Health; 2018:14.1-14.25:chap 14. Accessed December 12, 2023. https://www
.ncbi.nlm .nih.gov/books/NBK567998/ - 18.
- Vaxillaire M, Froguel P. Genetic basis of maturity-onset diabetes of the young. Endocrinol Metab Clin North Am. 2006;35(2):371-384. doi:10.1016/j.ecl.2006.02.009 [PubMed: 16632099] [CrossRef]
- 19.
- Gloyn AL, Pearson ER, Antcliff JF, et al. Activating mutations in the gene encoding the ATP-sensitive potassium-channel subunit Kir6.2 and permanent neonatal diabetes. N Engl J Med. 2004;350(18):1838-1849. doi:10.1056/NEJMoa032922 [PubMed: 15115830] [CrossRef]
- 20.
- Babenko AP, Polak M, Cavé H, et al. Activating mutations in the ABCC8 gene in neonatal diabetes mellitus. N Engl J Med. 2006;355(5):456-466. doi:10.1056/NEJMoa055068 [PubMed: 16885549] [CrossRef]
- 21.
- Sagen JV, Ræder H, Hathout E, et al. Permanent neonatal diabetes due to mutations in KCNJ11 encoding Kir6.2: patient characteristics and initial response to sulfonylurea therapy. Diabetes. 2004;53(10):2713-2718. doi:10.2337/diabetes.53.10.2713 [PubMed: 15448106] [CrossRef]
- 22.
- Salguero MV, Arosemena M, Pollin T, et al. Monogenic Forms of Diabetes. In: Lawrence JM, Casagrande SS, Herman WH, Wexler DJ, Cefalu WT, eds. Diabetes in America. eBook. National Institutes of Health; 2023. https://www
.ncbi.nlm .nih.gov/books/n/diaonline/monogenic - 23.
- Altshuler D, Hirschhorn JN, Klannemark M, et al. The common PPARgamma Pro12Ala polymorphism is associated with decreased risk of type 2 diabetes. Nat Genet. 2000;26(1):76-80. doi:10.1038/79216 [PubMed: 10973253] [CrossRef]
- 24.
- Gloyn AL, Weedon MN, Owen KR, et al. Large-scale association studies of variants in genes encoding the pancreatic β-cell KATP channel subunits Kir6.2 (KCNJ11) and SUR1 (ABCC8) confirm that the KCNJ11 E23K variant is associated with type 2 diabetes. Diabetes. 2003;52(2):568-572. doi:10.2337/diabetes.52.2.568 [PubMed: 12540637] [CrossRef]
- 25.
- Grant SF, Thorleifsson G, Reynisdottir I, et al. Variant of transcription factor 7-like 2 (TCF7L2) gene confers risk of type 2 diabetes. Nat Genet. 2006;38(3):320-323. doi:10.1038/ng1732 [PubMed: 16415884] [CrossRef]
- 26.
- Cauchi S, El Achhab Y, Choquet H, et al. TCF7L2 is reproducibly associated with type 2 diabetes in various ethnic groups: a global meta-analysis. J Mol Med (Berl). 2007;85(7):777-782. doi:10.1007/s00109-007-0203-4 [PubMed: 17476472] [CrossRef]
- 27.
- Udler MS, Florez JC. Diabetes genomics. In: Ginsburg GS, Willard HF, David SP, eds. Genomic and Personalized Medicine: Primary Care. 3rd ed. Academic Press; 2017:245-282.
- 28.
- McCarthy MI, Abecasis GR, Cardon LR, et al. Genome-wide association studies for complex traits: consensus, uncertainty and challenges. Nat Rev Genet. 2008;9(5):356-369. doi:10.1038/nrg2344 [PubMed: 18398418] [CrossRef]
- 29.
- Sladek R, Rocheleau G, Rung J, et al. A genome-wide association study identifies novel risk loci for type 2 diabetes. Nature. 2007;445(7130):881-885. doi:10.1038/nature05616 [PubMed: 17293876] [CrossRef]
- 30.
- Diabetes Genetics Initiative of Broad Institute of Harvard and MIT, Lund University, Novartis Institutes of BioMedical Research, et al. Genome-wide association analysis identifies loci for type 2 diabetes and triglyceride levels. Science. 2007;316(5829):1331-1336. doi:10.1126/science.1142358 [PubMed: 17463246] [CrossRef]
- 31.
- Zeggini E, Weedon MN, Lindgren CM, et al. Replication of genome-wide association signals in UK samples reveals risk loci for type 2 diabetes. Science. 2007;316(5829):1336-1341. doi:10.1126/science.1142364 [PMC free article: PMC3772310] [PubMed: 17463249] [CrossRef]
- 32.
- Scott LJ, Mohlke KL, Bonnycastle LL, et al. A genome-wide association study of type 2 diabetes in Finns detects multiple susceptibility variants. Science. 2007;316(5829):1341-1345. doi:10.1126/science.1142382 [PMC free article: PMC3214617] [PubMed: 17463248] [CrossRef]
- 33.
- Steinthorsdottir V, Thorleifsson G, Reynisdottir I, et al. A variant in CDKAL1 influences insulin response and risk of type 2 diabetes. Nat Genet. 2007;39(6):770-775. doi:10.1038/ng2043 [PubMed: 17460697] [CrossRef]
- 34.
- Zeggini E, Scott LJ, Saxena R, et al. Meta-analysis of genome-wide association data and large-scale replication identifies additional susceptibility loci for type 2 diabetes. Nat Genet. 2008;40(5):638-645. doi:10.1038/ng.120 [PMC free article: PMC2672416] [PubMed: 18372903] [CrossRef]
- 35.
- Voight BF, Scott LJ, Steinthorsdottir V, et al. Twelve type 2 diabetes susceptibility loci identified through large-scale association analysis. Nat Genet. 2010;42(7):579-589. doi:10.1038/ng.609 [PMC free article: PMC3080658] [PubMed: 20581827] [CrossRef]
- 36.
- Scott RA, Scott LJ, Mägi R, et al. An expanded genome-wide association study of type 2 diabetes in Europeans. Diabetes. 2017;66(11):2888-2902. doi:10.2337/db16-1253 [PMC free article: PMC5652602] [PubMed: 28566273] [CrossRef]
- 37.
- Xue A, Wu Y, Zhu Z, et al. Genome-wide association analyses identify 143 risk variants and putative regulatory mechanisms for type 2 diabetes. Nat Commun. 2018;9(1):2941. doi:10.1038/s41467-018-04951-w [PMC free article: PMC6063971] [PubMed: 30054458] [CrossRef]
- 38.
- Mahajan A, Taliun D, Thurner M, et al. Fine-mapping type 2 diabetes loci to single-variant resolution using high-density imputation and islet-specific epigenome maps. Nat Genet. 2018;50(11):1505-1513. doi:10.1038/s41588-018-0241-6 [PMC free article: PMC6287706] [PubMed: 30297969] [CrossRef]
- 39.
- Morris AP, Voight BF, Teslovich TM, et al. Large-scale association analysis provides insights into the genetic architecture and pathophysiology of type 2 diabetes. Nat Genet. 2012;44(9):981-990. doi:10.1038/ng.2383 [PMC free article: PMC3442244] [PubMed: 22885922] [CrossRef]
- 40.
- Scott RA, Lagou V, Welch RP, et al. Large-scale association analyses identify new loci influencing glycemic traits and provide insight into the underlying biological pathways. Nat Genet. 2012;44(9):991-1005. doi:10.1038/ng.2385 [PMC free article: PMC3433394] [PubMed: 22885924] [CrossRef]
- 41.
- Dupuis J, Langenberg C, Prokopenko I, et al. New genetic loci implicated in fasting glucose homeostasis and their impact on type 2 diabetes risk. Nat Genet. 2010;42(2):105-116. doi:10.1038/ng.520 [PMC free article: PMC3018764] [PubMed: 20081858] [CrossRef]
- 42.
- Knowles JW, Xie W, Zhang Z, et al. Identification and validation of N-acetyltransferase 2 as an insulin sensitivity gene. J Clin Invest. 2015;125(4):1739-1751. doi:10.1172/JCI74692 [PMC free article: PMC4409020] [PubMed: 25798622] [CrossRef]
- 43.
- Manning AK, Hivert MF, Scott RA, et al. A genome-wide approach accounting for body mass index identifies genetic variants influencing fasting glycemic traits and insulin resistance. Nat Genet. 2012;44(6):659-669. doi:10.1038/ng.2274 [PMC free article: PMC3613127] [PubMed: 22581228] [CrossRef]
- 44.
- Prokopenko I, Poon W, Mägi R, et al. A central role for GRB10 in regulation of islet function in man. PLoS Genet. 2014;10(4):e1004235. doi:10.1371/journal.pgen.1004235 [PMC free article: PMC3974640] [PubMed: 24699409] [CrossRef]
- 45.
- Saxena R, Hivert MF, Langenberg C, et al. Genetic variation in GIPR influences the glucose and insulin responses to an oral glucose challenge. Nat Genet. 2010;42(2):142-148. doi:10.1038/ng.521 [PMC free article: PMC2922003] [PubMed: 20081857] [CrossRef]
- 46.
- Soranzo N, Sanna S, Wheeler E, et al. Common variants at 10 genomic loci influence hemoglobin A(1C) levels via glycemic and nonglycemic pathways. Diabetes. 2010;59(12):3229-3239. doi:10.2337/db10-0502 [PMC free article: PMC2992787] [PubMed: 20858683] [CrossRef]
- 47.
- Strawbridge RJ, Dupuis J, Prokopenko I, et al. Genome-wide association identifies nine common variants associated with fasting proinsulin levels and provides new insights into the pathophysiology of type 2 diabetes. Diabetes. 2011;60(10):2624-2634. doi:10.2337/db11-0415 [PMC free article: PMC3178302] [PubMed: 21873549] [CrossRef]
- 48.
- Chen J, Spracklen CN, Marenne G, et al. The trans-ancestral genomic architecture of glycemic traits. Nat Genet. 2021;53(6):840-860. doi:10.1038/s41588-021-00852-9 [PMC free article: PMC7610958] [PubMed: 34059833] [CrossRef]
- 49.
- Lagou V, Mägi R, Hottenga JJ, et al. Sex-dimorphic genetic effects and novel loci for fasting glucose and insulin variability. Nat Commun. 2021;12(1):24. doi:10.1038/s41467-020-19366-9 [PMC free article: PMC7785747] [PubMed: 33402679] [CrossRef]
- 50.
- Wheeler E, Leong A, Liu CT, et al. Impact of common genetic determinants of hemoglobin A1c on type 2 diabetes risk and diagnosis in ancestrally diverse populations: a transethnic genome-wide meta-analysis. PLoS Med. 2017;14(9):e1002383. doi:10.1371/journal.pmed.1002383 [PMC free article: PMC5595282] [PubMed: 28898252] [CrossRef]
- 51.
- Lagou V, Jiang L, Ulrich A, et al. Random glucose GWAS in 493,036 individuals provides insights into diabetes pathophysiology, complications and treatment stratification. medRxiv. 2021. doi:10.1101/2021.04.17.21255471 [PMC free article: PMC10484788] [PubMed: 37679419] [CrossRef]
- 52.
- Below JE, Gamazon ER, Morrison JV, et al. Genome-wide association and meta-analysis in populations from Starr County, Texas, and Mexico City identify type 2 diabetes susceptibility loci and enrichment for expression quantitative trait loci in top signals. Diabetologia. 2011;54(8):2047-2055. doi:10.1007/s00125-011-2188-3 [PMC free article: PMC3761075] [PubMed: 21647700] [CrossRef]
- 53.
- Kooner JS, Saleheen D, Sim X, et al. Genome-wide association study in individuals of South Asian ancestry identifies six new type 2 diabetes susceptibility loci. Nat Genet. 2011;43(10):984-989. doi:10.1038/ng.921 [PMC free article: PMC3773920] [PubMed: 21874001] [CrossRef]
- 54.
- Palmer ND, McDonough CW, Hicks PJ, et al. A genome-wide association search for type 2 diabetes genes in African Americans. PLoS One. 2012;7(1):e29202. doi:10.1371/journal.pone.0029202 [PMC free article: PMC3251563] [PubMed: 22238593] [CrossRef]
- 55.
- Parra EJ, Below JE, Krithika S, et al. Genome-wide association study of type 2 diabetes in a sample from Mexico City and a meta-analysis of a Mexican-American sample from Starr County, Texas. Diabetologia. 2011;54(8):2038-2046. doi:10.1007/s00125-011-2172-y [PMC free article: PMC3818640] [PubMed: 21573907] [CrossRef]
- 56.
- Shu XO, Long J, Cai Q, et al. Identification of new genetic risk variants for type 2 diabetes. PLoS Genet. 2010;6(9):e1001127. doi:10.1371/journal.pgen.1001127 [PMC free article: PMC2940731] [PubMed: 20862305] [CrossRef]
- 57.
- Tsai FJ, Yang CF, Chen CC, et al. A genome-wide association study identifies susceptibility variants for type 2 diabetes in Han Chinese. PLoS Genet. 2010;6(2):e1000847. doi:10.1371/journal.pgen.1000847 [PMC free article: PMC2824763] [PubMed: 20174558] [CrossRef]
- 58.
- Unoki H, Takahashi A, Kawaguchi T, et al. SNPs in KCNQ1 are associated with susceptibility to type 2 diabetes in East Asian and European populations. Nat Genet. 2008;40(9):1098-1102. doi:10.1038/ng.208 [PubMed: 18711366] [CrossRef]
- 59.
- Yamauchi T, Hara K, Maeda S, et al. A genome-wide association study in the Japanese population identifies susceptibility loci for type 2 diabetes at UBE2E2 and C2CD4A-C2CD4B. Nat Genet. 2010;42(10):864-868. doi:10.1038/ng.660 [PubMed: 20818381] [CrossRef]
- 60.
- Yasuda K, Miyake K, Horikawa Y, et al. Variants in KCNQ1 are associated with susceptibility to type 2 diabetes mellitus. Nat Genet. 2008;40(9):1092-1097. doi:10.1038/ng.207 [PubMed: 18711367] [CrossRef]
- 61.
- Cho YS, Chen CH, Hu C, et al. Meta-analysis of genome-wide association studies identifies eight new loci for type 2 diabetes in east Asians. Nat Genet. 2011;44(1):67-72. doi:10.1038/ng.1019 [PMC free article: PMC3582398] [PubMed: 22158537] [CrossRef]
- 62.
- Go MJ, Hwang JY, Park TJ, et al. Genome-wide association study identifies two novel loci with sex-specific effects for type 2 diabetes mellitus and glycemic traits in a Korean population. Diabetes Metab J. 2014;38(5):375-387. doi:10.4093/dmj.2014.38.5.375 [PMC free article: PMC4209352] [PubMed: 25349825] [CrossRef]
- 63.
- Li H, Gan W, Lu L, et al. A genome-wide association study identifies GRK5 and RASGRP1 as type 2 diabetes loci in Chinese Hans. Diabetes. 2013;62(1):291-298. doi:10.2337/db12-0454 [PMC free article: PMC3526061] [PubMed: 22961080] [CrossRef]
- 64.
- Saxena R, Elbers CC, Guo Y, et al. Large-scale gene-centric meta-analysis across 39 studies identifies type 2 diabetes loci. Am J Hum Genet. 2012;90(3):410-425. doi:10.1016/j.ajhg.2011.12.022 [PMC free article: PMC3309185] [PubMed: 22325160] [CrossRef]
- 65.
- Saxena R, Saleheen D, Been LF, et al. Genome-wide association study identifies a novel locus contributing to type 2 diabetes susceptibility in Sikhs of Punjabi origin from India. Diabetes. 2013;62(5):1746-1755. doi:10.2337/db12-1077 [PMC free article: PMC3636649] [PubMed: 23300278] [CrossRef]
- 66.
- Tabassum R, Chauhan G, Dwivedi OP, et al. Genome-wide association study for type 2 diabetes in Indians identifies a new susceptibility locus at 2q21. Diabetes. 2013;62(3):977-986. doi:10.2337/db12-0406 [PMC free article: PMC3581193] [PubMed: 23209189] [CrossRef]
- 67.
- SIGMA Type 2 Diabetes Consortium, Williams AL, Jacobs SB, et al. Sequence variants in SLC16A11 are a common risk factor for type 2 diabetes in Mexico. Nature. 2014;506(7486):97-101. doi:10.1038/nature12828 [PMC free article: PMC4127086] [PubMed: 24390345] [CrossRef]
- 68.
- Hanson RL, Muller YL, Kobes S, et al. A genome-wide association study in American Indians implicates DNER as a susceptibility locus for type 2 diabetes. Diabetes. 2014;63(1):369-376. doi:10.2337/db13-0416 [PMC free article: PMC3868048] [PubMed: 24101674] [CrossRef]
- 69.
- Hara K, Fujita H, Johnson TA, et al. Genome-wide association study identifies three novel loci for type 2 diabetes. Hum Mol Genet. 2014;23(1):239-246. doi:10.1093/hmg/ddt399 [PubMed: 23945395] [CrossRef]
- 70.
- Ng MC, Shriner D, Chen BH, et al. Meta-analysis of genome-wide association studies in African Americans provides insights into the genetic architecture of type 2 diabetes. PLoS Genet. 2014;10(8):e1004517. doi:10.1371/journal.pgen.1004517 [PMC free article: PMC4125087] [PubMed: 25102180] [CrossRef]
- 71.
- Suzuki K, Akiyama M, Ishigaki K, et al. Identification of 28 new susceptibility loci for type 2 diabetes in the Japanese population. Nat Genet. 2019;51(3):379-386. doi:10.1038/s41588-018-0332-4 [PubMed: 30718926] [CrossRef]
- 72.
- Adeyemo AA, Zaghloul NA, Chen G, et al. ZRANB3 is an African-specific type 2 diabetes locus associated with beta-cell mass and insulin response. Nat Commun. 2019;10(1):3195. doi:10.1038/s41467-019-10967-7 [PMC free article: PMC6642147] [PubMed: 31324766] [CrossRef]
- 73.
- Wojcik GL, Graff M, Nishimura KK, et al. Genetic analyses of diverse populations improves discovery for complex traits. Nature. 2019;570(7762):514-518. doi:10.1038/s41586-019-1310-4 [PMC free article: PMC6785182] [PubMed: 31217584] [CrossRef]
- 74.
- DIAbetes Genetics Replication and Meta-analysis (DIAGRAM) Consortium, Asian Genetic Epidemiology Network Type 2 Diabetes (AGEN-T2D) Consortium, South Asian Type 2 Diabetes (SAT2D) Consortium, et al. Genome-wide trans-ancestry meta-analysis provides insight into the genetic architecture of type 2 diabetes susceptibility. Nat Genet. 2014;46(3):234-244. doi:10.1038/ng.2897 [PMC free article: PMC3969612] [PubMed: 24509480] [CrossRef]
- 75.
- Polfus LM, Darst BF, Highland H, et al. Genetic discovery and risk characterization in type 2 diabetes across diverse populations. HGG Adv. 2021;2(2):1000029. doi:10.1016/j.xhgg.2021.100029 [PMC free article: PMC8486151] [PubMed: 34604815] [CrossRef]
- 76.
- Vujkovic M, Keaton JM, Lynch JA, et al. Discovery of 318 new risk loci for type 2 diabetes and related vascular outcomes among 1.4 million participants in a multi-ancestry meta-analysis. Nat Genet. 2020;52(7):680-691. doi:10.1038/s41588-020-0637-y [PMC free article: PMC7343592] [PubMed: 32541925] [CrossRef]
- 77.
- Mahajan A, Spracklen CN, Zhang W, et al. Multi-ancestry genetic study of type 2 diabetes highlights the power of diverse populations for discovery and translation. Nat Genet. 2022;54(5):560-572. doi:10.1038/s41588-022-01058-3 [PMC free article: PMC9179018] [PubMed: 35551307] [CrossRef]
- 78.
- Loh M, Zhang W, Ng HK, et al. Identification of genetic effects underlying type 2 diabetes in South Asian and European populations. Commun Biol. 2022;5(1):329. doi:10.1038/s42003-022-03248-5 [PMC free article: PMC8991226] [PubMed: 35393509] [CrossRef]
- 79.
- Mägi R, Horikoshi M, Sofer T, et al. Trans-ethnic meta-regression of genome-wide association studies accounting for ancestry increases power for discovery and improves fine-mapping resolution. Hum Mol Genet. 2017;26(18):3639-3650. doi:10.1093/hmg/ddx280 [PMC free article: PMC5755684] [PubMed: 28911207] [CrossRef]
- 80.
- Hou K, Ding Y, Xu Z, et al. Causal effects on complex traits are similar for common variants across segments of different continental ancestries within admixed individuals. Nat Genet. 2023;55(4):549-558. doi:10.1038/s41588-023-01338-6 [PMC free article: PMC11120833] [PubMed: 36941441] [CrossRef]
- 81.
- Suzuki K, Hatzikotoulas K, Southam L, et al. Multi-ancestry genome-wide study in >2.5 million individuals reveals heterogeneity in mechanistic pathways of type 2 diabetes and complications. medRxiv. 2023. doi:10.1101/2023.03.31.23287839 [CrossRef]
- 82.
- Moltke I, Grarup N, Jørgensen ME, et al. A common Greenlandic TBC1D4 variant confers muscle insulin resistance and type 2 diabetes. Nature. 2014;512(7513):190-193. doi:10.1038/nature13425 [PubMed: 25043022] [CrossRef]
- 83.
- Voight BF, Kang HM, Ding J, et al. The Metabochip, a custom genotyping array for genetic studies of metabolic, cardiovascular, and anthropometric traits. PLoS Genet. 2012;8(8):e1002793. doi:10.1371/journal.pgen.1002793 [PMC free article: PMC3410907] [PubMed: 22876189] [CrossRef]
- 84.
- The 1000 Genomes Project Consortium. An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491(7422):56-65. doi:10.1038/nature11632 [PMC free article: PMC3498066] [PubMed: 23128226] [CrossRef]
- 85.
- McCarthy S, Das S, Kretzschmar W, et al. A reference panel of 64,976 haplotypes for genotype imputation. Nat Genet. 2016;48(10):1279-1283. doi:10.1038/ng.3643 [PMC free article: PMC5388176] [PubMed: 27548312] [CrossRef]
- 86.
- Taliun D, Harris DN, Kessler MD, et al. Sequencing of 53,831 diverse genomes from the NHLBI TOPMed Program. Nature. 2021;590(7845):290-299. doi:10.1038/s41586-021-03205-y [PMC free article: PMC7875770] [PubMed: 33568819] [CrossRef]
- 87.
- Grove ML, Yu B, Cochran BJ, et al. Best practices and joint calling of the HumanExome BeadChip: the CHARGE Consortium. PLoS One. 2013;8(7):e68095. doi:10.1371/journal.pone.0068095 [PMC free article: PMC3709915] [PubMed: 23874508] [CrossRef]
- 88.
- Pulit SL, Voight BF, de Bakker PIW. Multiethnic genetic association studies improve power for locus discovery. PLoS One. 2010;5(9):e12600. doi:10.1371/journal.pone.0012600 [PMC free article: PMC2935880] [PubMed: 20838612] [CrossRef]
- 89.
- Mahajan A, Wessel J, Willems SM, et al. Refining the accuracy of validated target identification through coding variant fine-mapping in type 2 diabetes. Nat Genet. 2018;50(4):559-571. doi:10.1038/s41588-018-0084-1 [PMC free article: PMC5898373] [PubMed: 29632382] [CrossRef]
- 90.
- SIGMA Type 2 Diabetes Consortium, Estrada K, Aukrust I, et al. Association of a low-frequency variant in HNF1A with type 2 diabetes in a Latino population. JAMA. 2014;311(22):2305-2314. doi:10.1001/jama.2014.6511 [PMC free article: PMC4425850] [PubMed: 24915262] [CrossRef]
- 91.
- Flannick J, Mercader JM, Fuchsberger C, et al. Exome sequencing of 20,791 cases of type 2 diabetes and 24,440 controls. Nature. 2019;570(7759):71-76. doi:10.1038/s41586-019-1231-2 [PMC free article: PMC6699738] [PubMed: 31118516] [CrossRef]
- 92.
- Steinthorsdottir V, Thorleifsson G, Sulem P, et al. Identification of low-frequency and rare sequence variants associated with elevated or reduced risk of type 2 diabetes. Nat Genet. 2014;46(3):294-298. doi:10.1038/ng.2882 [PubMed: 24464100] [CrossRef]
- 93.
- Fuchsberger C, Flannick J, Teslovich TM, et al. The genetic architecture of type 2 diabetes. Nature. 2016;536(7614):41-47. doi:10.1038/nature18642 [PMC free article: PMC5034897] [PubMed: 27398621] [CrossRef]
- 94.
- Albrechtsen A, Grarup N, Li Y, et al. Exome sequencing-driven discovery of coding polymorphisms associated with common metabolic phenotypes. Diabetologia. 2013;56(2):298-310. doi:10.1007/s00125-012-2756-1 [PMC free article: PMC3536959] [PubMed: 23160641] [CrossRef]
- 95.
- Flannick J, Thorleifsson G, Beer NL, et al. Loss-of-function mutations in SLC30A8 protect against type 2 diabetes. Nat Genet. 2014;46(4):357-363. doi:10.1038/ng.2915 [PMC free article: PMC4051628] [PubMed: 24584071] [CrossRef]
- 96.
- Backman JD, Li AH, Marcketta A, et al. Exome sequencing and analysis of 454,787 UK Biobank participants. Nature. 2021;599(7886):628-634. doi:10.1038/s41586-021-04103-z [PMC free article: PMC8596853] [PubMed: 34662886] [CrossRef]
- 97.
- Curtis D. Analysis of rare coding variants in 200,000 exome-sequenced subjects reveals novel genetic risk factors for type 2 diabetes. Diabetes Metab Res Rev. 2022;38(1):e3482. doi:10.1002/dmrr.3482 [PubMed: 34216101] [CrossRef]
- 98.
- Deaton AM, Parker MM, Ward LD, et al. Gene-level analysis of rare variants in 379,066 whole exome sequences identifies an association of GIGYF1 loss of function with type 2 diabetes. Sci Rep. 2021;11(1):21565. doi:10.1038/s41598-021-99091-5 [PMC free article: PMC8566487] [PubMed: 34732801] [CrossRef]
- 99.
- Nag A, Dhindsa RS, Mitchell J, et al. Human genetics uncovers MAP3K15 as an obesity-independent therapeutic target for diabetes. Sci Adv. 2022;8(46):eadd5430. doi:10.1126/sciadv.add5430 [PMC free article: PMC9668288] [PubMed: 36383675] [CrossRef]
- 100.
- Li Z, Li X, Zhou H, et al. A framework for detecting noncoding rare-variant associations of large-scale whole-genome sequencing studies. Nat Methods. 2022;19(12):1599-1611. doi:10.1038/s41592-022-01640-x [PMC free article: PMC10008172] [PubMed: 36303018] [CrossRef]
- 101.
- Wessel J, Majarian TD, Highland HM, et al. Rare non-coding variation identified by large scale whole genome sequencing reveals unexplained heritability of type 2 diabetes. medRxiv. 2020. doi:10.1101/2020.11.13.20221812 [CrossRef]
- 102.
- Schaid DJ, Chen W, Larson NB. From genome-wide associations to candidate causal variants by statistical fine-mapping. Nat Rev Genet. 2018;19(8):491-504. doi:10.1038/s41576-018-0016-z [PMC free article: PMC6050137] [PubMed: 29844615] [CrossRef]
- 103.
- Carrat GR, Hu M, Nguyen-Tu MS, et al. Decreased STARD10 expression is associated with defective insulin secretion in humans and mice. Am J Hum Genet. 2017;100(2):238-256. doi:10.1016/j.ajhg.2017.01.011 [PMC free article: PMC5294761] [PubMed: 28132686] [CrossRef]
- 104.
- Gaulton KJ, Ferreira T, Lee Y, et al. Genetic fine mapping and genomic annotation defines causal mechanisms at type 2 diabetes susceptibility loci. Nat Genet. 2015;47(12):1415-1425. doi:10.1038/ng.3437 [PMC free article: PMC4666734] [PubMed: 26551672] [CrossRef]
- 105.
- Liu CT, Raghavan S, Maruthur N, et al. Trans-ethnic meta-analysis and functional annotation illuminates the genetic architecture of fasting glucose and insulin. Am J Hum Genet. 2016;99(1):56-75. doi:10.1016/j.ajhg.2016.05.006 [PMC free article: PMC5005440] [PubMed: 27321945] [CrossRef]
- 106.
- Miguel-Escalada I, Bonàs-Guarch S, Cebola I, et al. Human pancreatic islet three-dimensional chromatin architecture provides insights into the genetics of type 2 diabetes. Nat Genet. 2019;51(7):1137-1148. doi:10.1038/s41588-019-0457-0 [PMC free article: PMC6640048] [PubMed: 31253982] [CrossRef]
- 107.
- Thurner M, van de Bunt M, Torres JM, et al. Integration of human pancreatic islet genomic data refines regulatory mechanisms at type 2 diabetes susceptibility loci. eLife. 2018;7:e31977. doi:10.7554/eLife.31977 [PMC free article: PMC5828664] [PubMed: 29412141] [CrossRef]
- 108.
- Viñuela A, Varshney A, van de Bunt M, et al. Genetic variant effects on gene expression in human pancreatic islets and their implications for T2D. Nat Commun. 2020;11(1):4912. doi:10.1038/s41467-020-18581-8 [PMC free article: PMC7528108] [PubMed: 32999275] [CrossRef]
- 109.
- Dornbos P, Singh P, Jang DK, et al. Evaluating human genetic support for hypothesized metabolic disease genes. Cell Metab. 2022;34(5):661-666. doi:10.1016/j.cmet.2022.03.011 [PMC free article: PMC9166611] [PubMed: 35421386] [CrossRef]
- 110.
- Forgetta V, Jiang L, Vulpescu NA, et al. An effector index to predict target genes at GWAS loci. Hum Genet. 2022;141(8):1431-1447. doi:10.1007/s00439-022-02434-z [PubMed: 35147782] [CrossRef]
- 111.
- Chiou J, Zeng C, Cheng Z, et al. Single-cell chromatin accessibility identifies pancreatic islet cell type- and state-specific regulatory programs of diabetes risk. Nat Genet. 2021;53(4):455-466. doi:10.1038/s41588-021-00823-0 [PMC free article: PMC9037575] [PubMed: 33795864] [CrossRef]
- 112.
- Su C, Gao L, May CL, et al. 3D chromatin maps of the human pancreas reveal lineage-specific regulatory architecture of T2D risk. Cell Metab. 2022;34(9):1394-1409.e4. doi:10.1016/j.cmet.2022.08.014 [PMC free article: PMC9664375] [PubMed: 36070683] [CrossRef]
- 113.
- Raulerson CK, Ko A, Kidd JC, et al. Adipose tissue gene expression associations reveal hundreds of candidate genes for cardiometabolic traits. Am J Hum Genet. 2019;105(4):773-787. doi:10.1016/j.ajhg.2019.09.001 [PMC free article: PMC6817527] [PubMed: 31564431] [CrossRef]
- 114.
- Wu Y, Broadaway KA, Raulerson CK, et al. Colocalization of GWAS and eQTL signals at loci with multiple signals identifies additional candidate genes for body fat distribution. Hum Mol Genet. 2019;28(24):4161-4172. doi:10.1093/hmg/ddz263 [PMC free article: PMC7202621] [PubMed: 31691812] [CrossRef]
- 115.
- Fulco CP, Nasser J, Jones TR, et al. Activity-by-contact model of enhancer-promoter regulation from thousands of CRISPR perturbations. Nat Genet. 2019;51(12):1664-1669. doi:10.1038/s41588-019-0538-0 [PMC free article: PMC6886585] [PubMed: 31784727] [CrossRef]
- 116.
- Wesolowska-Andersen A, Zhuo Yu G, Nylander V, et al. Deep learning models predict regulatory variants in pancreatic islets and refine type 2 diabetes association signals. eLife. 2020;9:e51503. doi:10.7554/eLife.51503 [PMC free article: PMC7007221] [PubMed: 31985400] [CrossRef]
- 117.
- Rottner AK, Ye Y, Navarro-Guerrero E, et al. A genome-wide CRISPR screen identifies CALCOCO2 as a regulator of beta cell function influencing type 2 diabetes risk. Nat Genet. 2023;55(1):54-65. doi:10.1038/s41588-022-01261-2 [PMC free article: PMC9839450] [PubMed: 36543916] [CrossRef]
- 118.
- Thomsen SK, Ceroni A, van de Bunt M, et al. Systematic functional characterization of candidate causal genes for type 2 diabetes risk variants. Diabetes. 2016;65(12):3805-3811. doi:10.2337/db16-0361 [PMC free article: PMC5402869] [PubMed: 27554474] [CrossRef]
- 119.
- Chimienti F, Devergnas S, Favier A, Seve M. Identification and cloning of a β-cell-specific zinc transporter, ZnT-8, localized into insulin secretory granules. Diabetes. 2004;53(9):2330-2337. doi:10.2337/diabetes.53.9.2330 [PubMed: 15331542] [CrossRef]
- 120.
- Nicolson TJ, Bellomo EA, Wijesekara N, et al. Insulin storage and glucose homeostasis in mice null for the granule zinc transporter ZnT8 and studies of the type 2 diabetes-associated variants. Diabetes. 2009;58(9):2070-2083. doi:10.2337/db09-0551 [PMC free article: PMC2731533] [PubMed: 19542200] [CrossRef]
- 121.
- Pound LD, Sarkar SA, Ustione A, et al. The physiological effects of deleting the mouse SLC30A8 gene encoding zinc transporter-8 are influenced by gender and genetic background. PLoS One. 2012;7(7):e40972. doi:10.1371/journal.pone.0040972 [PMC free article: PMC3400647] [PubMed: 22829903] [CrossRef]
- 122.
- Kleiner S, Gomez D, Megra B, et al. Mice harboring the human SLC30A8 R138X loss-of-function mutation have increased insulin secretory capacity. Proc Natl Acad Sci U S A. 2018;115(32):E7642-E7649. doi:10.1073/pnas.1721418115 [PMC free article: PMC6094147] [PubMed: 30038024] [CrossRef]
- 123.
- Dwivedi OP, Lehtovirta M, Hastoy B, et al. Loss of ZnT8 function protects against diabetes by enhanced insulin secretion. Nat Genet. 2019;51(11):1596-1606. doi:10.1038/s41588-019-0513-9 [PMC free article: PMC6858874] [PubMed: 31676859] [CrossRef]
- 124.
- Rusu V, Hoch E, Mercader JM, et al. Type 2 diabetes variants disrupt function of SLC16A11 through two distinct mechanisms. Cell. 2017;170(1):199-212.e20. doi:10.1016/j.cell.2017.06.011 [PMC free article: PMC5562285] [PubMed: 28666119] [CrossRef]
- 125.
- Huyghe JR, Jackson AU, Fogarty MP, et al. Exome array analysis identifies new loci and low-frequency variants influencing insulin processing and secretion. Nat Genet. 2013;45(2):197-201. doi:10.1038/ng.2507 [PMC free article: PMC3727235] [PubMed: 23263489] [CrossRef]
- 126.
- Eipper BA, Milgram SL, Husten EJ, Yun HY, Mains RE. Peptidylglycine alpha-amidating monooxygenase: a multifunctional protein with catalytic, processing, and routing domains. Protein Sci. 1993;2(4):489-497. doi:10.1002/pro.5560020401 [PMC free article: PMC2142366] [PubMed: 8518727] [CrossRef]
- 127.
- Oyarce AM, Eipper BA. Neurosecretory vesicles contain soluble and membrane-associated monofunctional and bifunctional peptidylglycine alpha-amidating monooxygenase proteins. J Neurochem. 1993;60(3):1105-1114. doi:10.1111/j.1471-4159.1993.tb03261.x [PubMed: 8436961] [CrossRef]
- 128.
- Thomsen SK, Raimondo A, Hastoy B, et al. Type 2 diabetes risk alleles in PAM impact insulin release from human pancreatic β-cells. Nat Genet. 2018;50(8):1122-1131. doi:10.1038/s41588-018-0173-1 [PMC free article: PMC6237273] [PubMed: 30054598] [CrossRef]
- 129.
- Udler MS, McCarthy MI, Florez JC, Mahajan A. Genetic risk scores for diabetes diagnosis and precision medicine. Endocr Rev. 2019;40(6):1500-1520. doi:10.1210/er.2019-00088 [PMC free article: PMC6760294] [PubMed: 31322649] [CrossRef]
- 130.
- Lango H, UK Type 2 Diabetes Genetics Consortium, Palmer CN, et al. Assessing the combined impact of 18 common genetic variants of modest effect sizes on type 2 diabetes risk. Diabetes. 2008;57(11):3129-3135. doi:10.2337/db08-0504 [PMC free article: PMC2570411] [PubMed: 18591388] [CrossRef]
- 131.
- Meigs JB, Shrader P, Sullivan LM, et al. Genotype score in addition to common risk factors for prediction of type 2 diabetes. N Engl J Med. 2008;359(21):2208-2219. doi:10.1056/NEJMoa0804742 [PMC free article: PMC2746946] [PubMed: 19020323] [CrossRef]
- 132.
- Lyssenko V, Jonsson A, Almgren P, et al. Clinical risk factors, DNA variants, and the development of type 2 diabetes. N Engl J Med. 2008;359(21):2220-2232. doi:10.1056/NEJMoa0801869 [PubMed: 19020324] [CrossRef]
- 133.
- Vassy JL, Hivert MF, Porneala B, et al. Polygenic type 2 diabetes prediction at the limit of common variant detection. Diabetes. 2014;63(6):2172-2182. doi:10.2337/db13-1663 [PMC free article: PMC4030114] [PubMed: 24520119] [CrossRef]
- 134.
- Hivert MF, Jablonski KA, Perreault L, et al. Updated genetic score based on 34 confirmed type 2 diabetes loci is associated with diabetes incidence and regression to normoglycemia in the Diabetes Prevention Program. Diabetes. 2011;60(4):1340-1348. doi:10.2337/db10-1119 [PMC free article: PMC3064108] [PubMed: 21378175] [CrossRef]
- 135.
- Khera AV, Chaffin M, Aragam KG, et al. Genome-wide polygenic scores for common diseases identify individuals with risk equivalent to monogenic mutations. Nat Genet. 2018;50(9):1219-1224. doi:10.1038/s41588-018-0183-z [PMC free article: PMC6128408] [PubMed: 30104762] [CrossRef]
- 136.
- Multhaup ML, Kita R, Krock B, et al. The Science Behind 23andMe’s Type 2 Diabetes Report. Updated April 2019. Accessed October 16, 2023. https://permalinks
.23andme .com/pdf/23_19-Type2Diabetes _March2019.pdf - 137.
- Dornbos P, Koesterer R, Ruttenburg A, et al. A combined polygenic score of 21,293 rare and 22 common variants improves diabetes diagnosis based on hemoglobin A1C levels. Nat Genet. 2022;54(11):1609-1614. doi:10.1038/s41588-022-01200-1 [PMC free article: PMC9995082] [PubMed: 36280733] [CrossRef]
- 138.
- Goodrich JK, Singer-Berk M, Son R, et al. Determinants of penetrance and variable expressivity in monogenic metabolic conditions across 77,184 exomes. Nat Commun. 2021;12(1):3505. doi:10.1038/s41467-021-23556-4 [PMC free article: PMC8190084] [PubMed: 34108472] [CrossRef]
- 139.
- Mirshahi UL, Colclough K, Wright CF, et al. Reduced penetrance of MODY-associated HNF1A/HNF4A variants but not GCK variants in clinically unselected cohorts. Am J Hum Genet. 2022;109(11):2018-2028. doi:10.1016/j.ajhg.2022.09.014 [PMC free article: PMC9674944] [PubMed: 36257325] [CrossRef]
- 140.
- Oram RA, Patel K, Hill A, et al. A type 1 diabetes genetic risk score can aid discrimination between type 1 and type 2 diabetes in young adults. Diabetes Care. 2016;39(3):337-344. doi:10.2337/dc15-1111 [PMC free article: PMC5642867] [PubMed: 26577414] [CrossRef]
- 141.
- Redondo MJ, Hagopian WA, Oram R, et al. The clinical consequences of heterogeneity within and between different diabetes types. Diabetologia. 2020;63(10):2040-2048. doi:10.1007/s00125-020-05211-7 [PMC free article: PMC8498993] [PubMed: 32894314] [CrossRef]
- 142.
- Dimas AS, Lagou V, Barker A, et al. Impact of type 2 diabetes susceptibility variants on quantitative glycemic traits reveals mechanistic heterogeneity. Diabetes. 2014;63(6):2158-2171. doi:10.2337/db13-0949 [PMC free article: PMC4030103] [PubMed: 24296717] [CrossRef]
- 143.
- Udler MS, Kim J, von Grotthuss M, et al. Type 2 diabetes genetic loci informed by multi-trait associations point to disease mechanisms and subtypes: a soft clustering analysis. PLoS Med. 2018;15(9):e1002654. doi:10.1371/journal.pmed.1002654 [PMC free article: PMC6150463] [PubMed: 30240442] [CrossRef]
- 144.
- DiCorpo D, LeClair J, Cole JB, et al. Type 2 diabetes partitioned polygenic scores associate with disease outcomes in 454,193 individuals across 13 cohorts. Diabetes Care. 2022;45(3):674-683. doi:10.2337/dc21-1395 [PMC free article: PMC8918228] [PubMed: 35085396] [CrossRef]
- 145.
- Kim H, Westerman KE, Smith K, et al. High-throughput genetic clustering of type 2 diabetes loci reveals heterogeneous mechanistic pathways of metabolic disease. Diabetologia. 2023;66(3):495-507. doi:10.1007/s00125-022-05848-6 [PMC free article: PMC10108373] [PubMed: 36538063] [CrossRef]
- 146.
- Laber S, Strobel S, Mercader JM, et al. Discovering cellular programs of intrinsic and extrinsic drivers of metabolic traits using LipocyteProfiler. Cell Genom. 2023;3(7):100346. doi:10.1016/j.xgen.2023.100346 [PMC free article: PMC10363917] [PubMed: 37492099] [CrossRef]
- 147.
- Deutsch AJ, Ahlqvist E, Udler MS. Phenotypic and genetic classification of diabetes. Diabetologia. 2022;65(11):1758-1769. doi:10.1007/s00125-022-05769-4 [PMC free article: PMC9522707] [PubMed: 35953726] [CrossRef]
- 148.
- Mansour Aly D, Dwivedi OP, Prasad RB, et al. Genome-wide association analyses highlight etiological differences underlying newly defined subtypes of diabetes. Nat Genet. 2021;53(11):1534-1542. doi:10.1038/s41588-021-00948-2 [PubMed: 34737425] [CrossRef]
- 149.
- Nair ATN, Wesolowska-Andersen A, Brorsson C, et al. Heterogeneity in phenotype, disease progression and drug response in type 2 diabetes. Nat Med. 2022;28(5):982-988. doi:10.1038/s41591-022-01790-7 [PubMed: 35534565] [CrossRef]
- 150.
- Ahlqvist E, Storm P, Käräjämäki A, et al. Novel subgroups of adult-onset diabetes and their association with outcomes: a data-driven cluster analysis of six variables. Lancet Diabetes Endocrinol. 2018;6(5):361-369. doi:10.1016/S2213-8587(18)30051-2 [PubMed: 29503172] [CrossRef]
- 151.
- Jordan DM, Verbanck M, Do R. HOPS: a quantitative score reveals pervasive horizontal pleiotropy in human genetic variation is driven by extreme polygenicity of human traits and diseases. Genome Biol. 2019;20(1):222. doi:10.1186/s13059-019-1844-7 [PMC free article: PMC6815001] [PubMed: 31653226] [CrossRef]
- 152.
- Beer NL, Tribble ND, McCulloch LJ, et al. The P446L variant in GCKR associated with fasting plasma glucose and triglyceride levels exerts its effect through increased glucokinase activity in liver. Hum Mol Genet. 2009;18(21):4081-4088. doi:10.1093/hmg/ddp357 [PMC free article: PMC2758140] [PubMed: 19643913] [CrossRef]
- 153.
- Gloudemans MJ, Balliu B, Nachun D, et al. Integration of genetic colocalizations with physiological and pharmacological perturbations identifies cardiometabolic disease genes. Genome Med. 2022;14(1):31. doi:10.1186/s13073-022-01036-8 [PMC free article: PMC8925074] [PubMed: 35292083] [CrossRef]
- 154.
- Scott RA, Freitag DF, Li L, et al. A genomic approach to therapeutic target validation identifies a glucose-lowering GLP1R variant protective for coronary heart disease. Sci Transl Med. 2016;8(341):341ra76. doi:10.1126/scitranslmed.aad3744 [PMC free article: PMC5219001] [PubMed: 27252175] [CrossRef]
- 155.
- Majithia AR, Flannick J, Shahinian P, et al. Rare variants in PPARG with decreased activity in adipocyte differentiation are associated with increased risk of type 2 diabetes. Proc Natl Acad Sci U S A. 2014;111(36):13127-13132. doi:10.1073/pnas.1410428111 [PMC free article: PMC4246964] [PubMed: 25157153] [CrossRef]
- 156.
- Flannick J, Johansson S, Njølstad PR. Common and rare forms of diabetes mellitus: towards a continuum of diabetes subtypes. Nat Rev Endocrinol. 2016;12(7):394-406. doi:10.1038/nrendo.2016.50 [PubMed: 27080136] [CrossRef]
- 157.
- Bonnefond A, Boissel M, Bolze A, et al. Pathogenic variants in actionable MODY genes are associated with type 2 diabetes. Nat Metab. 2020;2(10):1126-1134. doi:10.1038/s42255-020-00294-3 [PubMed: 33046911] [CrossRef]
- 158.
- Davies NM, Holmes MV, Davey Smith G. Reading Mendelian randomisation studies: a guide, glossary, and checklist for clinicians. BMJ. 2018;362:k601. doi:10.1136/bmj.k601 [PMC free article: PMC6041728] [PubMed: 30002074] [CrossRef]
- 159.
- Wittemans LBL, Lotta LA, Langenberg C. Prioritising risk factors for type 2 diabetes: causal inference through genetic approaches. Curr Diab Rep. 2018;18(7):40. doi:10.1007/s11892-018-1009-1 [PMC free article: PMC5960479] [PubMed: 29779155] [CrossRef]
- 160.
- Ruth KS, Day FR, Tyrrell J, et al. Using human genetics to understand the disease impacts of testosterone in men and women. Nat Med. 2020;26(2):252-258. doi:10.1038/s41591-020-0751-5 [PMC free article: PMC7025895] [PubMed: 32042192] [CrossRef]
- 161.
- Fall T, Xie W, Poon W, et al. Using genetic variants to assess the relationship between circulating lipids and type 2 diabetes. Diabetes. 2015;64(7):2676-2684. doi:10.2337/db14-1710 [PubMed: 25948681] [CrossRef]
- 162.
- White J, Swerdlow DI, Preiss D, et al. Association of lipid fractions with risks for coronary artery disease and diabetes. JAMA Cardiol. 2016;1(6):692-699. doi:10.1001/jamacardio.2016.1884 [PMC free article: PMC5642865] [PubMed: 27487401] [CrossRef]
- 163.
- Ridker PM, Danielson E, Fonseca FAH, et al. Rosuvastatin to prevent vascular events in men and women with elevated C-reactive protein. N Engl J Med. 2008;359(21):2195-2207. doi:10.1056/NEJMoa0807646 [PubMed: 18997196] [CrossRef]
- 164.
- Besseling J, Kastelein JJ, Defesche JC, Hutten BA, Hovingh GK. Association between familial hypercholesterolemia and prevalence of type 2 diabetes mellitus. JAMA. 2015;313(10):1029-1036. doi:10.1001/jama.2015.1206 [PubMed: 25756439] [CrossRef]
- 165.
- Spracklen CN, Horikoshi M, Kim YJ, et al. Identification of type 2 diabetes loci in 433,540 East Asian individuals. Nature. 2020;582(7811):240-245. doi:10.1038/s41586-020-2263-3 [PMC free article: PMC7292783] [PubMed: 32499647] [CrossRef]
- 166.
- Ge T, Irvin MR, Patki A, et al. Development and validation of a trans-ancestry polygenic risk score for type 2 diabetes in diverse populations. Genome Med. 2022;14(1):70. doi:10.1186/s13073-022-01074-2 [PMC free article: PMC9241245] [PubMed: 35765100] [CrossRef]
- 167.
- Manning AK, LaValley M, Liu CT, et al. Meta-analysis of gene-environment interaction: joint estimation of SNP and SNP x environment regression coefficients. Genet Epidemiol. 2011;35(1):11-18. doi:10.1002/gepi.20546 [PMC free article: PMC3312394] [PubMed: 21181894] [CrossRef]
- 168.
- Yang J, Loos RJ, Powell JE, et al. FTO genotype is associated with phenotypic variability of body mass index. Nature. 2012;490(7419):267-272. doi:10.1038/nature11401 [PMC free article: PMC3564953] [PubMed: 22982992] [CrossRef]
- 169.
- Wang Z, Emmerich A, Pillon NJ, et al. Genome-wide association analyses of physical activity and sedentary behavior provide insights into underlying mechanisms and roles in disease prevention. Nat Genet. 2022;54(9):1332-1344. doi:10.1038/s41588-022-01165-1 [PMC free article: PMC9470530] [PubMed: 36071172] [CrossRef]
- 170.
- Young AI, Wauthier FL, Donnelly P. Identifying loci affecting trait variability and detecting interactions in genome-wide association studies. Nat Genet. 2018;50(11):1608-1614. doi:10.1038/s41588-018-0225-6 [PubMed: 30323177] [CrossRef]
- 171.
- Takeuchi F, Isono M, Nabika T, et al. Confirmation of ALDH2 as a major locus of drinking behavior and of its variants regulating multiple metabolic phenotypes in a Japanese population. Circ J. 2011;75(4):911-918. doi:10.1253/circj.cj-10-0774 [PubMed: 21372407] [CrossRef]
- 172.
- Tcheandjieu C, Zhu X, Hilliard AT, et al. Large-scale genome-wide association study of coronary artery disease in genetically diverse populations. Nat Med. 2022;28(8):1679-1692. doi:10.1038/s41591-022-01891-3 [PMC free article: PMC9419655] [PubMed: 35915156] [CrossRef]
- 173.
- Klarin D, Lynch J, Aragam K, et al. Genome-wide association study of peripheral artery disease in the Million Veteran Program. Nat Med. 2019;25(8):1274-1279. doi:10.1038/s41591-019-0492-5 [PMC free article: PMC6768096] [PubMed: 31285632] [CrossRef]
- 174.
- Mishra A, Malik R, Hachiya T, et al. Stroke genetics informs drug discovery and risk prediction across ancestries. Nature. 2022;611(7934):115-123. doi:10.1038/s41586-022-05165-3 [PMC free article: PMC9524349] [PubMed: 36180795] [CrossRef]
- 175.
- Stanzick KJ, Li Y, Schlosser P, et al. Discovery and prioritization of variants and genes for kidney function in >1.2 million individuals. Nat Commun. 2021;12(1):4350. doi:10.1038/s41467-021-24491-0 [PMC free article: PMC8285412] [PubMed: 34272381] [CrossRef]
- 176.
- van Zuydam NR, Ladenvall C, Voight BF, et al. Genetic predisposition to coronary artery disease in type 2 diabetes mellitus. Circ Genom Precis Med. 2020;13(6):e002769. doi:10.1161/CIRCGEN.119.002769 [PMC free article: PMC7748049] [PubMed: 33321069] [CrossRef]
- 177.
- O’Connor MJ, Schroeder P, Huerta-Chagoya A, et al. Recessive genome-wide meta-analysis illuminates genetic architecture of type 2 diabetes. Diabetes. 2022;71(3):554-565. doi:10.2337/db21-0545 [PMC free article: PMC8893948] [PubMed: 34862199] [CrossRef]
- 178.
- Kong A, Steinthorsdottir V, Masson G, et al. Parental origin of sequence variants associated with complex diseases. Nature. 2009;462(7275):868-874. doi:10.1038/nature08625 [PMC free article: PMC3746295] [PubMed: 20016592] [CrossRef]
- 179.
- Groop L, Pociot F. Genetics of diabetes—are we missing the genes or the disease? Mol Cell Endocrinol. 2014;382(1):726-739. doi:10.1016/j.mce.2013.04.002 [PubMed: 23587769] [CrossRef]
- 180.
- Tavaglione F, Targher G, Valenti L, Romeo S. Human and molecular genetics shed lights on fatty liver disease and diabetes conundrum. Endocrinol Diabetes Metab. 2020;3(4):e00179. doi:10.1002/edm2.179 [PMC free article: PMC7576307] [PubMed: 33102799] [CrossRef]
- 181.
- Florez JC, Burtt N, de Bakker PI, et al. Haplotype structure and genotype-phenotype correlations of the sulfonylurea receptor and the islet ATP-sensitive potassium channel gene region. Diabetes. 2004;53(5):1360-1368. doi:10.2337/diabetes.53.5.1360 [PubMed: 15111507] [CrossRef]
- 182.
- Wessel J, Chu AY, Willems SM, et al. Low-frequency and rare exome chip variants associate with fasting glucose and type 2 diabetes susceptibility. Nat Commun. 2015;6:5897. doi:10.1038/ncomms6897 [PMC free article: PMC4311266] [PubMed: 25631608] [CrossRef]
- 183.
- Mahajan A, Sim X, Ng HJ, et al. Identification and functional characterization of G6PC2 coding variants influencing glycemic traits define an effector transcript at the G6PC2-ABCB11 locus. PLoS Genet. 2015;11(1):e1004876. doi:10.1371/journal.pgen.1004876 [PMC free article: PMC4307976] [PubMed: 25625282] [CrossRef]
- 184.
- Froguel P, Vaxillaire M, Sun F, et al. Close linkage of glucokinase locus on chromosome 7p to early-onset non-insulin-dependent diabetes mellitus. Nature. 1992;356(6365):162-164. doi:10.1038/356162a0 [PubMed: 1545870] [CrossRef]
- 185.
- Glaser B, Kesavan P, Heyman M, et al. Familial hyperinsulinism caused by an activating glucokinase mutation. N Engl J Med. 1998;338(4):226-230. doi:10.1056/NEJM199801223380404 [PubMed: 9435328] [CrossRef]
- 186.
- Ashcroft FM, Lloyd M, Haythorne EA. Glucokinase activity in diabetes: too much of a good thing? Trends Endocrinol Metab. 2023;34(2):119-130. doi:10.1016/j.tem.2022.12.007 [PubMed: 36586779] [CrossRef]
- 187.
- Zhu D, Li X, Ma J, et al. Dorzagliatin in drug-naïve patients with type 2 diabetes: a randomized, double-blind, placebo-controlled phase 3 trial. Nat Med. 2022;28(5):965-973. doi:10.1038/s41591-022-01802-6 [PubMed: 35551294] [CrossRef]
- 188.
- Kamata K, Mitsuya M, Nishimura T, Eiki J, Nagata Y. Structural basis for allosteric regulation of the monomeric allosteric enzyme human glucokinase. Structure. 2004;12(3):429-438. doi:10.1016/j.str.2004.02.005 [PubMed: 15016359] [CrossRef]
- 189.
- Raimondo A, Rees MG, Gloyn AL. Glucokinase regulatory protein: complexity at the crossroads of triglyceride and glucose metabolism. Curr Opin Lipidol. 2015;26(2):88-95. doi:10.1097/MOL.0000000000000155 [PMC free article: PMC4422901] [PubMed: 25692341] [CrossRef]
- 190.
- Florez JC, Pearson ER. A roadmap to achieve pharmacological precision medicine in diabetes. Diabetologia. 2022;65(11):1830-1838. doi:10.1007/s00125-022-05732-3 [PMC free article: PMC9522818] [PubMed: 35748917] [CrossRef]
- 191.
- TODAY Study Group, Zeitler P, Hirst K, et al. A clinical trial to maintain glycemic control in youth with type 2 diabetes. N Engl J Med. 2012;366(24):2247-2256. doi:10.1056/NEJMoa1109333 [PMC free article: PMC3478667] [PubMed: 22540912] [CrossRef]
- 192.
- Kahn SE, Haffner SM, Heise MA, et al. Glycemic durability of rosiglitazone, metformin, or glyburide monotherapy. N Engl J Med. 2006;355(23):2427-2443. doi:10.1056/NEJMoa066224 [PubMed: 17145742] [CrossRef]
- 193.
- GoDARTS and UKPDS Diabetes Pharmacogenetics Study Group, Wellcome Trust Case Control Consortium 2, Zhou K, et al. Common variants near ATM are associated with glycemic response to metformin in type 2 diabetes. Nat Genet. 2011;43(2):117-120. doi:10.1038/ng.735 [PMC free article: PMC3030919] [PubMed: 21186350] [CrossRef]
- 194.
- Zhou K, Yee SW, Seiser EL, et al. Variation in the glucose transporter gene SLC2A2 is associated with glycemic response to metformin. Nat Genet. 2016;48(9):1055-1059. doi:10.1038/ng.3632 [PMC free article: PMC5007158] [PubMed: 27500523] [CrossRef]
- 195.
- Li JH, Perry JA, Jablonski KA, et al. Identification of genetic variation influencing metformin response in a multiancestry genome-wide association study in the Diabetes Prevention Program (DPP). Diabetes. 2023;72(8):1161-1172. doi:10.2337/db22-0702 [PMC free article: PMC10382652] [PubMed: 36525397] [CrossRef]
- 196.
- Dawed AY, Yee SW, Zhou K, et al. Genome-wide meta-analysis identifies genetic variants associated with glycemic response to sulfonylureas. Diabetes Care. 2021;44(12):2673-2682. doi:10.2337/dc21-1152 [PMC free article: PMC8669535] [PubMed: 34607834] [CrossRef]
- 197.
- Umapathysivam MM, Araldi E, Hastoy B, et al. Type 2 diabetes risk alleles in peptidyl-glycine alpha-amidating monooxygenase influence GLP-1 levels and response to GLP-1 receptor agonists. medRxiv. 2023. doi:10.1101/2023.04.07.23288197 [CrossRef]
- 198.
- Geng Z, Li Q, Huang R, et al. KCNQ1 variant rs163184 is a potential biomarker of glycemic response to exenatide. Pharmacogenomics. 2022;23(6):355-361. doi:10.2217/pgs-2021-0154 [PubMed: 35311356] [CrossRef]
- 199.
- Kyriakidou A, Kyriazou AV, Koufakis T, et al. Clinical and genetic predictors of glycemic control and weight loss response to liraglutide in patients with type 2 diabetes. J Pers Med. 2022;12(3):424. doi:10.3390/jpm12030424 [PMC free article: PMC8955617] [PubMed: 35330424] [CrossRef]
- 200.
- Yu M, Wang K, Liu H, Cao R. GLP1R variant is associated with response to exenatide in overweight Chinese type 2 diabetes patients. Pharmacogenomics. 2019;20(4):273-277. doi:10.2217/pgs-2018-0159 [PubMed: 30883264] [CrossRef]
- 201.
- Cromer SJ, Lakhani CM, Mercader JM, et al. Association and interaction of genetics and area-level socioeconomic factors on the prevalence of type 2 diabetes and obesity. Diabetes Care. 2023;46(5):944-952. doi:10.2337/dc22-1954 [PMC free article: PMC10154653] [PubMed: 36787958] [CrossRef]
- 202.
- Marston NA, Kamanu FK, Nordio F, et al. Predicting benefit from evolocumab therapy in patients with atherosclerotic disease using a genetic risk score: results from the FOURIER trial. Circulation. 2020;141(8):616-623. doi:10.1161/CIRCULATIONAHA.119.043805 [PMC free article: PMC8058781] [PubMed: 31707849] [CrossRef]
- 203.
- Graham SE, Clarke SL, Wu KH, et al. The power of genetic diversity in genome-wide association studies of lipids. Nature. 2021;600(7890):675-679. doi:10.1038/s41586-021-04064-3 [PMC free article: PMC8730582] [PubMed: 34887591] [CrossRef]
- 204.
- Evangelou E, Warren HR, Mosen-Ansorena D, et al. Genetic analysis of over 1 million people identifies 535 new loci associated with blood pressure traits. Nat Genet. 2018;50(10):1412-1425. doi:10.1038/s41588-018-0205-x [PMC free article: PMC6284793] [PubMed: 30224653] [CrossRef]
- 205.
- Yengo L, Sidorenko J, Kemper KE, et al. Meta-analysis of genome-wide association studies for height and body mass index in approximately 700000 individuals of European ancestry. Hum Mol Genet. 2018;27(20):3641-3649. doi:10.1093/hmg/ddy271 [PMC free article: PMC6488973] [PubMed: 30124842] [CrossRef]
Drs. Kreienkamp, Voight, and Udler had no financial disclosures to report. Dr. Gloyn’s spouse holds stock options in Roche.
Publication Details
Author Information and Affiliations
Authors
Raymond J. Kreienkamp, MD, PhD,1 Benjamin F. Voight, PhD,2 Anna L. Gloyn, DPhil,3 and Miriam S. Udler, MD, PhD 4.
4.Affiliations
Diabetes Unit
Center for Human Genetic Research
Broad Institute
Programs in Metabolism and Medical & Population Genetics
Harvard Medical School
Department of Medicine
Department of Pediatrics, Division of Endocrinology
Boston, MA
Department of Systems Pharmacology and Translational Therapeutics
Department of Genetics
Philadelphia, PA
Department of Pediatrics, Division of Endocrinology & Metabolism
Department of Genetics
Diabetes Research Center
Stanford, CA
Diabetes Unit
Center for Human Genetic Research
Broad Institute
Programs in Metabolism and Medical & Population Genetics
Harvard Medical School
Department of Medicine
Boston, MA
Corresponding author.Publication History
Initial Posting: December 20, 2023.
Copyright
Diabetes in America is in the public domain of the United States. You may use the work without restriction in the United States.
Publisher
National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK), Bethesda (MD)
NLM Citation
Kreienkamp RJ, Voight BF, Gloyn AL, et al. Genetics of Type 2 Diabetes. 2023 Dec 20. In: Lawrence JM, Casagrande SS, Herman WH, et al., editors. Diabetes in America [Internet]. Bethesda (MD): National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK); 2023-.






