Summary
Clinical characteristics.
Hereditary fibrosing poikiloderma with tendon contractures, myopathy, and pulmonary fibrosis (POIKTMP) is characterized by the skin findings of poikiloderma (typically beginning in the first six months and mainly localized to the face), hypohidrosis with heat intolerance, mild lymphedema of the extremities, chronic erythematous and scaly skin lesions on the extremities, sclerosis of the digits, and mild palmoplantar keratoderma. Scalp hair, eyelashes, and/or eyebrows are typically sparse. Muscle contractures are usually seen in childhood and can be present as early as age two years. The majority of affected individuals develop progressive weakness of the proximal and distal muscles of all four limbs. Some adults develop progressive interstitial pulmonary fibrosis, which can be life threatening within three to four years after respiratory symptoms appear. Other features are exocrine pancreatic insufficiency, liver impairment, hematologic abnormalities, relative short stature, and cataract.
Diagnosis/testing.
The diagnosis of POIKTMP is established in a proband with early-onset poikiloderma with other findings, especially muscle contractures and/or muscle weakness and a heterozygous missense pathogenic variant in FAM111B identified by molecular genetic testing.
Management.
Treatment of manifestations: Dermatologic manifestations are treated with avoidance of excessive sun exposure and use of sunscreens with both UVA and UVB protection; avoidance of excessive heat exposure and control of fever, especially in early childhood; routine management of lymphedema; emollients, topical steroids for eczema-like lesions. For older individuals, pulsed dye laser may be an option for cosmesis of the telangiectatic component of the rash. Physical therapy and exercise to promote mobility and prevent contractures. Use of self-inflating manual-ventilation bag or mechanical insufflation-exsufflation device if needed for lung disease; noninvasive ventilation if needed. Pancreatic enzyme supplementation for pancreatic exocrine insufficiency.
Surveillance: Annual surveillance (or frequency as needed) includes dermatologic examination, physical therapy assessment for muscle weakness and/or contractures, assessment for orthopedic complications (contractures, especially of the Achilles tendon, and scoliosis), pulmonary function testing, serum transaminases (SGOT, SGPT), alkaline phosphatase, gamma-glutamyl transferase, blood ionogram including calcium, TSH, complete blood count with differential), and ophthalmologic examination.
Agents/circumstances to avoid: Excessive sun exposure (may exacerbate the rash), exposure to heat because of heat intolerance.
Genetic counseling.
POIKTMP is inherited in an autosomal dominant manner. In approximately 50% of affected individuals, the FAM111B pathogenic variant is inherited and in approximately 50% it is de novo. Each child of an individual with POIKTMP has a 50% chance of inheriting the FAM111B pathogenic variant. Once the FAM111B pathogenic variant has been identified in an affected family member, prenatal and preimplantation genetic testing are possible.
Diagnosis
Suggestive Findings
Hereditary fibrosing poikiloderma with tendon contractures, myopathy, and pulmonary fibrosis (POIKTMP) should be suspected in individuals with the following clinical and imaging findings.
Clinical findings
- Early-onset poikiloderma (characterized by erythema of the cheeks and face, skin atrophy, telangiectasias, and mottled pigmentation)
- Hypotrichosis with sparse scalp hair, sparse or absent eyelashes and/or eyebrows
- Hypohidrosis with heat intolerance
- Mild lymphedema of the extremities
- Multiple contractures, in particular triceps surae contractures in childhood
- Myopathy with diffuse progressive muscular weakness, scoliosis
- Restrictive pulmonary syndrome and/or pulmonary fibrosis
- Exocrine pancreatic insufficiency
- Liver impairment
- Hematologic abnormalities (e.g., eosinophilia, thrombocytopenia, and/or bone marrow hypocellularity)
- Other
- Relative short stature
- Cataract
- Nail dystrophy
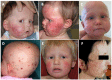
Figure 1.
Facial and scalp skin lesions A-E. Poikiloderma and alopecia of the scalp, eyebrows, and eyelashes in childhood
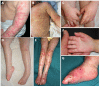
Figure 2.
Skin lesions of the upper and lower limbs A. Eczema-like and psoriasis-like dermatosis of the upper limbs
Imaging findings
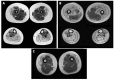
- See Figure 3. Muscle MRI typically shows severe diffuse fatty infiltration of the legs, especially:
- The vastus lateralis muscles (with relative sparing of the tibialis posterior); and
- The anterior compartment of thighs (with relative sparing of posterior compartment).
- These findings can confirm muscle involvement in asymptomatic individuals.
Establishing the Diagnosis
The diagnosis of POIKTMP is established in a proband with early-onset poikiloderma with other findings (especially muscle contractures and/or muscle weakness) and a heterozygous pathogenic missense variant in FAM111B identified on molecular genetic testing (Table 1).
Molecular genetic testing approaches can include a combination of gene-targeted testing (single-gene testing, multigene panel) and comprehensive genomic testing (exome sequencing, genome sequencing) depending on the phenotype.
Gene-targeted testing requires that the clinician determine which gene(s) are likely involved, whereas genomic testing does not. Individuals with the distinctive findings described in Suggestive Findings are likely to be diagnosed using gene-targeted testing (see Option 1), whereas those in whom the diagnosis of POIKTMP has not been considered are more likely to be diagnosed using genomic testing (see Option 2).
Option 1
Single-gene testing. Sequence analysis of FAM111B to detect small intragenic deletions/insertions and missense, nonsense, and splice site variants.
Gene-targeted deletion/duplication analysis may be performed; however, since POIKTMP likely occurs as the result of a dominant-negative genetic mechanism and since large intragenic deletions or duplications have not been reported, testing for intragenic deletions or duplications is unlikely to identify a disease-causing variant (see Molecular Genetics).
A multigene panel that includes FAM111B and other genes of interest (see Differential Diagnosis) may also be considered. Note: (1) The genes included in the panel and the diagnostic sensitivity of the testing used for each gene vary by laboratory and are likely to change over time. (2) Some multigene panels may include genes not associated with the condition discussed in this GeneReview (e.g., RECQL4, USB1, FERMT1); thus, clinicians need to determine which multigene panel provides the best opportunity to identify the genetic cause of the condition while limiting identification of pathogenic variants in genes that do not explain the underlying phenotype. (3) Methods used in a panel may include sequence analysis, deletion/duplication analysis, and/or other non-sequencing-based tests.
For an introduction to multigene panels click here. More detailed information for clinicians ordering genetic tests can be found here.
Option 2
Comprehensive genomic testing does not require the clinician to determine which gene is likely involved. Exome sequencing is most commonly used; genome sequencing is also possible.
For an introduction to comprehensive genomic testing click here. More detailed information for clinicians ordering genomic testing can be found here.
Table 1.
Molecular Genetic Testing Used in Hereditary Fibrosing Poikiloderma with Tendon Contractures, Myopathy, and Pulmonary Fibrosis
Clinical Characteristics
Clinical Description
To date, 31 individuals have been identified with a pathogenic variant in FAM111B [Mercier et al 2013, Mercier et al 2015, Seo et al 2016, Takeichi et al 2017, Goussot et al 2017, Kazlouskaya et al 2018, Zhang et al 2019, Chen et al 2019, Chasseuil et al 2019, Dokic et al 2020]. The following description of the phenotypic features associated with this condition is based on these reports.
Individuals with hereditary fibrosing poikiloderma with tendon contractures, myopathy, and pulmonary fibrosis (POIKTMP) can exhibit few or many of the associated clinical features. The severity of the features (e.g., skin or muscle abnormalities) can vary. Intrafamilial clinical variability has been observed [Khumalo et al 2006, Seo et al 2016].
Skin
Skin abnormalities are the earliest findings. Of note, skin lesions – particularly facial poikiloderma – improve with time.
Poikiloderma appears during early infancy, typically in the first six months. It is mainly localized to the face (Figure 1). Transient exacerbations of facial erythema are seen following sun exposure. Mottled pigmentation is also a constituent part of poikiloderma.
Hypohidrosis with heat intolerance is observed in most.
Lymphedema of the lower and/or upper extremities may be present in childhood and is usually mild; it can be complicated by episodes of cellulitis.
Chronic erythematous and scaly skin lesions described as eczema-like, ichthyosis-like, or psoriasis-like lesions are often observed on the extremities.
Sclerosis of the digits and mild palmoplantar keratoderma may also be observed.
Hair/nails. Sparse scalp hair and sparse or absent eyelashes and/or eyebrows of variable severity are found in almost all affected individuals. Four affected individuals had mild nail dysplasia.
Muscle
Muscle contractures are usually seen in childhood and can be present as early as age two years. The most commonly affected muscles are the triceps surae, leading to a shortening of the Achilles tendons and varus deformities of the feet. Contractures of upper limbs (biceps brachii and carpal extensors) are also observed.
Myopathy. The majority of affected individuals develop progressive weakness of the proximal and distal muscles of all four limbs; the first manifestations (observed in lower limbs) are proximal rather than distal. Variability of muscle weakness ranges from loss of ambulation in childhood to absence of symptoms in adulthood [Mercier et al 2013, Mercier et al 2015, Seo et al 2016, Takeichi et al 2017].
Muscle weakness is generally associated with muscle atrophy and sometimes thoracolumbar scoliosis.
Serum creatine kinase is either normal or slightly increased. When performed, electromyography may show a normal or myopathic pattern.
Lung
Recurrent bronchitis can be observed. Abnormal lung function with restrictive pulmonary disease is common.
Some adults develop progressive interstitial pulmonary fibrosis, manifest as progressive breathlessness and dry cough; it can be life threatening within three to four years after the first respiratory symptoms appear.
Gastrointestinal
Pancreatic exocrine insufficiency (common in affected individuals) typically begins in childhood. Manifestations include fatty stools and diarrhea leading to chronic malabsorption of fats and lipid-soluble vitamins if not treated. Diagnosis is confirmed based on fecal elastase deficiency.
Liver impairment (reported in a few affected individuals) manifests initially as mildly elevated transaminases, alkaline phosphatase, gamma-glutamyl transferase, and/or bilirubin. One individual had hepatomegaly and cholestasis [Mercier et al 2013, Mercier et al 2015]. Another individual reported has transaminitis and lymphocytic ductulitis [Dokic et al 2020].
Other Features
Relative short stature and/or poor weight gain associated with delayed puberty have been reported.
Hematologic findings include eosinophilia or mild thrombocytopenia in some. Bone marrow hypocellularity was reported in a multiplex family [Seo et al 2016].
Hypothyroidism was described in a girl age 14 years [Takeichi et al 2017].
Eye. Cataract was reported in a girl age 13 years [Mercier et al 2015].
Cognitive development and function are normal. Of note, one individual had schizophrenia [Mercier et al 2015], which could be an incidental association.
Histopathology
Muscle histology shows extensive fatty infiltration. Residual muscle tissue is composed of fragmented muscle fascicles with either normal fibers or atrophic fibers with central nuclei (Figure 4 A-D). No neuropathic features (i.e., normal ATPase pattern) or mitochondrial network abnormalities are found on histochemistry or immunolabeling. Western blot analysis can show a secondary reduction of calpain.
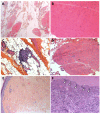
Figure 4
A-D. Muscle histology: A, B. Fatty tissue, fragmented muscle fascicles next to normal fascicles
Skin histology shows a characteristic pattern of epidermal atrophy with collagen sclerosis and elastic degeneration in the superficial and deep dermis. Elastic globes in the papillary dermis are associated with a diffuse mild collagen sclerosis (Figure 4 E-F).
Postmortem findings of one affected member of a South African family revealed diffuse fatty infiltration and fibrosis of organs including the lungs, esophagus, and pancreas [Khumalo et al 2006].
Genotype-Phenotype Correlations
POIKTMP is most frequently caused by heterozygosity for missense FAM111B pathogenic variants in the predicted trypsin-like cysteine/serine peptidase domain of the protein, in particular in the loop of the functional domain.
- Upstream variants located outside the loop (codons 421, 430) appear to be associated with a less severe phenotype, especially regarding muscle features [Mercier et al 2015, Seo et al 2016, Takeichi et al 2017].
- The phenotype of pathogenic variants in codon 621 appears to be less severe than the phenotype observed with pathogenic variants in codons 625, 627, and 628 [Khumalo et al 2006, Mercier et al 2013].
Further studies are needed to confirm these preliminary genotype-phenotype correlations.
Penetrance
To the authors' knowledge the penetrance of POIKTMP is 100%, with occurrence of skin features in early childhood.
Prevalence
The prevalence of POIKTMP is unknown. The condition is thought to be ubiquitous and is likely underdiagnosed.
Genetically Related (Allelic) Disorders
No phenotypes other than those discussed in this GeneReview are known to be associated with pathogenic variants in FAM111B.
Differential Diagnosis
Disorders with phenotypic similarity to hereditary fibrosing poikiloderma with tendon contractures, myopathy, and pulmonary fibrosis (POIKTMP) are summarized in Table 2. Notably, unlike POIKTMP, the disorders in Table 2 are not associated with muscle contractures, myopathy, or exocrine pancreatic insufficiency.
Table 2.
Genes and Disorders to Consider in the Differential Diagnosis of Hereditary Fibrosing Poikiloderma with Tendon Contractures, Myopathy, and Pulmonary Fibrosis
Hereditary sclerosing poikiloderma (HSP) of Weary (OMIM 173700), a disorder of unknown genetic cause, can also be considered in the differential diagnosis of POIKTMP. Like POIKTMP, HSP of Weary is associated with poikiloderma and sclerosis of palms and soles. Unlike POIKTMP, HSP of Weary is also known to be associated with linear sclerotic bands, subcutaneous calcifications, and valvular heart disease. HSP of Weary can be further distinguished from POIKTMP by the absence of muscle contractures, myopathy, and exocrine pancreatic insufficiency.
Management
Evaluations Following Initial Diagnosis
To establish the extent of disease and needs in an individual diagnosed with hereditary fibrosing poikiloderma with tendon contractures, myopathy, and pulmonary fibrosis (POIKTMP), the evaluations summarized in Table 3 (if not performed as part of the evaluation that led to the diagnosis) are recommended.
Table 3.
Recommended Evaluations Following Initial Diagnosis in Individuals with Hereditary Fibrosing Poikiloderma with Tendon Contractures, Myopathy, and Pulmonary Fibrosis
Treatment of Manifestations
Table 4.
Treatment of Manifestations in Individuals with Hereditary Fibrosing Poikiloderma with Tendon Contractures, Myopathy, and Pulmonary Fibrosis
Surveillance
Table 5.
Recommended Surveillance for Individuals with Hereditary Fibrosing Poikiloderma with Tendon Contractures, Myopathy, and Pulmonary Fibrosis
Agents/Circumstances to Avoid
Avoid the following:
- Excessive sun exposure, which may exacerbate the rash
- Exposure to heat because of heat intolerance secondary to hypohidrosis
Evaluation of Relatives at Risk
See Genetic Counseling for issues related to testing of at-risk relatives for genetic counseling purposes.
Therapies Under Investigation
Search ClinicalTrials.gov in the US and EU Clinical Trials Register in Europe for access to information on clinical studies for a wide range of diseases and conditions.
Genetic Counseling
Genetic counseling is the process of providing individuals and families with information on the nature, mode(s) of inheritance, and implications of genetic disorders to help them make informed medical and personal decisions. The following section deals with genetic risk assessment and the use of family history and genetic testing to clarify genetic status for family members; it is not meant to address all personal, cultural, or ethical issues that may arise or to substitute for consultation with a genetics professional. —ED.
Mode of Inheritance
Hereditary fibrosing poikiloderma with tendon contractures, myopathy, and pulmonary fibrosis (POIKTMP) is inherited in an autosomal dominant manner.
Risk to Family Members
Parents of a proband
- Approximately 50% of individuals diagnosed with POIKTMP have an affected parent.
- Approximately 50% of individuals diagnosed with POIKTMP have the disorder as the result of a de novo FAM111B pathogenic variant.
- If the proband appears to be the only affected family member (i.e., a simplex case), molecular genetic testing is recommended for the parents of the proband to confirm their genetic status and to allow reliable recurrence risk counseling.
- If the pathogenic variant identified in the proband is not identified in either parent and parental identity testing has confirmed biological maternity and paternity, the following possibilities should be considered:
- The proband has a de novo pathogenic variant.
- The proband inherited a pathogenic variant from a parent with germline (or somatic and germline) mosaicism. Note: Testing of parental leukocyte DNA may not detect all instances of somatic mosaicism and will not detect a pathogenic variant that is present in the germ cells only.
Sibs of a proband. The risk to the sibs of the proband depends on the genetic status of the proband’s parents:
- If a parent of the proband is affected and/or is known to have the pathogenic variant identified in the proband, the risk to the sibs of inheriting the pathogenic variant is 50%. Although all sibs who inherit a FAM111B pathogenic variant are expected to develop manifestations of the disorder, intrafamilial clinical variability has been observed (see Clinical Description).
- If the FAM111B pathogenic variant detected in the proband cannot be detected in the leukocyte DNA of either parent, the recurrence risk to sibs is estimated to be 1% because of the theoretic possibility of parental germline mosaicism [Rahbari et al 2016].
- If the parents have not been tested for the FAM111B pathogenic variant but are clinically unaffected, the risk to the sibs of a proband appears to be low. However, sibs of a proband with clinically unaffected parents are still presumed to be at increased risk for POIKTMP because of the theoretic possibility of parental germline mosaicism.
Offspring of a proband. Each child of an individual with POIKTMP has a 50% chance of inheriting the FAM111B pathogenic variant.
Other family members. The risk to other family members depends on the status of the proband's parents: if a parent has the FAM111B pathogenic variant, his or her family members may be at risk.
Related Genetic Counseling Issues
Family planning
- The optimal time for determination of genetic risk and discussion of the availability of prenatal/preimplantation genetic testing is before pregnancy.
- It is appropriate to offer genetic counseling (including discussion of potential risks to offspring and reproductive options) to young adults who are affected or at risk.
Prenatal Testing and Preimplantation Genetic Testing
Once the FAM111B pathogenic variant has been identified in an affected family member, prenatal and preimplantation genetic testing are possible.
Differences in perspective may exist among medical professionals and within families regarding the use of prenatal testing. While most centers would consider use of prenatal testing to be a personal decision, discussion of these issues may be helpful.
Resources
GeneReviews staff has selected the following disease-specific and/or umbrella support organizations and/or registries for the benefit of individuals with this disorder and their families. GeneReviews is not responsible for the information provided by other organizations. For information on selection criteria, click here.
- MedlinePlus
Molecular Genetics
Information in the Molecular Genetics and OMIM tables may differ from that elsewhere in the GeneReview: tables may contain more recent information. —ED.
Table A.
Hereditary Fibrosing Poikiloderma with Tendon Contractures, Myopathy, and Pulmonary Fibrosis: Genes and Databases
Table B.
OMIM Entries for Hereditary Fibrosing Poikiloderma with Tendon Contractures, Myopathy, and Pulmonary Fibrosis (View All in OMIM)
Molecular Pathogenesis
The FAM111B protein is predicted to contain a trypsin-like cysteine/serine peptidase domain. A role in DNA replication is suggested [Aviner et al 2015].
Hereditary fibrosing poikiloderma with tendon contractures, myopathy, and pulmonary fibrosis (POIKTMP) is most frequently caused by heterozygosity for missense FAM111B pathogenic variants in the predicted trypsin-like cysteine/serine peptidase domain of the protein, in particular in the loop of the functional domain.
Mechanism of disease causation. The mechanism of disease for POIKTMP is not known; however, the spectrum of pathogenic variants and functional studies suggest a dominant-negative mechanism [Mercier et al 2015; Author, unpublished data].
Table 6.
Notable FAM111B Pathogenic Variants
Chapter Notes
Author Notes
Areas of interest/inquiry:
- Prof Sandra Mercier, MD, PhD, clinician geneticist; Dr Sébastien Barbarot, MD, PhD, dermatologist: diagnosis and clinical follow up of patients with POIKTMP
- Sébastien Küry, DVM, PhD: FAM111B molecular diagnosis
Prof Sandra Mercier and Dr Sébastien Küry are part of the Inserm UMR 1087 / CNRS UMR 6291 research team where functional studies are underway to understand the pathophysiology of the disease and find therapeutic approaches.
Contact: [email protected]
Acknowledgments
We thank the families and patients for their cooperation and support.
Revision History
- 9 September 2021 (sm) Revision: nucleotide variant correction: c.1881A>T
- 5 August 2021 (sw) Comprehensive update posted live
- 13 October 2016 (bp) Review posted live
- 1 February 2016 (sm) Original submission
References
Literature Cited
- Aviner R, Shenoy A, Elroy-Stein O, Geiger T. Uncovering hidden layers of cell cycle regulation through integrative multi-omic analysis. PLoS Genet. 2015;11:e1005554. [PMC free article: PMC4595013] [PubMed: 26439921]
- Chasseuil E, McGrath JA, Seo A, Balguerie X, Bodak N, Chasseuil H, Denis-Musquer M, Goldenberg A, Goussot R, Irvine AD, Khumalo NP, King MC, Küry S, Lipsker D, Mallet S, Mayosi BM, Nanda A, Puzenat E, Salort-Campana E, Sidbury R, Shimamura A, Bézieau S, Mercier S, Barbarot S. Dermatological manifestations of hereditary fibrosing poikiloderma with tendon contractures, myopathy and pulmonary fibrosis (POIKTMP): a case series of 28 patients. Br J Dermatol. 2019;181:862–4. [PubMed: 30972747]
- Chen F, Zheng L, Li Y, Li H, Yao Z, Li M. Mutation in FAM111B causes hereditary fibrosing poikiloderma with tendon contracture, myopathy, and pulmonary fibrosis. Acta Derm Venereol. 2019;99:695–6. [PubMed: 30938824]
- Dokic Y, Albahrani Y, Phung T, Patel K, de Guzman M, Hertel P, Hunt R. Hereditary fibrosing poikiloderma with tendon contractures, myopathy, and pulmonary fibrosis: Hepatic disease in a child with a novel pathogenic variant of FAM111B. JAAD Case Rep. 2020;6:1217–20. [PMC free article: PMC7701006] [PubMed: 33294546]
- Goussot R, Prasad M, Stoetzel C, Lenormand C, Dollfus H, Lipsker D. Expanding phenotype of hereditary fibrosing poikiloderma with tendon contractures, myopathy, and pulmonary fibrosis caused by FAM111B mutations: Report of an additional family raising the question of cancer predisposition and a short review of early-onset poikiloderma. JAAD Case Rep. 2017;3:143–50. [PMC free article: PMC5358901] [PubMed: 28349113]
- Kazlouskaya V, Feldman EJ, Jakus J, Heilman E, Glick S. A case of hereditary fibrosing poikiloderma with tendon contractures, myopathy and pulmonary fibrosis (POIKTMP) with the emphasis on cutaneous histopathological findings. J Eur Acad Dermatol Venereol. 2018;32:e443–e445. [PubMed: 29578632]
- Khumalo NP, Pillay K, Beighton P, Wainwright H, Walker B, Saxe N, Mayosi BM, Bateman ED. Poikiloderma, tendon contracture and pulmonary fibrosis: a new autosomal dominant syndrome? Br J Dermatol. 2006;155:1057–61. [PubMed: 17034542]
- Mercier S, Küry S, Salort-Campana E, Magot A, Agbim U, Besnard T, Bodak N, Bou-Hanna C, Breheret F, Brunelle P, Caillon F, Chabrol B, Cormier-Daire V, David A, Eymard B, Faivre L, Figarella-Branger D, Fleurence E, Ganapathi M, Gherardi R, Goldenberg A, Hamel A, Igual J, Irvine AD, Israel-Biet D, Kannengiesser C, Laboisse C, Le Caignec C, Mahe JY, Mallet S, MacGowan S, McAleer MA, McLean I, Meni C, Munnich A, Mussini JM, Nagy PL, Odel J, O'Regan GM, Pereon Y, Perrier J, Piard J, Puzenat E, Sampson JB, Smith F, Soufir N, Tanji K, Thauvin C, Ulane C, Watson RM, Khumalo NP, Mayosi BM, Barbarot S, Bezieau S. Expanding the clinical spectrum of hereditary fibrosing poikiloderma with tendon contractures, myopathy and pulmonary fibrosis due to FAM111B mutations. Orphanet J Rare Dis. 2015;10:135. [PMC free article: PMC4608180] [PubMed: 26471370]
- Mercier S, Küry S, Shaboodien G, Houniet DT, Khumalo NP, Bou-Hanna C, Bodak N, Cormier-Daire V, David A, Faivre L, Figarella-Branger D, Gherardi RK, Glen E, Hamel A, Laboisse C, Le Caignec C, Lindenbaum P, Magot A, Munnich A, Mussini JM, Pillay K, Rahman T, Redon R, Salort-Campana E, Santibanez-Koref M, Thauvin C, Barbarot S, Keavney B, Bézieau S, Mayosi BM. Mutations in FAM111B cause hereditary fibrosing poikiloderma with tendon contracture, myopathy, and pulmonary fibrosis. Am J Hum Genet. 2013;93:1100–7. [PMC free article: PMC3853004] [PubMed: 24268661]
- Rahbari R, Wuster A, Lindsay SJ, Hardwick RJ, Alexandrov LB, Turki SA, Dominiczak A, Morris A, Porteous D, Smith B, Stratton MR, Hurles ME, et al. Timing, rates and spectra of human germline mutation. Nat Genet. 2016;48:126–33. [PMC free article: PMC4731925] [PubMed: 26656846]
- Seo A, Walsh T, Lee MK, Ho PA, Hsu EK, Sidbury R, King MC, Shimamura A. FAM111B mutation is associated with inherited exocrine pancreatic dysfunction. Pancreas. 2016;45:858–62. [PMC free article: PMC4841754] [PubMed: 26495788]
- Takeichi T, Nanda A, Yang HS, Hsu CK, Lee JY, Al-Ajmi H, Akiyama M, Simpson MA, McGrath JA. Syndromic inherited poikiloderma due to a de novo mutation in FAM111B. Br J Dermatol. 2017;176:534–6. [PubMed: 27406236]
- Zhang Z, Zhang J, Chen F, Zheng L, Li H, Liu M, Li M, Yao Z. Family of hereditary fibrosing poikiloderma with tendon contractures, myopathy and pulmonary fibrosis caused by a novel FAM111B mutation. J Dermatol. 2019;46:1014–8. [PubMed: 31392773]
Publication Details
Author Information and Affiliations
Publication History
Initial Posting: October 13, 2016; Last Revision: September 9, 2021.
Copyright
GeneReviews® chapters are owned by the University of Washington. Permission is hereby granted to reproduce, distribute, and translate copies of content materials for noncommercial research purposes only, provided that (i) credit for source (http://www.genereviews.org/) and copyright (© 1993-2024 University of Washington) are included with each copy; (ii) a link to the original material is provided whenever the material is published elsewhere on the Web; and (iii) reproducers, distributors, and/or translators comply with the GeneReviews® Copyright Notice and Usage Disclaimer. No further modifications are allowed. For clarity, excerpts of GeneReviews chapters for use in lab reports and clinic notes are a permitted use.
For more information, see the GeneReviews® Copyright Notice and Usage Disclaimer.
For questions regarding permissions or whether a specified use is allowed, contact: ude.wu@tssamda.
Publisher
University of Washington, Seattle, Seattle (WA)
NLM Citation
Mercier S, Küry S, Barbarot S. Hereditary Fibrosing Poikiloderma with Tendon Contractures, Myopathy, and Pulmonary Fibrosis. 2016 Oct 13 [Updated 2021 Sep 9]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024.



