
Submitting high-throughput sequence data to GEO
- Submission instructions

- Tutorial video
- Data file compression
- Single-cell studies
- NanoString GeoMx Digital Spatial Profiling (DSP)
- Organizing your submission
- Uploading your submission
- General information
Submission instructions Back to top
GEO accepts next generation sequence data that examine quantitative gene expression, gene regulation, epigenomics or other aspects of functional genomics using methods such as RNA-seq (including single-cell), miRNA-seq, ChIP-seq, RIP-seq, HiC-seq, methyl-seq, etc. We process all components of your study, including the samples, project description, processed data files, and we submit the raw data files to the Sequence Read Archive (SRA) on your behalf.
- Check that GEO accepts your data type.
- Gather raw files.
- Gather processed data files.
- Download metadata spreadsheet and fill in Metadata tab for your study. Use one spreadsheet per data type (e.g., ChIP-seq, RNA-seq).
- In the metadata spreadsheet file, list the MD5 checksum for all raw and processed data files in the 'MD5 Checksums' tab.
- Create single folder on your computer that contains all raw and processed data files for your experiment. If you have multiple data types, please use one folder per experiment.
- Transfer your data to GEO by FTP using these instructions.
- After FTP transfer has completed, submit metadata file(s) on the Submit to GEO page.
More information on required components:
-
Metadata spreadsheet
Metadata refers to descriptive information about the overall study, individual samples, all protocols, and references to processed and raw data file names. Information is supplied by completing all fields of a metadata template spreadsheet. Guidelines on the content of each field are provided within the spreadsheet.
Provide enough details so that users can get a general understanding of the study and samples from the GEO records. Please spell out acronyms and abbreviations. Submit a separate metadata spreadsheet for each data type.
Have you already submitted raw data to SRA and now want to submit to GEO?
If you already have your raw data in SRA, you do not need to submit it again to GEO. We need only processed data and a specialized metadata file in order to create GEO records and link them to your raw data records previously submitted to SRA.You will need to enter the PRJNA, SAMN and SRX or SRR for all samples with raw data already submitted to SRA. You can get that information for your SUB ID on the Submission Portal page.
-
Processed data files
GEO requires that submitters deposit the processed data that support the findings of their study. The processed data should have a quantitative component, such as gene abundances or other count data. Please do not submit alignment files (e.g., BAM, SAM, BED) as processed data, as these are considered intermediary files and do not include a quantitative component. When standard alignments are the only processed data available, please write to us to inquire about whether your data are suitable for submission to GEO.
Processed data format and content will depend on the data type: RNA-seq processed data can include raw and/or normalized counts (FPKM, TPM, etc) of sequencing reads for the features of interest (protein-coding genes, lncRNA, miRNA, circRNA, etc).
ChIP-Seq and ATAC-seq processed data can include peak files with quantitative data, tag density files, etc. Common formats include WIG, bigWig, bedGraph. Please leave files in native format and do not paste peak data into Excel.
Methylation data are often provided as average beta values.
Processed data guidelines:
- Processed data may be formatted either as a matrix table or individual files for each sample.
- If processed data for all samples is submitted in a matrix table, column headers should match the library name for each sample listed in the SAMPLES section of the metadata spreadsheet.
- Provide complete data with values for all features (e.g., genes) and all samples. Do not submit lists of genes identified with differential expression.
- Features (e.g., genes, transcripts) in processed data files should be traceable using public accession numbers or chromosome coordinates. The reference assembly used (e.g., hg19, mm9, GCF_000001405.13) should be provided in the metadata spreadsheet.
- If you provide WIG, bedGraph, GFF, or GTF files, please refer to the UCSC file format FAQ for format requirements.
-
Raw data files
Raw data are a required part of GEO submissions. The raw data files should be the original files containing reads and quality scores, as generated by the sequencing instrument. Edited files may not be processed correctly by SRA.
Raw data for high throughput sequencing studies submitted to GEO will be brokered to SRA for you.
Raw data can instead be submitted directly to SRA. After you have received the SRA accessions, please see above for instructions and specific template for this case. Please submit the metadata and processed data to GEO.
If your raw data files exceed 2 terabytes (TB), submit them directly to SRA. After you have received the SRA accessions, please see above for instructions and specific template for this case. Please submit the metadata and processed data to GEO.
If the R1 and R2 files for one library exceed 600 GB, split them into smaller files so that they will load efficiently. R1/R2 file pairs larger than 600GB will be delayed in loading.
Raw Data File Formats: Acceptable file formats include FASTQ, as well as other formats described in the SRA File Format Guide. Files that do not conform to supported format requirements will be deleted from our systems.
Raw file names should not include white space or special characters such as: /, &, #, % or any non-ascii characters. All raw files must have unique names.
Barcode/Multiplexed Data: For bulk RNA-seq studies, we require that raw data files be demultiplexed so that each barcoded sample ends up with a dedicated run file. Most single-cell sequencing studies studies should be submitted with multiplexed raw data files (e.g. 10x Genomics, Drop-Seq, InDrops).
Paired-end Experiments: We usually expect 2 files per run (3 or 4 files per run when sequences and qualities are included in separate files).
MD5 Checksums: We recommend that submitters provide MD5 checksums for their raw data files. The checksums are used to verify file integrity. Checksums can be calculated using the following methods:
- Unix: md5sum <file>
- OS X: md5 <file>
- Windows: Application required. Many are available for free download.
Tutorial video Back to top
Data file compression Back to top
-
Individual files can be compressed to speed transfer, but this is not required.
- Acceptable compression formats are gzip and bzip2 (i.e. files ending with a .gz or .bz2 extension).
- Never compress binary files (e.g., BAM, bigWig, bigBed, HDF5).
- DO NOT upload ZIP archives (files with a .zip extension). Zip archives with fastq files are often corrupted and will delay submission processing.
-
Do not submit raw data files in tar archives. GEO must be able to easily access raw data files for QC purposes.
- Exception: You can submit tar.gz archives of base-called fast5 files for raw data produced by Nanopore instruments.
Single-cell studies Back to top
Raw and processed data are required for single-cell data submissions. We are expecting the raw data for single-cell studies to be submitted in fastq format. Fastq files are preferred for 10X Genomics studies so that they can be correctly archived in SRA format.
Single-cell data should be multiplexed so that each GEO sample record (GSM) represents many individual cells. If your data are not already multiplexed and you have separate raw data files for each cell, please email GEO for guidance with your submission.
Processed data for single-cell studies should be cell-level data. Files can be submitted as Cell Ranger software output files (barcodes.tsv, features.tsv, matrix.mtx), H5 or HDF5 archives, or RDS objects. Processed data for single-cell TCR and BCR samples should include contig annotations and cell barcode information.
If you have used the Cell Ranger aggr pipeline and you are submitting H5/HDF5 archives, you must submit the aggregation.csv file.
When submitting multi-omics types of studies (ADT, HTO, TCR, BCR, GDO, CMO, LMO) and using 10X Genomics protocols and software you must submit the feature_reference.csv file so that the data can be correctly interpreted. List different *omics libraries on separate rows in the SAMPLES section of the metadata spreadsheet:
- sample1_GEX
- sample1_TCR
- sample1_ADT
- sample1_HTO
If the feature_reference.csv file is not available, please submit a supplementary file named "feature_README.txt" (download the template for this file) with the following information as applicable for the included samples:
| id | name | read | pattern | sequence | feature_type | multiplexed_sample | target_gene_id | target_gene_name | mhc_allele |
|---|---|---|---|---|---|---|---|---|---|
| ADT_1 | TotalSeq-A0106 anti-mouse CD11c | R2 | 5P(BC) | GTTATGGACGCTTGC | Antibody Capture | ||||
| HTO_1 | TotalSeq-A0301 anti-mouse Hashtag 1 | R2 | 5P(BC) | ACCCACCAGTAAGAC | Multiplexing Capture | unimmunized mouse 1 | |||
| GDO_1 | GF1B_ch4:132977695-132977714 | R2 | GCATAGCTCTTTAAAC(BC) | TTTGGCAGGGCGTCCCATCC | CRISPR Guide Capture | ENSG00000165702 | GFI1B |
NanoString GeoMx Digital Spatial Profiling (DSP) Back to top
Some NanoString GeoMx® Digital Spatial Profiling (DSP) formats use high throughput sequencing methods to produce the raw data. NanoString GeoMx DSP studies may analyze either RNA or Protein targets using hundreds to thousands of probes. Raw data should be submitted in fastq format. Processed data may be submitted in supplementary text files that include matrices or you can submit the DCC files which contain raw read counts along with RTS_IDs (probe barcodes). If DCC files are provided, you must also submit the PKC file that includes probe metadata describing the targets and is needed to interpret RTS_IDs in the DCC files.
Organizing your submission Back to top
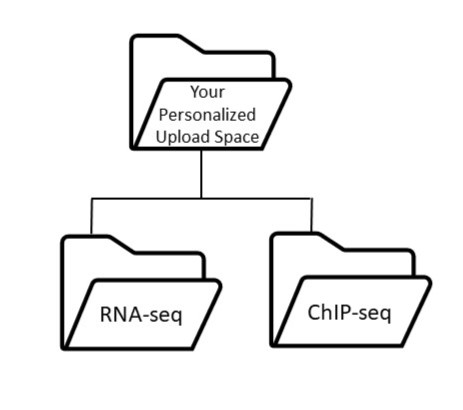
- If you have one dataset, place all data files in a single folder given a meaningful name (e.g., geo_sub_RNAseq).
- If you have multiple datasets, use a separate folder for each dataset (e.g., RNAseq, ChIPseq, HiC).
- Do not use subfolders within your dataset folder.
- All files must have unique names.
Here is an example of the folder structure:
Uploading your submission Back to top
Submit a new high-throughput sequencing submission
1. Transfer all your raw and processed data files to the GEO FTP server.
Submit a separate folder for each dataset you upload, e.g., RNAseq, ChIPseq, HiC.
Do not upload the metadata file(s) by FTP.
2. Submit metadata.
Upload your metadata file(s) after FTP transfer is complete (one metadata file for each dataset).
This step will place your submission into our processing queue for review.
Submit additional or replacement files for a submission in progress
You may use this option to upload files requested by curators or submit additional supplementary files for an existing record.
1. Transfer the replaced or additional data files for your current GEO submission to the GEO FTP server.
2. After the FTP transfer is complete, notify GEO using the Submit to GEO web form.
General information Back to top
|
Data provision and standards
GEO sequence submission procedures are designed to encourage provision of MINSEQE elements:
|
Administration
All standard GEO administration and processing procedures apply to sequence submissions. These include:
More information on these aspects is provided in our FAQ. |
Categories of sequence submissions processed by GEO Back to top
| GEO accepts | GEO does not accept |
Studies concerning quantitative gene expression, gene regulation, epigenetics, or other functional genomic studies. Examples include:
If you have questions about whether GEO can accept your data type, please e-mail GEO. |
For more information about submitting data to NCBI, please refer to the Submission Wizard. |
|---|