

GCP SARS-CoV-2 Public Dataset Program - quick start guide
- Introduction
- Open Bucket Contents
- Querying the Metadata to find Sequence of Interest
- Example queries that can be used in the BigQuery console
- Filtering based on genomic position on the reference sequence, NC_045512.2:
- Filter for runs with a specific mutation:
- Filtering based on read support:
- Filtering based on SARS-CoV-2 samples derived metagenomic/metatranscriptomic libraries:
- Filtering based on organism to find only runs which were not submitted as SARS-CoV-2, but where it was detected:
- Filter for all variations called from samples collected in the United States after '2022-06-01':
- Example queries that can be used in the BigQuery console
- Viewing and Navigating the Open Bucket
- Using the Sequence Data from the Command Line
- Notes on NCBI-generated variants
- Additional Resources
- Contact SRA
Introduction
The NCBI COVID-19 Genome Sequence Dataset (https://console.cloud.google.com/storage/browser/nih-sequence-read-archive) is hosted in Google Cloud Storage and is available for free use and download with no egress charges. It includes both sequence data and searchable metadata from the Sequence Read Archive. The data and metadata encompass Runs identified to contain Coronaviridae using a kmer-based approach to organismal content analysis (SRA Detection Tool). This set includes files in both the originally submitted data format as well as normalized SRA file formats for each Run. NCBI has also made annotated variation metadata for this set available in BigQuery to allow rapid, SQL-based searching either manually or programmatically in the cloud.
Open Bucket Contents
The run directory contains the normalized SRA sequence data, organized by Run accession and accessible via the SRA Toolkit (https://github.com/ncbi/sra-tools/wiki).
The sra-src directory contains the originally submitted sequence data files, also organized by Run accession.
The SARS_COV_2 directory contains the variation and VIGOR3 annotation information for these Runs in JSON format.
The vcf directory contains the NCBI-generated VCF files for these data, organized by Run accession.
Querying the Metadata to find Sequence of Interest
Annotated variation metadata (https://www.ncbi.nlm.nih.gov/sra/docs/sars-cov-2-variant-calling/) for this SARS-CoV-2 dataset has been deposited into BigQuery as a table called annotated_variations to provide the bioinformatics community with access to this information. This table can be used to answer questions directly or to identify runs of interest for further analysis. You can find a list of fields available in this table here: https://www.ncbi.nlm.nih.gov/sra/docs/sra-cloud-based-annotated-variations-table/.
After finding a set of Runs you would like to analyze, you can move on to download or dump the
sequence data in your format of choice. We generally recommend querying the metadata using BigQuery,
however the underlying data files are available in JSON format from the open bucket as well
(in the SARS_COV_2 directory; gs://nih-sequence-read-archive/SARS_COV_2/annotated_variations).
This public dataset is included in BigQuery's 1TB/mo of free tier processing
(https://cloud.google.com/free/docs/gcp-free-tier/#bigquery), meaning each user receives 1TB of free BigQuery.
processing each month that can be used to run queries on this metadata.
To get started using a BigQuery public dataset, you must first create or select a project. If you want to use more than the free tier of data, you must also enable billing. To query this data in the BigQuery web interface, log in to the Google Cloud Platform, then from the project selector page (https://console.cloud.google.com/projectselector2/home/dashboard), select or create a Google Cloud project. If you don't plan to keep the resources that you create during this process- you may create a project, then delete it when done, which removes all resources associated with the project.
Next, navigate to the BigQuery interface by either searching for it in the search bar at the top of the screen or navigating there directly: https://console.cloud.google.com/bigquery. Then click the Add Data button on the upper left side of the screen in the Explorer panel, select Pin a project by name, paste bigquery-public-data into the "Project name" box and click "Pin". This will bring you to a BigQuery window where the bigquery_public_data project will be shown in the left-hand Explorer section.
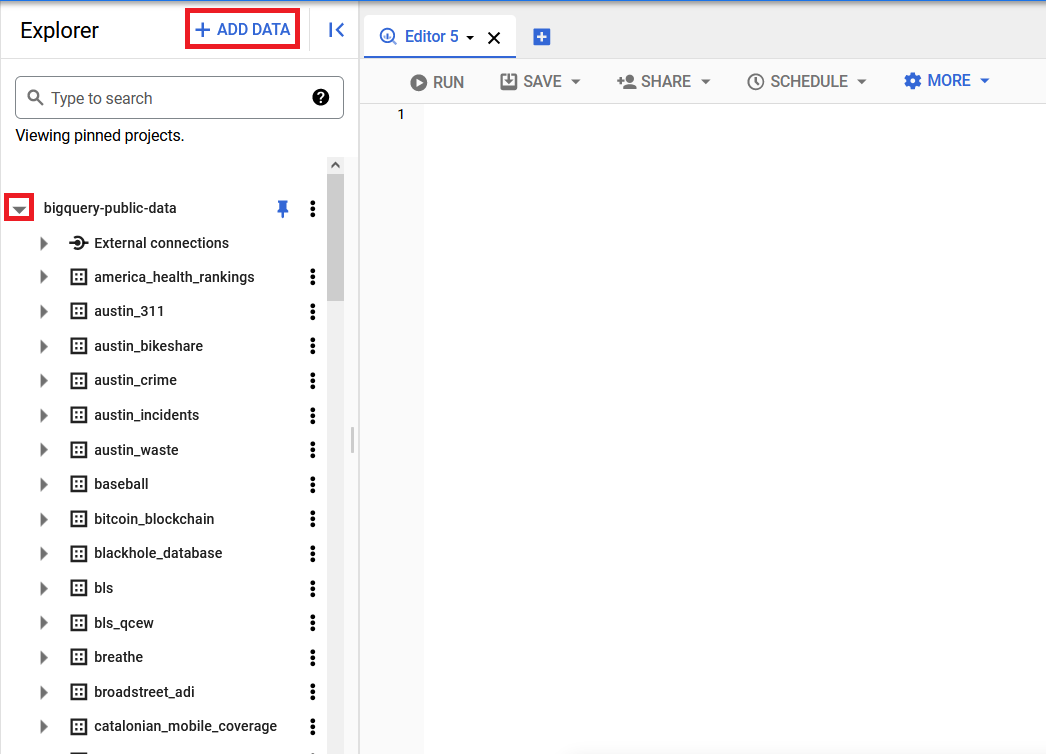
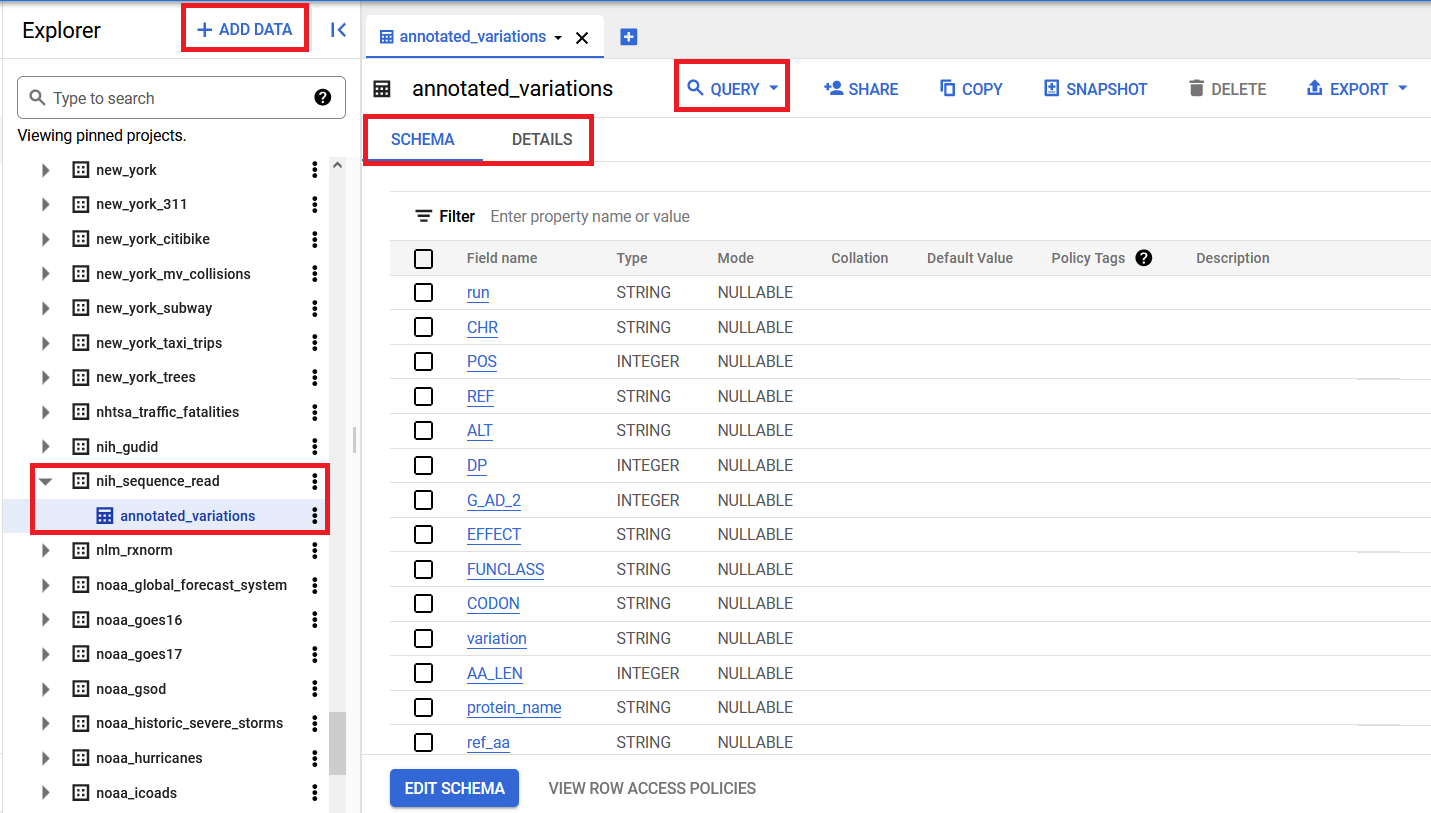
From there, click the triangular expand node to the left of the project entry to see the list of datasets available (you will likely need to click the More Results link when you reach the bottom of the list to show the full list), then scroll down to find the nih_sequence_read dataset. Click the triangular expand node to the left of the dataset to see the annotated_variations table. Clicking on the table name will open it in the main window. You may then click on the Schema, Details, and Preview tabs to further explore the contents of the table. The Schema tab gives information about the name of each field in the table and how that field is encoded (string, integer). The Details tab provides information about the data underlying the table, such as the last time it was updated and how many rows the table contains. The Preview tab shows you the first few rows of the table so you can see what the data actually look like. Finally, click the Query button to open up a new query edit window prefilled for that table, where you can type in your own query from scratch or copy/paste one of the examples below.
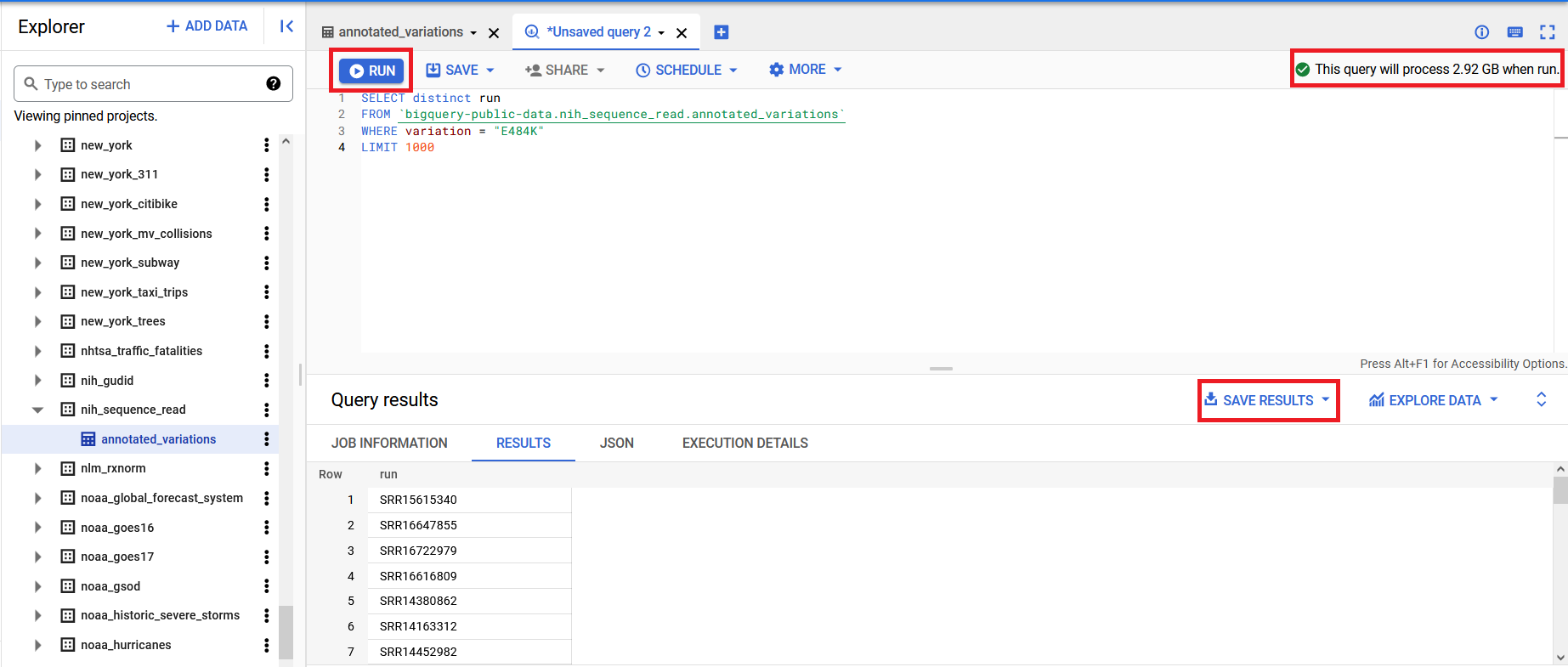
After entering a valid query in the window, you will see a message like This query will process XX when run in the upper right corner of the query window. This tells you how much data will be processed to run the query, which determines the cost of the query (https://cloud.google.com/bigquery/pricing) or how much of your allotted free tier data processing it will consume. When you are ready to proceed you can click the Run button in the upper left corner of the interface to run the query. The results will be displayed underneath the query window and you can save a local copy using the Save Results button displayed at the top of that section.
Example queries that can be used in the BigQuery console
Filtering based on genomic position on the reference sequence, NC_045512.2:
FROM `bigquery-public-data.nih_sequence_read.annotated_variations`
WHERE POS < 1000 and POS > 2000
Filter for runs with a specific mutation:
FROM `bigquery-public-data.nih_sequence_read.annotated_variations`
WHERE variation = "E484K"
Filtering based on read support:
FROM `bigquery-public-data.nih_sequence_read.annotated_variations`
WHERE DP <= 100 and G_AD_2 <49 AND G_AD_2/DP <=0.15
Queries of the annotated variation table can be combined with the SRA metadata table, which contains library and sample metadata for all public Runs in SRA:
Filtering based on SARS-CoV-2 samples derived metagenomic/metatranscriptomic libraries:
FROM `bigquery-public-data.nih_sequence_read.annotated_variations`
WHERE run in
(SELECT acc
FROM `nih-sra-datastore.sra.metadata`
WHERE organism = "Severe acute respiratory syndrome coronavirus 2"
and librarysource in ("METAGENOMIC","METATRANSCRIPTOMIC"))
Filtering based on organism to find only runs which were not submitted as SARS-CoV-2, but where it was detected:
FROM `bigquery-public-data.nih_sequence_read.annotated_variations`
WHERE run in
(SELECT acc
FROM `nih-sra-datastore.sra.metadata`
WHERE organism != "Severe acute respiratory syndrome coronavirus 2")
Filter for runs with a specific mutation, then list the count and geographic location where the sample was collected in order of decreasing frequency:
FROM `nih-sra-datastore.sra.metadata`
WHERE acc in
(SELECT distinct run FROM `bigquery-public-data.nih_sequence_read.annotated_variations`
WHERE variation = "E484K" )
GROUP BY geo_loc_name_country_calc
ORDER BY count DESC
Filter for all variations called from samples collected in the United States after '2022-06-01':
FROM `bigquery-public-data.nih_sequence_read.annotated_variations`
WHERE run in
(SELECT acc
FROM `nih-sra-datastore.sra.metadata`
WHERE collection_date_sam < '2022-06-01' and geo_loc_name_country_calc = 'USA')
The data in these tables is updated over time as more Runs containing SARS-CoV-2 are submitted to SRA, processed, and included. If you would like to pin the SRA Metadata table for future use, the process is similar to pinning the annotated variations table above. From the BigQuery interface, click the Add data button on the upper left side of the screen in the Explorer panel. Next, select Pin a project by name, paste nih-sra-datastore into the Project name box and click Pin. It will now appear on the left side of the page in the Explorer panel. The list of fields in this table is available here: SRA metadata table.
You can find more information on using public datasets here: https://cloud.google.com/bigquery/public-data.
A video tutorial with more in-depth background and examples of using BigQuery on SRA metadata is available here: https://youtu.be/DkNz-RCCm-M.
Viewing and Navigating the Open Bucket
For users who wish to browse the dataset or manually download a few files, the data files can be viewed on the web using the cloud console here: https://console.cloud.google.com/storage/browser/nih-sequence-read-archive.
For users who wish to browse the dataset or manually download a few files, the data files can be viewed on the web using the cloud console here: https://console.cloud.google.com/storage/browser/nih-sequence-read-archive.
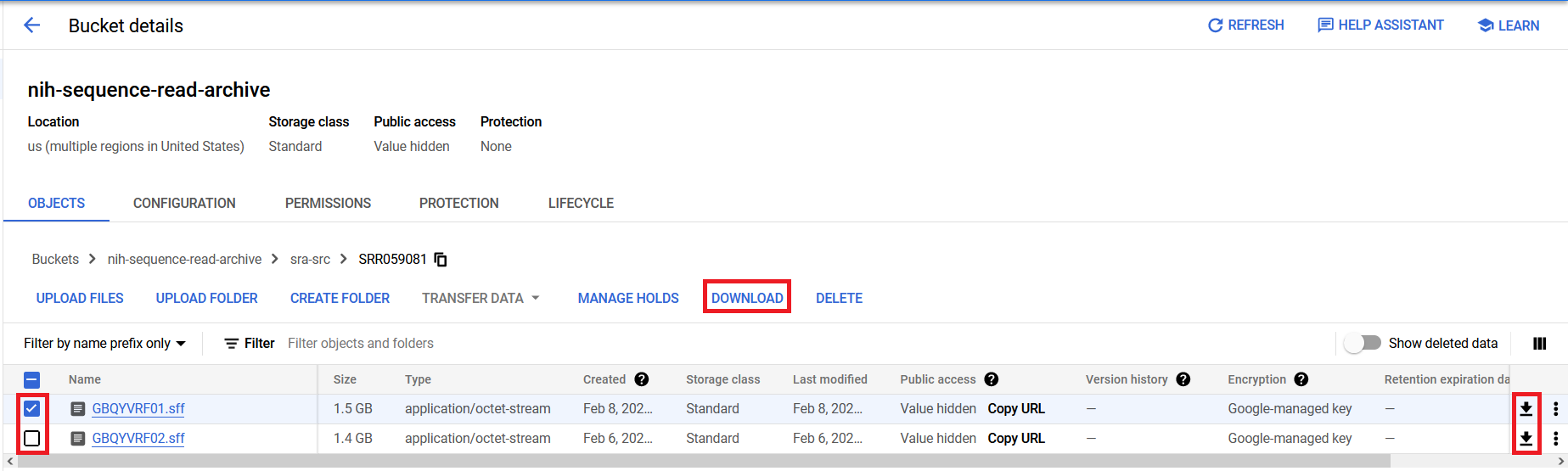
If you are interested in specific accessions, you can use the Filter box to filter the displayed contents of the directory.

The bucket/object path for any level of the directory structure can be copied to the clipboard by clicking the copy icon shown next to the path schema near the top of the bucket page.
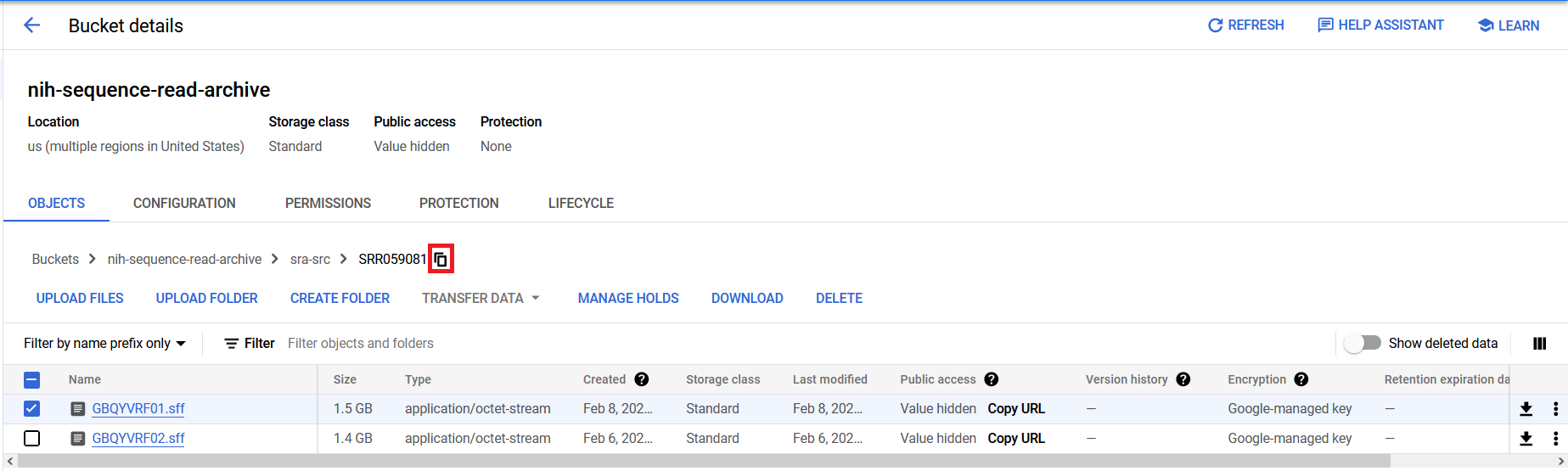
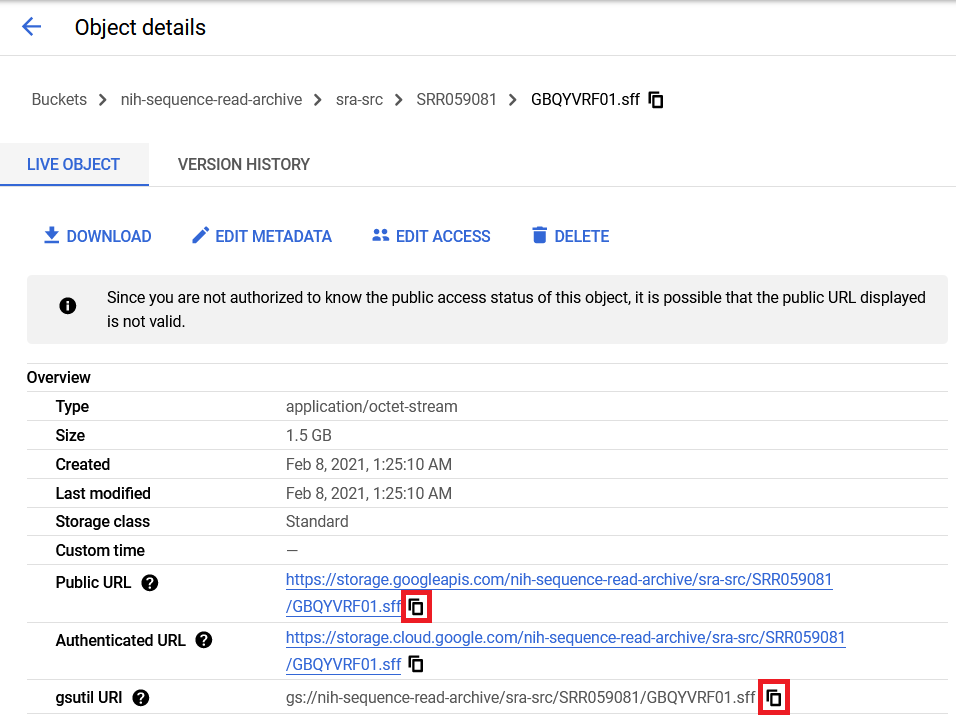
You can also click on any object in the bucket to go to the object details page that lists the public URL and gsutil URI, which can be used to move or download the data. Either of these paths can be copied by clicking on the copy icon next to their respective sections of the object details page.
Using the Sequence Data from the Command Line
For data in normalized SRA format (files in the run directory), the SRA Toolkit is required to access the data.
If you are working on a Google Cloud VM
(https://www.ncbi.nlm.nih.gov/sra/docs/SRA-Google-Cloud/), the toolkit
will automatically pull the files from cloud storage with no need to manually copy them (be sure to enable
report cloud instance identity from the GCP tab of the configuration utility during setup).
You can find instructions for installing and configuring the toolkit here:
https://github.com/ncbi/sra-tools/wiki/02.-Installing-SRA-Toolkit
https://github.com/ncbi/sra-tools/wiki/03.-Quick-Toolkit-Configuration
Once installed and configured you can access these data by accession using the SRA Toolkit. For instance, to generate fastq formatted files from the normalized data, the fasterq-dump tool can be used:
The default output will be separate forward and reverse fastq files for each Run
(if the data are paired-end), written to the current working directory. The toolkit accepts space
delimited sets of Run accessions directly on the command line or they can be provided in
a text file, one accession per line, using the --option-file flag.
If you prefer to move files to your own storage, the files from the open bucket can be copied programmatically using the gsutil package (https://cloud.google.com/storage/docs/gsutil) and the gsutil URI:
The gsutil URI can be generated by concatenating the bucket path (gs://nih-sequence-read-archive/)
with the object ID (eg: run/SRR10003816/SRR10003816):
Alternatively, these files can be downloaded via WGET or similar protocols. You can construct the HTTP address by replacing the GS:// prefix of the URI with:
https://storage.googleapis.com/
to generate a download URL, eg:
https://storage.googleapis.com/nih-sequence-read-archive/run/SRR10003816/SRR10003816
Notes on NCBI-generated variants
The details of variant calling for these runs can be found here:
https://www.ncbi.nlm.nih.gov/sra/docs/sars-cov-2-variant-calling/
The annotation was carried out using VIGOR3:
https://pubmed.ncbi.nlm.nih.gov/22669909/
Additional Resources
If you would like to download the raw underlying metadata or manually import it as a custom table in BigQuery, the location of the raw metadata files in JSON format for the annotated variations table can be found below:
gs://nih-sequence-read-archive/SARS_COV_2/annotated_variations/*
Full table definitions can be found here:
https://www.ncbi.nlm.nih.gov/sra/docs/sra-cloud-based-annotated-variations-table/
https://www.ncbi.nlm.nih.gov/sra/docs/sra-cloud-based-metadata-table/
Contact SRA
Contact SRA staff for assistance at [email protected]