|
Summary
Information for refSNPs
The NCBI genome map viewer can be configured to
supply additional information about elements in the master map by selecting
verbose mode in the display settings dialog box.
When the "SNP" map is selected as the
master map, the user is presented with a graphical summary of several
properties of the SNP in a display that looks like this:
In silico map results
The quality of the SNP is computed from mapping
and validation data. The current set of map icons and their meanings
are:
| Map |
Meaning |
 |
Marker mapped to unique position in the genome. Markers mapped
to two contigs on the same chromosome will also receive this icon, on the
assumption that the contigs might not have been joined during sequence
assembly. |
 |
Marker mapped to multiple positions in the genome. Markers
mapped to 2 to 10 different locations of the genome will receive this warning
icon. Markers mapped to more than 10 locations will not be annotated on human
genome sequence, nor will they be included in the genome map views. |
 |
Markers that have been WITHDRAWN in dbSNP as a duplicated gene
region or experimental artifact will be flagged with this WITHDRAWN icon.
Evidence for withdrawing the marker can be found in the dbSNP refSNP report for
the marker. |
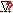 |
This
marker is not currently mapped to any genome location. |
|
Mouseover Text |
|
MAP COUNTS:
reports the number of hits to each of the following categories:
Total number of chromosomes [CH]
Total number of contigs [NT]
Total distinct locations [TOT] |
Gene region / functional classification
Markers placed on the sequence map are examined
for neighboring gene features (gene, mRNA, CDS, and exon). Markers that satisfy
the conditions below are considered a member of the set of markers associated
the the gene, and each is assigned a functional classification depending on its
particular location in the gene region:
| Gene |
Meaning |
 |
LOCUS: Any part
of the marker position on sequence map is within a 2kb interval 5' of the most
5' feature of gene (CDS, mRNA, gene), OR the marker position is within a 500
base interval 3' of the most 3' feature of the gene. Both strands of sequence
are examined for gene features, so a marker can potentially be a variation on
multiple genes at a single location. |
 |
TRANSCRIPT: Any
part of marker position overlaps with mRNA location (or overlaps with
UTR/intron and mRNA feature is missing), BUT marker position is not within the
coding region of the transcript. |
 |
CODING: Any part
of the marker position overlaps with a coding sequence (CDS) region (or
overlaps with exon region in the unlikely case an exon is annotated but CDS is
missing). |
 |
The marker is not
within the gene locus (as defined above) for any annotated gene. |
 |
The marker is not
within a transcript region for any annotated gene. |
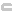 |
The marker is not
within a coding region for any annotated gene. |
|
Mouseover Text |
|
Mouseover on any
letter will show the set of gene symbols for each respective functional
category |
Marker heterozygosity
This 0 – 100% scale indicates the average
heterozygosity as the range avg_het±2[SE(avg_het)], where SE(avg_het) is
the standard error of the estimator. Thus the graph shows an approximate 95%
confidence interval for the marker.
| Het |
Meaning |
 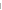 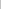 |
No allele
frequencies or measures of observed heterozygosity were submitted for this
marker. |
 |
Average
heterozygosity shown as a 95% confidence interval (0.26 – 0.30). The
estimate has a small standard error. |

|
Average
heterozygosity shown as a 95% confidence interval (0.00 – 0.40) The
estimate has a large standard error. |
|
Mouseover Text |
|
Mouseover on the
graph will report average heterozygosity and standard error in the form Het
(S.E.). E.g. Het (S.E.)=0.32 (0.008) |
Quality Information
This scale distinguishes validated markers
( ) from
unvalidated ones ( ) from
unvalidated ones ( ). Viewed as a vertically stacked histogram, the width
of each scale reports the likelihood that a marker is real as defined by the
submitter. This likelihood is based on validation experiments conducted by the
submitter for each batch of reported markers, where the success rate for a
batch is defined as 1 minus the false positive rate. Scale probabilities
are: ). Viewed as a vertically stacked histogram, the width
of each scale reports the likelihood that a marker is real as defined by the
submitter. This likelihood is based on validation experiments conducted by the
submitter for each batch of reported markers, where the success rate for a
batch is defined as 1 minus the false positive rate. Scale probabilities
are:
| Validation |
Meaning |
|
Marker has been
validated by independent resequencing. |
|
Unvalidated
marker: success rate > 95% |
|
Unvalidated
marker: success rate > 90% |
| |
Unvalidated
marker: success rate > 80% |
|
Mouseover Text |
|
The success rate
for the marker is reported. If several submissions have been clustered into a
single refSNP, then the reported figure is the maximum success rate for the
cluster. |
Other Information
| |
Meaning |
 |
Individual
genotypes have been submitted to dbSNP for this marker. |
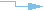 |
Links have been
established between the dbSNP marker and a submitter website or database. These
resources can provide additional information about the quality of the marker or
its phenotypic effects for the organism or gene. |
|