

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Feingold KR, Anawalt B, Blackman MR, et al., editors. Endotext [Internet]. South Dartmouth (MA): MDText.com, Inc.; 2000-.

ABSTRACT
In the U.S. alone, more than one million people are living with type 1 diabetes (TID) and approximately 80 people per day, or 30,000 individuals per year, are newly diagnosed (1, 2). Recent epidemiological studies demonstrate that the global T1D incidence is increasing at a rate of approximately 3-4% per year, notably among younger children (3, 4). Despite improvements in insulins, insulin delivery methods, and home glucose monitoring, the vast majority of those with T1D do not achieve recommended levels of glycemic control. This is particularly true in childhood and adolescence, where a recent U.S. study reported mean HbA1c values exceeding 9.5%, and a high frequency of both DKA and severe hypoglycemia (5). In addition to the increased risk of morbidity and mortality, TID places significant emotional and financial burdens on individuals, families, and society. These realities highlight the need for both better TID therapies and the continued push towards the prevention of TID. In recent decades, research efforts have described the natural history of type 1 diabetes and expanded the ability to identify individuals at risk for the disease even before clinical onset, via the recognition of genetic markers or TID-specific autoantibodies. The increasing ability to identify the at-risk population affords researchers the opportunity to intervene at progressively earlier stages in the disease. With the understanding that established islet autoimmunity, confirmed by the presence of multiple T1D autoantibodies, inevitably leads to clinical TID, investigative efforts are shifting towards the prevention or modification of autoimmunity. Furthermore, with the mounting evidence that any amount of residual C-peptide improves long term clinical outcomes in TID, some therapies aim to preserve remaining beta cell function in those with clinical disease. In this chapter, we review the epidemiology of TID, genetic and environmental risk factors, the scientific underpinnings of previous and current approaches towards disease-modifying therapy, and future directions of clinical trials. For complete coverage of all related areas of Endocrinology, please visit our on-line FREE web-text, WWW.ENDOTEXT.ORG.

EPIDEMIOLOGY OF DIABETES
T1D, or autoimmune diabetes, represents 5-10% of diabetes, and like autoimmunity in general, TID is increasing worldwide. The increase likely is attributable to environmental factors or epigenetic changes, as genetic changes don’t occur rapidly enough to explain such a dramatic increase. The SEARCH for Diabetes in Youth Study is a multicenter observational study investigating trends in incidence and prevalence of diabetes in American youth < age 20. SEARCH data suggests that the prevalence of TID among non-Hispanic white youth is ~1/300 in the US by age 20 years (6). Between 2002 and 2009, the incidence of TID among non-Hispanic white youth < age 20 years increased by an average of 2.7% per year (7). Similarly, the EURODIAB study evaluated TID incidence trends in 17 European countries from 1989-2003 in youth < age 15 years, and found an average annual incidence increase of 3.9%. This trend predicts a 70% increase in TID prevalence between 2005-2020 among European youth < 15 years old (8) with the peak of diagnosis between ages 10-14 (9). While incidence and prevalence are well documented in children, TID occurs in adults as well, at a frequency that is less certain; estimates are that 25-50% of all TID cases are diagnosed in adulthood. The uncertainty likely is due to a less dramatic clinical presentation than is typically seen in children who present with TID. The incidence of TID varies tremendously by geographic location, with higher rates generally seen in countries located farther from the equator. Worldwide incidence data was reported in 2000 by the DIAMOND project (10), a WHO-sponsored effort to address the public health implications of TID. The incidence of TID between 1990 and 1994 in 50 countries is shown in Figure 1. Between 1990 and 1994, the incidence of TID in individuals aged 0-14 years in both Finland and Sardinia was 37/100,000 individuals, whereas the incidence in both China and Venezuela was 0.1/100,000 individuals, a 350-fold difference. The increased incidence coupled with reduced early mortality has contributed to the increasing prevalence of disease.
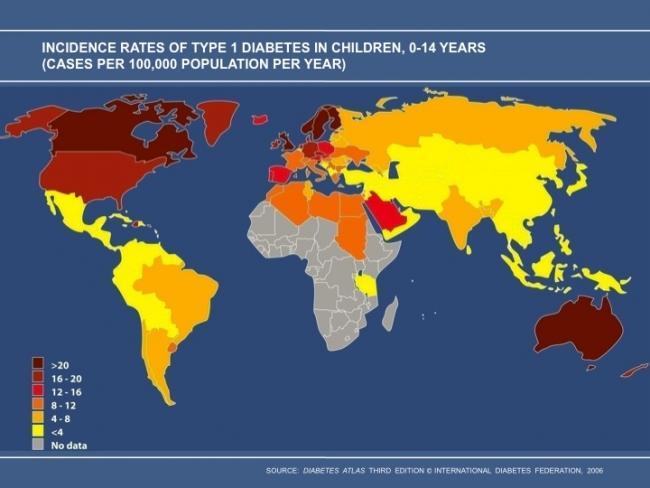
Figure 1.
Worldwide incidence of TID 1990-1994, used with permission from International Diabetes Federation.


WHAT IS THE RISK OF TYPE 1 DIABETES?
As is true for Cindy, 85% of individuals who develop TID have no family history of TID; nonetheless, a family history of the disease does increase an individual’s relative risk. The prevalence of TID in the US non-Hispanic white population by age 20 is ~0.3%, as compared with ~5% of those with a relative with TID, a 15-fold increase in relative risk. This relative risk is depicted in Figure 2.

Figure 2.
Among 300 people without a family member with diabetes, 1 will have TID. Among 300 people with a family member with diabetes, 15 will have TID
The risk of TID among family members varies depending on who the affected family member is, as shown in Table 1.
Table 1.
Prevalence of TID in Individuals with a Family History of TID.
The heritability pattern suggests that both genes and environment contribute to risk. Curiously, the risk of TID in offspring is higher if the father has TID (~6%) as compared to if the mother has TID (~2%) (11, 12). Moreover, the risk to a dizygotic twin is slightly higher (~10%) than is the risk to a non-twin sibling with similar HLA risk genes (~6%) (13, 14) suggesting that the intrauterine environment and/or similar early life exposures may be important. Lastly, the risk to a monozygotic twin is upwards of ~50%; surprisingly the second twin’s diagnosis may occur many decades after the index twin, highlighting the complexities of gene and environmental interactions that underlie the disease (15).


THE NATURAL HISTORY TYPE 1 DIABETES
It is now understood that TID is an immune-mediated disease that begins in the setting of genetic predisposition and then progresses along a predictable path: early islet autoimmunity (one autoantibody), established islet autoimmunity (two or more autoantibodies), abnormal glucose tolerance, clinical TID with some remaining beta cell function, and finally, little or no remaining beta cell function. This understanding comes from decades of effort by multiple investigators and from participation by thousands of patients with TID and their family members. George Eisenbarth’s description of TID as a chronic autoimmune disease, manifested by autoimmunity and a gradual linear fall in beta cell function until there is insufficient beta cell mass to suppress symptomatic hyperglycemia, has served for decades as the TID natural history paradigm (16). The “Eisenbarth” model has undergone refinements in recent years; namely, although autoimmunity and beta cell dysfunction do appear prior to diagnosis, these changes are often step-wise and non-linear. Furthermore, beta cell destruction may not be absolute. Nonetheless, the paradigm is largely correct and serves as the underlying rationale for TID trials.
The long pre-symptomatic natural history of TID presents an opportunity to intervene earlier than is done currently. Diabetes-specific autoantibodies can appear many years before clinical diagnosis and may reliably be used to predict disease progression. In 2015, JDRF, the Endocrine Society, and the American Diabetes Association proposed a new TID staging system which underscores that TID begins with islet autoimmunity rather than with symptomatic hyperglycemia (17). Stage 1 TID is defined as the presence of 2 or more autoantibodies with normoglycemia; stage 2 TID is 2 or more autoantibodies, impaired glucose tolerance and no symptoms; stage 3 TID is clinical disease. The staging system is depicted in figure 3.
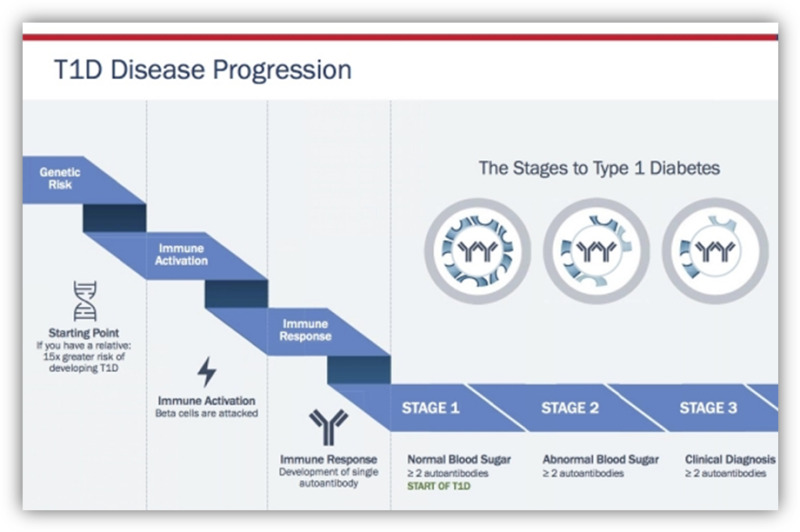
Figure 3.
New staging classification of Type 1 diabetes. Stages of Type 1 Diabetes. Adapted from internet image. https://beyondtype1.org/clinical-trials-and-the-type-1-diabetes-cure/final-trialnet-stages-of-diabetes-graph-2/ Used with permission.
HOW TO DETERMINE RISK OF TID
Risk of TID may be determined by the identification of autoantibodies, usually in those identified as having genetic risk through HLA testing or by family history. Autoantibodies are detectable years before the onset of clinical TID.
Determining Risk: Genes
With the knowledge that TID runs in families and with advances in technology, investigators have described the genetic risk of TID. TID risk is strongly linked to HLA class II DR3 and DR4 haplotypes, with the highest risk in those with the DR3/DR4 genotype. The importance of HLA genes to TID risk highlights the role of the adaptive immune system in the development of autoimmunity. Newer studies have discovered multiple other genes that also contribute to TID risk (18). They are largely genes known also to impact immune function; however, their contribution is dwarfed by the impact of HLA genes. Interestingly, recent work suggests that HLA genes primarily contribute to development of autoantibodies, while non-HLA genes and environmental factors may be more important in the progression from autoantibodies to clinically overt disease (19, 20). The description of non-HLA risk genes (such as the genes for insulin, a major TID autoantigen) highlights other potential pathways to disease and potential therapies.
Although the contribution of HLA class II risk genes overwhelms the contribution of non-HLA risk genes, the HLA contribution may be decreasing as the overall incidence of TID increases. This suggests that in a population with non-HLA genetic susceptibility, the environment may have become more conducive to the development of TID. This was reported in a 2004 Lancet article by Gillespie, et al., in which the investigators compared the frequency of HLA class II haplotypes in a UK cohort of 194 individuals diagnosed with TID between 1922-1946 (the Golden Years cohort) to a cohort of 582 individuals diagnosed between 1985-2002 (the BOX cohort) (21). In this comparison, shown in Figure 4, 47% of individuals in the Golden Years cohort were positive for the highest risk genotype DR3-DQ2/DR4-DQ8, compared to 35% of individuals in the BOX cohort.
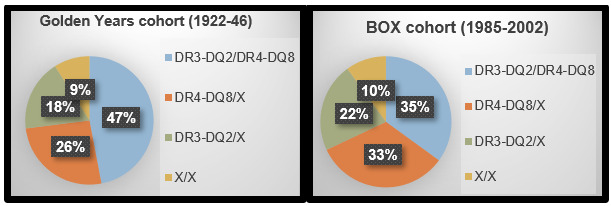
Figure 4.
Decreased contribution of high-risk HLA haplotypes over time. HLA class II haplotypes in Golden Years and BOX cohorts, adapted from Gillespie et.al Lancet 2004 (21).
Determining Risk: Family History and Islet Cell Autoantibodies
Natural history studies of relatives such as Diabetes Prevention Trial (DPT-1) and Diabetes TrialNet Pathway to Prevention have helped define the risk of TID in those with a family history of TID. Since 2000, Diabetes TrialNet has screened over 200,000 relatives of people with TID, aiming to enroll at-risk individuals in prevention trials. Among relatives of people with TID, ~5% will have at least one of five islet autoantibodies (22). TrialNet screens for islet cell antibodies (ICA), autoantibodies to insulin (IAA or mIAA), antibodies to a tyrosine phosphatase (IA-2; previously ICA512), antibodies to glutamic acid decarboxylase (GAD), and antibodies to a zinc transporter (ZnT8). With each additional autoantibody, the risk of TID increases predictably. Unsurprisingly, those with islet autoimmunity and abnormal glucose tolerance are at an even further increased risk of symptomatic T1D. The TrialNet strategy to identify islet autoimmunity among relatives of individuals with TID is shown in Figure 5. There are many other screening efforts ongoing outside of TrialNet. (23-25)
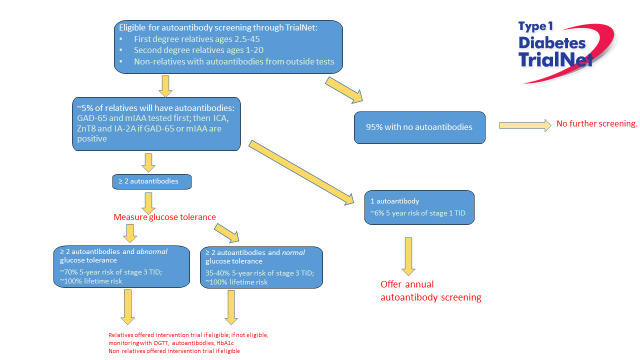
Figure 5.
Diabetes TrialNet process for identifying relatives with islet autoimmunity.
Natural history studies have shown not only that islet autoimmunity predicts TID risk, but also that islet autoantibodies usually appear early in life; 64% of babies destined to develop T1D before puberty will have antibodies by age 2 and 95% by age 5 (26). Furthermore, the data from both prospective birth cohort studies (27) and cross-sectional studies (28) (29) (30, 31) is remarkably consistent and suggests that the risk of progression from established autoimmunity to clinical TID is in the range of 40% after 5 years, 70% after 10 years, and 85% after 15 years. This risk over time is depicted in Figure 6. The key understanding from natural history studies is that essentially all individuals with confirmed islet autoimmunity will eventually develop clinical T1D at a rate of 11% per year.
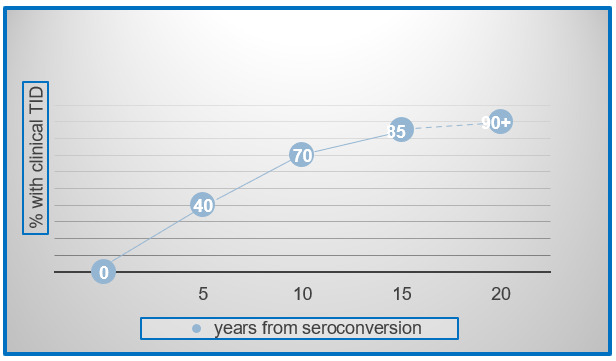
Figure 6.
Established islet autoimmunity inevitably progresses to clinical T1D. Extrapolated data from multiple studies in genetically at-risk individuals; Ziegler et al. JAMA 2013; DPT-1 Study Group Diabetes 1997; Sosenko et al. Diabetes Care 2014; Mahon et al. Pediatric Diabetes 2009
Identifying individuals with islet autoimmunity has two potential benefits; namely, the opportunity to monitor closely for disease progression, conferring a reduced risk of morbidity and mortality at the time of TID diagnosis, and the identification of individuals who are eligible for prevention trials. It is perhaps underappreciated that there is potentially a direct clinical benefit to identifying those with islet autoimmunity. Individuals with islet autoimmunity followed regularly until clinical diagnosis present with lower HbA1c and experience less DKA than those diagnosed in the community (Table 2) (32-36). For this reason, since 2009, the ADA has recommended that all individuals with a relative with T1D be counseled about the opportunity to be screened for diabetes autoantibodies in the context of a clinical research trial (37).
Table 2.
Individuals Diagnosed with T1D While Enrolled in a Clinical Trial have Less Morbidity at the Time of Diagnosis. (32-36)
| STUDY | HbA1c at time of TID diagnosis | % with DKA at time of TID diagnosis | ||
|---|---|---|---|---|
| Enrolled in study | Usual care | Enrolled in study | Usual care | |
| SEARCH | 25.5% | |||
| BABYDIAB | 8.6% | 11.0% | 3.3% | 29.1% |
| DPT-1 | 6.4% | 3.7% | ||
| DAISY | 7.2% | 10.9% | < 4% | |
| TEDDY < age 5 | 13.1% | |||
| SEARCH < age 5 | 36.4% | |||
| BABYDIAB < age 5 | 32.3% | |||
STRATEGIES TO BRING SCREENING FOR RISK TO CLINICAL PRACTICE
Screening relatives does identify a population of those at risk for clinical T1D; however, at least 85% who get T1D have no relatives with disease. Thus, to truly prevent all T1D, testing of the general population would have to occur. This could be done with current technology by testing all babies for genetic (HLA) risk at birth and then following with antibody testing. The Population Level Estimate of type 1 Diabetes risk Genes in children (PLEDGE) study enrolls newborns from the general population and offers one-time genetic testing and follow-up autoantibody testing at 2 and 4 years of age (38). The study aims to demonstrate feasibility and to develop evidence to support eventual inclusion of a T1D screening program in standard primary care.
Other studies, such as The Environmental Determinants of Diabetes in the Young (TEDDY) study, the Diabetes Autoimmunity Study in the Young (DAISY), and the Global Platform for the Prevention of Autoimmune Diabetes (GPPAD) are exploring similar methodologies to screen and monitor for risk (24, 39, 40). However, with an increasing number of individuals developing T1D even without the high-risk HLA types, such approaches may still miss some destined to develop disease.
An alternative risk detection strategy for those without a family history may be to perform point-of-care antibody testing in a routine pediatric visit. Since almost all who will develop diabetes before puberty will have antibodies by age 5; such testing could be done at age 4-5 and perhaps once again in the teenage years. This method will still miss those who develop T1D before this age, but would likely be a cost-effective approach to finding those at risk. If these at-risk subjects are monitored regularly until development of clinical disease they would benefit from reduced morbidity at time of diagnosis even if a prevention therapy were not yet available.
There are many ongoing projects aimed at screening members of the general population for diabetes autoantibodies even without prior HLA testing (23, 25, 41, 42).
As risk-screening programs employ varying assays and recruit from different populations, interpretation and translation of results is unclear. It is not yet known whether those found to be autoantibody positive through one program will experience the same rates of T1D progression and/or benefit from the same therapies as individuals who have participated in other screening and intervention efforts.

Source:(37)

PRENATAL INFLUENCES
The prenatal environment can have profound effects on the developing fetus. With the recognition that antibodies often develop early in life and that essentially all those with established islet autoimmunity (two or more autoantibodies) will eventually develop TID, investigators have looked to the prenatal period to search for factors that could contribute to disease development in utero. As shown in Table 3, decades of observational studies have yielded inconsistent results. Yet this remains an important area of investigation and one that may lead to primary prevention strategies for T1D. The Environmental Determinants of Islet Autoimmunity (ENDIA) study is an ongoing prospective birth cohort study in Australia that enrolled infants and unborn infants of first degree relatives with T1D. Biologic samples including blood, stool, and saliva will be collected longitudinally for investigation of factors including viral exposures during pregnancy and early childhood, maternal and fetal microbiome, delivery method, maternal and early infant nutrition, pregnancy and early childhood body weight, and both innate and adaptive immune function. In 2018, the ENDIA study completed target enrollment of ~1500 subjects, who will be followed regularly until the development of islet autoimmunity (43).
Table 3.
Potential Prenatal Influences on TID Risk
| Pre-natal or intrauterine exposure | Relative risk to offspring | Reference |
|---|---|---|
| Maternal age | Inconsistent data | (44-46) |
| Birth weight > 2 SD above norm (~4000g) | Inconsistent data | (47-51) |
| Birth weight < 2 SD below norm (~2500g) | Inconsistent data | (49-51) |
| Birth order: second and later borns | Inconsistent data | (46, 52, 53) |
| Birth interval < 3 years | Inconsistent data | (46, 54) |
| Caesarean delivery | Inconsistent data | (51, 55, 56) |
| Pre-eclampsia | Inconsistent data | (51, 57) |
| Pre-term delivery (<37 weeks gestation) | Inconsistent data | (51, 58) |
| Maternal vitamin D supplementation | Inconsistent data | (59-62) |
| Maternal antibiotic use | No association | (53, 63) |
| maternal BMI/pregnancy weight gain | No association | (51, 64) |
| Maternal omega 3 fatty acid supplementation | No association | (60, 65, 66) |

Source: (67)

Investigators also have studied the early childhood period for clues to the causes of islet autoimmunity and TID; these have included both observational studies and randomized clinical trials. Such influences might be divided into early nutritional exposures and early microbial/infectious exposures, both of which can affect development of the normal immune system.
The inconsistent findings relating to environmental factors reported from observational studies and clinical trials led to the design and implementation of a large international comprehensive evaluation of genetically at-risk babies using cutting edge technologies to study genetics, genomics (gene function), metabolomics, and the microbiome. The Environmental Determinants of Diabetes in the Young (TEDDY) is an international prospective birth cohort study that recruited almost 8,000 babies at increased risk for TID (based on HLA and family history) from Finland, Germany, Sweden, and the US from 2004-2010. Information on environmental exposures such as diet (including breastfeeding history), infections, vaccinations, and psychosocial stressors will be collected. Participants will be followed until the age of 15 for the development of islet autoimmunity or TID. The wealth of data from this study will provide a foundation for future randomized clinical trials (24). One interesting finding reported in December 2019 is that there are subtle differences in the gut microbiome—such as, persistent stool enterovirus B species--in children who develop islet autoimmunity compared to children who do not develop autoimmunity (68).
EARLY NUTRITIONAL EXPOSURES
Breastfeeding
The hypothesis that human breastmilk may protect against future TID development was presented as early as 1984 (69). Since then, there have been several prospective cohort studies to suggest that breastmilk lowers the risk of islet autoimmunity and TID, including the German BABYDIAB/BABYDIET study (70), the Colorado-based DAISY study (71), and the Norwegian MIDIA study (72), but others show no effect (73). Although the data on whether breastmilk is protective against TID isn’t clear, it certainly isn’t harmful. Given the well-established general benefits of breastfeeding, patients may safely be advised to follow the American Academy of Pediatrics’ guidelines related to infant feeding. The mechanism by which breastmilk may lower the risk of TID is uncertain, but one theory suggests that breastmilk has positive effects on the infant microbiome. The microbiome is discussed in greater detail below.
Cow’s Milk and Bovine Insulin Exposure
In contrast to considering breastfeeding as potentially beneficial in protecting against autoimmunity, it was hypothesized that early introduction of cow’s milk or cow protein might accelerate disease. This concept was tested in the Trial to Reduce IDDM in the Genetically at Risk (TRIGR) which asked whether weaning to hydrolyzed casein (which is free of bovine proteins including insulin) formula (n=1081) instead of regular cow’s milk formula (n=1078) in genetically at-risk infants could prevent or delay TID. Though the TRIGR pilot study was suggestive of benefit, no benefit was seen in the fully powered study (74) (75). Similarly, The Finnish Dietary Intervention Trial for the Prevention of Type 1 Diabetes of (FINDIA) suggested that weaning to hydrolyzed cow’s milk formula was not effective in reducing the appearance of autoantibodies, though they did report that a patented cow’s milk formula specifically removing bovine insulin appeared to be beneficial in this pilot study (76). While additional studies may be informative, current data does not support that weaning to hydrolyzed cow’s milk formula is protective against islet autoimmunity.
Gluten Exposure
Both BABYDIAB (77) and DAISY (78) were observational studies that suggested an association between introduction of gluten and islet autoimmunity. However, these studies had different results as to the timing of gluten introduction. Similarly, no effect was found in the BABYDIET study; a randomized controlled trial that asked whether delayed introduction of gluten to 6 vs 12 months would affect the risk of diabetes autoimmunity (79, 80).
Vitamin D and/or Omega 3 Fatty Acids
Vitamin D is an important component of a normal immune response; moreover, the higher incidence of TID in northern climates suggests that vitamin D deficiency could contribute to autoimmunity and TID. However, data from observational studies is mixed on whether vitamin D and/or omega 3 supplementation is beneficial or not (60, 81-86). A pilot randomized trial of omega 3 supplementation to pregnant mothers and infants failed to demonstrate a profound immunologic effect of treatment (87). With routine vitamin D supplementation recommended for infants (88), it is unlikely that a fully powered randomized trial would be feasible to assess the impact on autoimmunity.
MICROBIAL EXPOSURES
The Hygiene Hypothesis
Parallel to the rising incidence of TID and other autoimmune diseases, there has been a worldwide trend towards urbanization, increased standard of living, smaller family sizes, less crowded living conditions, safer water and food supplies, less cohabitation with animals, wide use of antibiotics, childhood vaccination, etc. While these trends are generally considered improvements in human existence, the so-called “hygiene hypothesis,” proposed by Strachan in 1989 (89) suggests a possible downside; that is, that early microbial exposures might have a protective effect via the early education of the immune system and the development of normal tolerance to self-antigens. Data cited in support of the hygiene hypothesis comes from comparisons between eastern Finland and Russian Karelia (Figure 7) (90-92).
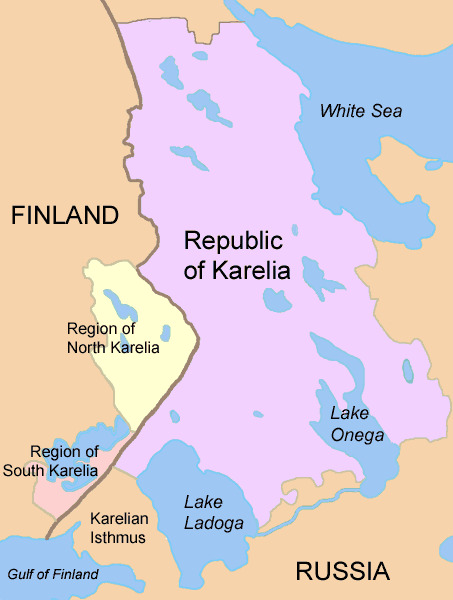
Figure 7.
Border between Finland and Russian Karelia, with a 6-fold difference in the incidence of TID, from "Karelia today”. The countries share a common border and ancestry and thus have similar geography, climate, vitamin D levels, and prevalence of HLA risk haplotypes. However, Finland has 6-fold higher incidence of TID. This markedly higher rate of TID is accompanied by a much lesser rate of infectious disease. In Finland as compared to Karelia 2% vs 24% had hepatitis A; 5% vs 24% had toxoplasma gondii; and 5% vs. 73% for helicobacter pylori. There is an ongoing study aiming to better understand the mechanisms that may underlie these differences.
The Microbiome
Another possible interface between microbial exposure and human disease is through the microbiome; that is the gut flora established within the first 3 years of life (93). It has been hypothesized that perturbations in normal early microbiome development might pre-dispose to disease whether through direct modulation of innate immunity or via alteration of intestinal permeability and the downstream effects on adaptive immunity. Interestingly, it appears that the gut microbiome is less diverse and less “protective” in individuals with islet autoimmunity or recent onset TID (94-96). Whether this difference is cause, effect, or correlation isn’t known. Nonetheless, multiple factors might affect the early intestinal microbiome, some of which also have been shown to correlate with risk of islet autoimmunity and TID. For example, breastfeeding can alter the intestinal microbiome of the infant by increasing the number and diversity of beneficial microbiota (97, 98). As previously discussed, multiple prospective observational studies suggest that breastfeeding protects against future development of islet autoimmunity and TID, but there’s no evidence to connect this directly to the infant microbiome.
Viral Infections
A viral etiology for initiation of autoimmunity is an attractive idea; a beta cell trophic virus could contribute to disease by directly killing beta cells, by leading to a chronic infection which triggers an immune response, or by molecular mimicry in which self-antigens are erroneously recognized as viral epitopes targeted for destruction. Notably, these possible mechanisms would not necessarily point to a particular virus; any virus widespread in a population could theoretically lead to autoimmunity in genetically susceptible individuals if encountered at a vulnerable time in immune system or beta cell development. With the notable exception of congenital rubella which is associated with type 1 diabetes (99), other data relating viruses to initiation of autoimmunity is less conclusive. While some studies have reported viral “footprints” in islets from individuals who have died from TID, these have not been consistently confirmed. Similarly, many studies have focused on enteroviruses, including coxsackie B, due to observations suggesting seasonal variation in antibody development that is reminiscent of the timing of such infections (100) (101), yet this remains controversial. Aside from a viral role in the initiation of autoimmunity, others have proposed that acute viral infections may impact the transition from islet autoimmunity to clinical TID due to increased insulin demand during infections. Patients commonly report an acute viral illness preceding the diagnosis of TID, and the clinical onset of TID more commonly presents in the fall and winter months in both the northern and southern hemispheres (102); but this does not imply a causal relationship.
Vaccinations
In recent decades, an increasing number of parents in Western countries have declined routine childhood vaccination of their children, which has created a situation with significant personal and public health consequences. Multiple high-quality studies have thoroughly investigated vaccinations and TID, and none have found any association with islet autoimmunity or TID (103-107)


DISEASE-MODIFYING THERAPY FOR PRECLINICAL TID
As previously discussed, the ability to recognize autoimmunity (via the detection of autoantibodies) in subjects even before the clinical onset of T1D affords the possibility of designing trials specifically for the high-risk population. One might consider established islet autoimmunity not only a marker of impending T1D, but a condition in its own right. Just as hypertension warrants treatment to prevent stroke and myocardial infarction, in the future, TID may be treated in its earliest stages to prevent symptomatic hyperglycemia. Some potential strategies are discussed in the following section.
Many TID studies have tested antigen-based therapies. With this type of therapy, the concept is that administration of a specific antigen could shift the immune response towards tolerance of the antigen. For example, in allergy desensitization therapy, small amounts of antigen are repeatedly administered to ‘teach’ the immune system to be tolerant of the foreign protein so that the immune system no longer reacts. In TID, the aim is to administer self-antigens in order to tolerize the immune system to beta-cell-derived proteins and downregulate the immune attack. Theoretically this can be done through oral, nasal, subcutaneous, or parenteral administration of antigen, with or without repeated dosing. Conceptually, antigen therapy should be more effective early in the disease process (i.e., to prevent progression from islet immunity to symptomatic disease rather than in those already clinically diagnosed) and thus most studies have targeted the at-risk population.
Perhaps the most rigorously tested antigen therapy for pre-clinical T1D is insulin, as in the GGAP-03 POInt, DPT-1, TrialNet oral insulin, DIPP, and INIT II trials, described next. The JDRF-funded GGAP-03 POInT Trial, a primary intervention dose-finding study, is evaluating whether or not early exposure to oral insulin, even before those with high genetic risk develop autoantibodies, may confer greater benefit. Results are expected in early 2025. Preliminary results from the pre-POINT pilot trial suggest that higher doses of oral insulin may elicit greater immunologic response (109). In the Diabetes Prevention Trial (DPT-1), 372 family members of T1D probands who were positive for both ICA and mIAA were assigned to receive either daily oral insulin or placebo (110). While this trial did not meet its primary endpoint, post-hoc analysis showed a delay in disease onset in participants with the highest levels of insulin autoantibodies. Specifically, those with a mIAA titer ≥80nU/ml showed a 4.5 year delay in disease onset and those with a mIAA titer ≥300nU/ml showed a 10 year delay in disease onset (111, 112). In response to these intriguing findings, Diabetes TrialNet launched a larger study to determine whether or not these results could be replicated While the fully-powered TrialNet study showed no benefit to oral insulin in the primary cohort of more than 300 individuals, an independently-randomized cohort of 55 antibody positive individuals who had low first phase insulin response at baseline had a significant delay in disease progression in those treated with oral insulin (113). This intriguing finding raised the possibility that oral insulin may benefit those who are closer to clinical diagnosis; that is, those with more active disease.
In addition to studying oral insulin, the DPT-1 evaluated the effect of parenteral insulin on individuals who were considered to have the highest risk for T1D. These participants were ICA positive with abnormal beta-cell function (dysglycemia on an OGTT or low first phase insulin response on IVGTT). These 339 high risk participants were assigned to either close observation or low dose subcutaneous ultra-Lente insulin in addition to annual four-day continuous insulin infusions. While the therapy was found to be ineffective in preventing the progression to T1D, there was no excessive hypoglycemia, and a subset analysis found a temporary decrease in the immune response to beta cell proteins (114).
To date, trials with intranasal insulin have proven safe but ineffective in preserving insulin secretion. The Type 1 Diabetes Prediction and Prevention Study (DIPP), a randomized controlled trial evaluating the effects of intranasal insulin in children with high-risk genotypes and autoantibody positivity, was negative. When intranasal insulin was administered soon after the detection of autoantibodies, there was no delay in the progression to T1D (115). Similarly, the Intranasal Insulin Trial II (INIT II), which tested a different dose and dosing schedule of nasal insulin in a phase II prevention trial, showed that intranasal insulin was safe and induced an immune response, but this did not alter the progression to T1D. Participants were first-degree relatives of T1D probands with autoantibody positivity (116, 117).
Another approach to antigen therapy is to use a plasmid to transfer DNA into cells, where it encodes for a given antigen, a technique that should decrease the anti-inflammatory response from intravenous, subcutaneous, oral, or nasal antigen delivery. This technique is being tested in the TrialNet TOPPLE T1D Study, a phase 1 trial launched in 2021 to evaluate the safety of a plasmid therapy called NNC0361-0041 in adults with recent-onset T1D. NNC0361-0041 encodes for four different human proteins: pre-proinsulin (PPI), transforming growth factor β1 (TGF-β1), interleukin-10 (IL-10), and interleukin-2 (IL-2) (118). In preclinical trials in NOD mice, the treatment was well-tolerated and led to beta cell preservation. If this phase 1 trial shows no safety concerns, then a larger study of the same treatment is planned to assess whether or not NNC0361-0041 can slow disease progression in the at-risk human population.
Antigen therapy may be more effective in both new-onset and at-risk populations when combined with other immune-modulating agents. Such combination trials are currently underway. In late 2020, enrollment was completed for a phase 1b/2a study assessing the safety and tolerability of different doses of an oral therapy called AG019 administered alone or in association with teplizumab infusions (see below) in individuals with recent-onset T1D. AG019 consists of live Lactococcus lactis bacteria, genetically modified to secrete human proinsulin and human interleukin 10. Results are pending (119).
While some trials have tested antigen-based therapies to treat islet immunity and prevent progression to clinical disease, others are building on successful studies of immunomodulating therapy in individuals with recently diagnosed TID. Examples include abatacept (Orencia; CTLA4 Ig) and teplizumab (Anti-CD3), both of which have been shown to slow loss of beta cell function post diagnosis. (See Recent Clinical Trials with Compelling Results and Figure 8). TrialNet recently completed enrollment of a placebo controlled trial testing abatacept in individuals with Stage 1 TID with results expected in late 2021 (120). In 2019, TrialNet published results of its placebo-controlled trial testing teplizumab in 76 individuals with Stage 2 TID. The trial demonstrated that a two-week course of teplizumab delayed the onset of clinical type 1 diabetes by two years and halved the rate of clinical diagnoses (121). This trial was highly significant in that it was the first ever to show that clinical type 1 diabetes can be delayed in children and adults at high risk. The latest findings from this trial, published in March of 2021, show ongoing delay of diabetes in the teplizumab treated group, with a median time to diagnosis of approximately 60 months (5 years) vs. approximately 27 months (2.3 years) in the placebo group (122). Teplizumab has been granted Breakthrough Therapy Designation by the FDA, and the manufacturer of teplizumab is pursuing full FDA approval.


Table 4.
Clinical Preconceptions are Not Always Correct
| AGE OF DIAGNOSIS: TID IS DIAGNOSED IN CHILDHOOD AND T2D IS DIAGNOSED IN ADULTHOOD. At least 25% of people with TID are diagnosed as adults. T1D is not “juvenile” diabetes. |
| WEIGHT: PEOPLE WITH TID ARE THIN, AND PEOPLE WITH T2D ARE OVERWEIGHT. At least 50% of people living with TID in the US are overweight or obese, a statistic which mirrors the general US population. Excess weight doesn’t prevent autoimmunity! |
| CLINICAL PRESENTATION: THE ONSET OF TID IS DRAMATIC, AND INSULIN IS IMMEDIATELY REQUIRED FOR TREATMENT. While this is generally true, the presentation of TID tends to be less abrupt in adults (in whom beta cell destruction is more gradual). Moreover, insulin isn’t always required immediately, especially in adults or in overweight individuals, where treatments to improve insulin sensitivity such as weight loss and/or metformin, may be sufficient to control blood glucose for a limited period of time. |
| RESIDUAL INSULIN SECRETION: PEOPLE WITH TID HAVE AN ABSOLUTE INSULIN DEFICIENCY. At the time of diagnosis, essentially all people with TID have clinically significant amounts of C-peptide. Furthermore, among those with > 40 years of TID, 6-16% have a non-fasting C-peptide level ≥0.017 nmol/L. |
| AUTOIMMUNITY: IF YOU DON’T FIND ANTIBODIES, IT’S NOT TID. There are five well-characterized antibodies associated with TID; most commercial laboratories don’t measure all five, so the results can be misleading. In addition, up to 10% of those with newly-diagnosed TID may not have antibodies. While these individuals may have a monogenic form of diabetes (http: |
IMPORTANCE OF BETA CELL PRESERVATION IN LIGHT OF RISKS OF THERAPY
The preservation of residual beta cell function, as measured by C-peptide, has repeatedly been demonstrated to be clinically important in those with T1D, warranting ongoing efforts to develop therapies to prevent beta cell destruction both in individuals with islet autoimmunity and in those with new-onset disease. In addition to its primary finding that intensive insulin therapy results in better outcomes (125, 126), the landmark Diabetes Control and Complications Trial (DCCT) showed that among intensively treated subjects, those who had ≥ 0.20 nmol/l stimulated C-peptide initially or sustained over a year had fewer complications, including 79% risk reduction in progression of retinopathy (127, 128). Importantly, these benefits were seen in the face of markedly less severe hypoglycemia. Subjects in the intensive insulin therapy group with ≥ 0.20 nmol/l C-peptide had about the same frequency of severe hypoglycemia as those in the standard care group; a 62% relative reduction as compared to those who received intensive therapy without this level of C-peptide. Subsequent analyses have demonstrated that even lower levels of preserved beta cell function in DCCT subjects were protective against complications (129). Importantly, a beneficial effect of preserved insulin secretion was also recently reported in those with type 2 diabetes. Endogenous insulin deficiency was strongly associated with hypoglycemia and a limited ability to control HbA1c in Type 2 subjects in the ACCORD study (130). Together, these data strongly support the concept that preserved insulin secretion coupled with intensive insulin therapy can reduce diabetes complications while averting the severe hypoglycemia that has been a limiting factor in attaining glycemic control.
Islet transplant studies confirm a positive association between C-peptide secretion and a lower risk of hypoglycemia. Subjects eligible for islet transplantation are largely individuals suffering from severe hypoglycemic unawareness. Vantyghem et al. showed that while significant beta cell function was required to improve mean glucose, lower glucose excursions, and result in insulin independence in transplant patients, only minimal beta cell function was needed to abrogate severe hypoglycemic events (131).
Additionally, post islet-cell transplant patients with higher as compared to absent or minimal C-peptide levels are more likely to maintain fasting blood glucose values in the 60-140mg/dL (3.3 – 7.8 mmol/l) range, HbA1c values <6.5% (47.4 mmol/mol), and insulin independence after transplantation (132). The DCCT showed similar metabolic benefits in those with residual C-peptide. In this trial, patients with C-peptide ≥ 0.2nmol/l had lower fasting glucose and HbA1c values. A 9-year longitudinal analysis showed that for every 1 nmol/l increase in baseline stimulated C-peptide, there was an associated 1% reduction in HbA1c among intensively treated DCCT participants (133). Such positive clinical outcomes in those with preserved C-peptide reinforce the significance of efforts to protect beta cell function.
Of course, the benefits of beta cell preservation must be weighed against the intrinsic risks of therapies used to preserve C-peptide. Two therapies in particular highlight the challenges of balancing benefits with risk. First, one of the initial immunomodulatory therapies used in T1D was cyclosporine, a general immunosuppressant. Treatment with cyclosporine induced remission from insulin dependence in children with recently diagnosed TID, with half of participants not requiring insulin after a full year of treatment (134). Unfortunately, the risks of using this drug were deemed to outweigh the benefits. Continuous effectiveness required continuous therapy, which induced nephrotoxicity (134).
More recently, studies with autologous hematopoietic stem cell transplant (HSCT) in the new onset population have further highlighted the risks of more aggressive approaches to treatment. Although the pooled data from HSCT trials suggests that this therapy imparts a high diabetes remission rate, the remission is not durable, and there are significant risks associated with the treatment, including neutropenic fever, serious infection, gonadal failure, and even death (135).
Importantly, there are dozens of immunotherapeutic agents or combinations of agents that are safely used in current clinical practice in other autoimmune diseases. For example, adults and children with juvenile idiopathic arthritis (JIA) are routinely treated with immunotherapy, an approach that has markedly transformed the lives of many living with this disease. Similarly, the aim for T1D is to use disease modifying therapies prudently and safely to truly improve the lives of those living with T1D. Possible approaches may include short term therapy aimed at inducing a long-term effect (tolerance), intermittent therapy, or limited doses of chronic therapy. Some of these methodologies are described below.
CLINICAL TRIALS WITH COMPELLING RESULTS IN NEW-ONSET T1D
Selecting therapies for clinical trials is based on multiple factors. We can now take advantage of the tremendous advances in understanding the disease process and basic and applied immunology. As illustrated in Figure 8, there are now therapies that target specific mechanisms underlying disease. Trials are considered in the context of what is known about safety of the therapy and efficacy in animal models, pilot studies, and other autoimmune diseases. Using these approaches, we have succeeded in altering disease course without the excessive risk previously described.
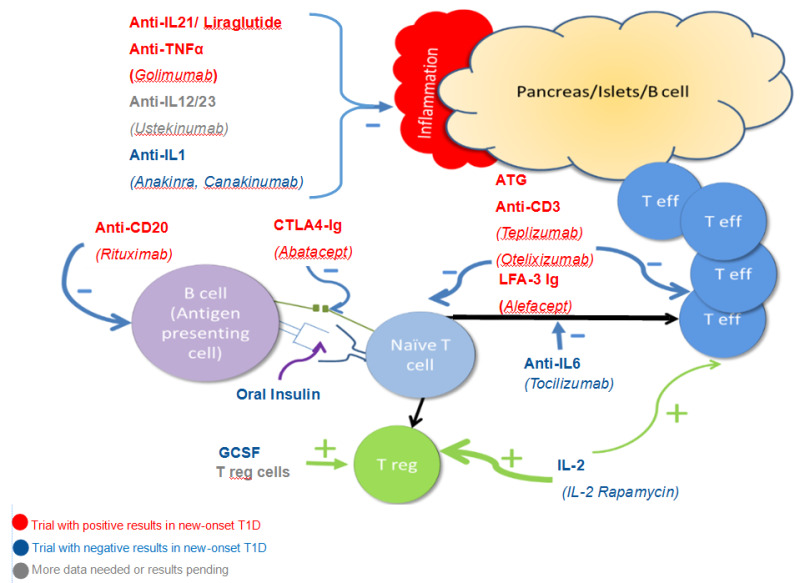
Figure 8.
Major pathways leading to beta cell destruction and potential mechanisms underlying the use of selected therapies. Both CD4 and CD8 T effector cells infiltrate and impair/destroy beta cells along with inflammatory cytokines such as IL 21, IL-1 and IL12/23. Anti-IL21/Liraglutide, Golimumab, Ustekinumab, Anakinra, and Canakinumab are aimed at blocking these inflammatory pathways. Activation of Teff cells depends upon presentation of antigen to naïve T cells which result in both Teff turning the immune response “on” and Treg cells turning the immune response “off”. Rituximab decreases B cells and therefore decreases the presentation of antigen to the immune system. Abatacept blocks co-stimulation and oral insulin (and other antigen therapy including the use of antigen specific dendritic cells) alters the response to self-antigen. The aim in both cases is to deviate the response to Treg cells or keep Teff cells from fully activating. ATG and anti-CD3 agents modulate and/or deplete activated T cells. Alefacept has a similar mechanism although primarily aimed at memory T cells. By blocking IL-6, Tocilizumab should change the balance of immune activation towards T regulatory cells. Similarly, GSCF, IL-2 (at the “right dose”), and infusion of Treg cells should preferentially increase Treg cells.
It is well established that T1D is the result of an immune-cell mediated destruction of the pancreatic beta cells. Many research efforts have thus targeted T-cells as well as the cells with which they interact. As in secondary prevention trials, anti-inflammatory agents, antigen therapies, and immunomodulatory drugs have all been used in tertiary prevention studies, which are designed to stop further beta cell destruction in the new onset population, therefore preventing complications. In addition, cellular therapies have been tested in this population. Excitingly, several therapies have now been shown to safely alter the disease course, particularly in the period soon after drug administration, allowing treated subjects to retain more C-peptide than controls 1-4 years later (Figure 9). Thus, while not yet ready for clinical use by endocrinologists, it is likely that immunotherapy with these or other agents will become a part of T1D new onset clinical care in the future.
Otelixizumab and Teplizumab (anti-CD3)
Some success in beta cell preservation has been shown with Teplizumab (hOKT3gl Ala-Ala) and Otelixizumab (ChAglyCD3), both of which are humanized Anti-CD3 monoclonal antibodies directed against the CD3 portion of the T-cell receptor. These drugs are distinct from OKT3, an anti-T cell agent with significant short term adverse effects. A study with Otelixizumab showed preserved insulin secretion for up to four years after 80 new-onset participants were treated with a single 6-day course of drug (136, 137). At 6, 12, and 18 months, the treatment group showed more residual beta cell function and a delay in the rise in insulin requirements as compared to the placebo group.
Similarly, in 2002, Herold et al. reported that a single 14-day course of Teplizumab given within the first 2 months of diagnosis resulted in more residual beta cell function at 12 months as compared to untreated individuals (138). While the effect of the therapy appeared most pronounced early on, follow-up of study participants continued to show differences in insulin production between treated and control subjects at 2 and 5 years after drug administration (139). In the AbATE Trial, a second course of Teplizumab was given 12 months after the first. In this study, C-peptide loss was delayed by an average of 15.9 months in treated subjects versus control subjects at 2 years (140). Finally, the Protégé Trial was a large phase III, placebo controlled randomized trial. While this study failed to meet its primary endpoint, post-hoc analysis found preserved beta cell function in a subset of the recent onset individuals who received Teplizumab as compared with placebo (141). As previously discussed, TrialNet found that 14 consecutive daily infusions of Teplizumab successfully delayed the progression from stage 2 T1D to stage 3 TID in family members by up to 3 years (122). Additionally, in 2019 Provention Bio launched Recent-Onset Type 1 Diabetes Trial Evaluating Efficacy and Safety of Teplizumab (PROTECT), a phase 3 trial (n=300) comparing two courses of 12 daily infusions of either teplizumab or placebo. The two courses are administered either 6 or 12 months apart. Results of the PROTECT study will provide additional safety and efficacy data for use of teplizumab in T1D.
Rituximab (anti-CD20)
In addition to anti-T cell therapies, investigators have studied anti-B-cell agents. A placebo controlled, double masked, randomized trial with Rituximab (anti-CD20) found that a single course of drug preserved C-peptide for 8.2 months in the drug-treated group compared to the placebo-treated group (142). The precise mechanism of action of Rituximab remains unclear, although it is believed that this therapy may reduce the production of pro-inflammatory cytokines or inhibit B lymphocyte antigen presentation, thus inhibiting the cascade of events leading to T-lymphocyte activation. Other anti-B-cell agents are being considered for study.
ATG-GCSF
In 2019, TrialNet completed a 3-arm study (n=82) of ATG compared to ATG and granulocyte colony stimulating factor (GCSF) compared to placebo. GCSF was combined with ATG to test whether GCSF may facilitate the return of T-regs following ATG-induced lymphocyte depletion. The 2-year C-peptide AUC was significantly higher in ATG treated subjects compared to placebo treated. Interestingly, GCSF did not provide additional benefit compared to ATG alone (143). Given the demonstrated benefit of low-dose ATG in stage 3 T1D, TrialNet may study this therapy in those with earlier stage disease.
Abatacept (CTLA4 Ig)
Abatacept works through co-stimulatory blockade; that is, the interruption of the interactions between different components of the immune system that propagate an immune response. A placebo-controlled, double-masked, randomized trial in the new onset population showed that when Abatacept therapy was provided continuously over 2 years, treated individuals benefited from a 9.6-month delay in beta cell destruction (144). Like the anti-B cell and anti-T cell therapies, the effect of Abatacept therapy on insulin secretion was most pronounced soon after initiation of drug. Importantly, while continued loss of beta cell function occurred over the remaining treatment period, when the drug was withdrawn, no acceleration of disease progression was seen (145). These findings set the stage for testing a shorter course of therapy in those with early stage T1D (stage 1 or stage 2). TrialNet is now studying Abatacept therapy in this population with the aim to prevent or slow onset of clinical disease (120).
Alefacept (LFA-3 Ig)
The T1Dal study assessed the use of Alefacept (LFA-3 Ig) in the new onset population in a placebo-controlled, double-masked, randomized trial. It was expected that Alefacept would target the memory cells of the immune response and mechanistic studies indicated that this was the case. Unfortunately, there was insufficient drug available to fully complete the study. As such, while there was a trend, the difference in C-peptide secretion measured at 2h between treated and control subjects was not statistically significant at 1 year. However, Alefacept therapy did preserve the 4h C-Peptide AUC at 1 year with lower insulin use, and also reduced hypoglycemic events, suggesting at least some efficacy (146). Moreover, further data found a positive effect of therapy 2 years after randomization (147).
Cytokine and Anti-cytokine Therapies
IL-1: It has been recognized for many years that the cytokine IL-1, a key factor in the inflammatory response, can injure beta cells. However, in recently diagnosed patients, two Phase 2 trials with different anti-IL-1 therapies (Anakinra and Canakinumab) failed to preserve beta cell function (148).
IL-2: IL-2 is necessary for immune cell proliferation, but the amount of IL-2 needed to promote T regulatory cells differs from that needed to promote T effector cells. A pilot study using IL-2 in T1D subjects aimed to exploit this difference and even exaggerate it by combining the therapy with Rapamycin, which selectively blocks T effector cells, thus resulting in an augmentation of T regulatory cells. Indeed, a marked increase in T regulatory cells was seen. Unfortunately, a transient decrease in beta cell function was also observed, leading to the trial’s early termination (149). It was suggested that (150) the decrease in beta cell function may have been due to IL-2 simulation of eosinophils and natural killer cells and it has thus been postulated that giving a lower dose or alternative form of IL-2 may more selectively augment Tregs. This was suggested by a small (n=24) study which defined an IL-2 dose range that was both safe and able to induce Treg expansion (151).
IL-6 is another important cytokine in the immune cascade. It promotes a particular type of T effector cell (Th17 cells), and some patients with T1D have an exaggerated response to IL-6. Tocilizumab blocks the IL-6 receptor and is effective (and approved for clinical use) in adult and pediatric arthritis patients. The Tocilizumab (TCZ) in New-onset Type 1 Diabetes (EXTEND) trial was a randomized trial in adults and children (n=136) with new onset T1D, completed in 2020. While the study confirmed the safety of tocilizumab, it did not demonstrate efficacy in new onset T1D, as measured by 2-hour C-peptide AUC in response to standardized MMTT (150).
IL12 and IL23 may indirectly contribute to the etiopathology of T1D, as they are involved in the production of IFN λ and IL-17, key cytokines in the generation of Th1 and Th17 effector cells. Ustekinumab is a monoclonal antibody that blocks a subunit common to IL12 and IL23 and is currently approved for treatment of psoriasis, psoriatic arthritis, ulcerative colitis and Crohn’s disease. Its efficacy to preserve C-peptide is being tested in a Canadian Phase 2/3 study in adults with recently diagnosed T1D (152, 153).
Anti-TNFα: The results of the T1GER Study, which assessed the effects of the anti-TNFα medication golimumab on beta cell function in 84 youth with new-onset T1D, were published in November, 2020. Participants aged 6-21 received either subcutaneous golimumab or placebo via injection in a 2:1 randomization for 52 weeks. At week 52, endogenous insulin production was significantly higher in the treatment group (0.64±0.42 pmol per milliliter vs. 0.43±0.39 pmol per milliliter, P<0.001) and exogenous insulin use was significantly lower. There was no significant difference in mean HbA1c or number of hypoglycemic events between groups, although there were more hypoglycemic events that met adverse event criteria in the treatment group. The promising results of this trial may warrant further investigation of anti-TNFα agents (154).
Anti-IL-21: A recent trial funded by Novo Nordisk investigated combination therapy with anti-interleukin (IL)-21 antibody and liraglutide (to improve β-cell function) as a means of enabling β-cell survival. 308 participants were randomly assigned to receive either anti-IL-21 plus liraglutide, anti-IL-21, liraglutide, or placebo (77 assigned to each group). Compared with placebo (ratio to baseline 0·61, 39% decrease), the decrease in MMTT-stimulated C-peptide concentration from baseline to week 54 was significantly smaller with combination treatment (0·90, 10% decrease; estimated treatment ratio 1·48, 95% CI 1·16-1·89; p=0·0017), but not with anti-IL-21 alone (1·23, 0·97-1·57; p=0·093) or liraglutide alone (1·12, 0·87-1·42; p=0·38). It is important to note, however, that 26 weeks after cessation of therapy, both the liraglutide monotherapy group and the combination therapy group showed increased C-peptide loss, perhaps suggesting that while liraglutide may transiently augment insulin secretion in the peri-diagnostic period, it is not beneficial to long-term beta cell function or survival (155).
OTHER APPROACHES
Cellular Therapy
Several clinical trials have tested administration of cells as compared to pharmaceutical agents with the aim of preserving beta cells. These include administration of antigen specific dendritic cells which are thought to restore immune tolerance by exploiting the role of dendritic cells in presenting antigen to the immune system (156). Autologous mesenchymal stromal cells (MSCs) are considered to have immunomodulatory properties and have also been examined and shown preliminary safety and proof of concept information in a pilot study (157). Other investigators have infused participants with T-regulatory cells (Tregs). These cells, which can come from saved umbilical cord blood or by expanding the patient’s own cells, should increase the number of Tregs, thereby altering the immune balance with T-effector cells and preventing further beta cell injury. Small studies to date have had conflicting results (158-160);
Therapies Directed at Components of the Innate Immune System
General anti-inflammatory agents have been tested as single agents in stage 3 TID and may be used in combination with other therapies in the future. For example, alpha-1-antitrypsin (A1AT) is a serum protease inhibitor that broadly suppresses pro-inflammatory cytokines such as IL-1, TNF-α, and IL-6. It has been tested in stage 3 TID, where it appears safe and well-tolerated (161). Bacillus Calmette-Guerin (BCG) has been proposed as a “vaccine” for those with T1D, citing the concept that BCG stimulation of innate immunity would alter the cytokine attack on beta cells. Notably, BCG is widely used, particularly in Europe, as a vaccine to prevent tuberculosis. Despite this broad usage, there is no epidemiological evidence that BCG administration has impacted the incidence of T1D. Moreover, a large, placebo controlled randomized trial demonstrated that BCG has no effect on insulin secretion, insulin requirements, or HbA1c in individuals with new onset T1D (162). Finally, the tyrosine kinase inhibitor imatinib (Gleevac), developed to treat leukemia, has several effects supporting its use in autoimmunity and T1D. The initial proposed mechanism of action is that the therapy reduces innate inflammation (163). However, other studies suggest it may also directly improve beta cell secretion (164). In a recent multicenter, randomized, double-blind, placebo-controlled study, 64 newly diagnosed adults were treated with either a 26-week course of imatinib or placebo in a 2:1 ratio. The study met its primary endpoint, showing preserved c-peptide secretion in the treatment group at 12 months. However, this effect was not sustained out to 24 months. Additionally, during the 24-month follow-up, 71% of participants who received imatinib had a grade 2 severity or worse adverse event. Imatinib might offer a novel means to alter the course of type 1 diabetes, but care must be taken to monitor for toxicities. Further trials to define an ideal dose and duration of therapy and to evaluate safety and efficacy in children or the at-risk population should be considered (165).
LESSONS FROM TRIALS WITH DISEASE MODIFYING THERAPIES
The trials that have successfully altered the course of disease by changing the rate of loss of C-peptide, even if for a brief period of time, have taught us much about the immune system and the natural history of T1D. First, it appears that the time of administration in the course of T1D may determine the effectiveness of a therapy as there appears to be a window during which agents may elicit the greatest effect upon the autoimmune process. Interestingly, in the cases of rituximab, otelixizumab/teplizumab, alefacept, ATG, golimumab, anti-IL-21, and abatacept, each of which has a different mechanism of action, treatment effected a marked delay in beta cell destruction/dysfunction initially, but thereafter, rates of decline in C-peptide paralleled those of the placebo groups (136, 140, 142-144, 155, 165, 166). Collectively, these observations suggest a difference in immune activity soon after diagnosis as compared with later on in the disease course (see Figure 9).
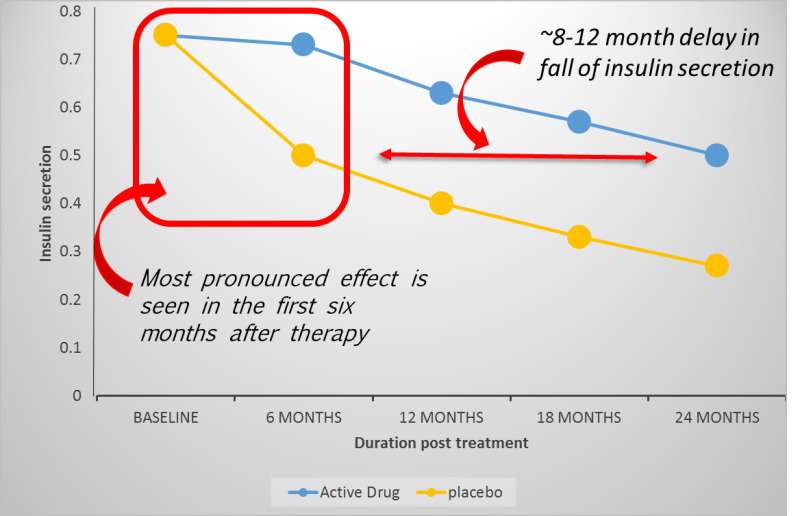
Figure 9.
Because of the time-dependent nature of the therapeutic response, the traditional approach of testing therapies in those with new-onset T1D before moving them “upstream” for use in treating autoimmunity may not be optimal. Several medications or combinations of medications are more likely to be effective earlier in disease. Thus, demonstration of efficacy in new onset trials should not be required before testing whether therapies can effectively treat islet autoimmunity.
The results of several trials have demonstrated that not all T1D patients are alike, and they vary in their response to therapy. For instance, in the Abate trial, 45% of subjects treated with teplizumab appeared to respond to the drug, showing almost no change in C-peptide secretion at two years, whereas 55% were deemed “non-responders” as their C-peptide secretion was not distinguishable from controls. Post-hoc analysis suggests that responders had lower A1C levels, less exogenous insulin use, and fewer Th-1-like T cells than non-responders (140). Next, post-hoc analysis from the Protégé trial revealed that C-peptide preservation was better in teplizumab treated patients who were aged 8-17, randomized within 6 weeks of diagnosis, had mean C-peptide AUC > 0.2nmol/l, A1c< 7.5%, and insulin dose < 0.4 units/kg/day (167). Last, as previously discussed, upon initial analysis of DPT-1 data, oral insulin did not appear to prevent T1D in the at-risk population. However, subsequent analysis showed a marked delay in diabetes development among those participants who had high titer anti-insulin autoantibodies (111). These results suggest that individualized therapies, which take into account a patient’s unique characteristics, are not only a possibility, but may be a necessity.
Participant age also appears to play a role in response to therapy, suggesting that optimal disease modifying agents may differ between pediatric and adult populations. Pre-teen children have less C-peptide at diagnosis than older children and adults. All age groups of children have a markedly different rate of fall of C-peptide than adults in the first year after diagnosis (168). Additionally, prior to diagnosis, children progress much faster through the preclinical stages of disease. Specifically, children with early autoimmunity (1 antibody) are more likely to develop established autoimmunity (2+antibodies) than adults; and children with established autoimmunity with or without abnormal glucose tolerance progress more rapidly to clinical diabetes than adults (169). Historically, the FDA has required that therapies first be tested in the adult population before they may be approved for use in the pediatric population. However, this approach may prevent researchers from identifying therapies that may only be viable in pediatric populations. Changing this paradigm was the focus of a recent American Diabetes Association consensus conference on disease modifying therapy (169).
In the next few years, not only will new agents be tested, but the community will build on these results by using them in selected individuals (personalized medicine), in combination trials, and at different stages of disease. Each step takes us closer to clinical use of a disease modifying agent.


RESIDUAL INSULIN SECRETION
Traditional teaching holds that all subjects with T1D will eventually lose all of their beta cells. This statement is no longer true; multiple lines of research demonstrate that a proportion of those even with longstanding T1D may have residual beta cell function. The Joslin Medalist study showed that 67% of 411 T1D subjects at least 50 years from diagnosis had at least minimal (0.03 nmol/l) random serum C-peptide levels. Of these individuals, 2.6% had random serum C-peptide ≥ 0.20 nmol/l. Post-mortem analysis of pancreata from these same subjects revealed that insulin positive cells were noted in 9/9 pancreases studied (170). Since many of the Joslin Medalists were diagnosed at a time when life expectancy was markedly reduced in those with T1D, it was felt that this was a unique population, not representative of the majority of people with T1D and that the preservation of C-peptide itself may have contributed to their long-term survival. However, multiple studies have now confirmed that C-peptide is present in a significant proportion of individuals with T1D. At the time of diagnosis, essentially all individuals (both youth and adults) have clinically significant levels of C-peptide (123, 168, 171). Two years after diagnosis, more than 66% of individuals retain these high levels (168). Unfortunately, with increasing duration of disease, the proportion of those with detectable C-peptide falls (124, 168, 170). However, as recently reported by Davis et al. (124), about 6-7% of those even more than 40 years from diagnosis have measurable C-peptide and more sensitive assays can actually detect C-peptide in a greater proportion of individuals. Moreover, like the pancreata from the Joslin cohort, studies from those who have had T1D for at least 4 years have shown that residual (insulin-positive) β-cells can be found in ~ 40% of T1D pancreases upon autopsy (172). Careful studies of post-mortem samples using new technologies have suggested that insulin-positive cells may be scattered in the exocrine tissue, raising the tantalizing possibility that new beta cells could emerge. Longitudinal studies of those long from diagnosis with low levels of C-peptide are underway to better understand variation over time.
There are two important take-aways from these new data. First, the presence of C-peptide does NOT rule out a T1D diagnosis. Yet, this data should not be over-interpreted; most individuals will eventually lose essentially all of their C-peptide secretion. The Davis study showed that 93% of those diagnosed as children had absent or extremely low levels of C-peptide >20 years from diagnosis (124).
To date, there are no therapies that have regenerated beta cells in humans. It is abundantly clear that mouse and human beta cells are markedly different, and therapies touted to grow cells in mice have not had such effects in humans. Instead of regeneration, replacement of dead or dysfunctional beta cells may be a viable option. Beta cell replacement is currently done through either whole pancreas or islet transplantation in conjunction with immune therapies to suppress the alloimmune (tissue rejection) and autoimmune (initial disease process) response. While outside the scope of this chapter, it has been recently recommended that those with severe hypoglycemic unawareness be referred for islet transplant (173). Other efforts to replace beta cells include placing them in capsules to allow viability and function while blocking immune cells from entering the capsules. These efforts remain experimental.

FUTURE CONSIDERATIONS
Despite advances in glucose monitoring and insulin delivery, the daily psychological and financial burden of disease on individuals, their families, and society together with the persistence of complications and reduced life span demand a paradigm shift.
As of 2021, we know much about the natural history of disease. We know that antibodies can develop early in life and that essentially all of those with established islet autoimmunity will develop clinically overt disease. We also know that identifying these individuals is of significant clinical benefit. Those with islet autoimmunity followed carefully until diagnosis have markedly less morbidity at the time of diagnosis and lower HbA1c values. Family members of T1D probands should be made aware of their disease risk and should be offered autoantibody screening and enrollment in monitoring trials. Correspondingly, patients with TID should be informed of the opportunity to have their relatives screened for TID risk in the setting of a clinical research study.
While the interaction of humans with their environment must contribute to disease; how this occurs is still being elucidated. It is likely that there are many different paths by which individual gene/environment interactions result in T1D; suggesting that dissecting this heterogeneity will provide better insights and therapies.
Whatever the primary cause, we know that the immune system is involved in disease progression. There have been successes in delaying beta cell destruction. Looking ahead, we will likely see the development of more targeted immunotherapies as well as more trials with combination therapies. Advances in treating childhood cancers have relied upon combining multiple approaches; this will be mimicked in T1D as well. More studies will be done in those with islet autoimmunity and variations in dose and route of administration of drugs will be tested in the search for greater efficacy. With newer and safer drugs, studies are likely to test chronic intermittent treatment for both islet cell autoimmunity and in new-onset TID to prevent further beta cell loss. Future studies will reflect the heterogeneity of TID. As medicine in general becomes more personalized, TID disease modifying therapies will target those most likely to benefit, whether because they are more likely to respond to therapy, or because their underlying disease is predicted to be worse.
Yet, there are non-scientific barriers to the use of disease modifying therapies for either islet cell autoimmunity or new-onset TID. One barrier is the lack of familiarity with these therapies amongst clinicians. Immune-modulating medications are used routinely by rheumatologists; whereas endocrinologists and others who care for people with TID are generally less comfortable with these therapies. This lack of familiarity exaggerates the risks and minimizes the benefits of immune-modulating medications. If we consider islet cell autoimmunity a silent disease in the same way that we consider hypertension a silent disease, then it makes sense to prevent the consequences of that disease, such as hyperglycemia in the case of islet cell autoimmunity, or cardiovascular disease in the case of hypertension. Similarly, if we consider new-onset TID in the same way we consider JIA, our goal in TID is to preserve beta cell function, just as in JIA, the goal is to preserve joint function.
With a shift in mindset and training, and in anticipation of successful clinical trials, one can envision a not-too-distant future in which endocrinologists might use immune modulating therapies to treat their patients who have islet cell autoimmunity and/or new-onset TID.
Table 5.
How to Keep Informed About Research Opportunities
| TrialNet http://www | Offers free autoantibody screening to relatives of individuals with type 1 diabetes. If autoantibody positive, participants may be eligible for a diabetes prevention or preservation trial. Offers New-onset trials to preserve beta cell function in those with new onset T1D (typically within 100 days of diagnosis) |
| ClinicalTrials.gov https://clinicaltrials.gov/ | Offers a complete registry of clinical trials being conducted in the US and worldwide. Provides an online search tool that allows users to search for clinical trials for which they might be eligible. |
| JDRF’s Clinical Trial Finder https://www.jdrf.org/impact/research/clinical-trials/ | JDRF is a global organization funding T1D research aimed at improving the lives of those living with the disease. JDRF has created a search tool that matches potential participants with enrolling trials. |
| Immune Tolerance Network http://www.immunetolerance.org/ | Offers clinical trials aimed at developing new therapeutic approaches for many immune-mediated diseases, including T1D. |
REFERENCES
- 1.
- JDRF Fact Sheet (accessed 2021 Dec 14) Available from: https://www
.jdrf.org /t1d-resources/about/facts/. - 2.
- Centers for Disease Control and Prevention, National Diabetes Statistics Report, 2014. 2014; (accessed 2021 Dec 14) Available from: http://www
.cdc.gov/diabetes /pubs/statsreport14 /national-diabetes-report-web.pdf. - 3.
- Patterson C., et al. Trends in childhood type 1 diabetes incidence in Europe during 1989-2008:Evidence of non-uniformity over time in rates of increase. Diabetologia. 2012;55(8):2142–2147. [PubMed: 22638547]
- 4.
- Bell R., et al. Diabetes in non-Hispanic white youth:prevalence, incidence, and clinical characteristics: the SEARCH for diabetes in youth study. Diabetes Care. 2009;32 Suppl 2:S102–S111. [PMC free article: PMC2647694] [PubMed: 19246575]
- 5.
- Miller K.M., et al. Current State of Type 1 Diabetes Treatment in the U.S.: Updated Data From the T1D Exchange Clinic Registry. Diabetes Care. 2015;38(6):971–978. [PubMed: 25998289]
- 6.
- Liese A.D., D'Agostino R.B. Jr, Hamman R. The burden of diabetes mellitus among US youth; prevalence estimates from the SEARCH for diabetes in youth study. Pediatrics. 2006;118(4):1510–18. [PubMed: 17015542]
- 7.
- Lawrence, J., et al., Trends in incidence of type 1 diabetes among non-Hispanic white youth in the U.S., 2002-2009 Diabetes, 2014. 63(11): p. 3938-3945. [PMC free article: PMC4207387] [PubMed: 24898146]
- 8.
- Patterson C., et al. Incidence trends for childhood type 1 diabetes in Europe during 1989-2003 and predicted new cases 2005-20: a multicentre prospective registration study. Lancet. 2009;373(9680):2027–2033. [PubMed: 19481249]
- 9.
- Incidence and trends of childhood Type 1 diabetes worldwide 1990-1999. Diabet. Med. 2006;23(8):857–866. [PubMed: 16911623]
- 10.
- Karvonen M., et al. Incidence of childhood type 1 diabetes worldwide. Diabetes Mondiale (DiaMond) Project Group. Diabetes Care. 2000;23(10):1516–1526. [PubMed: 11023146]
- 11.
- Warram J.H., et al. Differences in risk of insulin-dependent diabetes in offspring of diabetic mothers and diabetic fathers. N Engl J Med. 1984;311(3):149–52. [PubMed: 6738600]
- 12.
- Pociot F., et al. A nationwide population-based study of the familial aggregation of type 1 (insulin-dependent) diabetes mellitus in Denmark. Danish Study Group of Diabetes in Childhood. Diabetologia. 1993;36(9):870–875. [PubMed: 8405760]
- 13.
- Nistico L., et al. Emerging effects of early environmental factors over genetic background for type 1 diabetes susceptibility: evidence from a Nationwide Italian Twin Study. J Clin Endocrinol Metab. 2012;97(8):E1483–E1491. [PubMed: 22569240]
- 14.
- Kyvik K., Green A., Beck-Nielsen H. Concordance rates of insulin dependent diabetes mellitus: a population based study of young Danish twins. BMJ. 1995;311(7010):913–917. [PMC free article: PMC2550917] [PubMed: 7580548]
- 15.
- Redondo M., et al. Concordance for islet autoimmunity among monozygotic twins. N Engl J Med. 2008;359(26):2849–2850. [PubMed: 19109586]
- 16.
- Eisenbarth G.S. Type I diabetes mellitus. A chronic autoimmune disease. N. Engl. J. Med. 1986;314(21):1360–1368. [PubMed: 3517648]
- 17.
- Insel R.A., et al. Staging presymptomatic type 1 diabetes: a scientific statement of JDRF, the Endocrine Society, and the American Diabetes Association. Diabetes Care. 2015;38(10):1964–74. [PMC free article: PMC5321245] [PubMed: 26404926]
- 18.
- Concannon P., Rich S.S., Nepom G.T. Genetics of type 1A diabetes. N. Engl. J. Med. 2009;360(16):1646–1654. [PubMed: 19369670]
- 19.
- Knip M. Can we predict type 1 diabetes in the general population? Diabetes Care. 2002;25(3):623–5. [PubMed: 11874959]
- 20.
- Steck A.K., et al. Predictors of Progression From the Appearance of Islet Autoantibodies to Early Childhood Diabetes: The Environmental Determinants of Diabetes in the Young (TEDDY). Diabetes Care. 2015;38(5):808–13. [PMC free article: PMC4407751] [PubMed: 25665818]
- 21.
- Gillespie K.M., et al. The rising incidence of childhood type 1 diabetes and reduced contribution of high-risk HLA haplotypes. Lancet. 2004;364(9446):1699–1700. [PubMed: 15530631]
- 22.
- Mahon J.L., et al. The TrialNet Natural History Study of the Development of Type 1 Diabetes: objectives, design, and initial results. Pediatr. Diabetes. 2008 [PubMed: 18823409]
- 23.
- JDRF. T1Detect: Learn why you should be screened. (accessed 2021 Dec 14); Available from: https://www
.jdrf.org /t1d-resources/t1detect/. - 24.
- Hagopian W.A., et al. TEDDY--The Environmental Determinants of Diabetes in the Young: an observational clinical trial. Ann N Y Acad Sci. 2006;1079:320–6. [PubMed: 17130573]
- 25.
- Combined Antibody Screening for Celiac and Diabetes Evaluation (CASCADE). 2020.
- 26.
- Parikka V. N.-S.K., Saarinen M, Simell T, Ilonen J, Hyöty H, Veijola R, Knip M, Simell O., Early seroconversion and rapidly increasing autoantibody concentrations predict prepubertal manifestation of type 1 diabetes in children at genetic risk. Diabetologia. 2012;55(7):1926–36. [PubMed: 22441569]
- 27.
- Vehik K., et al. Methods, quality control and specimen management in an international multicentre investigation of type 1 diabetes: TEDDY. Diabetes Metab Res Rev. 2013;29(7):557–67. [PMC free article: PMC3992860] [PubMed: 23674484]
- 28.
- Ziegler A.G., et al. Seroconversion to Multiple Islet Autoantibodies and Risk of Progression to Diabetes in Children. Journal of the American Medical Association. 2013;309(23):2473–9. [PMC free article: PMC4878912] [PubMed: 23780460]
- 29.
- DPT-1 Study Group. Demographics of relatives screened and ICA positive in the diabetes prevention trial-type 1 diabetes (DPT-1). Diabetes. 1997;46 Suppl 1:142A.
- 30.
- Sosenko J., et al. Use of the Diabetes Prevention Trial- Type 1 Risk Score (DPTRS) for improving the accuracy of the risk classification of type 1 diabetes. Diabetes Care. 2014;37(4):979–984. [PMC free article: PMC3964487] [PubMed: 24550217]
- 31.
- Mahon J., et al. The TrialNet Natural History Study of the Development of Type 1 Diabetes: Objectives, design, and initial results. Pediatr Diabetes. 2009;10(2):97–104. [PubMed: 18823409]
- 32.
- Winkler C., et al. Markedly reduced rate of diabetic ketoacidosis at onset of type 1 diabetes in relatives screened for islet autoantibodies. Pediatric Diabetes. 2012;13(4):308–313. [PubMed: 22060727]
- 33.
- Triolo T., et al. Diabetic subjects diagnosed through the Diabetes Prevention Trial-Type 1 (DPT-1) are often asymptomatic with normal A1C at diabetes onset. Diabetes Care. 2009;32:769–773. [PMC free article: PMC2671125] [PubMed: 19407074]
- 34.
- Elding Larsson H., et al. Reduced prevalence of diabetic ketoacidosis at diagnosis of type 1 diabetes in young children participating in longitudinal follow-up. Diabetes Care. 2011;34(11):2347–52. [PMC free article: PMC3198296] [PubMed: 21972409]
- 35.
- Rewers A., et al. Presence of diabetic ketoacidosis at diagnosis of diabetes mellitus in youth: the Search for Diabetes in Youth Study. Pediatrics. 2008;121:e1258–e1266. [PubMed: 18450868]
- 36.
- Barker J., et al. Clinical characteristics of children diagnosed with type 1 diabetes through intensive screening and follow-up. Diabetes Care. 2004;27(6):1399–404. [PubMed: 15161795]
- 37.
- American Diabetes Association. I., American Diabetes Association Standards of Medical Care in Diabetes- 2021. Diabetes Care. 2021;44 Supplement 1:S18.
- 38.
- General Population Level Estimation for Type 1 Diabetes Risk in Children 0-5 Years Old During Routine Care Delivery (PLEDGE). 2021 October 27, 2020 (accessed 2021 Dec 14); Available from: https:
//clinicaltrials .gov/ct2/show/NCT04477928?term =PLEDGE+study&cond =t1d&draw =2&rank=1. - 39.
- Rewers M., et al. Newborn screening for HLA markers associated with IDDM: diabetes autoimmunity study in the young (DAISY). Diabetologia. 1996;39(7):807–12. [PubMed: 8817105]
- 40.
- Munchen, T.U., GPPAD-POInT (Global Platform of Autoimmune Diabetes - Primary Oral Insulin Trial). [PMC free article: PMC6609035] [PubMed: 31256036]
- 41.
- Barbara Davis Center for Diabetes, Autoimmunity Screening for Kids. 2019, March 13 (accessed 2021 Dec 14); Available from: https://www
.askhealth.org/. - 42.
- Puff R., et al. (Early diagnosis, early care--"Fr1da" screening of children for type 1 diabetes). MMW Fortschr Med. 2016;158(4):65–6. [PubMed: 27119891]
- 43.
- Megan AS Penno, J.J.C., Maria E Craig, et al. ENDIA Study Group, Environmental determinants of islet autoimmunity (ENDIA): a pregnancy to early life cohort study in children at-risk of type 1 diabetes, BMC Pediatr, 2013. 13-124: p. 1471-2431. [PMC free article: PMC3751791] [PubMed: 23941366]
- 44.
- Cardwell C., et al. Maternal age at birth and childhood type 1 diabetes: a pooled analysis of 30 observational studies. Diabetes. 2010;59(2):486–494. [PMC free article: PMC2809958] [PubMed: 19875616]
- 45.
- Flood T., Brink S., Gleason R. Increased incidence of type 1 diabetes in children of older mothers. Diabetes Care. 1982;5(6):571–573. [PubMed: 6985452]
- 46.
- Warram J.H., Martin B.C., Krolewski A.S. Risk of IDDM in children of diabetic mothers decreases with increasing maternal age at pregnancy. Diabetes. 1991;40(12):1679–1684. [PubMed: 1756908]
- 47.
- Cardwell C., et al. Birthweight and the risk of childhood-onset type 1 diabetes: a meta-analysis of observational studies using individual patient data. Diabetologia. 2010;53(4):641–651. [PubMed: 20063147]
- 48.
- Stene L., et al. Birth weight and childhood onset type 1 diabetes: population-based cohort study. BMJ. 2001;322(7291):889–892. [PMC free article: PMC30582] [PubMed: 11302899]
- 49.
- Dahlquist G., Bennich S., Kallen B. Intrauterine growth pattern and risk of childhood onset insulin dependent (type 1) diabetes: population based case-control study. BMJ. 1996;313(7066):1174–1177. [PMC free article: PMC2352527] [PubMed: 8916747]
- 50.
- Harder T., et al. Birth weight, early weight gain, and subsequent risk of type 1 diabetes: systematic review and meta-analysis. Am J Epidemiol. 2009;169(12):1428–1436. [PubMed: 19363100]
- 51.
- Robertson L., Harrild K. Maternal and neonatal risk factors for childhood type 1 diabetes: a Matched case-control study. BMC Public Health. 2010;10:281. [PMC free article: PMC2885337] [PubMed: 20507546]
- 52.
- Cardwell C., et al. Birth order and childhood type 1 diabetes risk: a pooled analysis of 31 observational studies. Int J Epidemiol. 2011;40(2):363–374. [PubMed: 21149280]
- 53.
- Virtanen S.M., et al. Microbial exposure in infancy and subsequent appearance of type 1 diabetes mellitus-associated autoantibodies: a cohort study. JAMA Pediatr. 2014;168(8):755–63. [PubMed: 24957949]
- 54.
- Cardwell C., et al. Interbirth interval is associated with childhood type 1 diabetes risk. Diabetes. 2012;61(3):702–707. [PMC free article: PMC3282800] [PubMed: 22315303]
- 55.
- Cardwell C., et al. Caesarean section is associated with an increased risk of childhood-onset type 1 diabetes mellitus: a meta-analysis of observational studies. Diabetologia. 2008;51(5):726–735. [PubMed: 18292986]
- 56.
- Khashan A., et al. Mode of obstetrical delivery and type 1 diabetes: a sibling design study. Pediatrics. 2014;134(3):e806–e813. [PubMed: 25092933]
- 57.
- Dahlquist G., Kallen B. Maternal-child blood goup incompatibility and other perinatal events increase the risk for early-onset type 1 (insulin-dependent) diabetes mellitus. Diabetologia. 1992;35(7):671–675. [PubMed: 1644246]
- 58.
- Zhang L., et al. Preterm birth and risk of type 1 and type 2 diabetes: systematic review and meta-analysis. Obes. Rev. 2014;15(10):804–811. [PubMed: 25073871]
- 59.
- Granfors M., et al. No association between use of multivitamin supplement containing vitamin D during pregnancy and risk of Type 1 Diabetes in the child. Pediatr Diabetes. 2016;17(7):525–530. [PubMed: 26552946]
- 60.
- Stene L.C., Joner G. Use of cod liver oil during the first year of life is associated with lower risk of childhood-onset type 1 diabetes: a large, population-based, case-control study. Am. J. Clin. Nutr. 2003;78(6):1128–1134. [PubMed: 14668274]
- 61.
- Fronczak C., et al. In utero dietary exposures and risk of islet autoimmunity in children. Diabetes Care. 2003;26(12):3237–3242. [PubMed: 14633808]
- 62.
- Marjamaki L., et al. Maternal intake of vitamin D during pregnancy and risk of advanced beta cell autoimmunity and type 1 diabetes in offspring. Diabetologia. 2010;53(8):1599–607. [PubMed: 20369220]
- 63.
- Kikkinen A., et al. Use of antimicrobials and risk of type 1 diabetes in a population-based mother-child cohort. Diabetalogia. 2006;49(1):66–70. [PubMed: 16344923]
- 64.
- Arkkola T., et al. Relationship of maternal weight status and weight gain rate during pregnancy to the development of advanced beta cell autoimmunity in the offspring: a prospective birth cohort study. Pediatr Diabetes. 2011;12(5):478–84. [PubMed: 21129139]
- 65.
- Niinisto S., et al. Fatty acid status in infancy is associated with the risk of type 1 diabetes-associated autoimmunity. Diabetologia. 2017;60(7):1223–1233. [PubMed: 28474159]
- 66.
- Sorenson I., et al. Serum long chain n-3 fatty acids (EPA and DHA) in the pregnant mother are independent of tisk of type 1 diabetes in the offspring. Diabetes Metab Res Rev. 2012;28(5):431–438. [PubMed: 22396195]
- 67.
- Ross A., et al. The 2011 report on dietary reference intakes for calcium and vitamin D from the Institute of Medicine: what clinicians need to know. J Clin Endocrinol Metab. 2011;96(1):53–38. [PMC free article: PMC3046611] [PubMed: 21118827]
- 68.
- Vehik. Kendra; Lynch, K.W., Matthew; et al. TEDDY Study Group, Prospective virome analyses in young children at increased genetic risk for type 1 diabetes. Nature Medicine, 2019. 12. [PMC free article: PMC6898786] [PubMed: 31792456]
- 69.
- Borch-Johnsen K., et al. Relation between breast-feeding and incidence rates of insulin-dependent diabetes mellitus: A hypothesis. Lancet. 1984;2(8411):1083–1086. [PubMed: 6150150]
- 70.
- Chimel R., et al. Early infant feeding and risk of developing islet autoimmunity and type 1 diabetes. Acta Diabetol. 2014 [PubMed: 25038720]
- 71.
- Frederiksen B., et al. Infant exposures and development of type 1 diabetes mellitus: The Diabetes Autoimmunity Study in the Young (DAISY). JAMA Pediatr. 2013;167(9):808–815. [PMC free article: PMC4038357] [PubMed: 23836309]
- 72.
- Lund-Blix N., et al. Infant feeding in relation to islet autoimmunity and type 1 diabetes in genetically susceptible children: the MIDIA Study. Diabetes Care. 2015;38(2):257–263. [PubMed: 25422170]
- 73.
- Couper J.J., et al. Lack of association between duration of breast-feeding or introduction of cow's milk and development of islet autoimmunity. Diabetes. 1999;48(11):2145–2149. [PubMed: 10535447]
- 74.
- Knip M., et al. Hydrolyzed infant formula and early beta-cell autoimmunity: a randomized clinical trial. JAMA. 2014;311(22):2279–87. [PMC free article: PMC4225544] [PubMed: 24915259]
- 75.
- Knip M. Writing group for teh TRIGR Study Group, Effect of Hydrolyzed Infant Formula vs Conventional Formula on Risk of Type 1 Diabetes. JAMA. 2018 Jan.319(1):38–48. [PMC free article: PMC5833549] [PubMed: 29297078]
- 76.
- Vaarala O., et al. Removal of Bovine Insulin From Cow's Milk Formula and Early Initiation of Beta-Cell Autoimmunity in the FINDIA Pilot Study. Arch Pediatr Adolesc Med. 2012;166(7):608–14. [PubMed: 22393174]
- 77.
- Ziegler A.G., et al. Early infant feeding and risk of developing type 1 diabetes-associated autoantibodies. JAMA. 2003;290(13):1721–1728. [PubMed: 14519706]
- 78.
- Norris J.M., et al. Timing of initial cereal exposure in infancy and risk of islet autoimmunity. JAMA. 2003;290(13):1713–1720. [PubMed: 14519705]
- 79.
- Hummel S, P.M., Hummel M. Primary dietary intervention study to reduce the risk of islet autoimmunity in children at increased risk for type 1 diabetes: the BABYDIET study. Diabetes Care. 2011;34(6):1302–1305. [PMC free article: PMC3114350] [PubMed: 21515839]
- 80.
- Beyerlein A., et al. Timing of gluten introduction and islet autoimmunity in young children: updated results from the BABYDIET study. Diabetes Care. 2014;37(9):e194–e195. [PubMed: 25147260]
- 81.
- Brekke H., Ludvigsson J. Vitamin D supplementation and diabetes-related autoimmunity in the ABIS study. Pediatr Diabetes. 2007;8(1):11–14. [PubMed: 17341286]
- 82.
- Vitamin D supplement in early childhood and risk for Type I (insulin-dependent) diabetes mellitus. The EURODIAB Substudy 2 Study Group. Diabetologia. 1999;42(1):51–54. [PubMed: 10027578]
- 83.
- Hypponen E., et al. Intake of vitamin D and risk of type 1 diabetes: a birth-cohort study. Lancet. 2001;358(9292):1500–1503. [PubMed: 11705562]
- 84.
- Zipitis C., Akobeng A. Vitamin D supplementation in early childhood and risk of type 1 diabetes: a systematic review and meta-analysis. Arch Dis Child. 2008;93(6):512–517. [PubMed: 18339654]
- 85.
- Simpson M., et al. No association of vitamin D intake or 25-hydroxyvitamin D levels in childhood with risk of islet autoimmunity and type 1 diabetes: the Diabetes Autoimmunity Study in the Young (DAISY). Diabetologia. 2011;54(11):2779–2788. [PMC free article: PMC3478880] [PubMed: 21858504]
- 86.
- Norris J., et al. Omega-3 polyunsaturated fatty acid intake and islet autoimmunity in children at increased risk for type 1 diabetes. JAMA. 2007;298(12):1420–1428. [PubMed: 17895458]
- 87.
- Chase H., et al. Effect of docosahexaenoic acid supplementation on inflammatory cytokine levels in infants at high risk for type 1 diabetes. Pediatr Diabetes. 2014 [PMC free article: PMC4291300] [PubMed: 25039804]
- 88.
- Wagner C., Greer F. Prevention of rickets and vitamin D deficiency in infants, children, and adolescents. Pediatrics. 2008;122(5):1142–1152. [PubMed: 18977996]
- 89.
- Strachan D.P. Hay fever, hygiene, and household size. BMJ. 1989;299(6710):1259–1260. [PMC free article: PMC1838109] [PubMed: 2513902]
- 90.
- Karelia map. 2015; (accessed 2021 Dec 14) Available from: https://commons
.wikimedia .org/wiki/File:Karelia_today .png#/media /File:Karelia_today.png. - 91.
- Kondrashova A., et al. The 'Hygiene hypothesis' and the sharp gradient in the incidence of autoimmune and allergic diseases between Russian Karelia and Finland. APMIS. 2013;121(6):478–93. [PubMed: 23127244]
- 92.
- Seiskari T., et al. Allergic senstization and microbial load--a comparison between Finland and Russian Karelia. Clin Exp Immunol. 2007;148(1):47–52. [PMC free article: PMC1868862] [PubMed: 17302731]
- 93.
- Yatsunenko T., et al. Human gut microbiome viewed across age and geography. Nature. 2012;486(7402):222–227. [PMC free article: PMC3376388] [PubMed: 22699611]
- 94.
- Giongo A., et al. Toward defining the autoimmune microbiome for type 1 diabetes. ISME J. 2011;5(1):82–91. [PMC free article: PMC3105672] [PubMed: 20613793]
- 95.
- Brown C.T., et al. Gut microbiome metagenomics analysis suggests a functional model for the development of autoimmunity for type 1 diabetes. PLoS One. 2011;6(10):e25792. p. [PMC free article: PMC3197175] [PubMed: 22043294]
- 96.
- de Goffau M.C., et al. Fecal microbiota composition differs between children with beta-cell autoimmunity and those without. Diabetes. 2013;62(4):1238–44. [PMC free article: PMC3609581] [PubMed: 23274889]
- 97.
- Bezirtzoglou E., Tsiotsias A., Welling G.W. Microbiota profile in feces of breast- and formula-fed newborns by using flourescence in situ hybridization (FISH). Anaerobe. 2011;2011(17):6. [PubMed: 21497661]
- 98.
- Stark P., Lee A. The microbial ecology of the large bowel of breast-fed and formula-fed infants during the first year of life. J Med Microbiol. 1982;15(2):189–203. [PubMed: 7143428]
- 99.
- Menser M., Forrest J., Bransby R. Rubella infection and diabetes mellitus. Lancet. 1978;1:57–60. [PubMed: 74564]
- 100.
- Oikarinen S., et al. Enterovirus RNA in blood is linked to the development of type 1 diabetes. Diabetes. 2011;60(1):276–9. [PMC free article: PMC3012181] [PubMed: 20943747]
- 101.
- Kimpimaki T., et al. The first signs of beta-cell autoimmunity appear in infancy in genetically susceptible children from the general population: the Finnish Type 1 Diabetes Prediction and Prevention Study. J Clin Endocrinol Metab. 2001;86(10):4782–8. [PubMed: 11600541]
- 102.
- Moltchanova E., et al. Seasonal variation of diagnosis of type 1 diabetes mellitus in children worldwide. Diabet Med. 2009;26(7):673–678. [PubMed: 19573115]
- 103.
- Duderstadt S., et al. Vaccination and risk of type 1 diabetes mellitus in active component U.S. military, 2002-2008. Vaccine. 2012;30(4):813–819. [PubMed: 22075092]
- 104.
- Graves P., et al. Lack of association between early childhood immunizations and beta-cell autoimmunity. Diabetes Care. 1999;22(10):1694–1697. [PubMed: 10526737]
- 105.
- DeStefano F., et al. Childhood vaccinations, vaccination timing, and risk of type 1 diabetes mellitus. Pediatrics. 2001;108(6):E112. p. [PubMed: 11731639]
- 106.
- Hviid A., et al. Childhood vaccination and type 1 diabetes. N Engl J Med. 2004;350(14):1398–1404. [PubMed: 15070789]
- 107.
- Elding Larsson H., et al. Pandemrix(R) vaccination is not associated with increased risk of islet autoimmunity or type 1 diabetes in the TEDDY study children. Diabetologia. 2018;61(1):193–202. [PMC free article: PMC5774660] [PubMed: 28990147]
- 108.
- Breastfeeding and the use of human milk. Pediatrics. 2012;129(3):e827–e841. [PubMed: 22371471]
- 109.
- Bonifacio E., et al. Effects of high-dose oral insulin on immune responses in children at high risk for type 1 diabetes: the Pre-POINT randomized clinical trial. JAMA. 2015;313(15):1541–9. [PubMed: 25898052]
- 110.
- Skyler JS. Effects of oral insulin in relatives of patients with type 1 diabetes: The Diabetes Prevention Trial--Type 1. Diabetes Care. 2005;28(5) K.J., Wolfsdorf J et al. [PubMed: 15855569]
- 111.
- Skyler J. Update on worldwide efforts to prevent type 1 diabetes. Ann N Y Acad Sci. 2008;1150:190. [PMC free article: PMC2928677] [PubMed: 19120293]
- 112.
- Vehik K., et al. Long-term outcome of individuals treated with oral insulin: diabetes prevention trial-type 1 (DPT-1) oral insulin trial. Diabetes Care. 2011;34(7):1585–1590. [PMC free article: PMC3120180] [PubMed: 21610124]
- 113.
- Writing Committee for the Type 1 Diabetes TrialNet Oral Insulin Study, G., et al., Effect of Oral Insulin on Prevention of Diabetes in Relatives of Patients With Type 1 Diabetes: A Randomized Clinical Trial. JAMA, 2017. 318(19): p. 1891-1902. [PMC free article: PMC5798455] [PubMed: 29164254]
- 114.
- Greenbaum C.J., et al. Parenteral insulin suppresses T cell proliferation to islet antigens. Pediatr Diabetes. 2011;12(3 Pt 1):150–5. [PMC free article: PMC2957543] [PubMed: 20522167]
- 115.
- Nanto-Salonen K., et al. Nasal insulin to prevent type 1 diabetes in children with HLA genotypes and autoantibodies conferring increased risk of disease: a double-blind, randomised controlled trial. Lancet. 2008;372(9651):1746–55. [PubMed: 18814906]
- 116.
- Health, M., Trial of Intranasal Insulin in Children and Young Adults at Risk of Type 1 Diabetes (INITII). ClinicalTrialsgov (Internet) Bethesda (MD): National Library of Medicine (US) 2000- (accessed 2021 Dec 14) Available from: http:
//clinicaltrials .gov/show/NCT00336674 NLM Identifier: NCT00336674. - 117.
- Team, P.E. ADC 2018: Update on the Intranasal Insulin Trial II (INIT II) to Prevent Type 1 Diabetes. 2018 (accessed 2021 Dec 14); Available from: https://www
.practiceupdate .com/content/adc-2018-update-on-the-intranasal-insulin-trial-ii-init-ii-to-prevent-type-1-diabetes /73021#:~:text=The %20estimated%205%2Dyear %20risk,people%20(38 %25)%20developed%20diabetes. - 118.
- Clinicaltrials.gov. A multiple ascending dose trial investigating safety, tolerability and pharmacokinetics of NNC0361-0041 (TOPPLE T1D); NCT04279613. 2020 (accessed 2021 Dec 14); Available from: https:
//clinicaltrials .gov/ct2/show/NCT04279613?term =TOPPLE&draw =2&rank=1. - 119.
- ClinicalTrials.gov. A study to assess the safety and tolerability of different doses of AG019 administered alone or in combination with teplizumab in participants with recently diagnosed type 1 diabetes mellitus (T1D); NCT03751007, 2018 (accessed 2021 Dec 14); Available from: https:
//clinicaltrials .gov/ct2/show/NCT03751007?term =AG019&draw =2&rank=1. - 120.
- Clinicaltrials.gov. CTLA4-Ig (Abatacept)for Prevention of Abnormal Glucose Tolerance and Diabetes in Relatives At -Risk for Type 1; NCT01773707. 2017; Available from: https:
//clinicaltrials .gov/ct2/show/NCT01773707?term =NCT01773707&draw =2&rank=1. - 121.
- Herold K.C., et al. An Anti-CD3 Antibody, Teplizumab, in Relatives at Risk for Type 1 Diabetes. N Engl J Med. 2019;381(7):603–613. [PMC free article: PMC6776880] [PubMed: 31180194]
- 122.
- Sims E.K., et al. Teplizumab improves and stabilizes beta cell function in antibody-positive high-risk individuals. Science Translational Medicine. 2021;13(583) [PMC free article: PMC8610022] [PubMed: 33658358]
- 123.
- Greenbaum C.J., et al. Preservation of beta-cell function in autoantibody-positive youth with diabetes. Diabetes Care. 2009;32(10):1839–44. [PMC free article: PMC2752937] [PubMed: 19587365]
- 124.
- Davis A.K., et al. Prevalence of detectable C-Peptide according to age at diagnosis and duration of type 1 diabetes. Diabetes Care. 2015;38(3):476–81. [PubMed: 25519448]
- 125.
- DCCT Group. The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. The Diabetes Control and Complications Trial Research Group. N. Engl. J. Med. 1993;329(14):977–986. [PubMed: 8366922]
- 126.
- Nathan D.M., et al. Intensive diabetes treatment and cardiovascular disease in patients with type 1 diabetes. N Engl J Med. 2005;353(25):2643–2653. [PMC free article: PMC2637991] [PubMed: 16371630]
- 127.
- Steffes M.W., et al. Beta-cell function and the development of diabetes-related complications in the diabetes control and complications trial. Diabetes Care. 2003;26(3):832–836. [PubMed: 12610045]
- 128.
- Lachin J.M., et al. Impact of C-peptide preservation on metabolic and clinical outcomes in the Diabetes Control and Complications Trial. Diabetes. 2014;63(2):739–48. [PMC free article: PMC3900540] [PubMed: 24089509]
- 129.
- Jeyam A., et al. Clinical Impact of Residual C-Peptide Secretion in Type 1 Diabetes on Glycemia and Microvascular Complications. Diabetes Care. 2021;44(2):390–398. [PubMed: 33303639]
- 130.
- Chow L., et al. Biomarkers related to severe hypoglycemia and lack of good glycaemic control in ACCORD. Diabetologia. 2015;58(6):1160–1166. [PMC free article: PMC4416999] [PubMed: 25652389]
- 131.
- Vantyghem M., et al. Continuous glucose monitoring after islet transplantation in type 1 diabetes: An excellent graft function (B-score greater than 7) is required to abrogate severe hypoglycemia, whereas minimal function is necessary to suppress severe hypoglycemia (B-score greater than 3). J Clin Endocrinol Metab. 2012;97(11):E2078–E2083. [PMC free article: PMC3485599] [PubMed: 22996144]
- 132.
- Barton F.B., et al. Improvement in outcomes of clinical islet transplantation: 1999-2010. Diabetes Care. 2012;35(7):1436–1445. [PMC free article: PMC3379615] [PubMed: 22723582]
- 133.
- Palmer J.P., et al. C-peptide is the appropriate outcome measure for type 1 diabetes clinical trials to preserve beta-cell function: report of an ADA workshop, 21-22 October 2001. Diabetes. 2004;53(1):250–264. [PubMed: 14693724]
- 134.
- Bougneres P.F., et al. Factors associated with early remission of type I diabetes in children treated with cyclosporine. N Engl J Med. 1988;318(11):663–70. [PubMed: 3125434]
- 135.
- D'Addio F, V.V., Ben NM, et al. Autologous nonmyeloablative hematopoietic stem cell transplantation in new-onset type 1 diabetes: a multicenter analysis. Diabetes. 2014;63(9) [PubMed: 24947362]
- 136.
- Keymeulen B., et al. Insulin needs after CD3-antibody therapy in new-onset type 1 diabetes. N. Engl. J. Med. 2005;352(25):2598–2608. [PubMed: 15972866]
- 137.
- Keymeulen B., et al. Four-year metabolic outcome of a randomised controlled CD3-antibody trial in recent-onset type 1 diabetic patients depends on their age and baseline residual beta cell mass. Diabetologia. 2010;53(4):614–623. [PubMed: 20225393]
- 138.
- Herold K.C., et al. Anti-CD3 monoclonal antibody in new-onset type 1 diabetes mellitus. N. Engl. J. Med. 2002;346(22):1692–1698. [PubMed: 12037148]
- 139.
- Herold K.C., et al. A single course of anti-CD3 monoclonal antibody hOKT3gamma1(Ala-Ala) results in improvement in C-peptide responses and clinical parameters for at least 2 years after onset of type 1 diabetes. Diabetes. 2005;54(6):1763–1769. [PMC free article: PMC5315015] [PubMed: 15919798]
- 140.
- Herold K.C., et al. Teplizumab (anti-CD3 mAb) treatment preserves C-peptide responses in patients with new-onset type 1 diabetes in a randomized controlled trial: metabolic and immunologic features at baseline identify a subgroup of responders. Diabetes. 2013;62(11):3766–74. [PMC free article: PMC3806618] [PubMed: 23835333]
- 141.
- Sherry N., et al. Teplizumab for treatment of type 1 diabetes (Protege study): 1-year results from a randomised, placebo-controlled trial. Lancet. 2011;378(9790):487–97. [PMC free article: PMC3191495] [PubMed: 21719095]
- 142.
- Pescovitz M.D., et al. Rituximab, B-Lymphocyte Depletion, and Preservation of Beta-Cell Function. N. Engl. J. Med. 2009;361(22):2143–2152. [PMC free article: PMC6410357] [PubMed: 19940299]
- 143.
- Haller M.J., et al. Low-Dose Anti-Thymocyte Globulin Preserves C-Peptide, Reduces HbA1c, and Increases Regulatory to Conventional T-Cell Ratios in New-Onset Type 1 Diabetes: Two-Year Clinical Trial Data. Diabetes. 2019;68(6):1267–1276. [PMC free article: PMC6610026] [PubMed: 30967424]
- 144.
- Orban T., et al. Co-stimulation modulation with abatacept in patients with recent-onset type 1 diabetes: a randomised, double-blind, placebo-controlled trial. Lancet. 2011;378(9789):412–9. [PMC free article: PMC3462593] [PubMed: 21719096]
- 145.
- Orban T., et al. Costimulation modulation with abatacept in patients with recent-onset type 1 diabetes: follow-up 1 year after cessation of treatment. Diabetes Care. 2014;37(4):1069–75. [PMC free article: PMC3964491] [PubMed: 24296850]
- 146.
- Rigby M.R., et al. Targeting of memory T cells with alefacept in new-onset type 1 diabetes (T1DAL study): 12 month results of a randomised, double-blind, placebo-controlled phase 2 trial. Lancet Diabetes Endocrinol. 2013;1(4):284–94. [PMC free article: PMC3957186] [PubMed: 24622414]
- 147.
- Rigby M.R., et al. Alefacept provides sustained clinical and immunological effects in new-onset type 1 diabetes patients. J Clin Invest. 2015;125(8):3285–96. [PMC free article: PMC4623571] [PubMed: 26193635]
- 148.
- Moran A., et al. Interleukin-1 antagonism in type 1 diabetes of recent onset: two multicentre, randomised, double-blind, placebo-controlled trials. Lancet. 2013;381(9881):1905–15. [PMC free article: PMC3827771] [PubMed: 23562090]
- 149.
- Long S.A., et al. Rapamycin/IL-2 combination therapy in patients with type 1 diabetes augments Tregs yet transiently impairs beta-cell function. Diabetes. 2012;61(9):2340–8. [PMC free article: PMC3425404] [PubMed: 22721971]
- 150.
- Greenbaum C. Sanda, S for the ITN058AI EXTEND Study Team, IL-6 receptor blockade does not slow β cell loss in new-onset type 1 diabetes. JCI Insight. 2021;6(21) [PMC free article: PMC8663550] [PubMed: 34747368]
- 151.
- Hartemann A, e.a., Low-dose interleukin 2 in patients with type 1 diabetes: a phase 1/2 randomised, double-blind, placebo-controlled trial. Lancet Diabetes Endocrinol. 2013 Dec;1(4):295-305, 2013. [PubMed: 24622415]
- 152.
- Clinicaltrials.gov. Pilot Clinical Trial of Ustekinumab in Patients With New-onset T1D (USTID); NCT02117765. 2016; (accessed 2021 Dec 2014) Available from: https:
//clinicaltrials .gov/ct2/show/NCT02117765. - 153.
- Clinicaltrials.gov. Clinical phase II/III trial of ustekinumab to treat type 1 diabetes (UST1D2) (UST1D2); NCT03941132. 2019; (accessed 2021 Dec 14) Available from: https:
//clinicaltrials .gov/ct2/show/NCT03941132?term =NCT03941132&draw =2&rank=1. - 154.
- Quattrin T.H. M.J., Steck, A.K., Felner, E.I., Li, Y., Xia, Y., Leu, J.H., Zoka, R., Hedrick, J.A., Rigby, M.R., Vercruysse, F.; T1GER Study Investigators Golimumab and beta-cell function in youth with new-onset type 1 diabetes. NEJM. 2020;383(21):2007–2017. [PubMed: 33207093]
- 155.
- von Herrath M., et al. Anti-interleukin-21 antibody and liraglutide for the preservation of beta-cell function in adults with recent-onset type 1 diabetes: a randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Diabetes Endocrinol. 2021;9(4):212–224. [PubMed: 33662334]
- 156.
- Giannoukakis N., et al. Phase I (safety) study of autologous tolerogenic dendritic cells in type 1 diabetic patients. Diabetes Care. 2011;34(9):2026–2032. [PMC free article: PMC3161299] [PubMed: 21680720]
- 157.
- Carlsson P., et al. Preserved beta-cell function in type 1 diabetes by mesenchymal stromal cells. Diabetes. 2015;64(2):587–592. [PubMed: 25204974]
- 158.
- Haller M., et al. Autologous umbilical cord blood transfusion in young children with type 1 diabetes fails to preserve c-peptide. Diabetes Care. 2011;34(12):2567–2569. [PMC free article: PMC3220832] [PubMed: 22011412]
- 159.
- Marek-Trzonkowska N., et al. Administration of CD4+CD25highC. Diabetes Care. 2012;35(9):1817–1820. [PMC free article: PMC3425004] [PubMed: 22723342]
- 160.
- Marek-Trzonkowska N., et al. Therapy of type 1 diabetes with CD4(+)CD25(high)CD127-regulatory T cells prolongs survival of pancreatic islets - results of one year follow-up. Clin Immunol. 2014;153(1):23–30. [PubMed: 24704576]
- 161.
- Gottlieb PA. A.A., Michels AW α1−Antitrypsin therapy downregulates toll-like receptor-induced IL-1β responses in monocytes and myeloid dendritic cells and may improve islet function in recently diagnosed patients with type 1 diabetes. J Clin Endocrinol Metab. 2014;99:E1418–E1426. [PMC free article: PMC4121034] [PubMed: 24527714]
- 162.
- Allen H.F., et al. Effect of Bacillus Calmette-Guerin vaccination on new-onset type 1 diabetes. A randomized clinical study. Diabetes Care. 1999;22(10):1703–1707. [PubMed: 10526739]
- 163.
- Louvet C1. S.G., Lang J, Lee MR, Martinier N, Bollag G, Zhu S, Weiss A, Bluestone JA, Tyrosine kinase inhibitors reverse type 1 diabetes in nonobese diabetic mice. Proc Natl Acad Sci U S A. 2008;105(48):18895–900. [PMC free article: PMC2596241] [PubMed: 19015530]
- 164.
- Hagerkvist R., et al. Amelioration of diabetes by imatinib mesylate (Gleevec): Role of beta-cell NF-kappaB activation and anti-apoptotic preconditioning. FASEB J. 2007;21(2):618–628. [PubMed: 17135364]
- 165.
- Gitelman S., et al. Imatinib therapy for patients with recent-onset type 1 diabetes: a multicentre, randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Diabetes and Endocrinology. 2021;9(8):502–514. [PMC free article: PMC8494464] [PubMed: 34214479]
- 166.
- Pescovitz M.D., et al. Rituximab, B-lymphocyte depletion, and preservation of beta-cell function. N Engl J Med. 2009;361(22):2143–52. [PMC free article: PMC6410357] [PubMed: 19940299]
- 167.
- Hagopian W., et al. Teplizumab preserves C-peptide in recent-onset type 1 diabetes: two-year results from the randomized, placebo-controlled Protege trial. Diabetes. 2013;62(11):3901–8. [PMC free article: PMC3806608] [PubMed: 23801579]
- 168.
- Greenbaum C.J., et al. Fall in C-peptide during first 2 years from diagnosis: evidence of at least two distinct phases from composite Type 1 Diabetes TrialNet data. Diabetes. 2012;61(8):2066–73. [PMC free article: PMC3402330] [PubMed: 22688329]
- 169.
- Wherrett D.K., et al. Defining pathways for development of disease-modifying therapies in children with type 1 diabetes: a consensus report. Diabetes Care. 2015;38(10):1975–85. [PMC free article: PMC4876737] [PubMed: 26404927]
- 170.
- Keenan H.A., et al. Residual insulin production and pancreatic ss-cell turnover after 50 years of diabetes: Joslin Medalist Study. Diabetes. 2010;59(11):2846–53. [PMC free article: PMC2963543] [PubMed: 20699420]
- 171.
- Dabelea D., et al. Clinical evolution of beta cell function in youth with diabetes: the SEARCH for Diabetes in Youth study. Diabetologia. 2012;55(12):3359–68. [PMC free article: PMC4492685] [PubMed: 22990715]
- 172.
- Gianani R., et al. Dimorphic histopathology of long-standing childhood-onset diabetes. Diabetologia. 2010;53:690–698. [PubMed: 20062967]
- 173.
- Choudhary P., et al. Evidence-informed clinical practice recommendations for treatment of type 1 diabetes complicated by problematic hypoglycemia. Diabetes Care. 2015;38(6):1016–1029. [PMC free article: PMC4439532] [PubMed: 25998294]
{kind=link}
- *
Contributed equally
- †
Contributed equally
- ABSTRACT
- EPIDEMIOLOGY OF DIABETES
- WHAT IS THE RISK OF TYPE 1 DIABETES?
- THE NATURAL HISTORY TYPE 1 DIABETES
- HOW TO DETERMINE RISK OF TID
- STRATEGIES TO BRING SCREENING FOR RISK TO CLINICAL PRACTICE
- PRENATAL INFLUENCES
- EARLY NUTRITIONAL EXPOSURES
- MICROBIAL EXPOSURES
- DISEASE-MODIFYING THERAPY FOR PRECLINICAL TID
- IMPORTANCE OF BETA CELL PRESERVATION IN LIGHT OF RISKS OF THERAPY
- CLINICAL TRIALS WITH COMPELLING RESULTS IN NEW-ONSET T1D
- OTHER APPROACHES
- LESSONS FROM TRIALS WITH DISEASE MODIFYING THERAPIES
- RESIDUAL INSULIN SECRETION
- FUTURE CONSIDERATIONS
- REFERENCES
- Folic acid supplementation and malaria susceptibility and severity among people taking antifolate antimalarial drugs in endemic areas.[Cochrane Database Syst Rev. 2022]Folic acid supplementation and malaria susceptibility and severity among people taking antifolate antimalarial drugs in endemic areas.Crider K, Williams J, Qi YP, Gutman J, Yeung L, Mai C, Finkelstain J, Mehta S, Pons-Duran C, Menéndez C, et al. Cochrane Database Syst Rev. 2022 Feb 1; 2(2022). Epub 2022 Feb 1.
- Short-term intensive insulin therapy could be the preferred option for new onset Type 2 diabetes mellitus patients with HbA1c > 9.[J Diabetes. 2017]Short-term intensive insulin therapy could be the preferred option for new onset Type 2 diabetes mellitus patients with HbA1c > 9.Weng J. J Diabetes. 2017 Oct; 9(10):890-893. Epub 2017 Aug 22.
- Continuous Subcutaneous Insulin Infusion (CSII) Pumps for Type 1 and Type 2 Adult Diabetic Populations: An Evidence-Based Analysis.[Ont Health Technol Assess Ser....]Continuous Subcutaneous Insulin Infusion (CSII) Pumps for Type 1 and Type 2 Adult Diabetic Populations: An Evidence-Based Analysis.Medical Advisory Secretariat. Ont Health Technol Assess Ser. 2009; 9(20):1-58. Epub 2009 Oct 1.
- Review Diabetes technology and treatments in the paediatric age group.[Int J Clin Pract Suppl. 2011]Review Diabetes technology and treatments in the paediatric age group.Shalitin S, Peter Chase H. Int J Clin Pract Suppl. 2011 Feb; (170):76-82.
- Review The Management of Type 1 Diabetes.[Endotext. 2000]Review The Management of Type 1 Diabetes.Subramanian S, Baidal D. Endotext. 2000
- Changing the Course of Disease in Type 1 Diabetes - EndotextChanging the Course of Disease in Type 1 Diabetes - Endotext
Your browsing activity is empty.
Activity recording is turned off.
See more...