

NCBI RefSeq Functional Elements
Overview
NCBI provides RefSeq and Gene records for non-genic functional elements that have been described in the literature and are experimentally validated. Elements in scope include experimentally verified gene regulatory regions (e.g., enhancers, silencers, locus control regions), known structural elements (e.g., insulators, DNase I hypersensitive sites, matrix/scaffold-associated regions), well-characterized DNA replication origins, and clinically-significant sites of DNA recombination and genomic instability. Priority is given to genomic regions that are implicated in human disease or are otherwise of significant interest to the research community. Currently, the scope of this project is restricted to human and mouse. Our current scope does not include functional elements predicted from large-scale epigenomic mapping studies, nor elements that exist solely based on disease-associated variation.
Each RefSeq Functional Element (RefSeqFE) sequence has a corresponding record in NCBI's Gene database (see example in Figure 1). NCBI Gene records for Functional Elements differ from conventional genes in that they have the Gene type 'biological region.' All Functional Element Gene records include a list and a graphical view of annotated feature types, a brief summary of the function of the region, a list of related INSDC accessions, and a comprehensive bibliography of relevant publications. A link to the orthologous human or mouse record is provided where appropriate.
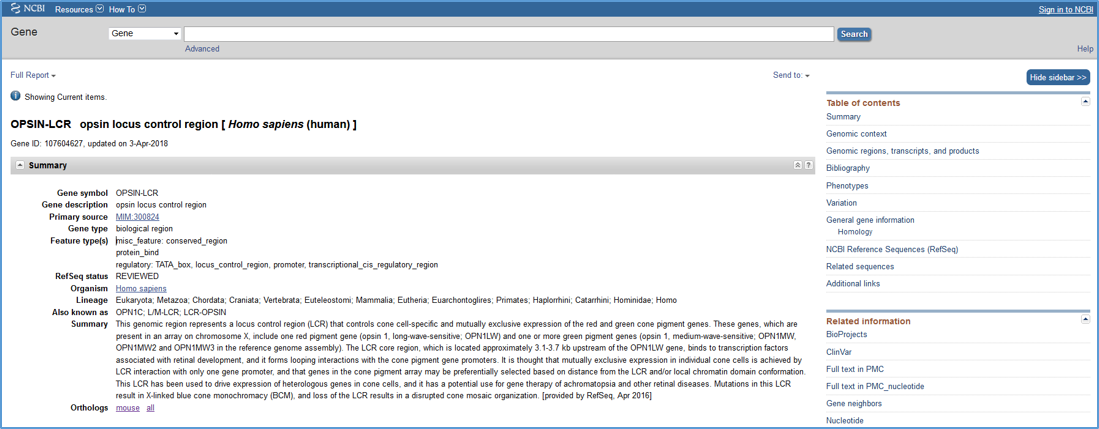
Figure 1. An example of an NCBI Gene record for a biological region (only the Summary section is shown here). Note that the 'Gene type' is 'biological region' and 'Feature type(s)' are listed.
RefSeq Functional Element Records
RefSeq Functional Element sequences are represented as follows:
- As genomic RefSeqs with an NG_ prefix and the 'RefSeqFE' keyword (e.g., NG_046887.1)
- As DNA sequences encompassing the genomic range of one or more experimentally-validated functional elements.
- Based on the plus strand of the current human or mouse reference genome assembly, unless otherwise indicated.
- With 100-nt padding on each end for extra genomic context.
- Elements considered to be functionally related and closely situated in the genome are included together on the same NG_ record (e.g., an enhancer and contained protein-binding sites; multiple nearby enhancer and/or promoter fragments).
- Experimentally-validated features are annotated on each sequence record through manual curation by NCBI RefSeq staff as described below.
- Manually curated RefSeq Functional Element records have a REVIEWED status (see About RefSeq for status descriptions).
- Records generated through automatic bulk processing, such as the validated dataset from the VISTA Enhancer Browser, have a PROVISIONAL status.
RefSeq Functional Element Feature Annotation
RefSeq Functional Element sequences include manually curated features in accordance with International Nucleotide Sequence Database Collaboration (INSDC) standards. Features that are supported by direct experimental evidence include at least one '/experiment' qualifier with an evidence code (ECO ID) from the Evidence & Conclusion Ontology, and at least one citation from PubMed. It is important to note that annotated sequence ranges may be approximate depending on the experimental evidence type, and that features may include extraneous sequences that are not necessary for function. Feature annotation can be viewed on the RefSeq Nucleotide flat file (Figure 2), in the graphical view in Gene records (Figure 3), and in NCBI genome browsers (see Access via NCBI Graphical Displays below).
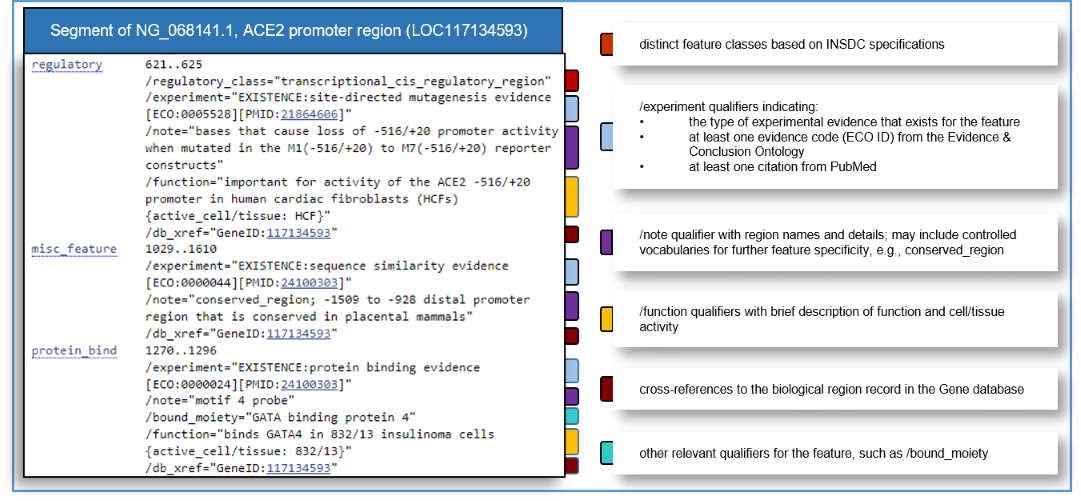
Figure 2. Example of a RefSeq Functional Element NG_ flat file and descriptions of feature annotation and common qualifiers.
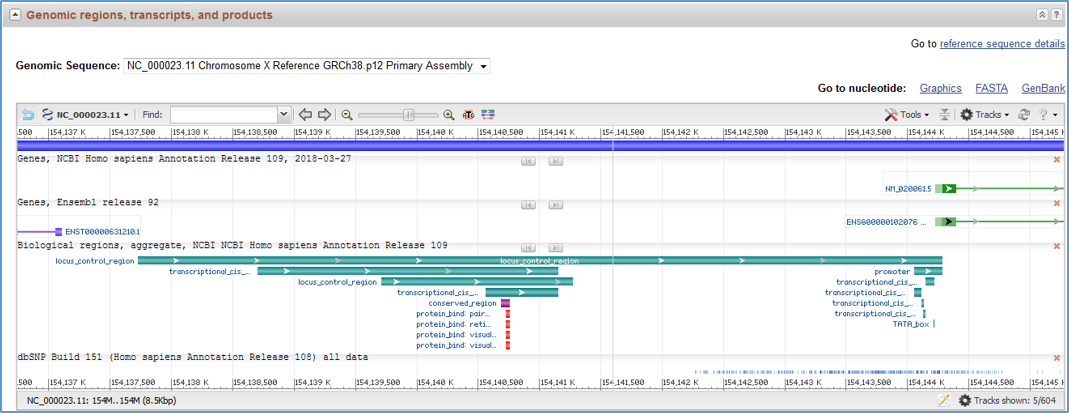
Figure 3. Feature annotation in the Gene graphical display. Additional track sets, including conventional gene annotation, repeat region and variation tracks, may be exposed using the Tracks button (see NCBI Sequence Viewer Documentation).
Feature Annotation Glossary
Features are annotated on RefSeq Functional Element NG_ records based on review of the scientific literature. Annotated features are in accord with INSDC Feature Table specifications, where some INSDC feature keys have specific feature classes, e.g., the 'misc_recomb' and 'regulatory' feature keys. In addition, RefSeq-specific controlled vocabulary terms are sometimes used to provide further feature specificity, e.g., for 'misc_feature,' or 'misc_recomb' or 'regulatory' features that are not defined by a specific feature class. The feature keys, feature classes and controlled vocabularies can be mapped to equivalent terms in the Sequence Ontology (SO), where those SO terms are used as SO_types for genome-annotated features in column 3 of NCBI GFF3 files (see the feature table below). The following feature types are used for RefSeq Functional Elements, with equivalent SO IDs shown in parentheses:
misc_feature
Used for functionally significant features that currently lack a more specific INSDC feature key. Controlled vocabularies are provided for additional feature specificity and to facilitate bulk search and retrieval. In GenBank flat files, controlled vocabulary terms are used at the beginning of a '/note' qualifier and are separated from any additional '/note' text by a semi-colon. Underscores replace spaces for the same terms in ASN.1 and GFF3 formats.
Flat file qualifier example: /note="conserved region; ultraconserved element uc.328"
RefSeq controlled vocabularies for 'misc_feature':
- biological_region (SO:0001411; Special note: This is a parental feature spanning all other feature annotation on each record. It is a 'misc_feature' denoted by '/note="Region: biological region"' in GenBank flat files, a 'Region' feature in ASN.1 format and a 'biological_region' in GFF3 format.
- CAGE_cluster (SO:0001917)
- conserved_region (SO:0000330)
- nucleotide_cleavage_site (SO:0002204)
- nucleotide_motif (SO:0000714)
- repeat_instability_region (SO:0002202)
- replication_start_site (SO:0002203)
- sequence_alteration (SO:0001059)
- sequence_comparison (SO:0002072)
- sequence_feature (SO:0000110)
- transcription_start_site (SO:0000315)
misc_recomb
Used for genomic regions known to undergo recombination events. See INSDC's Controlled vocabulary for recombination_class for details on 'recombination_class' types.
Flat file qualifier example: recombination_class="non_allelic_homologous"
INSDC 'recombination_class' types used for RefSeq Functional Elements:
- chromosome_breakpoint (SO:0001021)
- meiotic (SO:0002155)
- mitotic (SO:0002154)
- non_allelic_homologous (SO:0002094)
- other
RefSeq controlled vocabularies for recombination_class="other":
- recombination_hotspot (SO:0000298)
misc_structure (SO:0000002)
Used for miscellaneous structural regions that are considered functionally important, including G-quadruplex and cruciform structures. Additional details are provided in flat file '/note' qualifiers.
mobile_element (SO:0001037)
Used for mobile elements, including transposable elements, retrotransposons and endogenous retroviruses, that are described in the literature as being functionally significant and/or represent genomic landmarks for a region. The repeat family (e.g., SINE:AluSg) is indicated in the '/mobile_element_type' qualifier. An '/inference' qualifier may be provided with a reference to a publicly accessible algorithm, e.g., RepeatMasker:4.0.5. Note that the vast majority of mobile elements located within the span of a RefSeq Functional Element NG_ will not be annotated. Mobile elements can be viewed in NCBI graphical displays by exposing the 'Repeats identified by RepeatMasker' track when viewing annotation on the genome.
protein_bind (SO:0000410)
Used where there is experimental evidence of direct protein binding to a DNA fragment, e.g., electrophoretic mobility shift assay (EMSA) or DNase I footprint evidence for binding of a specific protein, family, or complex. Predicted binding sites that lack experimental validation, or which are validated solely by chromatin immunoprecipitation, are not annotated. The annotated range is based on the experimental fragment described in the literature (e.g., an EMSA probe) and is typically longer than the core binding motif. The bound protein name (or protein family name) is provided in the '/bound_moiety' qualifier, where the protein name is the HUGO Gene Nomenclature Committee (HGNC) official full name for the encoding gene (e.g., CTCF is 'CCCTC-binding factor').
regulatory
See INSDC's Controlled vocabulary for regulatory_class for details on 'regulatory_class' types. Typically, the annotated region corresponds to an experimentally-defined fragment that was found to be sufficient for function, e.g., a fragment used in a reporter assay. Annotated sequences may therefore include extraneous sequences that are not necessary for function.
Specific notes:
Short feature motifs (i.e., CAAT_signal, TATA_box, GC_signal) that lack direct experimental validation may occasionally be annotated when described in a publication as a significant genomic landmark.
DNase_I_hypersensitive_site ranges are determined on a case-by-case basis based on examination of published experimental evidence. They typically include generous padding equivalent to at least one nucleosome plus linker span (~200 nt) in addition to the determined core site. Closely situated sites that are difficult to resolve may be combined into a single annotated feature, as indicated in the '/note' qualifier.
INSDC 'regulatory_class' types used for RefSeq Functional Elements:
- CAAT_signal (SO:0000172)
- DNase_I_hypersensitive_site (SO:0000685)
- enhancer (SO:0000165)
- enhancer_blocking_element (SO:0002190)
- GC_signal (SO:0000173)
- imprinting_control_region (SO:0002191)
- insulator (SO:0000627)
- locus_control_region (SO:0000037)
- matrix_attachment_region (SO:0000036)
- promoter (SO:0000167)
- response_element (SO:0002205)
- replication_regulatory_region (SO:0001682)
- silencer (SO:0000625)
- TATA_box (SO:0000174)
- transcriptional_cis_regulatory_region (SO:0001055)
- other
RefSeq controlled vocabularies for regulatory_class="other":
- epigenetically_modified_region (SO:0001720)
- micrococcal_nuclease_hypersensitive_site (SO:0005836)
repeat_region
Used for tandem repeats, microsatellites, and other low-complexity repeats that are described in the literature as being functionally significant and/or represent genomic landmarks for a region. Note that not all low-complexity regions located within the span of a RefSeq Functional Element NG_ will be annotated. An '/inference' qualifier may be provided with a reference to a publicly accessible algorithm, e.g., RepeatMasker:4.0.5.
INSDC repeat types used for RefSeq Functional Elements:
- minisatellite (SO:0000643)
- microsatellite (SO:0000289)
- direct_repeat (SO:0000314)
- dispersed_repeat (SO:0000658)
- inverted_repeat (SO:0000294)
- tandem_repeat (SO:00007050)
rep_origin (SO:0000296)
Used for DNA replication origins that are well-supported and reproducibly observed in the literature. A '/direction' qualifier may be included when it is known that the origin fires in one or both directions.
stem_loop (SO:0000313)
Used prior to August 2020 for stem-loop regions that are considered functionally important. Note that stem-loop structures are formed by complementary pairing on the same strand and frequently correspond to cruciform-like structures in double-stranded DNA, thus these are now represented as misc_structure features.
Cell or Tissue Type Activity
Most RefSeqFE features include formatted cell/tissue type activity derived from the experimental evidence for the feature. Cell/tissue types are generally named as described in supporting publications, and may be appended with specific conditions, developmental states or additives needed for feature activity. Cell/tissue type activities are indicated in '/function' qualifiers in GenBank flat files, or in equivalent 'function=' attributes in GFF3 or bigBed files for genome-annotated features.
General '/function' qualifier format for most curated features:
/function="free-text describing feature function {active_cell/tissue: cell type 1 | cell type 2}"
NG_055003.1 examples:
- /function="up-regulates the human A-gamma-globin promoter in transiently transfected K562 cells and in K562 and MEL cell colony assays {active_cell/tissue: K562 | MEL}"
- /function="necessary for Hbb-y promoter activity and binds BKLF and EKLF in SCFA-induced MEL cells {active_cell/tissue: MEL(+SCFA)}"
- /function="necessary for expression of the Hbb-y gene in embryonic yolk sac {active_cell/tissue: yolk sac(E10.5)}"
Specific formatting for VISTA enhancer features:
/function="enhancer in: tissue 1[score] | tissue 2[score]"
NG_053556.1 example:
- /function="enhancer in: neural tube[8/9] | forebrain[9/9]"
Formatting includes:
- Cell/tissue types listed within curly brackets and preceded by the string 'active_cell/tissue: ' for most cases, or preceded by 'enhancer in: ' for VISTA enhancer features.
- Space-flanked pipe delimitation for multiple cell/tissue types.
- Optional parentheses following a cell/tissue type to indicate specific conditions or developmental states. A plus sign indicates an additive needed for activity, e.g., a chemical used for cellular induction or a co-transfected factor necessary for activity. If a feature is active with and without an additive or condition, or if specific conditions for activity are not readily apparent from descriptions in cited publications, only the root cell/tissue name is provided.
- Optional square brackets following a cell/tissue type to indicate activity scores. These are currently specific to VISTA enhancer features given the general non-quantitative nature of RefSeqFE features, which are based on a diverse range of evidence types from different sources.
Limitations and notes for use:
- Cell/tissue type specificity may not be limited to the listed cell/tissue type(s), which are derived from publications cited for experimental evidence. This does not exclude possible feature activity in other cell/tissue types that were not tested in the cited evidence.
- Lack of activity in a tested cell/tissue type is not included.
- Markup for cell/tissue activity conditions may not be fully comprehensive, thus users should refer to cited publications for full details.
- Not all features are in scope for cell/tissue activity markup, e.g., repeat or recombination features.
- A minority of in-scope RefSeqFE features currently lack formatted markup, which will be gradually backfilled.
Cell or tissue type activity data can be accessed as follows:
- From '/function' qualifiers on either individual RefSeqFE NG_ accessions obtained from the Nucleotide database (see below) or from bulk download of weekly updated GenBank flat files obtained from the RefSeq FTP site, as described below.
- From 'function=' attributes in column 9 of GFF3 files downloaded from the Genomes FTP site, as described below. Examples of RefSeqFE cell/tissue type activity data extraction from GFF3 files are included below.
- From a dedicated custom metadata column (#14) in 'FEfeats_AR##.bb' bigBed files produced from March 2023 onwards, obtained from the RefSeq FTP site as described below. Relevant cell/tissue types are provided in a pipe-delimited list per feature. Examples of RefSeqFE cell/tissue type activity data extraction from bigBedToBed-converted files are included below.
- From metadata popup boxes that display '/function' qualifier texts for individual features in NCBI graphical displays and the RefSeqFE track hub.
Interaction Data
Non-genic biological regions are linked to target genes and other biological regions when there is experimental evidence for interactions. Interaction data is provided for regulatory interactions, typically between a gene regulatory element and a target gene, and for recombination partner interactions between recombination-type biological regions. Biological region ranges are based on parental 'biological_region' features that may include several underlying gene regulatory or other non-genic functional features, while target gene ranges are based on NCBI 'gene' feature annotations. Each interaction is supported by experimental data in the literature, and publications are provided in our bigInteract files described below.
Interaction data can be accessed as follows:
-
By download from the RefSeq FTP site in bigInteract file format. Pairwise interactions with genomic coordinates are provided in the 'FEregintxns_AR##.inter.bb' and 'FErecombpartners_AR##.inter.bb' files, where '##' represents the NCBI annotation release identifier. These files are available at this link for human, or at this link for mouse. We recommend the use of data from the most recent annotation release directory. Separate bigInteract files are provided for regulatory interactions (both human and mouse) and recombination partners (human only). These files also include a custom metadata column (#19) containing a comma-delimited list of PubMed IDs with experimental support for each interaction.
-
By download from the RefSeq FTP site in bigBed file format. Interactions are provided for biological regions in a custom metadata column (#13) in the 'FEbiolregions_AR##.bb' files as pipe-delimited lists of loci known to interact with the given biological region. These files are available from the same FTP directories indicated for the bigInteract files above. Additional information about 'FEbiolregions_AR##.bb' files can be found below.
-
By download from the RefSeq FTP site in GenBank flat file format ('[human/mouse].biological_region.gbff.gz' files). RefSeq accessions for biological regions with validated functional interactions contain a '/function' qualifier listing loci that are known to interact with the biological region. The qualifier is present on the 'misc_feature' denoted as '/note="Region: biological region"'. For example, on NG_042043.1 the '/function' qualifier appears as:
/function="regulatory_interactions: H19 | Igf2 | LOC105311846"
The weekly-updated files are available at this link. Alternatively, individual RefSeq accessions may be queried, viewed and downloaded in flat file format in the Nucleotide database, as described below. -
By download from the Genomes FTP site in GFF3 file format. Relevant 'biological_region' features have a 'function' attribute in column 9, where interacting loci are indicated as described for GenBank flat files above. GFF3 file availability is described below.
-
By graphical visualization from the RefSeq Functional Elements track hub (RefSeqFE Hub). Interaction data may be viewed in the regulatory interactions and recombination partner tracks, as described for our track hub below and shown in Figure 5.
Data Access
RefSeq Functional Element records can be accessed via the following NCBI resources:
Access via Gene
RefSeq Functional Element records can be found in Gene by searching in a variety of ways, including by record names or symbols, associated PubMed IDs, accession IDs, annotated chromosome and base locations, organism, text words, and properties (e.g., genetype biological region[prop]). Additional options can be found in the Gene Advanced Search Builder. Results can be further filtered by selecting side facets in search results pages. See the Gene Help document for more information on querying Gene.
Example queries:
To find all RefSeq Functional Element records in human: genetype biological region[prop] AND homo sapiens[orgn]
To find named recombination regions in human: genetype biological region[prop] AND homo sapiens[orgn] AND recombination region[gene/protein name]
To find human records that include a locus control region feature: genetype biological region[prop] AND homo sapiens[orgn] AND feattype locus control region[prop]
To find VISTA enhancer records in mouse: genetype biological region[prop] AND mus musculus[orgn] AND VISTA*[gene/protein name]
To find human Functional Element records associated with coronavirus biology:
coronavirus related[filter] AND genetype biological region[properties]
Or find subsets of those records involved in particular processes with:
Access via Nucleotide
The Nucleotide database displays RefSeq Functional Element records in GenBank flat file format by default. See the Entrez Sequences Help document for details on Nucleotide record display options, including instructions on how to retrieve FASTA sequences for specific features annotated on each RefSeq.
RefSeq Functional Element records can be queried in the Nucleotide database in several ways, including by names, symbols, accession ID, organism or associated publications. See the Nucleotide Advanced Search Builder for further options. Queries by the 'RefSeqFE' keyword, by BioProject ID (see Access via BioProject below) and by Feature key are particularly useful for retrieving Functional Element RefSeqs.
Example queries:
To retrieve all human Functional Element RefSeqs using the 'RefSeqFE' keyword:
homo sapiens[orgn] AND RefSeqFE[keyword]
Or alternatively by using the PRJNA343958 BioProject accession:
homo sapiens[orgn] AND PRJNA343958[bioproject]
To retrieve mouse Functional Element RefSeqs with an annotated enhancer feature: regulatory enhancer[feature key] AND mus musculus[orgn] AND RefSeqFE[keyword]
To retrieve human Functional Element RefSeqs with an annotated rep_origin feature: rep origin[feature key] AND homo sapiens[orgn] AND RefSeqFE[keyword]
To retrieve human Functional Element RefSeqs associated with coronavirus biology:
coronavirus related[prop] AND biomol genomic[prop]
Or find subsets of those records involved in particular processes with:
Access via BLAST
RefSeq Functional Element sequences are in NCBI's Nucleotide database, thus matching RefSeqs can be retrieved through Nucleotide BLAST sequence searches when the following options are selected in the 'Choose Search Set' area:
- Standard databases (nr etc.) -- radio button selection
- Nucleotide collection (nr/nt) -- pull-down menu selection
- Organism -- entering an organism name (human or mouse) is optional but will yield organism-specific results with faster searching
Access via BioProject
All RefSeq Functional Elements are represented in BioProject accession PRJNA343958. Sequence records can be retrieved from links within the 'Project Data' section. Nucleotide database queries can be appended with that BioProject accession for retrieval of RefSeq Functional Element records (see Access via Nucleotide example queries above).
Access via NCBI Graphical Displays
All genome-annotated features from RefSeq Functional Elements can be viewed by turning on the 'Biological regions' track available in the 'Genes/Products' track group and 'NCBI Other Features' category in NCBI graphical displays, including the Genome Data Viewer, Sequence Viewer, Variation Viewer and graphical images in Gene records. Note that the 'Biological regions' track may be viewable by default depending on the NCBI browser. For example, it will be on view in Gene records when RefSeq features are annotated on the reference genome, or in the Genome Data Viewer or Variation Viewer when the 'Genes' track set is selected under 'NCBI Recommended Track Sets', but it may be necessary to turn on the track if other track sets or the default tracks are selected. The track can be turned on under Tracks -> Configure Tracks -> Genes/Products -> Category: NCBI Other Features, where selection of the most recent 'Biological regions, aggregate' track is recommended; see the Figure 4 track configuration interface. Note that the 'Biological regions' track does not include overlapping or nearby conventional gene annotations, which can be found in the 'Genes' track (Figure 3). Similarly, users should refer to 'Variation' type tracks to see overlapping variation features, e.g., dbSNP, ClinVar or dbVar tracks. If the RefSeq has not yet been annotated on the genome, only the RefSeq (NG_ accession) sequence will be available for graphical viewing. For RefSeq graphical images, individual feature types, if not already viewable by default, may be viewed by turning on desired track types in the 'Features' track group (e.g., 'regulatory' Features, 'protein_bind' Features).
Graphical view example for HBB-LCR, GeneID:109580095: https://go.usa.gov/xf9ea
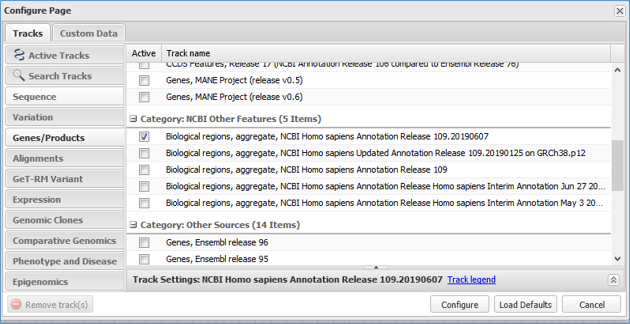
Figure 4. NCBI genome browser track configuration dialog box. The most recent 'Biological regions' track can be selected in the 'Genes/Products' track group and 'NCBI Other Features' category.
Access via the RefSeq Functional Elements Track Hub
The RefSeq Functional Elements track hub (RefSeqFE Hub) includes tracks for biological regions, features, regulatory interactions and recombination partners. The hub is in UCSC track hub format and can be viewed on a compatible genome browser, including the UCSC Genome Browser (all tracks and metadata, see Figure 5 example), the NCBI Genome Data Viewer (biological region and feature tracks only; metadata is best viewed in the existing 'Biological regions' track described above) or the Ensembl genome browser (biological region and feature tracks only).
Use the following URL to connect to the RefSeqFE Hub: https://ftp.ncbi.nlm.nih.gov/refseq/FunctionalElements/trackhub/hub.txt
Please see additional information in the track hub document. Briefly, the following tracks are represented:
-
RefSeq Functional Element biological regions (FE_biol_regions): Biological regions with metadata from the 'FEbiolregions_AR##.bb' bigBed file described below. See the biological regions track document for more details.
-
RefSeq Functional Element features (FE_features): Functional features with metadata from the 'FEfeats_AR##.bb' bigBed file described below. See the features track document for more details.
-
RefSeq Functional Element recombination partners (Recomb_partners; currently human only): Recombination interactions from the 'FErecombpartners_AR##.inter.bb' bigInteract file described above. See the recombination partners track document for more details.
-
RefSeq Functional Element regulatory interactions (Reg_interactions): Regulatory interactions from the 'FEregintxns_AR##.inter.bb' bigInteract file described above. See the regulatory interactions track document for more details.
Note on track display in other genome browsers: While not all commonly used genome browsers are compatible with UCSC-formatted track hubs, many browsers, including those mentioned above, may offer options to view RefSeq Functional Element tracks by remote connection from file URLs, or as custom tracks based on downloaded files saved locally. These browsers will vary in their ability to accept bigBed or bigInteract files, or to display color-coding or metadata, and some may require the conversion of binary files to non-binary formats. Please check the relevant genome browser documentation for futher information.
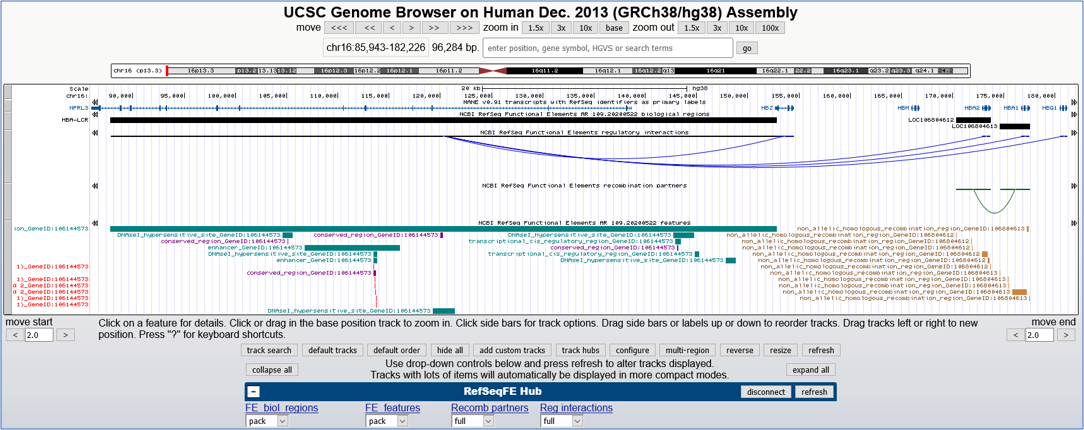
Figure 5. UCSC Genome Browser image displaying tracks from the RefSeqFE Hub at the human alpha-globin locus.
Access via FTP
RefSeq Functional Elements are available for FTP download in the NCBI FTP site, including the following specific subsites:
RefSeq FTP
All RefSeq records including those not yet annotated on a genome assembly. Includes a FunctionalElements directory with the following content:
Weekly updated RefSeq accession files:
- [human/mouse].biological_region.fna.gz -- RefSeq accessions for genomic biological regions (NG_ prefix) in FASTA format
- [human/mouse].biological_region.gbff.gz -- RefSeq accessions for genomic biological regions (NG_ prefix) in GenBank flatfile format
These files can also be found in species-specific 'biological_region' directories for human and mouse.
Files and subdirectories necessary to support the RefSeq Functional Elements track hub described above. See UCSC track hub documentation for an explanation of track hub structure and the necessary components. The following data files are provided within the data subdirectory and the species-specific annotation release (AR##) subdirectories therein:
-
Genome-annotated biological region and feature files in bigBed format:
- FEbiolregions_AR##.bb -- biological regions with metadata, provided as binary indexed BED 9+81 files where columns 1-9 are in standard BED format, with eight additional custom columns representing:
- Column 10: the Gene database identifier cross-reference
- Column 11: the locus name
- Column 12: the locus summary
- Column 13: known interacting loci, regulatory interactions or recombination partners as described above
- Column 14: the RefSeq accession ID
- Column 15: symbol aliases; empty if not applicable 2
- Column 16: name aliases; empty if not applicable 2
- Column 17: supporting publications from PubMed; includes all publications associated with the biological region 2
1 BED 9+5 format for biological region bigBed files predating February 2024
2 only present in biological region bigBed files produced from February 2024 onwardsNote that the locus symbol is used for the name (column 4). These files use UCSC-style chromosome notation (column 1), except for human T2T-CHM13v2.0 assembly files, which use GenBank accession notation.
- FEfeats_AR##.bb -- functional features with metadata, provided as binary indexed BED 9+51 files where columns 1-9 are in standard BED format, with five additional custom columns representing:
- Column 10: database cross-references, including to the Gene record and other database records if applicable
- Column 11: functional and descriptive information including experimental evidence and publication support
- Column 12: supporting publications from PubMed; publications with evidence for the specific feature 2
- Column 13: the RefSeq accession ID 3
- Column 14: cell or tissue type activity 4
- Column 15: dbSNP cross-reference for functionally validated genetic variants corresponding to the RefSeqFE feature, if applicable 5
1 BED 9+3 format for the human
FEfeats_AR109.20200522.bband mouseFEfeats_AR108.20200622.bbfiles, BED 9+4 format for subsequent annotation release files predating March 2023
2 not included in the humanFEfeats_AR109.20200522.bband mouseFEfeats_AR108.20200622.bbfiles; publications can be extracted from column 11
3 this is column 12 in the humanFEfeats_AR109.20200522.bband mouseFEfeats_AR108.20200622.bbfiles
4 only present in feature bigBed files produced from March 2023 onwards
5 only present in feature bigBed files produced from October 2023 onwards, human only, including for T2T-CHM13v2.0 assembly featuresNote that the name (column 4) is in the format
<SO_type>_<GeneID>for most features, where the 'SO_type' is the 'GFF3 column 3 SO_type' in the feature table below. Protein binding site features are labeled in the formatprotein_binding_site_<bound_moiety>_<GeneID>, where the bound_moiety is the HGNC official full name for the encoding gene. These files use UCSC-style chromosome notation (column 1), except for human T2T-CHM13v2.0 assembly files, which use GenBank accession notation. - FEbiolregions_AR##.bb -- biological regions with metadata, provided as binary indexed BED 9+81 files where columns 1-9 are in standard BED format, with eight additional custom columns representing:
-
Pairwise interaction data files in bigInteract format, provided as binary indexed BED 5+14 files where columns 1-18 are in standard bigInteract format, and column 19 is a custom column listing supporting publications:
- FErecombpartners_AR##.inter.bb -- recombination interactions as described above
- FEregintxns_AR##.inter.bb -- regulatory interactions as described above
These files use UCSC-style chromosome notation (columns 1, 9 and 14), except for human T2T-CHM13v2.0 assembly files, which use GenBank accession notation.
All of the bigBed and bigInteract files can also be found in species-specific 'biological_region' directories and annotation release (AR##) subdirectories therein for human and mouse.
Gene FTP
All RefSeqs and associated Gene data, and genomic context if annotated on a genome assembly. See the Gene README file for a description of the Gene FTP directory contents.
Genomes FTP
Genome-annotated feature data for human and mouse provided in GFF3 format. See the Feature Annotation Glossary above for descriptions of RefSeq Functional Element feature types. These GFF3 files use RefSeq accession chromosome notation (column 1). Effective as of human Updated Annotation Release 109.20200522 and mouse Updated Annotation Release 108.20200622, RefSeq Functional Element features are indicated by the 'RefSeqFE' source in column 2 of NCBI GFF3 files.
For extracting specific feature types from GFF3 files, please note that equivalent Sequence Ontology (SO) terms are used as SO_types in column 3. Alternatively, specific feature types may be extracted from GFF3 files based on feature key, feature class or controlled vocabulary attributes in column 9. The following table shows how features are indicated in columns 3 and 9 of GFF3 files:
Feature Table
| INSDC feature | Feature class or controlled vocabulary | SO ID | GFF3 column 3 SO_type | GFF3 column 9 specific attribute(s) |
|---|---|---|---|---|
| misc_feature | biological_region | SO:0001411 | biological_region | gbkey=Region |
| misc_feature | CAGE_cluster | SO:0001917 | CAGE_cluster | feat_class=CAGE_cluster |
| misc_feature | conserved_region | SO:0000330 | conserved_region | feat_class=conserved_region |
| misc_feature | nucleotide_cleavage_site | SO:0002204 | nucleotide_cleavage_site | feat_class=nucleotide_cleavage_site |
| misc_feature | nucleotide_motif | SO:0000714 | nucleotide_motif | feat_class=nucleotide_motif |
| misc_feature | repeat_instability_region | SO:0002202 | repeat_instability_region | feat_class=repeat_instability_region |
| misc_feature | replication_start_site | SO:0002203 | replication_start_site | feat_class=replication_start_site |
| misc_feature | sequence_alteration | SO:0001059 | sequence_alteration | feat_class=sequence_alteration |
| misc_feature | sequence_comparison | SO:0002072 | sequence_comparison | feat_class=sequence_comparison |
| misc_feature | sequence_feature | SO:0000110 | sequence_feature | feat_class=sequence_feature |
| misc_feature | transcription_start_site | SO:0000315 | TSS | feat_class=transcription_start_site |
| misc_recomb | chromosome_breakpoint | SO:0001021 | chromosome_breakpoint | recombination_class=chromosome_breakpoint |
| misc_recomb | meiotic | SO:0002155 | meiotic_recombination_region | recombination_class=meiotic |
| misc_recomb | mitotic | SO:0002154 | mitotic_recombination_region | recombination_class=mitotic |
| misc_recomb | non_allelic_homologous | SO:0002094 | non_allelic_homologous_recombination_region | recombination_class=non_allelic_homologous |
| misc_recomb | recombination_hotspot | SO:0000298 | recombination_feature | recombination_class=recombination_hotspot |
| misc_structure | n/a | SO:0000002 | sequence_secondary_structure | gbkey=misc_structure |
| mobile_element | n/a | SO:0001037 | mobile_genetic_element | gbkey=mobile_element |
| protein_bind | n/a | SO:0000410 | protein_binding_site | gbkey=protein_bind or bound_moiety= |
| regulatory | CAAT_signal | SO:0000172 | CAAT_signal | regulatory_class=CAAT_signal |
| regulatory | DNase_I_hypersensitive_site | SO:0000685 | DNaseI_hypersensitive_site1 | regulatory_class=DNase_I_hypersensitive_site |
| regulatory | enhancer | SO:0000165 | enhancer | regulatory_class=enhancer |
| regulatory | enhancer_blocking_element | SO:0002190 | enhancer_blocking_element | regulatory_class=enhancer_blocking_element |
| regulatory | GC_signal | SO:0000173 | GC_rich_promoter_region | regulatory_class=GC_signal |
| regulatory | imprinting_control_region | SO:0002191 | imprinting_control_region | regulatory_class=imprinting_control_region |
| regulatory | insulator | SO:0000627 | insulator | regulatory_class=insulator |
| regulatory | locus_control_region | SO:0000037 | locus_control_region | regulatory_class=locus_control_region |
| regulatory | matrix_attachment_region | SO:0000036 | matrix_attachment_site | regulatory_class=matrix_attachment_region |
| regulatory | epigenetically_modified_region | SO:0001720 | epigenetically_modified_region | regulatory_class=epigenetically_modified_region |
| regulatory | micrococcal_nuclease_hypersensitive_site | SO:0005836 | regulatory_region | regulatory_class=micrococcal_nuclease_hypersensitive_site |
| regulatory | promoter | SO:0000167 | promoter | regulatory_class=promoter |
| regulatory | response_element | SO:0002205 | response_element | regulatory_class=response_element |
| regulatory | replication_regulatory_region | SO:0001682 | replication_regulatory_region | regulatory_class=replication_regulatory_region |
| regulatory | silencer | SO:0000625 | silencer | regulatory_class=silencer |
| regulatory | TATA_box | SO:0000174 | TATA_box | regulatory_class=TATA_box |
| regulatory | transcriptional_cis_regulatory_region | SO:0001055 | transcriptional_cis_regulatory_region | regulatory_class=transcriptional_cis_regulatory_region |
| repeat_region | minisatellite | SO:0000643 | minisatellite | satellite=minisatellite |
| repeat_region | microsatellite | SO:0000289 | microsatellite | satellite=microsatellite |
| repeat_region | direct_repeat | SO:0000314 | direct_repeat | rpt_type=direct |
| repeat_region | dispersed_repeat | SO:0000658 | dispersed_repeat | rpt_type=dispersed |
| repeat_region | inverted_repeat | SO:0000294 | inverted_repeat | rpt_type=inverted |
| repeat_region | n/a | SO:0000001 | region | gbkey=repeat_region |
| repeat_region | n/a | SO:0000657 | repeat_region | gbkey=repeat_region |
| repeat_region | tandem_repeat | SO:0000705 | tandem_repeat | rpt_type=tandem |
| rep_origin | n/a | SO:0000296 | origin_of_replication | gbkey=rep_origin |
| stem_loop2 | n/a | SO:0000313 | stem_loop2 | gbkey=stem_loop2 |
1 Denoted as 'DNAseI_hypersensitive_site' in older annotation releases (ARs) up to human AR 109.20201120 and mouse AR 109
2 No longer used for Functional Element features as of August 2020, replaced with misc_structure
Feature and Metadata Extraction Examples
Annotated RefSeq Functional Element features can be extracted from NCBI-provided GFF3 files or from the 'FEfeats_AR##.bb' bigBed files following their conversion to a non-binary format using the bigBedToBed application provided in the UCSC binary utilities directory. The following data extraction examples are based on the Unix command line.
Feature extraction from GFF3 files:
The GFF3 file from NCBI's latest human or mouse annotation release can be downloaded to a local directory (see the human and mouse Genomes FTP links above), with substitution of the GFF3 file names (if applicable) in the following commands. Please refer to the feature table above to determine appropriate strings for extraction of desired feature types.
-
Extraction of all RefSeq Functional Element features using
awk, excluding parental 'biological_region' features, for GFF3 files produced from human AR 109.20200522 and mouse AR 108.20200622 onwards. This command uses the 'RefSeqFE' source to extract all relevant features:zgrep -v "^#" GCF_000001405.40_GRCh38.p14_genomic.gff.gz | awk 'BEGIN{FS="\t";OFS"\t"}$2=="RefSeqFE"&&$3!="biological_region"' -
Extraction of all RefSeq Functional Element features using
awk, excluding parental 'biological_region' features, for GFF3 files prior to human AR 109.20200522 and mouse AR 108.20200622. This command takes advantage of the fact that RefSeq Functional Element features are not stranded (except for 'stem_loop' features), while all other NCBI-annotated features are stranded, including 'gene', 'exon' and 'CDS' features:zgrep -v "^#" GCF_000001405.40_GRCh38.p14_genomic.gff.gz | awk 'BEGIN{FS="\t";OFS"\t"}$7=="."||$3=="stem_loop"' -
Extraction of all regulatory_class features using
awk:zgrep -v "^#" GCF_000001405.40_GRCh38.p14_genomic.gff.gz | awk 'BEGIN{FS="\t";OFS"\t"}$9~/regulatory_class=/'Alternatively,
$9~/regulatory_class=/can be substituted with$9~/gbkey=regulatory/ -
Extraction of enhancer features based on the column 3 SO_type using
awk:zgrep -v "^#" GCF_000001405.40_GRCh38.p14_genomic.gff.gz | awk 'BEGIN{FS="\t";OFS"\t"}$3=="enhancer"' -
Extraction of enhancer features based on the column 9 specific attribute using
awk:zgrep -v "^#" GCF_000001405.39_GRCh38.p13_genomic.gff.gz | awk 'BEGIN{FS="\t";OFS"\t"}$9~/regulatory_class=enhancer/&&$9!~/regulatory_class=enhancer_blocking_element/' -
Extraction of enhancer features based on the column 9 specific attribute using
grep:zgrep -v "^#" GCF_000001405.40_GRCh38.p14_genomic.gff.gz | grep regulatory_class=enhancer | grep regulatory_class=enhancer_blocking_element -v -
Extraction of features from human records associated with coronavirus biology, here selecting relevant GeneIDs based on flat file 'coronavirus related' attributes found in the
human.biological_region.gbff.gzfile described above:zgrep -v "^#" GCF_000001405.40_GRCh38.p14_genomic.gff.gz | awk -F '\t' '$2=="RefSeqFE"&&$3!="biological_region"' | fgrep -f <(zgrep -A30 "coronavirus related" human.biological_region.gbff.gz | grep -o "GeneID:[0-9]*" | sort -u) -w -
Extraction of features with dbSNP cross-references for functionally validated genetic variants corresponding to the RefSeqFE feature:
zgrep -v "^#" GCF_000001405.40_GRCh38.p14_genomic.gff.gz | awk -F '\t' '$2=="RefSeqFE"&&$9~/Dbxref=dbSNP:/'
Metadata extraction from GFF3 files:
-
To extract feature types, feature IDs and cell/tissue types for all features containing cell/tissue activity data:
zgrep -v "^#" GCF_000001405.40_GRCh38.p14_genomic.gff.gz | awk -F '\t' '$2=="RefSeqFE"&&$3!="biological_region"&&$9~/active_cell\/tissue|VISTA/' | cut -f3,9 | sed 's/ID=id-\([^;]*\).*active_cell\/tissue: \(.*\)}.*/\1\t\2/; s/ID=id-\([^;]*\).*function=enhancer in: \([^;]*\);.*/\1\t\2/' -
To extract features that are active in a cell/tissue type of interest, HepG2 cells in this example:
zgrep -v "^#" GCF_000001405.40_GRCh38.p14_genomic.gff.gz | awk -F '\t' '$2=="RefSeqFE"&&$3!="biological_region"&&$9~/active_cell\/tissue|VISTA/&&$9~/HepG2/'
Feature extraction from bigBed files:
The 'FEfeats_AR##.bb' file from NCBI's latest human or mouse annotation release can be downloaded to a local directory (see file availability from the RefSeq FTP site above).
-
File conversion to non-binary format using the
bigBedToBedapplication provided in the UCSC binary utilities directory:bigBedToBed FEfeats_RS_2023_03_GRCh38.p14.bb FEfeats_RS_2023_03_GRCh38.p14.bed -
Extraction of enhancer features from the bigBedToBed-converted file based on the column 4 name using
awk:awk -F '\t' '$4~/enhancer_GeneID/' FEfeats_RS_2023_03_GRCh38.p14.bed -
Extraction of features from the bigBedToBed-converted file for human functional elements associated with coronavirus biology, here selecting relevant GeneIDs based on flat file 'coronavirus related' attributes found in the 'human.biological_region.gbff.gz' file described above:
cat FEfeats_RS_2023_03_GRCh38.p14.bed | fgrep -f <(zgrep -A30 "coronavirus related" human.biological_region.gbff.gz | grep -o "GeneID:[0-9]*" | sort -u) -w
Caution for extraction of specific features:
Some feature strings may also be present in names or other free-text attributes in GFF3 and bigBedToBed-converted files. The use of explicit terms with associated strings is therefore recommended to avoid the extraction of non-specific features. Explicit features can be extracted by using awk based on the SO_type in column 3 of GFF3 files in the format $3=="<feature_name>", while full attribute strings may be necessary for extractions based on GFF3 column 9 attributes. For example, the use of regulatory_class=enhancer with awk or grep as in the GFF3 examples above, where it is additionally necessary to exclude 'enhancer_blocking_element' features given that the 'enhancer' string is also found within regulatory_class=enhancer_blocking_element. In bigBedToBed-converted files, 'enhancer' features can be distinguished from 'enhancer_blocking_element' features by specifying $4~/enhancer_GeneID/ in an awk command given that an <SO_type>_<GeneID> naming format is used in column 4.
Metadata extraction from bigBedToBed-converted files:
Specific types of metadata may also be extracted from either the 'FEbiolregions_AR##.bb' or 'FEfeats_AR##.bb' files following their conversion to non-binary format. See above for a description of the custom metadata columns within these files.
-
Extraction of basic enhancer features (columns 1-4) together with just functional and descriptive information (column 11) from a converted non-binary
FEfeats_AR109.20201120.bedfile:awk -F '\t' '$4~/enhancer_GeneID/' FEfeats_RS_2023_10_GRCh38.p14.bed | cut -f1-4,11 -
To extract the basic enhancer features (columns 1-4) along with just publications from column 12:
awk -F '\t' '$4~/enhancer_GeneID/' FEfeats_RS_2023_10_GRCh38.p14.bed | cut -f1-4,12 -
Using
sedto extract the basic enhancer features (columns 1-4) along with just publications from column 11 for the humanFEfeats_AR109.20200522.bband mouseFEfeats_AR108.20200622.bbfiles, which lack a standalone supporting publications column:awk -F '\t' '$4~/enhancer_GeneID/' FEfeats_AR109.20200522.bed | cut -f1-4,11 | sed 's/\(_GeneID:[0-9]*\t\).*\[PMID/\1PMID/; s/\(PMID:[0-9]*\)\].*/\1/' -
To extract feature types, feature IDs and cell/tissue types for all features containing cell/tissue activity data (applies to feature files produced from March 2023 onwards):
awk -F '\t' '$14!=""' FEfeats_RS_2023_10_GRCh38.p14.bed | cut -f4,14 -
To extract features that are active in a cell/tissue type of interest, K562 cells in this example:
awk -F '\t' '$14~/K562/' FEfeats_RS_2023_10_GRCh38.p14.bed -
To retrieve the full list of cell/tissue types represented in a given bigBedToBed-converted feature file, here removing appended conditions and scores to display root names only:
awk -F '\t' '$14!=""' FEfeats_RS_2023_10_GRCh38.p14.bed | cut -f14 | sed 's/ | /\n/g; s/(.*//; s/\[.*//; s/}.*//; s/ $//' | sort | uniq -
Extraction of GeneIDs, locus summaries and RefSeq accessions from a similarly converted non-binary 'FEbiolregions_AR##.bed' file:
cut -f10,12,14 FEbiolregions_RS_2023_10_GRCh38.p14.bed -
Extraction of locus symbols, locus names and interacting loci for biological regions with regulatory interaction data:
awk -F '\t' '$13~/regulatory_interactions/' FEbiolregions_RS_2023_10_GRCh38.p14.bed | cut -f4,11,13 -
Extraction of target genes with regulatory interactions from a bigBedToBed-converted 'FEregintxns_AR##.inter.bb' file:
awk -F '\t' '$4~/target_gene/' FEregintxns_RS_2023_10_GRCh38.p14.inter.bed | cut -f4 | sed 's/:/\t/g; s/_t/\tt/; s/_r/\tr/' | awk '{if ($3~/target/) {print $1} else {print $2}}' | sort | uniq -
Extraction of features with dbSNP cross-references for functionally validated genetic variants corresponding to the RefSeqFE feature, where the SNPs can be independently extracted from column 15 (human files produced from October 2023 onwards):
awk -F '\t' '$15~/rs/' FEbiolregions_RS_2023_10_GRCh38.p14.bed
Similar command line strategies may also be used to extract feature-specific metadata from column 9 of GFF3 files, where several semi-colon-delimited functional and descriptive attributes are present.
All of the above examples are just a sampling. The extraction of numerous other combinations of features or metadata is also possible, including by using other computer programming languages, tools or data parsers. We encourage users to explore our data based on individual data needs.
References
Please use the following citation for RefSeq Functional Elements:
- Farrell CM, Goldfarb T, Rangwala SH, Astashyn A, Ermolaeva OD, Hem V, Katz KS, Kodali VK, Ludwig F, Wallin CL, Pruitt KD, Murphy TD. RefSeq Functional Elements as experimentally assayed nongenic reference standards and functional interactions in human and mouse. Genome Res. 2022 Jan;32(1):175-188. doi: 10.1101/gr.275819.121. Epub 2021 Dec 7. PMID: 34876495; PMCID: PMC8744684.
Feedback
We welcome user comments and suggestions. Please use the yellow vertical Feedback tab on the bottom right of the page to enter your comments, or contact us through the NLM Support Center interface, or by e-mailing [email protected].