

Abstract
Aims
Atherosclerosis is the dominant pathologic basis of many cardiovascular diseases. Large genome-wide association studies have identified that single-nucleotide polymorphisms proximal to Krüppel-like factor 14 (KLF14), a member of the zinc finger family of transcription factors, are associated with higher cardiovascular risks. Macrophage dysfunction contributes to atherosclerosis development and has been recognized as a potential therapeutic target for treating many cardiovascular diseases. Herein, we address the biologic function of KLF14 in macrophages and its role during the development of atherosclerosis.
Methods and results
KLF14 expression was markedly decreased in cholesterol loaded foam cells, and overexpression of KLF14 significantly increased cholesterol efflux and inhibited the inflammatory response in macrophages. We generated myeloid cell-selective Klf14 knockout (Klf14LysM) mice in the ApoE-/- background for the atherosclerosis study. Klf14LysMApoE-/- and litter-mate control mice (Klf14fl/flApoE-/-) were placed on the Western Diet for 12 weeks to induce atherosclerosis. Macrophage Klf14 deficiency resulted in increased atherosclerosis development without affecting the plasma lipid profiles. Klf14-deficient peritoneal macrophages showed significantly reduced cholesterol efflux resulting in increased lipid accumulation and exacerbated inflammatory response. Mechanistically, KLF14 upregulates the expression of a key cholesterol efflux transporter, ABCA1 (ATP-binding cassette transporter A1), while it suppresses the expression of several critical components of the inflammatory cascade. In macrophages, activation of KLF14 by its activator, perhexiline, a drug clinically used to treat angina, significantly inhibited the inflammatory response and increased cholesterol efflux in a KLF14-dependent manner in macrophages without triggering hepatic lipogenesis.
Conclusions
This study provides insights into the anti-atherosclerotic effects of myeloid KLF14 through promoting cholesterol efflux and suppressing the inflammatory response. Activation of KLF14 may represent a potential new therapeutic approach to prevent or treat atherosclerosis.
Keywords: Krüppel-like factor • Atherosclerosis• Cholesterol efflux • Inflammation
Graphical Abstract
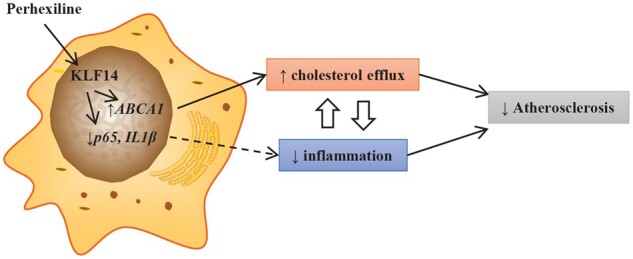
Time for primary review: 24 days
1. Introduction
Atherosclerosis, a chronic and progressive vascular disease, is an important cause of cardiovascular diseases worldwide. It is characterised by an accumulation of excess cholesterol and a chronic inflammatory process in the arterial wall, which triggers acute coronary syndrome when the atherosclerotic plaque ruptures.1 High low-density lipoprotein cholesterol (LDL-C) level is the strongest and best-studied risk factor for atherosclerosis development.2 However, despite advances in therapeutics aimed at lowering LDL-C level, residual cardiovascular risks remain in coronary heart disease patients.3
Among the protective mechanisms against atherosclerosis, reverse cholesterol transport (RCT) represents an important protective pathway, in which excessive cholesterol from peripheral tissues, including foam cells in the atherosclerotic plaques, is moved by high-density lipoprotein (HDL) particles to the liver and finally excreted into the faeces.4 As decreased HDL-cholesterol (HDL-C) level is an independent, inverse predictor of cardiovascular risk, raising HDL level was initially proposed to be an operative way to promote atherosclerosis regression. However, many pharmacological approaches to raise HDL levels have not demonstrated an additional beneficial effect on major cardiovascular events in clinical trials. The debate on the HDL hypothesis led to studies showing that low cholesterol efflux capacity of HDL particles is more relevant to cardiovascular disease risks compared with the decreased HLD-C levels.5 Furthermore, cholesterol efflux is the first and rate-limiting step in RCT, in which free cholesterol inside the arterial foam cells is transported by the ATP-binding cassette transporter A1 and G1 (ABCA1 and ABCG1) to circulating cholesterol acceptors. ABCA1 controls the cellular cholesterol efflux to lipid poor ApoA-I/small HDL particles, and ABCG1 facilitates cholesterol efflux to mature HDL particles.4,6–8 In macrophage, liver X receptors (LXRs), LXRα and LXRβ, play pivotal roles in regulating the expression of ABCA1 and ABCG1. Synthetic LXR agonists attenuate atherosclerosis by upregulating ABC transporters and inhibiting inflammation in animal models.9 However, activation of LXRs triggers liver steatosis and hypertriglyceridaemia, limiting the clinical application of LXR agonists for atherosclerosis treatment. The current challenge is to understand the pathways regulating ABCA1 and ABCG1 expression in macrophages and identify novel therapeutic approaches for atherosclerosis treatment.
Krüppel-like factor 14 (KLF14) was identified by genome-wide association studies (GWASs) as showing direct correlations with both cardiovascular disease and HDL-C level.10 KLF14 belongs to a large zinc-finger transcription family, the Krüppel-like factor family, which regulates a vast number of genes and is involved in many biological processes.11,12 Previously, we identified the biological function of KLF14 and its transcriptional activator-perhexiline, a therapeutic agent currently in clinical to treat angina in Australia and New Zealand, in lipid metabolism. We demonstrated that KLF14 has strong anti-inflammatory effect in vascular endothelial cells by directly suppressing the expression of nuclear factor-κB p6513. We showed that administration of perhexiline significantly inhibited atherosclerotic plaque development, which could not be fully explained by the effect of KLF14 on raising HDL-C because the HDL-C levels remain intrinsically very low in ApoE−/− mice after 12 weeks of western diet challenge.14 Considering that macrophages are key integrators of the inflammatory response and cholesterol accumulation in the atherosclerotic plaques, in this study, we explore the protective effects of macrophage KLF14 on atherosclerosis. Here, we report that deficiency of KLF14 in macrophages increased the inflammatory response and decreased cholesterol efflux by regulating the expression of IL1β, p65, and ABCA1. Consequently, atherosclerotic plaque lesions were also increased in macrophage-selective Klf14-deficient (Klf14LysMApoE−/−) mice.
2. Methods
2.1 Animals
All animal experiments in this study are approved by the Institutional Animal Care and Use Committee (IACUC) and meet the standards for the animal care and use as established by the Animal Welfare Act and the NIH Guide for the Care and Use of Laboratory Animals. Klf14 floxed mice were previous generated in our laboratory.14Klf14fl/fl mice were crossed with LysM-Cre transgenic mice (Stock No: 004781 from the Jackson Laboratory) to obtain myeloid cell-selective Klf14 knockout (KO) mice (Klf14LysM). Subsequently, Klf14LysM mice were crossed with ApoE−/− mice (Stock No: 002052 from the Jackson Laboratory) to generate Klf14LysMApoE−/− mice for the atherosclerosis study. The litter-mate LysM-Cre-negative Klf14fl/flApoE−/− mice were used as controls. LXRα knockout mice used for peritoneal macrophages isolation were commercially available (Stock No. 013762 from Jackson Laboratory).
2.2 Atherosclerosis model and analysis of atherosclerotic lesions
Klf14 fl/fl ApoE −/− mice and Klf14LysMApoE−/− mice (both female and male) were used for the atherosclerosis study. At the end of the experiments, animals were fasted for 4 h and then euthanized by CO2 overdose. Two quantitative methods were used to analyse the areas of atherosclerotic lesions, as described in our previous study.14 First, en face analysis of the atherosclerotic lesions uses the whole aorta trees: periaortic adipose tissue was carefully removed, and the aortic trees were stained with Oil Red O (ORO) solution and opened longitudinally. The percentage of plaque areas stained by ORO to the total luminal surface area was quantified. Second, three to five locations of the cross-sections (at 80-μm intervals) from the aortic sinus region were examined. The largest plaques of the three valve leaflets were used for morphological analysis. ImageJ analysis software was used to quantify the indicated areas (http://imagej.nih.gov/ij/). All morphometric analyses were performed in a double-blinded manner.
2.3 Statistics
Data are presented as mean ± SEM. Each experiment was performed with at least three experimental replicates and independently repeated at least three times with similar results. Statistical analyses were performed using GraphPad Prism 8 software (GraphPad Software, Inc.). When data passed tests for normality and equal variance, statistical comparisons and analyses between two groups were performed by two-tailed, unpaired Student’s t-test. Comparisons among three groups or more were analysed with one-way ANOVA with Dunnett’s correction or two-way ANOVA with Tukey correction. If the data did not pass those tests, Mann–Whitney was used to compare two groups, and Kruskal–Wallis test followed by a two-stage step-up method of Benjamini, Krieger, and Yekutieli were used for >2 groups. P < 0.05 was considered statistically significant.
3. Results
3.1 The expression of KLF14 was significantly reduced under pro-atherogenic conditions
The macrophage is one of the most critical cell types that respond to pro-inflammatory stimuli and lipid accumulation during atherosclerosis development.15 To understand whether KLF14 expression was regulated specifically in macrophages under pathological conditions, we first used a chronic inflammatory animal model by subjecting wild-type (WT) C57BL/6J male mice to challenge with Western diet (WD) for 12 weeks, and isolated primary peritoneal macrophages (PMs) at the end of the treatment. The mRNA level of Klf14 was significantly reduced in PMs from WD-fed mice compared to a standard chow diet (CD)-fed mice (Figure 1A). Next, we induced foam cells16,17 using PMs isolated from CD-fed WT C57BL/6J mice by acetylated LDL (acLDL) loading, confirmed by an increase of intracellular cholesterol content (Supplementary material online, Figure S1), and found that Klf14 expression was also significantly decreased (Figure 1B). Consistent with our previous findings in endothelial cells,13Klf14 expression was also reduced upon pro-inflammatory stimuli (LPS, IL-1β, and TNFα) in THP-1-derived macrophages (Figure 1C and Supplementary material online, Figure S2). These results indicate that lipid loading and inflammation may lead to Klf14 downregulation and, therefore, accelerate atherosclerosis progression.
Figure 1.

KLF14 expression was significantly decreased under pathological stimuli. (A) PMs were isolated from C57BL/6J mice fed a regular chow diet (CD) or western diet (WD), and Klf14 mRNA levels were measured [three biological replicates for three independent experiments (triplet wells)]. (B) PMs were isolated from C57BL/6J mice and treated with acLDL (50 µg/mL) for 24 h, and Klf14 mRNA levels were measured [three biological replicates for three independent experiments (triplet wells)]. (C) THP-1-derived macrophages were treated with PBS, LPS (100 ng/mL), IL-1β (20 ng/mL), or TNFα (20 ng/mL) for 4 h. KLF14 mRNA levels were measured [three independent experiments (triplet wells)]. Data are presented as the mean ± SEM and analysed by two-tailed Student’s t-test for (A,B) and one-way ANOVA with Dunnett’s correction for (C).
3.2 Klf14 overexpression significantly alleviated lipid accumulation by promoting cholesterol efflux in macrophages
To understand the potential biological functions of KLF14 in macrophages, we first utilised a gain-of-function strategy to determine how KLF14 regulates cholesterol metabolism in these cells. Oxidised LDL (oxLDL) treatment significantly induced lipid accumulation in control THP-1-derived macrophages (AdLacZ-treated cells as control), while KLF14 overexpression significantly decreased lipid accumulation (Figure 2A), suggesting that KLF14 may be protective against lipid accumulation. Next, we determined the effect of KLF14 on cholesterol efflux and found that overexpression of KLF14 significantly increased cholesterol efflux to both ApoA-I and HDL particles as acceptors in THP-1-derived macrophages (approximately 1.5-fold for ApoA-I and 1.6-fold for HDL particles) (Figure 2B). In cholesterol loaded foam cells, the cholesterol transporters ABCA1 and ABCG1 play critical roles in the regulation of the net cholesterol efflux to lipid poor ApoA-I and HDL particles.6–8 Indeed, the expression of ABCA1 was increased by KLF14 overexpression at both the mRNA (Figure 2C) and protein levels (Figure 2D) compared with AdLacZ-treated macrophages. Our findings suggest that macrophage KLF14 regulates cholesterol efflux by modulation of ABCA1 expression, contributing to the decreased lipid accumulation in macrophages.
Figure 2.

KLF14 overexpression showed decreased lipid accumulation, increased cholesterol efflux, and ABCA1 expression in macrophages. THP-1 cells were differentiated to macrophages by treatment with PMA (100 ng/mL) for 72 h. Adenovirus-mediated overexpression was used as indicated. (A) AdLacZ- or AdKLF14-overexpressing macrophages were treated with PBS or oxLDL (25 µg/mL) for 24 h. Neutral lipid in the cells was stained by ORO staining and quantified by ORO area/total cell area [six technical replicates for the PBS group and nine technical replicates for the oxLDL group] Bar = 20µm. (B) Cholesterol efflux was measured in AdLacZ- or AdKLF14-overexpressing macrophages with ApoA-I or HDL as acceptors [four independent experiments (triplet wells)]. (C) The mRNA level and (D) protein levels of ABCA1 were determined by western blotting [three independent experiments (triplet wells)]. Data are presented as the mean ± SEM and analysed by two-tailed Student’s t-test for (B–D) or two-way ANOVA with Tukey correction for (A).
3.3 Klf14-deficient PMs showed increased lipid accumulation and decreased cholesterol efflux and ABCA1 expression
Next, we asked whether Klf14 is necessary for protecting against lipid accumulation in macrophages. We confirmed the genomic knockout of Klf14 in PMs by using primers flanking the single exon of the Klf14 gene in Klf14LysMApoE−/− mice (Supplementary material online, Figure S3A). In PMs from Klf14LysMApoE−/− mice, both mRNA and protein levels of KLF14 were significantly decreased (Supplementary material online, Figure S3B and C). In PMs isolated from Klf14LysMApoE−/− mice, Klf14 deficiency significantly increased intracellular lipid content upon oxLDL treatment compared to PMs from Klf14fl/flApoE−/− (Figure 3A), indicating a loss of the protective effect against foam cell formation in Klf14 deficient macrophages. Then, we determined if Klf14 deficiency also leads to a decreased cholesterol efflux in macrophages. Under basal conditions (without LXR agonist-induced expression of ABCA1 and ABCG1), Klf14-deficient PMs showed significantly lower cholesterol efflux to ApoA-I as acceptor (approximately reduced by 39.1%), but not to HDL particles (Figure 3B), indicating an important regulatory effect of KLF14 on the expression of ABCA1 in macrophages. Upon treatment with TO901317 (TO), an LXR agonist, to induce the expression of ABCA1 and ABCG1, cholesterol efflux was significantly increased in PMs from litter-mate control mice. However, this effect was significantly blocked in PMs isolated from Klf14 deficient mice (Figure 3B), indicating an important role of KLF14 in modulating cholesterol efflux.
Figure 3.

Klf14-deficient PMs showed increased lipid accumulation, decreased cholesterol efflux, and ABCA1 expression. PMs were isolated from Klf14fl/flApoE−/− and Klf14LysMApoE−/− mice and given indicated stimuli. (A) PMs were treated with PBS or oxLDL (25 µg/mL) for 24 h. Neutral lipid in the cell was stained by LipidSpotTM and quantified by LipidSpot signal/cell counts [PMs differentiated from 3–5 mice, four technical replicates]. Blue: DAPI. Green: LipidSpot. Bar = 20 µm. (B) The cholesterol efflux mediated by ApoA-I and HDL was determined in control and Klf14-deficient PMs under treatment with DMSO or TO901317 (TO, 0.1 µM) [PMs differentiated from 3–5 mice, six independent experiments (triplet wells)]. (C) Abca1 and Abcg1 expressions were determined in PMs treated with PBS or cAMP (3 mM) for 24 h [PMs differentiated from 3–5 mice, three independent experiments (triplet wells)]. (D) Klf14-deficient and control PMs were treated with DMSO or TO901317 (TO, 0.1 µM) for 24 h. Protein levels of ABCA1 were determined and quantified [PMs differentiated from 3–5 mice, three independent experiments (triplet wells)]. Data are presented as the mean ± SEM and analysed by two-way ANOVA with Tukey correction. n.s., not significant.
In PMs isolated from control mice, the mRNA level of Abca1 was upregulated by cAMP-mediated transcriptional regulation, but the expression of Abca1 was significantly blocked in Klf14-deficient PMs (Figure 3C). However, the expression of Abcg1 increased in Klf14-deficient PMs after cAMP induction, which may be caused by a compensatory effect18 (Figure 3C). Klf14 deficiency also significantly impaired the up-regulation of ABCA1 expression by activating the LXR pathway with TO901317 (Figure 3D), which is consistent with the impaired cholesterol efflux to ApoA-I in Figure 3C. Considering that increased lipid uptake also contributes to lipid accumulation, the expression of receptors involved in LDL or oxLDL uptake was determined. The expression of Ldlr and Cd36 was higher in Klf14-deficient PMs, while Sra1, Srb1, Lox1 were not changed (Supplementary material online, Figure S4A). However, Dil-labelled oxLDL uptake showed no difference between Klf14-deficient and control PMs (Supplementary material online, Figure S4B), indicating that KLF14 may not regulate lipid uptake in macrophages. Taken together, these data indicate that KLF14 protects against lipid accumulation and maintains cholesterol efflux by regulating Abca1 expression in macrophages.
3.4 Klf14 overexpression inhibits the pro-inflammatory response in macrophages
Cholesterol crystals (CCs) can be found in early atherosclerotic lesions, and CCs-induced inflammation is critical in all stages of the atherosclerotic process. Consistent with previous studies,19–21 CCs treatment triggered an inflammatory response in macrophages. In THP1-derived macrophages, KLF14 overexpression significantly reduced the levels of IL-1β, IL-6, TNFα released into supernatant (Figure 4A) and downregulated the mRNA expression of pro-inflammatory chemokines, such as IL1β, IL6, and MCP1 (Figure 4B). Intriguingly, we found that the expression of inflammasome components (NLRP3 and Pro-Caspase1) and a key effector in the NF-κB pathway (p65) were significantly suppressed by KLF14 (Figure 4C). These findings demonstrate that KLF14 suppressed the inflammatory signalling cascade in response to CCs stimulation, suggesting a protective role of KLF14 in atherosclerosis development.
Figure 4.

KLF14 overexpression inhibits inflammatory responses in macrophages. THP-1-derived macrophages were transfected with AdLacZ or AdKLF14 for 24 h. After 72 h of differentiation in the presence of 100 nM PMA, THP-1-derived macrophages were treated with cholesterol crystals (CCs, 200 µg/mL) for 24 h, and cell lysates or supernatants were collected at the endpoint. (A,B) The cytokines release into the supernatant (A) and the mRNA level of the indicated cytokines (B) were determined and quantified [three independent experiments (triplet wells)]. (C) Protein levels of NLRP3, p65, IL-1β, Pro-IL1β, and Pro-Caspase1 were determined and quantified [three independent experiments (triplet wells)]. Data are presented as the mean ± SEM and analysed by two-tailed Student’s t-test for (C) or two-way ANOVA with Tukey correction for (A,B).
3.5 Klf14 deficiency increases the inflammatory response, but not apoptosis in macrophages
Conversely, and in agreement with the findings above, Klf14-deficient PMs from Klf14LysMApoE−/− mice showed significantly higher response to inflammatory stimuli, evidenced by higher mRNA levels of Il1β, Il1α, Cxcl1, G-csf upon stimulus with IL-1β (Figure 5A). Considering that bone marrow-derived macrophages (BMDMs) and PMs may have different responses to inflammatory stimuli,22 we also repeated this experiment using BMDMs isolated and cultured from Klf14LysMApoE−/− and control mice and found that BMDMs showed similar results to the PMs (Supplementary material online, Figure S6). CCs activated the NLRP3 inflammasome and the NF-κB pathways, represented by increased expression of NLRP3 and p65 (Figure 5B). Under basal conditions (in the absence of CCs), Klf14 deficiency led to a significant increase of NLRP3 and p65 expression, while, in the presence of CCs, Klf14 deficiency aggravated the pro-inflammatory response by further increasing the p65 protein level (Figure 5B, lane 3 and 4). In accordance, IL-1β released from Klf14-deficient PMs was also higher after treatment with CCs+ATP (Figure 5C). Although Klf14 deficiency significantly induced M1 macrophage polarisation when challenged with LPS+INFγ (M1 induction) (Supplementary material online, Figure S7A), Klf14 deficiency did not affect the expression of M2 markers (Fizz1, Ym1) when cells were treated with IL-4 (M2 induction) (Supplementary material online, Figure S7B) and anti-inflammatory cytokine (IL-10) release under co-treatment with CCs or oxLDL (Supplementary material online, Figure S7C). The results show that macrophage Klf14 deficiency increases the inflammatory response.
Figure 5.

Klf14 deficiency increases the inflammatory response in macrophages. (A,B) PMs were isolated from Klf14fl/flApoE−/− and Klf14LysMApoE−/− mice. (A) PMs were treated with IL-1β (20 ng/mL) for 4 h. The mRNA level of pro-inflammatory cytokines was determined [PMs differentiated from 3–5 mice, 3 independent experiments (triplet wells)] (B) PMs were treated with PBS or cholesterol crystals (CCs, 200 µg/mL) for 24 h. Protein levels of NLRP3 and p65 were determined and quantified. [PMs differentiated from 3–5 mice, 3 independent experiments (triplet wells)] (C) PMs were treated with PBS, CCs only (200 µg/mL, 24 h) or CCs+ATP (ATP at 5 mM, CCs 24 h + ATP 1 hr). IL-1β released into the supernatant was measured by ELISA [PMs differentiated from 3–5 mice, three independent experiments (triplet wells)]. (D) ChIP assay was conducted in THP-1 cells overexpressing KLF14-flag. Amplification of potential binding site in IL1β promoter and the control region of RPL30 were done by qPCR [three independent experiments]. (E) −946/+45bp of the wild-type and mutated human IL1β promoter-driven luciferase was transfected in AD293 cells. LPS (100 ng/mL) was used for the induction of inflammation [5–6 technical replicates, repeated by three independent experiments]. Data are presented as the mean ± SEM and analysed by two-way ANOVA with Tukey correction.
Additionally, previous reports showed that free cholesterol accumulation in the macrophages could induce macrophage apoptosis,23 leading to higher plaque vulnerability in advanced atherosclerosis.24 Therefore, we determined whether Klf14 deficiency affects macrophage apoptosis. The full-length caspase-3, an effector caspase in apoptosis, was not significantly changed in Klf14-deficient PMs than the control under basal condition or cholesterol crystals (CCs) treatment (Supplementary material online, Figure S5). Cleaved caspase-3 was not detectable. To determine the activity of caspase-3, we also probed for a downstream target of active caspase-3, PARP1. PARP1 cleavage was increased by CCs stimulation, illustrated by the ratio of cleaved PARP1/full-length PARP1. There was no difference between the two groups (Supplementary material online, Figure S5), indicating that Klf14-deficiency did not increase apoptosis of macrophages.
Interestingly, we identified two potential KLF binding sites (−442 to −445 and −492 to −496) upstream of the human IL1β transcription start site. Chromatin immunoprecipitation (ChIP) assay performed in the THP-1 derived macrophages using anti-Flag antibody or anti-IgG (negative control), the genomic fragment was applied for PCR amplification using a primer set for this region and we found that KLF14 specifically bound to IL1β promoter region, but not to the control RPL30 promoter (Figure 5D). Furthermore, we generated a luciferase reporter construct containing the -946 to +45 region of the human IL1β promoter. KLF14 significantly inhibited the luciferase activity in both baseline and LPS stimulated conditions (Figure 5E). The mutation of the −442 to −445 region did not affect the regulatory effect of KLF14, while the mutation of −492 to −496 abolished the inhibitory effect of KLF14 on IL1β transcription, indicating that −492 to −496 (CACCC) is the functional binding site for KLF14 on the human IL1β promoter region (Figure 5E). Collectively, we demonstrated that KLF14 inhibits the inflammatory response via suppressing p65 expression13 and direct binding to the IL1β promoter to inhibit its transcription.
3.6 Myeloid-selective Klf14 deficiency accelerates atherosclerosis in the ApoE−/− hyperlipidemic mouse model
To investigate the role of myeloid KLF14 in atherosclerosis development, both male and female Klf14LysMApoE−/− and litter-mate control Klf14fl/flApoE−/− mice were placed on an atherogenic WD for 12 weeks. There were no differences in lipid profiles between Klf14LysMApoE−/− and Klf14fl/flApoE−/− control mice at the end of treatment, including triglycerides, total cholesterol, LDL-C, and HDL-C (Supplementary material online, Figure S8A). Nonetheless, both male and female Klf14LysMApoE−/− mice showed significantly accelerated atherosclerosis development (Figure 6A), shown by en face Oil Red O (ORO) staining of the aortic tree. Although male and female Klf14fl/flApoE−/− control mice have similar plaque burden at the endpoint, male Klf14LysMApoE−/− mice were more susceptible to atherogenesis, as evidenced by a 79% increase in the atherosclerotic plaque area in the male knockout group, compared to a 25% increase in the female knockout group. We also observed a significantly higher plaque area at the aortic root in male Klf14LysMApoE−/− mice than litter-mate control male mice, but no difference between female groups (Figure 6B).
Figure 6.

Myeloid Klf14 deficiency accelerates atherosclerosis development in mice. Both male and female Klf14LysMApoE−/− mice and litter-mate control mice were placed on WD for 12 weeks. (A) Aortic trees were isolated from those mice, and En face Oil Red O (ORO) staining was used to evidence atherosclerotic plaques. n = 8 mice for female Klf14fl/flApoE−/− group, n = 14 mice for female Klf14LysMApoE−/− group, n = 11 mice for male Klf14fl/flApoE−/− group, n = 10 mice for male Klf14LysMApoE−/− group. Bar = 5 mm. (B) Haematoxylin and eosin (HE) staining was used to evidence the atherosclerotic plaque at the aortic root from male Klf14fl/flApoE−/− and Klf14LysMApoE−/− mice. Bar = 100 µm. n = 8 mice for female Klf14fl/flApoE−/− group, n = 13 mice for female Klf14LysMApoE−/− group, n = 8 mice for male Klf14fl/flApoE−/− group, n = 7 mice for male Klf14LysMApoE−/− group. (C) Mac2 immunofluorescence staining was used to detect macrophage infiltration at the aortic root plaque from male mice. Bar = 50 µm. n = 9 mice for Klf14fl/flApoE−/−group, n = 6 mice for male Klf14LysMApoE−/−group. (D) The cholesterol crystals content was determined using a polarised light microscope at the aortic root plaque from male mice after 12 weeks of WD treatment. Bar = 50 µm. n = 10 mice for Klf14fl/flApoE−/−group, n = 7 mice for male Klf14LysMApoE−/−group. (E,F) ORO staining at the aortic root from male Klf14fl/flApoE−/− and Klf14LysMApoE−/− mice after (E) 12 weeks of WD treatment or (F) on CD. For (E), n = 7 mice for Klf14fl/flApoE−/−group, n = 6 mice for male Klf14LysMApoE−/−group. For (F), n = 7 biological replicates for the Klf14fl/flApoE−/− group, n = 5 biological replicates for the Klf14LysMApoE−/− group. Bar = 50 µm. Data are presented as the mean ± SEM and analysed by Mann–Whitney test for (F) and two-tailed Student’s t-test for other panels. n.s., not significant.
Atherosclerotic plaque components are highly heterogeneous. First, we observed a significantly greater Mac2 (a macrophage marker) positive area in Klf14LysMApoE−/− male mice, indicating increased macrophage infiltration (Figure 6C). Accordingly, we found that macrophage migration was significantly higher in Klf14-deficient macrophages under pro-inflammatory stimulation with LPS (Supplementary material online, Figure S9). Second, male Klf14LysMApoE−/− mice on WD showed significantly higher CCs contents in the plaque determined by polarised light microscopy, suggesting impaired cholesterol efflux in Klf14-deficient macrophages caused more free cholesterol accumulation in the atheroma (Figure 6D). Unexpectedly, the content of neutral triglycerides and lipids staining with ORO staining showed no significant difference at the aortic root between Klf14-deficient and litter-mate control mice (Figure 6E). Considering that a 12-week WD challenge induced atheroma may be too advanced to show a difference in cholesterol ester or neutral lipids, we next investigated the lipid accumulation using both Klf14LysMApoE−/− and Klf14fl/flApoE−/− control mice on a CD. ApoE−/− mice fed a CD present spontaneous atherosclerosis, which is considered relatively early-stage atherosclerosis compared to the WD-fed ApoE−/− mice.25 Although there was no difference in lipid profiles (Supplementary material online, Figure S8B), Klf14LysMApoE−/− male mice on CD showed a significantly higher ORO staining area in the atherosclerosis plaques (Figure 6F), indicating that myeloid Klf14 deficiency increases lipid accumulation in the plaque in the early stages of atherosclerosis development. There were no significant differences in lesion area and macrophage infiltration in this spontaneous atherosclerotic model (Supplementary material online, Figure S10A,B). Plaque stability and vulnerability were analysed by Masson trichrome staining (plaque fibrosis) and the area of necrotic cores. There was no difference between male myeloid-Klf14 deficient mice and controls after treatment (Supplementary material online, Figure S11A,B). This is inconsistent with the previous finding that apoptosis did not change in CCs-treated macrophages (Supplementary material online, Figure S5).
All similar analyses were also performed in aortic sinus cross-sections from female mice with no significant differences (for CD studies, Supplementary material online, Figure S12A–C; and for WD studies, Supplementary material online, Figure S12D–F). Contrary to the finding in males, CC area and macrophage infiltration were decreased in female Klf14LysMApoE−/− mice compared to control mice (Supplementary material online, Figure S12G and H). Female Klf14fl/flApoE−/− control mice have greater plaque area at the aortic root compared to male Klf14fl/flApoE−/− mice (P = 0.0064), while this difference disappeared in Klf14LysMApoE−/− mice due to a significantly increased atherosclerotic plaque burden in male mice (Figure 6B).
These results indicate that myeloid KLF14 protects against atherosclerosis development without affecting systemic lipid profiles and suggests a potential sex difference in myeloid KLF14 functions between males and females.
3.7 Perhexiline induces KLF14-dependent expression of ABCA1 in macrophages
We previously found that perhexiline is a bona fide KLF14 inducer, which directly upregulates the transcription of KLF14 in hepatocytes and endothelial cells.13,14 Perhexiline, developed initially to treat angina,26,27 has been identified as a carnitine palmitoyltransferase I (CPT1) inhibitor. In agreement with the previous results,14 perhexiline increased the expression of KLF14 and ABCA1 in a dose-dependent manner in THP-1-derived macrophages (Figure 7A and 7B). Additionally, perhexiline induced Klf14 and Abca1, and inhibited Il1β (Figure 7C) in mouse PMs. Similar to KLF14 overexpression, perhexiline could also significantly increase cholesterol efflux (Figure 7D). Perhexiline significantly increased ABCA1 expression in PMs from control mice, but this effect was largely abolished in the Klf14-deficient PMs (Figure 7E and 7F), indicating that perhexiline exerted its effect in a KLF14-dependent manner. We also treated J774 macrophages or THP-1 cell-derived macrophages with Etomoxir (Sigma), a well-known CPT1 inhibitor, which failed to upregulate the expression of Klf14, Abca1, and Abcg1 (Supplementary material online, Figure S13A and B). Considering that perhexiline also inhibits NOX227, using siRNA-mediated knockdown of Nox2 in PMs, we found that NOX2 was not required for the effect of perhexiline on inducing the expression of Klf14 and Abca1 (Supplementary material online, Figure S13C). These results indicate that perhexiline regulating the expression of Klf14 and Abca1 is independent of the inhibitory effects of perhexiline on CPT1 or NOX2.
Figure 7.

Perhexiline increases Abca1 expression in a KLF14-dependent manner. (A,B) In THP-1 differentiated macrophages, perhexiline treatment increases KLF14 and ABCA1 expression at both the mRNA (A) and protein levels (B) in a dose-dependent manner [three independent experiments (triplet wells)]. (C) PMs were isolated from WT mice and treated with 15 µM perhexiline for 24 h. The expression of Klf14, Abca1, and IL1β was determined by qRT–PCR [PMs differentiated from 3–5 mice, three independent experiments (triplet wells)]. (D) The cholesterol efflux mediated by HDL or ApoA-I was determined in THP-1-derived macrophages treated with DMSO or Perhexline (Px, 15 μM) for 24 h [three independent experiments (triplet wells) for the left panel, six independent experiments (triplet wells) for the right panel] (E,F) PMs from Klf14fl/flApoE−/− and Klf14LysMApoE−/− mice were treated with DMSO or 15 µM perhexiline for 24 h. The expression of ABCA1 was determined at the (E) mRNA or (F) protein level and quantified [PMs differentiated from 3–5 mice, three independent experiments (triplet wells)]. (G) PMs of WT or Lxr KO group were treated with DMSO or perhexiline (15 µM) for 24 h. The Klf14 and Abca1 mRNA levels were determined by qRT–qPCR [PMs differentiated from 3–5 mice, 4–6 independent experiments (triplet wells per experiment)]. (H) HepG2 cells were treated with DMSO, TO901317 (TO, 0.1 µM), or perhexiline (Px, 15 µM) for 24 h. SREBF1 and FASN mRNA levels were determined by RT–qPCR [three independent experiments (triplet wells)]. Data are presented as the mean ± SEM and analysed by one-way ANOVA with Dunnett’s correction for (A,B) and (H), two-tailed Student’s t-test for (C,D), or two-way ANOVA with Tukey correction for (E–G). n.s., not significant.
3.8 LXRs are dispensable for the induction of Abca1 by perhexiline
LXRα and LXRβ are two critical nuclear receptors that control cholesterol efflux through regulating the expression of ABCA1 and ABCG1.28,29 Synthetic LXR agonists attenuate atherosclerosis via the upregulation of the ABC cholesterol transporters in both mouse and rabbit atherosclerosis models.30,31 To understand whether LXRs are mediators of the KLF14 function in macrophages, we isolated PMs from LXRα knockout mice and then knocked down Lxrβ by using Lxrβ siRNAs to generate LXRα/β knockout/knockdown PMs (referred to as LXR KO, with the LXRα/β mRNA levels shown in Supplementary material online, Figure S14A and B). In PMs isolated from WT control mice, TO901317 significantly induced the expression of Abca1 (Supplementary material online, Figure S14C) but did not affect the expression of Klf14 (Supplementary material online, Figure S15), indicating that Klf14 is not regulated by LXR activation in macrophages. We found that perhexiline significantly increased Klf14 expression in PMs of both LXR KO and WT group, indicating that the induction of Klf14 by perhexiline is independent of LXRs (Figure 7G). While LXRs deficiency largely abolished the induction of Abca1 by TO901317 (Supplementary material online, Figure S14C), LXR KO did not affect the induction of Abca1 by perhexiline (Figure 7G), indicating that LXRs are dispensable for the effect of perhexiline on Abca1 in macrophages.
In addition, LXR agonists induce apparent side effects such as increased triglycerides and fatty liver, partly through upregulation of the expression of LXR target genes in the liver, like sterol regulatory element-binding transcription factor 1 (SREBF1) and fatty acid synthase (FASN), which limited their clinical use.32 Therefore, we also determined whether perhexiline affects the levels of lipogenic genes. In HepG2 cells, TO901317 treatment significantly increased the expression of SREBF1 and FASN. However, perhexiline treatment did not affect the expression of the two lipogenic genes (Figure 7H), indicating that perhexiline might induce macrophage cholesterol efflux with lower liver side effects compared to LXR agonists. Indeed, using adenovirus-mediated overexpression of KLF14 or LacZ in C56BL/6J mice,14 we did not observe any upregulation of Lxrα, Lxrβ, Srebf1, or Srebf2 in the liver (Supplementary material online, Figure S16), indicating KLF14 itself does not induce liver lipogenesis. Collectively, these data indicate that perhexiline, through upregulation of KLF14 in macrophages, may be an effective drug to induce cholesterol efflux for the treatment of atherosclerosis.
4. Discussion
Human genetic studies have identified several risk alleles leading to the decreased expression level of KLF14 in association with increased CAD risks.10,33 We previously demonstrated that hepatocyte-selective KLF14 deficiency gives rise to lower HDL-C level in plasma as well as a decrease in serum cholesterol efflux capacity due to reduced ApoA-I expression in the liver.14 Although the KLF14 inducer, perhexiline, inhibits atherosclerosis development in ApoE−/− mice, we noticed that HDL-C levels are very low in ApoE−/− mice after 12-week WD challenge,14 suggesting a yet undefined mechanism underlying the protective effects of KLF14 and perhexiline. Here, our findings demonstrate the anti-atherosclerotic effects of macrophage KLF14 through promoting cholesterol efflux and suppressing the inflammatory response.
Uptake and accumulation of cholesterol by macrophages promotes the formation of foam cells and drives the development of atherosclerotic lesions. Epidemiological studies have consistently shown that HDL-C levels are inversely related to atherosclerotic cardiovascular disease. One of the underlying mechanisms by which HDL and ApoA-I reduce lesion formation is reverse cholesterol transport. This process is regulated by the quality and quantity of HDL particles and the efficiency of cholesterol efflux from the peripheral tissues to lipid-poor ApoA-I or lipidated HDL particles.4,34 In the present study, we found that KLF14 also plays a critical role in macrophage biology. We demonstrate for the first time that mice with macrophage-selective Klf14-deficiency had increased atherosclerosis development due to impaired cholesterol efflux and increased inflammatory response, while KLF14 overexpression or induction by perhexiline significantly increased cholesterol efflux in macrophages. These findings agree with genetic studies that the T/T allele carriers in rs4731702, compared to the C/C allele carriers, show higher expression of KLF14 in an eQTL study24 and have lower cardiovascular risks. The present study builds upon and expands our previous study by demonstrating that KLF14 induction by perhexiline inhibits atherosclerosis by synergistically 1) increasing HDL-C in the circulation by modulating ApoA-I expression in the liver, 2) enhancing cholesterol efflux through modulating ABCA-1 expression in macrophages, and 3) suppressing the inflammatory response via inhibition of the expression of p65 and IL1β in macrophages.
Atherosclerosis-relevant stimuli, including cholesterol crystals, can trigger inflammasome activation and redundant inflammatory signalling pathways.21 In vitro, this requires a two-step activation: (1) NF-κB activators,35 adenovirus-mediated penetration,36 or other stimuli-induced priming processes upregulating NLRP3 expression, and (2) a CC-induced NLRP3 inflammasome activation.21 Besides, primary macrophages could also be ‘in vivo primed’ by hypercholesterolaemia in ApoE−/− mice.37 In our experiments, the priming process was accomplished by adenovirus-mediated penetration in the overexpression experiments and hypercholesterolaemia-induced ‘in vivo priming’. Furthermore, the expression of NLRP3 was regulated by KLF14, which may also affect the priming process during NLRP3 activation.
Despite examining cholesterol efflux and inflammation separately in our study, cholesterol efflux and inflammation homeostasis are two processes that reciprocally affect each other. Lower cholesterol efflux leads to cholesterol accumulation in the macrophages, which triggers the inflammasome activation and the release of IL-1β.18,20,21,38,39 On the other hand, higher inflammation decreases cholesterol efflux.40 Additionally, we provide evidence that KLF14 directly binds to the promoter of the IL1β gene to suppress its expression, suggesting that Klf14 deficiency leads to loss of direct inhibition of IL1β transcription and initiates the pro-inflammatory cascades which disrupt inflammation homeostasis and exacerbate cholesterol accumulation in macrophages. Although IL-1β has manifold effects on the cardiovascular system, the Canakinumab Anti-Inflammatory Thrombosis Outcome Study (CANTOS) has demonstrated that administration of canakinumab to neutralise IL-1β activity improves outcomes in individuals with post-acute myocardial infarction.41 Our findings indicate that KLF14 blocks multiple inflammatory pathways and presents protective effects on atherosclerosis.
W. Xie et al. also demonstrate that KLF14 plays an anti-atherogenic role via a miR-27a-dependent downregulation of lipoprotein lipase and subsequent inhibition of pro-inflammatory cytokine secretion.42 Nonetheless, a study from X. Wei et al. showed a contradictory result indicating that KLF14 induces inflammation in macrophages and increases atherosclerosis development using adenovirus-mediated gene silencing both in vivo and in vitro.43 However, we used Cre-loxP system-mediated myeloid cell-selective gene knockout in vivo and primary cell culture in vitro. The uncomparable methods maybe generate the seemingly opposite results. A recently published paper showed that endothelial KLF14 may induce macrophage M2 polarization,44 indicating a potential role of KLF14-mediating cell–cell interaction.
Previous genetic studies showed sex-specific metabolic phenotypes of KLF14 with female risk allele carriers having lower hip-to-waist ratio but higher type 2 diabetes risk.45 However, the KLF14 risk allele does not show such sex differences on cardiovascular and lipid profile phenotypes.10 Accordingly, we found that atherosclerosis was increased in both female and male mice, though Klf14LysMApoE−/− male mice showed higher susceptibility to WD-induced atherosclerosis compared to their female counterparts. The ApoE−/− mice model does not exhibit consistent sex differences during atherosclerosis development,46–48 which does not reflect the sex differences observed in humans49 and may not be an appropriate model to study sex differences in the biological functions of KLF14. The effects and underlying mechanisms of KLF14 in sex differences will be further investigated.
Perhexiline was used as a potent anti-anginal50 drug in the clinic even before its mechanism was fully understood.27 Later on, perhexiline has been administrated as a CPT1 inhibitor to increase metabolic efficiency in heat failure patients.51–53 Interestingly, perhexiline improves cardiac energetics and symptom status without significantly altering cardiac substrate utilization,53 suggesting possible unanticipated mechanisms. Perhexiline was also found to inhibit neutrophil NADPH oxidase.54 In our study, we found that perhexiline induces ABCA1 expression independent of CPT1 or NOX2, addressing a new mechanism of action of perhexiline.
ABCA1 is a target gene of LXR,9 and ABCA1 deficiency in patients results in Tangier disease and familial HDL deficiency.55,56 Although preclinical studies showed strong inhibition of atherosclerosis by LXR agonists,30,31,57 the clinical use of LXR agonists was halted due to their side effects, which is triggered mainly through upregulation of lipogenic genes in the liver.32 We did not detect a direct protein–protein interaction between LXR and KLF14 using co-immunoprecipitation (data not shown). Then, we determined that KLF14 induces cholesterol efflux is independent of LXRs using LXR KO PMs. We also found that increased KLF14 expression induced neither LXRα/β expression nor lipogenic gene (SREBF1 or FASN) expression, indicating that KLF14 upregulates the cholesterol efflux in macrophages without triggering lipogenesis in hepatocytes in an LXR-independent manner, which is confirmed in adenovirus-mediated overexpression of KLF14 in the liver. These findings define KLF14 as a promising therapeutic target for treating atherosclerosis.
5. Limitations
Although the ApoE−/− mouse model is one of the most commonly used mouse models for atherosclerosis studies, macrophage-specific ApoE deficiency itself demonstrated biology on cholesterol efflux and inflammation.58,59 Therefore, although we carefully designed and controlled our study, the deletion of ApoE in macrophages may reduce or enhance the observed phenotypes. LysM-Cre is expressed in all myeloid cells, including macrophages and neutrophils.60,61 We also determined the neutrophil infiltration in the plaque by using anti-Mouse Gr-1 monoclonal antibody (RB6-8C5), which has been widely used to detect neutrophils.62–64 In both male and female animals, Gr1+ cells were minimal and showed no significant difference (Supplementary material online, Figure S17) in this study. Gr-1 antibodies react with both the Ly6G (exclusively expressed on granulocyte) and Ly6C (monocyte/macrophage)62. LysM-Cre mice, and perhexiline treatment may regulate the biological functions of KLF14 in other subset of myeloid cells such as neutrophils and dendritic cells, causing another limitation to address the contribution of macrophage KLF14 to atherosclerosis in vivo. Other approaches should be necessary to study the contribution of macrophages and neutrophils to vascular inflammation and atherosclerosis in the future.
6. Conclusions
Our results uncovered a pivotal role of myeloid KLF14 in protecting against atherosclerosis development by regulating cholesterol efflux and inflammation. These findings provide new insights into a previously unrecognised effect of KLF14 in myeloid cells and extend our understanding of the underlying mechanism of perhexiline as a KLF14 activator to support its potential use for atherosclerosis treatment.
Supplementary material
Supplementary material is available at Cardiovascular Research online.
Author contributions
Y.G. and Y.E.C. conceived, designed, and supervised the study. H.W., W.H., Y.L., and Y.G. performed the experiments. H.L., L.C., M.T.G.-B., A.S., and J.Z. provided the technical support and contributed to the discussion of the project and article. H.W. and Y.G. analysed the data and wrote the article.
Supplementary Material
Acknowledgements
We gratefully acknowledge L.Q., O.M.D., C.L., and Y.W. from the University of Michigan for their mentoring and expert suggestions to improve this study. We also thank D.H., L.W., T.Z., Y.F., W.X., and Z.C. for their expert technical assistance in the experiments and data acquisition.
Conflict of interest: none declared.
Funding
This work was supported in whole or in part by the National Institutes of Health [HL068878, HL134569, and HL137214 to Y.E.C., HL138139 to J.Z.] and by the American Heart Association [15SDG24470155 to Y.G. and 20PRE35170017 to H.W.].
Data availability
The expanded Methods section and other supporting data are available in the Supplementary material online. The primary data for this article will be shared on reasonable request to the corresponding authors.
Translational Perspective
Here, using both gain- and loss-of-function strategies, we show that KLF14 regulates cholesterol efflux by regulating the expression of ABCA1 and inhibits inflammatory response in macrophages. These findings, along with our previous data, put activation of KLF14 forward as a prospective therapeutic target for atherosclerotic cardiovascular disease.
References
- 1. Libby P, Ridker PM, Hansson GK.. Progress and challenges in translating the biology of atherosclerosis. Nature 2011;473:317–325. [DOI] [PubMed] [Google Scholar]
- 2. Goldstein JL, Brown MS.. A century of cholesterol and coronaries: from plaques to genes to statins. Cell 2015;161:161–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Sampson UK, Fazio S, Linton MF.. Residual cardiovascular risk despite optimal LDL cholesterol reduction with statins: the evidence, etiology, and therapeutic challenges. Curr Atheroscler Rep 2012;14:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Tall AR, Yvan-Charvet L, Terasaka N, Pagler T, Wang N.. HDL, ABC transporters, and cholesterol efflux: implications for the treatment of atherosclerosis. Cell Metab 2008;7:365–375. [DOI] [PubMed] [Google Scholar]
- 5. Rohatgi A, Khera A, Berry JD, Givens EG, Ayers CR, Wedin KE, Neeland IJ, Yuhanna IS, Rader DR, de Lemos JA, Shaul PW.. HDL cholesterol efflux capacity and incident cardiovascular events. N Engl J Med 2014;371:2383–2393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wang N, Lan D, Chen W, Matsuura F, Tall AR.. ATP-binding cassette transporters G1 and G4 mediate cellular cholesterol efflux to high-density lipoproteins. Proc Natl Acad Sci USA 2004;101:9774–9779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kennedy MA, Barrera GC, Nakamura K, Baldan A, Tarr P, Fishbein MC, Frank J, Francone OL, Edwards PA.. ABCG1 has a critical role in mediating cholesterol efflux to HDL and preventing cellular lipid accumulation. Cell Metab 2005;1:121–131. [DOI] [PubMed] [Google Scholar]
- 8. Brooks-Wilson A, Marcil M, Clee SM, Zhang LH, Roomp K, van Dam M, Yu L, Brewer C, Collins JA, Molhuizen HO, Loubser O, Ouelette BF, Fichter K, Ashbourne-Excoffon KJ, Sensen CW, Scherer S, Mott S, Denis M, Martindale D, Frohlich J, Morgan K, Koop B, Pimstone S, Kastelein JJ, Genest J Jr, Hayden MR.. Mutations in ABC1 in Tangier disease and familial high-density lipoprotein deficiency. Nat Genet 1999;22:336–345. [DOI] [PubMed] [Google Scholar]
- 9. Costet P, Luo Y, Wang N, Tall AR.. Sterol-dependent transactivation of the ABC1 promoter by the liver X receptor/retinoid X receptor. J Biol Chem 2000;275:28240–28245. [DOI] [PubMed] [Google Scholar]
- 10. Teslovich TM, Musunuru K, Smith AV, Edmondson AC, Stylianou IM, Koseki M, Pirruccello JP, Ripatti S, Chasman DI, Willer CJ, Johansen CT, Fouchier SW, Isaacs A, Peloso GM, Barbalic M, Ricketts SL, Bis JC, Aulchenko YS, Thorleifsson G, Feitosa MF, Chambers J, Orho-Melander M, Melander O, Johnson T, Li X, Guo X, Li M, Shin Cho Y, Jin Go M, Jin Kim Y, Lee J-Y, Park T, Kim K, Sim X, Twee-Hee Ong R, Croteau-Chonka DC, Lange LA, Smith JD, Song K, Hua Zhao J, Yuan X, Luan J, Lamina C, Ziegler A, Zhang W, Zee RYL, Wright AF, Witteman JCM, Wilson JF, Willemsen G, Wichmann H-E, Whitfield JB, Waterworth DM, Wareham NJ, Waeber G, Vollenweider P, Voight BF, Vitart V, Uitterlinden AG, Uda M, Tuomilehto J, Thompson JR, Tanaka T, Surakka I, Stringham HM, Spector TD, Soranzo N, Smit JH, Sinisalo J, Silander K, Sijbrands EJG, Scuteri A, Scott J, Schlessinger D, Sanna S, Salomaa V, Saharinen J, Sabatti C, Ruokonen A, Rudan I, Rose LM, Roberts R, Rieder M, Psaty BM, Pramstaller PP, Pichler I, Perola M, Penninx BWJH, Pedersen NL, Pattaro C, Parker AN, Pare G, Oostra BA, O’Donnell CJ, Nieminen MS, Nickerson DA, Montgomery GW, Meitinger T, McPherson R, McCarthy MI, McArdle W, Masson D, Martin NG, Marroni F, Mangino M, Magnusson PKE, Lucas G, Luben R, Loos RJF, Lokki M-L, Lettre G, Langenberg C, Launer LJ, Lakatta EG, Laaksonen R, Kyvik KO, Kronenberg F, König IR, Khaw K-T, Kaprio J, Kaplan LM, Johansson Å, Jarvelin M-R, Cecile J. W. Janssens A, Ingelsson E, Igl W, Kees Hovingh G, Hottenga J-J, Hofman A, Hicks AA, Hengstenberg C, Heid IM, Hayward C, Havulinna AS, Hastie ND, Harris TB, Haritunians T, Hall AS, Gyllensten U, Guiducci C, Groop LC, Gonzalez E, Gieger C, Freimer NB, Ferrucci L, Erdmann J, Elliott P, Ejebe KG, Döring A, Dominiczak AF, Demissie S, Deloukas P, de Geus EJC, de Faire U, Crawford G, Collins FS, Chen Y-D. I, Caulfield MJ, Campbell H, Burtt NP, Bonnycastle LL, Boomsma DI, Boekholdt SM, Bergman RN, Barroso I, Bandinelli S, Ballantyne CM, Assimes TL, Quertermous T, Altshuler D, Seielstad M, Wong TY, Tai E-S, Feranil AB, Kuzawa CW, Adair LS, Taylor Jr HA, Borecki IB, Gabriel SB, Wilson JG, Holm H, Thorsteinsdottir U, Gudnason V, Krauss RM, Mohlke KL, Ordovas JM, Munroe PB, Kooner JS, Tall AR, Hegele RA, Kastelein JJP, Schadt EE, Rotter JI, Boerwinkle E, Strachan DP, Mooser V, Stefansson K, Reilly MP, Samani NJ, Schunkert H, Cupples LA, Sandhu MS, Ridker PM, Rader DJ, van Duijn CM, Peltonen L, Abecasis GR, Boehnke M, Kathiresan S.. Biological, clinical and population relevance of 95 loci for blood lipids. Nature 2010;466:707–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tetreault MP, Yang Y, Katz JP.. Kruppel-like factors in cancer. Nat Rev Cancer 2013;13:701–713. [DOI] [PubMed] [Google Scholar]
- 12. McConnell BB, Yang VW.. Mammalian Kruppel-like factors in health and diseases. Physiol Rev 2010;90:1337–1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hu W, Lu H, Zhang J, Fan Y, Chang Z, Liang W, Wang H, Zhu T, Garcia-Barrio MT, Peng D, Chen YE, Guo Y.. Kruppel-like factor 14, a coronary artery disease associated transcription factor, inhibits endothelial inflammation via NF-kappaB signaling pathway. Atherosclerosis 2018;278:39–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Guo Y, Fan Y, Zhang J, Lomberk GA, Zhou Z, Sun L, Mathison AJ, Garcia-Barrio MT, Zhang J, Zeng L, Li L, Pennathur S, Willer CJ, Rader DJ, Urrutia R, Chen YE.. Perhexiline activates KLF14 and reduces atherosclerosis by modulating ApoA-I production. J Clin Invest 2015;125:3819–3830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Libby P, Ridker PM, Maseri A.. Inflammation and atherosclerosis. Circulation 2002;105:1135–1143. [DOI] [PubMed] [Google Scholar]
- 16. Jones NL, Reagan JW, Willingham MC.. The pathogenesis of foam cell formation: modified LDL stimulates uptake of co-incubated LDL via macropinocytosis. Arterioscler Thromb Vasc Biol 2000;20:773–781. [DOI] [PubMed] [Google Scholar]
- 17. Henson DA, St Clair RW, Lewis JC.. Morphological characterization of beta-VLDL and acetylated-LDL binding and internalization by cultured pigeon monocytes. Exp Mol Pathol 1989;51:243–263. [DOI] [PubMed] [Google Scholar]
- 18. Yvan-Charvet L, Ranalletta M, Wang N, Han S, Terasaka N, Li R, Welch C, Tall AR.. Combined deficiency of ABCA1 and ABCG1 promotes foam cell accumulation and accelerates atherosclerosis in mice. J Clin Invest 2007;117:3900–3908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Grebe A, Latz E.. Cholesterol crystals and inflammation. Curr Rheumatol Rep 2013;15:313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Janoudi A, Shamoun FE, Kalavakunta JK, Abela GS.. Cholesterol crystal induced arterial inflammation and destabilization of atherosclerotic plaque. Eur Heart J 2016;37:1959–1967. [DOI] [PubMed] [Google Scholar]
- 21. Duewell P, Kono H, Rayner KJ, Sirois CM, Vladimer G, Bauernfeind FG, Abela GS, Franchi L, Nunez G, Schnurr M, Espevik T, Lien E, Fitzgerald KA, Rock KL, Moore KJ, Wright SD, Hornung V, Latz E.. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 2010;464:1357–1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bisgaard LS, Mogensen CK, Rosendahl A, Cucak H, Nielsen LB, Rasmussen SE, Pedersen TX.. Bone marrow-derived and peritoneal macrophages have different inflammatory response to oxLDL and M1/M2 marker expression—implications for atherosclerosis research. Sci Rep 2016;6:35234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Devries-Seimon T, Li Y, Yao PM, Stone E, Wang Y, Davis RJ, Flavell R, Tabas I.. Cholesterol-induced macrophage apoptosis requires ER stress pathways and engagement of the type A scavenger receptor. J Cell Biol 2005;171:61–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Tabas I, Seimon T, Timmins J, Li G, Lim W.. Macrophage apoptosis in advanced atherosclerosis. Ann N Y Acad Sci 2009;1173:E40–E45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Desurmont C, Caillaud JM, Emmanuel F, Benoit P, Fruchart JC, Castro G, Branellec D, Heard JM, Duverger N.. Complete atherosclerosis regression after human ApoE gene transfer in ApoE-deficient/nude mice. Arterioscler Thromb Vasc Biol 2000;20:435–442. [DOI] [PubMed] [Google Scholar]
- 26. Hirshleifer I. Perhexiline maleate in the treatment of angina pectoris. Curr Ther Res Clin Exp 1969;11:99–105. [PubMed] [Google Scholar]
- 27. Ashrafian H, Horowitz JD, Frenneaux MP.. Perhexiline. Cardiovasc Drug Rev 2007;25:76–97. [DOI] [PubMed] [Google Scholar]
- 28. Schwartz K, Lawn RM, Wade DP.. ABC1 gene expression and ApoA-I-mediated cholesterol efflux are regulated by LXR. Biochem Biophys Res Commun 2000;274:794–802. [DOI] [PubMed] [Google Scholar]
- 29. Venkateswaran A, Laffitte BA, Joseph SB, Mak PA, Wilpitz DC, Edwards PA, Tontonoz P.. Control of cellular cholesterol efflux by the nuclear oxysterol receptor LXR alpha. Proc Natl Acad Sci USA 2000;97:12097–12102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Verschuren L, de Vries-van der Weij J, Zadelaar S, Kleemann R, Kooistra T.. LXR agonist suppresses atherosclerotic lesion growth and promotes lesion regression in ApoE3Leiden mice: time course and mechanisms. J Lipid Res 2009;50:301–311. [DOI] [PubMed] [Google Scholar]
- 31. Honzumi S, Shima A, Hiroshima A, Koieyama T, Terasaka N.. Synthetic LXR agonist inhibits the development of atherosclerosis in New Zealand White rabbits. Biochim Biophys Acta 2011;1811:1136–1145. [DOI] [PubMed] [Google Scholar]
- 32. Cha JY, Repa JJ.. The liver X receptor (LXR) and hepatic lipogenesis. The carbohydrate-response element-binding protein is a target gene of LXR. J Biol Chem 2007;282:743–751. [DOI] [PubMed] [Google Scholar]
- 33. Chen X, Li S, Yang Y, Yang X, Liu Y, Liu Y, Hu W, Jin L, Wang X.. Genome-wide association study validation identifies novel loci for atherosclerotic cardiovascular disease. J Thromb Haemost 2012;10:1508–1514. [DOI] [PubMed] [Google Scholar]
- 34. Heinecke JW. The not-so-simple HDL story: a new era for quantifying HDL and cardiovascular risk? Nat Med 2012;18:1346–1347. [DOI] [PubMed] [Google Scholar]
- 35. Bauernfeind FG, Horvath G, Stutz A, Alnemri ES, MacDonald K, Speert D, Fernandes-Alnemri T, Wu J, Monks BG, Fitzgerald KA, Hornung V, Latz E.. Cutting edge: NF-kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J Immunol 2009;183:787–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Barlan AU, Griffin TM, McGuire KA, Wiethoff CM.. Adenovirus membrane penetration activates the NLRP3 inflammasome. J Virol 2011;85:146–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sheedy FJ, Grebe A, Rayner KJ, Kalantari P, Ramkhelawon B, Carpenter SB, Becker CE, Ediriweera HN, Mullick AE, Golenbock DT, Stuart LM, Latz E, Fitzgerald KA, Moore KJ.. CD36 coordinates NLRP3 inflammasome activation by facilitating intracellular nucleation of soluble ligands into particulate ligands in sterile inflammation. Nat Immunol 2013;14:812–820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Yvan-Charvet L, Welch C, Pagler TA, Ranalletta M, Lamkanfi M, Han S, Ishibashi M, Li R, Wang N, Tall AR.. Increased inflammatory gene expression in ABC transporter-deficient macrophages: free cholesterol accumulation, increased signaling via toll-like receptors, and neutrophil infiltration of atherosclerotic lesions. Circulation 2008;118:1837–1847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Westerterp M, Murphy AJ, Wang M, Pagler TA, Vengrenyuk Y, Kappus MS, Gorman DJ, Nagareddy PR, Zhu X, Abramowicz S, Parks JS, Welch C, Fisher EA, Wang N, Yvan-Charvet L, Tall AR.. Deficiency of ATP-binding cassette transporters A1 and G1 in macrophages increases inflammation and accelerates atherosclerosis in mice. Circ Res 2013;112:1456–1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Feingold KR, Grunfeld C.. The acute phase response inhibits reverse cholesterol transport. J Lipid Res 2010;51:682–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C, Fonseca F, Nicolau J, Koenig W, Anker SD, Kastelein JJP, Cornel JH, Pais P, Pella D, Genest J, Cifkova R, Lorenzatti A, Forster T, Kobalava Z, Vida-Simiti L, Flather M, Shimokawa H, Ogawa H, Dellborg M, Rossi PRF, Troquay RPT, Libby P, Glynn RJ, Group CT.. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N Engl J Med 2017;377:1119–1131. [DOI] [PubMed] [Google Scholar]
- 42. Xie W, Li L, Gong D, Zhang M, Lv YC, Guo DM, Zhao ZW, Zheng XL, Zhang DW, Dai XY, Yin WD, Tang CK.. Kruppel-like factor 14 inhibits atherosclerosis via mir-27a-mediated down-regulation of lipoprotein lipase expression in vivo. Atherosclerosis 2019;289:143–161. [DOI] [PubMed] [Google Scholar]
- 43. Wei X, Yang R, Wang C, Jian X, Li L, Liu H, Yang G, Li Z.. A novel role for the Kruppel-like factor 14 on macrophage inflammatory response and atherosclerosis development. Cardiovasc Pathol 2017;27:1–8. [DOI] [PubMed] [Google Scholar]
- 44. Su D. Up-regulation of MiR-145-5p promotes the growth and migration in LPS-treated HUVECs through inducing macrophage polarization to M2. J Recept Signal Transduct Res 2020;40:1–8. [DOI] [PubMed] [Google Scholar]
- 45. Small KS, Todorcevic M, Civelek M, El-Sayed Moustafa JS, Wang X, Simon MM, Fernandez-Tajes J, Mahajan A, Horikoshi M, Hugill A, Glastonbury CA, Quaye L, Neville MJ, Sethi S, Yon M, Pan C, Che N, Vinuela A, Tsai PC, Nag A, Buil A, Thorleifsson G, Raghavan A, Ding Q, Morris AP, Bell JT, Thorsteinsdottir U, Stefansson K, Laakso M, Dahlman I, Arner P, Gloyn AL, Musunuru K, Lusis AJ, Cox RD, Karpe F, McCarthy MI.. Regulatory variants at KLF14 influence type 2 diabetes risk via a female-specific effect on adipocyte size and body composition. Nat Genet 2018;50:572–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Zhang G, Li C, Zhu N, Chen Y, Yu Q, Liu E, Wang R.. Sex differences in the formation of atherosclerosis lesion in ApoE(-/-) mice and the effect of 17beta-estrodiol on protein S-nitrosylation. Biomed Pharmacother 2018;99:1014–1021. [DOI] [PubMed] [Google Scholar]
- 47. Smith DD, Tan X, Tawfik O, Milne G, Stechschulte DJ, Dileepan KN.. Increased aortic atherosclerotic plaque development in female apolipoprotein E-null mice is associated with elevated thromboxane A2 and decreased prostacyclin production. J Physiol Pharmacol 2010;61:309–316. [PMC free article] [PubMed] [Google Scholar]
- 48. Robinet P, Milewicz DM, Cassis LA, Leeper NJ, Lu HS, Smith JD.. Consideration of sex differences in design and reporting of experimental arterial pathology studies—statement from ATVB Council. Arterioscler Thromb Vasc Biol 2018;38:292–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Man JJ, Beckman JA, Jaffe IZ.. Sex as a biological variable in atherosclerosis. Circ Res 2020;126:1297–1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Brown MJ, Horowitz JD, Mashford ML.. A double-blind trial of perhexiline maleate in the prophylaxis of angina pectoris. Med J Aust 1976;1:260–263. [DOI] [PubMed] [Google Scholar]
- 51. Kennedy JA, Unger SA, Horowitz JD.. Inhibition of carnitine palmitoyltransferase-1 in rat heart and liver by perhexiline and amiodarone. Biochem Pharmacol 1996;52:273–280. [DOI] [PubMed] [Google Scholar]
- 52. Lee L, Campbell R, Scheuermann-Freestone M, Taylor R, Gunaruwan P, Williams L, Ashrafian H, Horowitz J, Fraser AG, Clarke K, Frenneaux M.. Metabolic modulation with perhexiline in chronic heart failure: a randomized, controlled trial of short-term use of a novel treatment. Circulation 2005;112:3280–3288. [DOI] [PubMed] [Google Scholar]
- 53. Beadle RM, Williams LK, Kuehl M, Bowater S, Abozguia K, Leyva F, Yousef Z, Wagenmakers AJ, Thies F, Horowitz J, Frenneaux MP.. Improvement in cardiac energetics by perhexiline in heart failure due to dilated cardiomyopathy. JACC Heart Fail 2015;3:202–211. [DOI] [PubMed] [Google Scholar]
- 54. Kennedy JA, Beck-Oldach K, McFadden-Lewis K, Murphy GA, Wong YW, Zhang Y, Horowitz JD.. Effect of the anti-anginal agent, perhexiline, on neutrophil, valvular and vascular superoxide formation. Eur J Pharmacol 2006;531:13–19. [DOI] [PubMed] [Google Scholar]
- 55. Oram JF. Tangier disease and ABCA1. Biochim Biophys Acta 2000;1529:321–330. [DOI] [PubMed] [Google Scholar]
- 56. Geller AS, Polisecki EY, Diffenderfer MR, Asztalos BF, Karathanasis SK, Hegele RA, Schaefer EJ.. Genetic and secondary causes of severe HDL deficiency and cardiovascular disease. J Lipid Res 2018;59:2421–2435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Joseph SB, McKilligin E, Pei L, Watson MA, Collins AR, Laffitte BA, Chen M, Noh G, Goodman J, Hagger GN, Tran J, Tippin TK, Wang X, Lusis AJ, Hsueh WA, Law RE, Collins JL, Willson TM, Tontonoz P.. Synthetic LXR ligand inhibits the development of atherosclerosis in mice. Proc Natl Acad Sci USA 2002;99:7604–7609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Fazio S, Babaev VR, Murray AB, Hasty AH, Carter KJ, Gleaves LA, Atkinson JB, Linton MF.. Increased atherosclerosis in mice reconstituted with apolipoprotein E null macrophages. Proc Natl Acad Sci USA 1997;94:4647–4652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Mazzone T, Reardon C.. Expression of heterologous human apolipoprotein E by J774 macrophages enhances cholesterol efflux to HDL3. J Lipid Res 1994;35:1345–1353. [PubMed] [Google Scholar]
- 60. Clausen BE, Burkhardt C, Reith W, Renkawitz R, Forster I.. Conditional gene targeting in macrophages and granulocytes using LysMcre mice. Transgenic Res 1999;8:265–277. [DOI] [PubMed] [Google Scholar]
- 61. Shan B, Wang X, Wu Y, Xu C, Xia Z, Dai J, Shao M, Zhao F, He S, Yang L, Zhang M, Nan F, Li J, Liu J, Liu J, Jia W, Qiu Y, Song B, Han JJ, Rui L, Duan SZ, Liu Y.. The metabolic ER stress sensor IRE1alpha suppresses alternative activation of macrophages and impairs energy expenditure in obesity. Nat Immunol 2017;18:519–529. [DOI] [PubMed] [Google Scholar]
- 62. Fleming TJ, Fleming ML, Malek TR.. Selective expression of Ly-6G on myeloid lineage cells in mouse bone marrow. RB6-8C5 mAb to granulocyte-differentiation antigen (Gr-1) detects members of the Ly-6 family. J Immunol 1993;151:2399–2408. [PubMed] [Google Scholar]
- 63. Matsushima H, Geng S, Lu R, Okamoto T, Yao Y, Mayuzumi N, Kotol PF, Chojnacki BJ, Miyazaki T, Gallo RL, Takashima A.. Neutrophil differentiation into a unique hybrid population exhibiting dual phenotype and functionality of neutrophils and dendritic cells. Blood 2013;121:1677–1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Drechsler M, Megens RT, van Zandvoort M, Weber C, Soehnlein O.. Hyperlipidemia-triggered neutrophilia promotes early atherosclerosis. Circulation 2010;122:1837–1845. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The expanded Methods section and other supporting data are available in the Supplementary material online. The primary data for this article will be shared on reasonable request to the corresponding authors.