

Abstract
Background
Hereditary spastic paraplegia is a rare familial hereditary neurodegenerative disease caused by multiple autosomal dominant mutations. More than 50 mutant genes have been reported to be associated with this disease.
Methods
In this study, we have reported a rare insertion mutation site in PRRT2 that caused a familial disorder of hereditary spastic paraplegia accompanied by polyneuropathy.
Results
We used second‐generation sequencing of samples of the proband's familial genome and found an insertion mutation of C/CC in NM_001256443:c.641dupC that was localized to the second exon of PRRT2. This functional mutation can cause an amino acid sequence change (arginine >proline) and dysfunctional neuronal transmembrane proteins, which might have been related to the onset of hereditary spastic paraplegia accompanied by polyneuropathy in the family reported in this study.
Conclusion
The discovery of this mutation site provides an important theoretical basis for specific gene‐based diagnosis and treatment of hereditary spastic paraplegia.
Keywords: hereditary spastic paraplegia, insertion mutation, polyneuropathy, PRRT2
1. INTRODUCTION
Hereditary spastic paraplegia (HSP), also known as familial spastic paraplegia, is a rare familial hereditary neurodegenerative disease with a morbidity rate of 1.2–9.6 per 100,000 population. The genetic patterns of this disease include autosomal dominant inheritance, autosomal recessive inheritance, and X‐linked recessive inheritance. 1 Although more than 50 causative genes have been identified, 2 the pathogenesis of HSP remains unclear. The pathological changes in HSP, such as bilateral corticospinal tract axonal degeneration and demyelination, are mainly seen in the spinal cord, particularly in the thoracic spinal cord. HSP is characterized by chronic progressive weakness and spastic lower limb paralysis. Optic atrophy, retinitis pigmentosa, extrapyramidal symptoms, cerebellar ataxia, sensory impairment, dementia, mental retardation, hearing loss, muscle atrophy, and autonomic nerve dysfunction may be present. Independent mutations in multiple genes can cause HSP; thus, the diagnosis and treatment of HSP are challenging. Therefore, further advancements in HSP gene mapping will be beneficial for the diagnosis and development of effective treatments based on genetic characteristics.
In this study, we found a new mutation at the proline‐rich transmembrane protein 2 (PRRT2) locus using second‐generation sequencing. This mutation caused familial HSP with peripheral neuropathy.
2. MATERIALS AND METHODS
2.1. Patients and families
We present a Han Chinese family with HSP who were enrolled in our study, which was conducted at the Department of Neurology, First Affiliated Hospital of Dalian Medical University, China (Figure 1). Peripheral blood samples were collected for our investigation. The study was approved by the Ethics Committee of the First Affiliated Hospital of Dalian Medical University and was performed in accordance with the recommendations of the Declaration of Helsinki. All study participants provided written informed consent.
FIGURE 1.
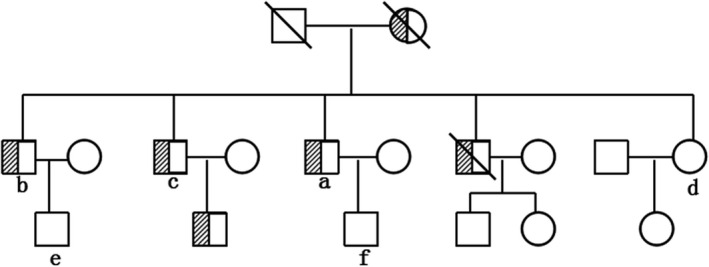
Pedigree of the reported family with HSP. Filled symbols with slashes represent affected family members. Empty symbols represent normal family members
2.2. Mutation analysis
Whole‐exome sequencing was performed on DNA extracted from peripheral blood samples. 3 , 4 , 5 , 6 After fragmenting the genomic DNA, ligating the paired‐end adaptors, amplifying the DNA, and purifying the amplified products, all human exons and the 50 bp regions of their adjacent introns were captured using the xGen Exome Research Panel (Integrated DNA Technologies). The DNA library was constructed following amplification, purification, and capture and then sequenced on a HiSeq sequencing platform (Illumina).
The sequence data were aligned to the hg19 human reference genome. NextGENe v2.3.4 software was used for variant calling and to determine the coverage and mean read depth of the target regions. The mean read depth was 158.56×, reaching at least 20× for 99.5% of the target sequences. We used NextGENe v2.3.4 and the laboratory's own scripts to obtain the annotation information, including the conserved nucleotide bases and amino acids, predictions of the biological functions, and frequency of the normal populations (1000 Genomes Project, ExAC, dbSNP, and locus‐specific databases), as well as data from the Human Genome Mutation Database, ClinVar, and Online Mendelian Inheritance in Man.
The variants were screened as follows: (1) preference to variants related to the diseases, small indels, canonical splice sites, and missense variants; (2) minor allele frequency (MAF) in normal populations <5% (except for known MAFs with ≥5% pathogenicity); (3) preference to variants in Human Genome Mutation Database and ClinVar; and (4) preference to variants in Online Mendelian Inheritance in Man. The pathogenic variants were obtained according to the standards and guidelines for the interpretation of sequence variants published by the American College of Medical Genetics and Genomics in 2015 using Human Genome Variation Society nomenclature. Finally, the identified variants were annotated using the Consensus Coding Sequences Database (20130630) from the National Center for Biotechnology Information. The quality control data parameters for whole‐exome sequencing are presented in Table 1.
TABLE 1.
Quality control data of whole‐exome sequencing
| Total | 5612.64 |
| Raw_data (Mb) | 5464.71 |
| Clean_data (Mb) | 99.8 |
| Aligned (%) | 11395561 |
| Initial bases on target | 11391533 |
| Base covered on target | 100.00% |
| Coverage of target region | 4383.77 |
| Total effective yield (Mb) | 2446.64 |
| Effective sequence on target (Mb) | 55.80% |
| Fraction of effective bases on target | 214.7 |
| Average sequencing depth on target | 99.90% |
| Fraction of target covered with at least 4X | 99.90% |
| Fraction of target covered with at least 10X | 99.80% |
| Fraction of target covered with at least 20X duplication rate (%) | 18.87 |
3. CASE DESCRIPTION
3.1. Case A (proband)
3.1.1. Disease history
The proband was a 54‐year‐old male who had experienced numbness and weakness of the lower limbs (which began with numbness in both feet, particularly the left foot) for the past 10 years. The numbness gradually increased to the base of the thigh, and there was a sense of weakness in both legs. Although the degree of weakness and numbness had increased over the past 10 years, the patient was still able to walk unaided.
3.1.2. Physical examination of the nervous system
The patient was conscious and showed a spastic gait, arcuate feet, fluent speech, and normal intelligence. The cranial nerves were normal. The muscle strength of both upper limbs was scored as 5 and that of the lower limbs was scored as 4. The upper limb muscle tension was normal, and the lower limb muscle tension was enhanced. Deep and superficial sensations were decreased under the hip joints. The upper limb tendons showed overactivity, and the lower limb tendon reflex was absent. The Babinski and Chaddock signs were both bilaterally positive.
3.1.3. Additional examinations
No specific abnormalities were observed in the results of blood biochemistry tests and magnetic resonance imaging (MRI) examination of the cervical and thoracic segments.
3.2. Case B (elder brother)
The elder brother of the proband was 60 years old and had an abnormal gait of both lower limbs for the past 40 years. For the past 25 years, the limb weakness had become aggravated and was accompanied by numbness. Defecation was normal. A physical examination of the nervous system yielded the same findings as that of the proband.
3.3. Case C (second‐eldest brother)
The second eldest brother of the proband was 56 years old and had lower limb weakness without numbness for the past 10 years. Defecation was normal. A physical examination of the nervous system yielded the same findings as that of the proband. The clinical features of the three patients are presented in Table 2.
TABLE 2.
Clinical features of family members carrying the NM_001256443:c.641dupC mutation
| Feature | Patient | ||
|---|---|---|---|
| a | b | c | |
| Age, years | 54 | 60 | 56 |
| Age at onset | 44 | 20 | 46 |
| Spastic gait | + | Wheelchair from last 5 years | + |
| Urinary urgency | − | − | − |
| Scoliosis | − | + | − |
| Upper limbs | |||
| Weakness | − | − | − |
| Sensory loss | − | − | − |
| Lower limbs | |||
| Increased tone | + | + | + |
| Hyperreflexia | + | + | + |
| Weakness | + | + | + |
| Extensor plantar response | + | + | + |
| Sensory loss | + | + | + |
| Decreased vibration sense | + | + | + |
(+), present; (−), absent.
3.4. Family pedigree of HSP
Figure 1 shows the pedigree of this family with HSP. Six members were confirmed to have the same clinical symptoms as the proband. The remaining family members (n = 11 members) were normal.
3.5. Genetic analysis
Genomic DNA was extracted from peripheral blood samples of the family members. An insertion mutation with C/CC in TRRP2 was identified in three family members showing clinical symptoms of HSP. Furthermore, the mutation was not found in three family members with undeveloped disease (Figure 2).
FIGURE 2.
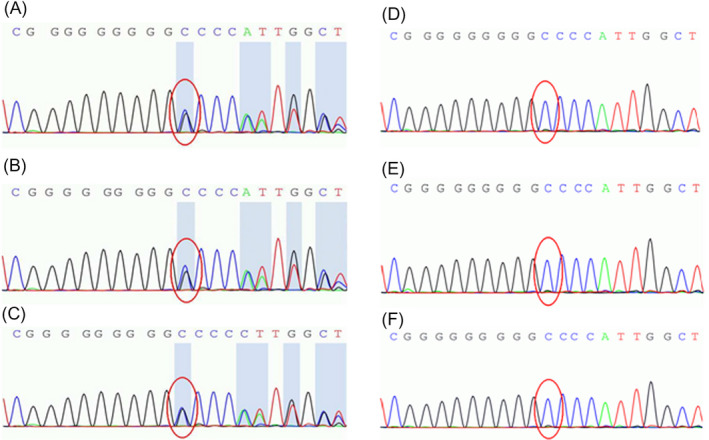
Results of second‐generation sequencing to detect the insertion mutation with C/CC in TRRP2. (A, B, C): C/CC insertion mutation (NM_001256443:c.641dupC) in three family members with clinical symptoms. (D, E, F): No C/CC insertion mutation was found in three family members with undeveloped disease
4. DISCUSSION
Here, we reported a genetic mutation at the PRRT2 locus that caused familial HSP with peripheral neuropathy. The C/CC insertion mutation in the second exon region of NM_001256443:c.641dupC can cause an amino acid sequence change (arginine >proline) that may result in functional alterations of neuronal transmembrane proteins and lead to the onset of HSP in probands and their siblings.
PRRT2 is located at 16p11.2. Although the gene product has been mainly confirmed in the brain and spinal cord tissues, 7 , 8 its specific function remains unclear. Several studies have indicated that PRRT2 is the major pathogenic gene in some paroxysmal diseases of the nervous system, such as paroxysmal kinesigenic dyskinesias (PKDs), benign familial infantile seizures, and infantile convulsions with paroxysmal choreoathetosis. These diseases are often manifested with an infantile and childhood onset that presents with an episodic and benign course, and antiepileptic drugs such as carbamazepine are effective. However, there are no reports of the association between PRRT2 and the incidence of HSP.
In our reported family, three patients with HSP showed heterozygous C/CC insertion mutations at NM_001256443:c.641dupC, whereas three of the asymptomatic family members did not have the insertion mutation. These results were highly suggestive of an association between NM_001256443:c.641dupC of PRRT2 and HSP accompanied by polyneuropathy.
Interestingly, we also found that although the proband's 26‐year‐old son did not show symptoms of HSP, he had experienced paroxysmal movement in the right limbs induced by involuntary jitter since the age of 15. Each seizure lasted no more than 2 min. The electroencephalogram and cranial MRI results were normal. He was administered carbamazepine and phenytoin for 3 years and has not suffered seizures since drug withdrawal. This suggested that he had PKD. Unfortunately, the son refused to undergo genetic analysis. His father (proband) had an insertion mutation in NM_001256443:c.641dupC, and the associated PKD mutation is also believed to be located in the second exon of PRRT2. Based on HapMap data, the two mutation sites in the genes of individuals of the Han community from Asia are interlinked; hence, it cannot be excluded that PKD may be induced by an NM_001256443:c.641dupC linkage mutation.
Paroxysmal kinesigenic dyskinesias mainly occurs in children and adolescents, with a male to female ratio of 4:1 and a benign course. The frequency of seizures gradually decreases with age. The clinical characteristic of PKD is transient dyskinesia induced by a sudden change in motion state. Unilateral and bilateral injuries can be involved along with bilateral alternating or simultaneous attacks. Tiredness of the facial and jaw muscles could result in dysarthria. Seizures usually last for less than 1 min, and the patient remains conscious during the course of the attack. Electroencephalogram and cranial MRI results are usually normal. 9 , 10 Antiepileptic drugs, especially carbamazepine, can effectively control seizures. 11
In summary, the insertion mutation found in PRRT2 may provide critical information for the gene and protein analysis of HSP accompanied by polyneuropathy. This will provide an important theoretical basis for the specific gene‐based diagnosis and treatment of HSP.
CONFLICT OF INTERESTS
The present study does not have any conflict of interest.
ACKNOWLEDGMENTS
The authors thank the patient’s family for their participation.
Wang and Dong contributed equally to this work and should be considered as equal first coauthors.
DATA AVAILABILITY STATEMENT
Due to the nature of this research, the study participants did not agree that their data could be publicly shared, so supporting data are unavailable.
REFERENCES
- 1. Bruno MK, Hallett M, Gwinn‐Hardy K, et al. Clinical evaluation of idiopathic paroxysmal kinesigenic dyskinesia: new diagnostic criteria. Neurology. 2004;63(12):2280‐2287. [DOI] [PubMed] [Google Scholar]
- 2. Finsterer J, Löscher W, Quasthoff S, Wanschitz J, Auer‐Grumbach M, Stevanin G. Hereditary spastic paraplegias with autosomal dominant, recessive, X‐linked, or maternal trait of inheritance. J Neurol Sci. 2012;318(1–2):1‐18. [DOI] [PubMed] [Google Scholar]
- 3. Zhang R, Chen S, Han P, et al. Whole exome sequencing identified a homozygous novel variant in CEP290 gene causes Meckel syndrome. J Cell Mol Med. 2020;24(2):1906‐1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Dai Y, Liang S, Dong X, et al. Whole exome sequencing identified a novel DAG1 mutation in a patient with rare, mild and late age of onset muscular dystrophy‐dystroglycanopathy. J Cell Mol Med. 2019;23(2):811‐818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zheng Y, Xu J, Liang S, Lin D, Banerjee S. Whole exome sequencing identified a novel heterozygous mutation in HMBS gene in a Chinese patient with acute intermittent porphyria with rare type of mild anemia. Front Genet. 2018;20(9):129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Han P, Wei G, Cai K, et al. Identification and functional characterization of mutations in LPL gene causing severe hypertriglyceridaemia and acute pancreatitis. J Cell Mol Med. 2020;24(2):1286‐1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Guo XN, Lu Q, Zhou XQ, Liu Q, Zhang X, Cui LY. Re‐evaluation of PRRT2 mutations in paroxysmal disorders. J Neurol. 2014;261(5):951‐953. [DOI] [PubMed] [Google Scholar]
- 8. Heron SE, Grinton BE, Kivity S, et al. PRRT2 mutations cause benign familial infantile epilepsy and infantile convulsions with choreoathetosis syndrome. Am J Hum Genet. 2012;90(1):152‐160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Li HF, Chen WJ, Ni W, et al. PRRT2 mutation correlated with phenotype of paroxysmal kinesigenic dyskinesia and drug response. Neurology. 2013;80(16):1534‐1535. [DOI] [PubMed] [Google Scholar]
- 10. Schlipf NA, Schüle R, Klimpe S, et al. Amplicon‐based high‐throughput pooled sequencing identifies mutations in CYP7B1 and SPG7 in sporadic spastic paraplegia patients. Clin Genet. 2011;80(2):148‐160. [DOI] [PubMed] [Google Scholar]
- 11. Silveira‐Moriyama L, Gardiner AR, Meyer E, et al. Clinical features of childhood‐onset paroxysmal kinesigenic dyskinesia with PRRT2 gene mutations. Dev Med Child Neurol. 2013;55(4):327‐334. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Due to the nature of this research, the study participants did not agree that their data could be publicly shared, so supporting data are unavailable.