

Abstract
Gene expression and genetic regulatory networks in multi-cellular organisms control complex physiological processes ranging from cellular differentiation to development to aging. Traditional methods to investigate gene expression relationships rely on using bulk, pooled-population assays (e.g. RNA-sequencing and RT-PCR) to compare gene expression levels in hypo- or hyper-morphic mutant animals (e.g. gain-of-function or knockout). This approach is limited, especially in complex gene networks, as these genetic mutations may affect the expressions of related genes in unforseen ways. In contrast, we developed a microfluidic-based pipeline to discover gene relationships in a single genetic background. The microfluidic device provides efficient reagent exchange and the ability to track individual animals. By automating a robust microfluidic reagent exchange strategy, we adapted and validated single molecule fluorescent in situ hybridization (smFISH) on-chip and combined this technology with live-imaging of fluorescent transcriptional reporters. Together, this multi-level information enabled us to quantify a gene expression relationship with single-animal resolution. While this microfluidic-based pipeline is optimized for live-imaging and smFISH C. elegans studies, the strategy is highly-adaptable to other biological models as well as combining other live and end-point biological assays, such as behavior-based toxicology screening and immunohistochemistry.
Introduction
Gene expression and gene regulation play pivotal roles in all living organisms. Ranging from cellular differentiation to development to aging, gene expression presents unique profiles at each stage.1–3 In addition, quantifying gene expression can reveal the intra-populational biological variability.4–6 For instance, even within isogenic populations, organisms live to be different ages due to the natural stochasticity of gene expression.7, 8 The expression profiles of certain genes, such as for some individual microRNAs, have even been identified as early-life predictors of longevity.7 Gene expression becomes much more complex when considering gene networks where one gene’s expression may influence another’s. For example, genes from the conserved TGFβ and serotonin pathways have a dynamic interaction where one gene regulates the gene-expression variability of the systems while the other regulates the dynamic range of the gene-expression responses.9 These gene interaction networks can even act across multiple tissues. For instance, investigations using the roundworm, Caenorhabditis elegans, have discovered a food-sensing gene that is typically expressed in one pair of neurons; however, when presented with pathogenic bacteria, its expression shifts to other neurons.10 These explorations have sparked many questions such as, how do specific tissues have unique gene expressions? Are some tissues’ expression more influential to the organism? What types of relationships are present in these gene networks? Addressing these questions requires methods that can measure gene expression with tissue-specificity and inter-individuality in the context of a whole organism.
Common methods to study population-level expression profiles in model organisms, such as C. elegans, Drosophila, and zebrafish, include RNA-sequencing (RNA-seq), real-time quantitative reverse transcription PCR (qRT-PCR), and cDNA microarrays. These studies provide the largest datasets and are able to capture the entire transcriptome. However, importantly, they rely on extracting mRNA from large populations of pooled animals, losing the identities of individual samples. Even in single-cell RNA-seq, individual cells’ expression profiles are used to only predict its original tissue, and the animal identity is lost.11, 12 While there have been advancements to adapt these bulk techniques to single animals, such as qRT-PCR in single worms13, they still lose cellular resolution and are labor-intensive, which can limit large population studies.
With techniques such as fluorescent transcriptional reporters and single molecule fluorescent in situ hybridization (smFISH), imaging and studying gene expression within individuals has become much more accessible. Fluorescent transcriptional reporters are often used in small model organisms to quantify protein expression with tissue-specificity. Here, animals are genetically modified to express a fluorescent marker under the same regulatory control of a gene of interest; typically, the promoter region of the gene is used to drive the expression of a fluorescent protein.14 Although not an exact measurement of the target protein, live imaging of these animal strains can reveal the relative expression levels with tissue-specificity.15 Further, for transparent organisms such as C. elegans, imaging can be done in live animals, and thus the experiment can be coupled with other manipulations, such as environmental perturbations. To reflect meaningful promoter activity and measure the subtleties of gene expression, transcriptional reporters need to be single copy integrants. One drawback for this technique is that creating new reporter strains is difficult, time-consuming, and can often be a bottleneck for large-scale studies on many genes or genetic networks simultaneously. Another disadvantage is the typical long half-life of fluorophores (~24 hours)16, 17, which can limit the temporal resolution of studies with dynamic phenotypes.
An orthogonal approach to study gene expression in situ within a whole organism, at the single-organism level, is smFISH. In this process, gene-specific, fluorescently labeled probes are delivered into a formaldehyde-fixed and ethanol-permeabilized sample, and these probes hybridize to the target mRNA molecules.18, 19 Each probe set consists of 20–40 fluorescently labeled short nucleic acids with different sequences that bind specifically to the target mRNA. A fluorescent punctum only becomes resolvable when 20–30 probes hybridize to the same mRNA molecule; this strategy ensures specificity, and the sequence-specific probes prevent false-positive signals from non-specific binding.18, 20 Through fluorescent microscopy, we are able to image these hybridized probes as individual puncta, corresponding to individual target mRNA molecules, which enables us to quantify and localize a specific gene’s expression. In contrast to transcriptional reporters, smFISH investigates mRNA expression, which is innately different than protein expression. An advantage of the technique over transgenic approaches, such as transcriptional reporters, is that smFISH can be performed on native samples without genetic modification, and thus is more generalizable. Using new smFISH multiplexing and barcoding techniques, some have been able to characterize gene expression on a transcriptome-level in mounted cells and even thin tissue slices.21, 22 However, these multiplexing techniques have not been achieved in whole animals. In addition, for subcellular features such as the axonal process or synapse of a neuron, localization of the smFISH puncta can be difficult; here, fluorescent transcriptional reporters are more appropriate as these smaller features are easily resolvable in live animals. Along with long incubation times (i.e. days) and multiple manual reagent exchanges, which makes keeping inter-individual identities extremely challenging in large populations, a major limitation of smFISH is its need for fixed samples; thus, smFISH can only be used as an endpoint assay.
Live-imaging of transcriptional reporters provides a real-time, relative gene expression readout, while smFISH can efficiently profile a snapshot of the animal’s true mRNA expression but only at an endpoint. To study complex gene networks in multiple tissues with high spatial resolution in many individuals, we present a platform that combines the advantages of live-imaging and smFISH in the same animals. In this work, we engineered a microfluidic-based pipeline to capture large populations of C. elegans in individual, trackable traps and investigate multi-level gene expression (i.e. protein and mRNA) (Fig. 1). Using this microfluidic platform, we can robustly load and isolate individual animals and effectively deliver different reagents over multiple cycles; the ability to track an individual over multiple steps allows us to relate multiple gene expressions and uncover their relationship. We automated and characterized the on-chip reagent exchange, validating that the smFISH process and reagent transport with our on-chip method is more robust and much faster than the off-chip approach. Current methods to study gene expression relationships rely on multiple strains of animals with knockout or gain-of-function mutations to examine how these influence each other. Here, we use our microfluidic platform to image a transcriptional reporter and then perform smFISH for another gene on the same population of animals while tracking individuality. By relating their expressions as single animals, it enabled us to discover a relationship between these genes in a single genetic background. Furthermore, this platform is highly adaptable to study other genes as well as introduce multiplexed approaches by using spectrally distinct smFISH probes for multiple genes.
Fig. 1.
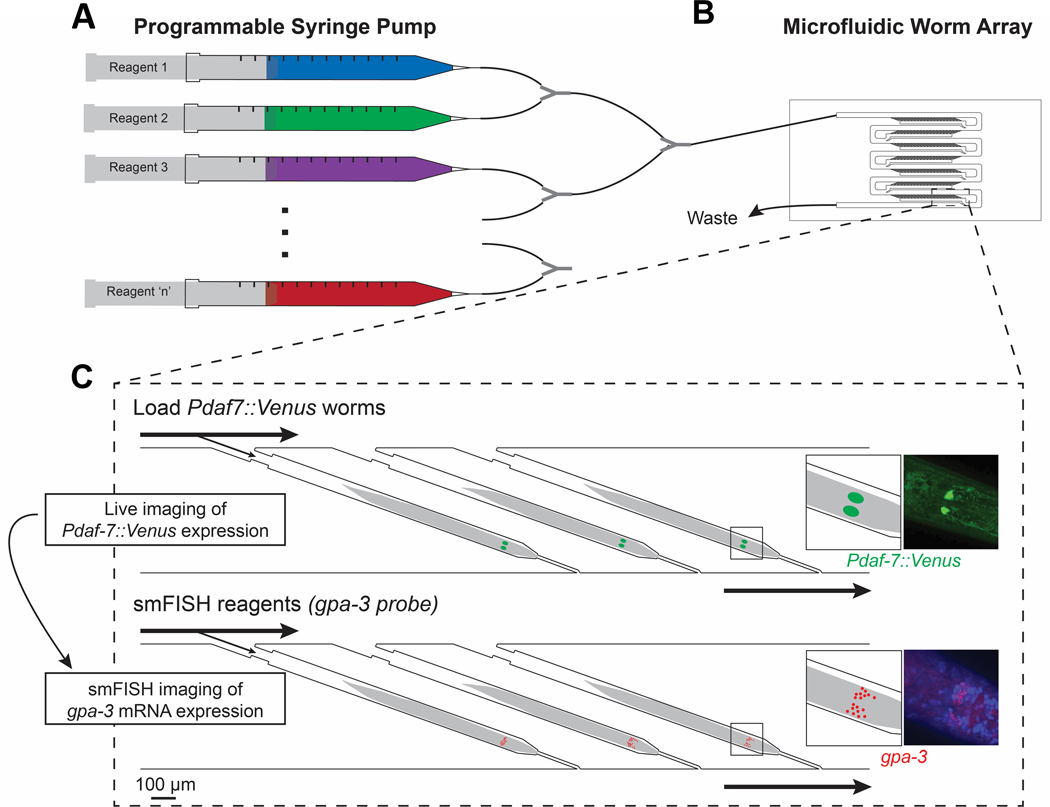
Schematic and overview of microfluidic-based pipeline. (A) Programmable syringe pump can consistently deliver multiple reagents over multiple cycles. (B) Microfluidic device has individual, trackable traps that can efficiently load animals and consistently deliver reagents. (C) Individual worms can be tracked over multiple steps to obtain multi-level gene expression information. Here Pdaf-7::Venus worms are imaged for daf-7 expression and then smFISH is performed to quantify gpa-3 mRNA counts.
Materials and methods
C. elegans strains and culture
The C. elegans strains used in this work were N2 strain (QLN2) and Pdaf-7::Venus strain (QL89). The daf-7 reporter, QL89, was previously constructed and validated (drcSI7[pdaf-7::Venus:unc-119] II; unc-119 (ed3) III).9 The N2 strain (wildtype) was used in the pipeline characterization and smFISH validation. The daf-7 reporter was used for all experiments with live imaging. All worms were grown following standard protocols on NGM agar plates with Escherichia coli (E. coli) OP50 lawns and maintained at 20 °C. For all studies, hermaphrodite worms were synchronized to Day 1 adults.
Microfluidic device fabrication
Microfluidic device fabrication has been previously described.23 Briefly, the master mold of the microfluidic device was fabricated with SU-8 2050 (Microchem), a negative photoresist, by UV photolithography. The microfluidic devices were fabricated in polydimethylsiloxane (PDMS, Dow Corning Sylgard 184) by traditional soft lithography. Prior to the micromolding process, the surfaces of the master mold wafers were treated with tridecafluoro-1,1,2,2-tetrahydrooctyl-1-trichlorosilane vapor (Sigma-Aldrich). A uniform layer of PDMS (10:1 ratio between the elastomer and curing agent) was added on the master mold to a height of ~5 mm. The PDMS was cured at 70 °C overnight (~16 hours) and peeled off. We found that the exact ratio between PDMS components (i.e. 10:1 rather than a higher ratio like 15:1) and the long curing time is crucial to prevent the device from swelling during the overnight ethanol incubation step in the smFISH process. Devices were cut into shapes, two holes (one inlet and one outlet) were punched with 19-gauge needles (McMaster-Carr) per device, and each device was bonded to a cover glass by plasma bonding.
Device Operation
For each experiment, the microfluidic device was first filled with S-Basal solution to remove air bubbles. The worms cultured on NGM agar plates were washed off using S-Basal solution and transferred into a 15 mL tube. After allowing the animals to settle to the bottom of the tube, the supernatant was removed. Animals were loaded into a syringe and delivered into the device manually using a flow rate of ~3 mL/hr for 1 minute, as described in the previous literature.23 After the animals were loaded into the microfluidic device, reagents were delivered using a programmable syringe pump. For device characterization, we flowed alternating cycles of fluorescein isothiocyanate (FITC)-Dextran at 150 μL/min. For all smFISH experiments, we flowed in the new reagent at 15 μL/min for 3 mins then 10 μL/hr for the various incubation times necessary. These reagents can be found below.
Single molecule fluorescent in situ hybridization
Probe design and the protocol to perform smFISH in C. elegans have been previously described.18 In this study, we ordered custom Stellaris smFISH probes for targeting gpa-3 labeled with Cal Fluor Red 610. For each reagent exchange done off-chip, animals were gently spun down for 10 s using a tabletop centrifuge, the supernatant was removed, and the new reagent was added. For each reagent exchange done on-chip, we flowed in the new reagent at 15 μL/min for 3 mins then 10 μL/hr for the various incubation times necessary.
All reagents for on- and off-chip experiments were the same. For all N2 experiments, animals were fixed off-chip; for all Pdaf-7::Venus (QL89) experiments, animals were fixed on-chip. For fixation, animals were exposed to 3.7% formaldehyde for 45 minutes. After fixation, they were washed and incubated in 70% ethanol to permeabilize overnight at 4 °C. For smFISH probe hybridization, animals were washed in wash buffer (10% formamide, 2X SSC, in nuclease-free water) for 5 minutes. Next, they were incubated in a solution of the custom probes (1.25 μM) in hybridization buffer (1 g dextran sulfate, 40 μL RNase-free BSA (50 mg/mL), 10% formamide, 9 mL nuclease-free water) at 30 °C overnight. Samples were washed with wash buffer for 30 mins then DAPI stain (5 ng/mL) for 30 mins. Prior to imaging, animals were washed with or suspended in GLOX antifade solution.
Imaging, image processing, and analysis
For device characterization we used FITC-Dextran (1 mg/mL) in S-Basal. The molecular transport was recorded with whole field illumination on a fluorescence dissecting scope (Leica, MZ16F). We quantified the dynamics and intensities using a custom MATLAB code. The plots and statistics were created and performed using GraphPad Prism and MATLAB.
For all other imaging experiments, we used a spinning disk confocal microscope (PerkinElmer UltraVIEW VoX) equipped with a Hamamatsu FLASH 4 sCMOS camera and a 60x oil immersion objective. All smFISH images were analyzed using a standard software, FISH-quant24, to identify and count the puncta. In order to have accurate comparisons between the on- and off-chip results, we matched our sample size for each experiment. For the experiments using the Pdaf-7::Venus strain, animals were loaded into the device and washed with 50 mM tetramisole for immobilization. Animals were immediately fixed for smFISH after live imaging. To quantify the daf-7 expression, we used a custom MATLAB code to draw an ROI that encompasses the two ASI cell bodies in a maximum projection and quantifies the average fluorescence intensity after average background subtraction. In order to have consistent quantification, we only included worms with the correct orientation where the ASI neurons did not overlap. The plots and statistics were created and performed using GraphPad Prism and MATLAB.
Results and discussion
Experimental design of the microfluidic-based pipeline
To collect multi-level gene expression information from the same worm in a large population of worms, we developed a pipeline based on a microfluidic array device23; this protocol loads animals individually into separate traps and delivers reagents driven by a programmable syringe pump (Fig. 1A,B). By retaining inter-individuality, this platform enabled us to study multiple pieces of information from single worms. Here, we first completed live imaging of Pdaf-7::Venus animals to measure daf-7 gene expression as an example of a live phenotype. We then fixed the same worms and performed smFISH to measure gpa-3 expression (Fig. 1C). This experiment required many complex reagent exchanges, which would have been difficult to perform on the laboratory-scale with conventional tools: (1) live animals needed to be immobilized in tetramisole for in vivo fluorescence microscopy, (2) after in vivo imaging, animals must be quickly fixed in formaldehyde to ensure preservation of the transcriptional states in each cell, (3) animals were permeabilized in ethanol overnight, (4) animals were washed in wash buffer, (5) smFISH probes were delivered overnight, (6) excess probes were washed with wash buffer, (7) DAPI stain was added, and (8) anti-fade solution was delivered for smFISH imaging. Each step added more complexity, which makes tracking an individual animal off-chip more challenging. With our pipeline, we could obtain this rich, individual dataset to study the relationship between the two genes; in this paper, we investigated the relationship between daf-7 and gpa-3. Further, this approach is generalizable to multiple live reporters and smFISH probes as well as other microfluidic array-based platforms.
Characterization of reagent delivery into individual animals using the microfluidic pipeline
Along with many other end-point assays, smFISH required multiple reagent exchange steps. Since there is natural variability between isogenic animals, consistent reagent delivery across different animals was essential to minimize experimental noise and have accurate quantification of gene expression. Thus, it was important for us to characterize the device and its reagent transport. As a model for the smFISH fluorescent probes, we used fluorescein isothiocyanate (FITC)-Dextran. This reagent has a similar molecular weight and charge to the smFISH probes, providing us with a visual, fluorescent representation of the transport. To quantify the extent and the dynamics of the reagent exchange on chip, we measured FITC intensity in different areas on the chip over multiple cycles of reagent exchange (Fig. 2). By design of the device, the worms are loaded and trapped by the narrow width of the resistance channel at the end of each trap. The channel is high enough to leave space above the worms for sufficient fluidic exchange, even when the worm is properly loaded. Each cycle of exchange was 120s of FITC-Dextran and 60s of S Basal, a nonfluorescent buffer. We quantified both the intensity in the fluidic channels (characterizing fluid delivery) and the intensity on the worm (characterizing the reagent delivery into the worm). We showed that the reagent exchanges over multiple cycles was efficient and had consistent dynamics across all traps (Fig. 2A). The time between starting reagent delivery and its transport into the worm traps was consistent as well as its dynamics. Further, over the multiple exchanges of FITC-Dextran, the exposure to the fluorescent molecules led to its delivery and accumulation into the fixed and permeabilized animals on-chip (Fig. 2B). This implied that the smFISH probes would also be transported into the organisms, further demonstrating that reagent delivery into the whole organisms was successfully achieved. We then compared reagent delivery efficiency with the traditional off-chip technique.
Fig. 2.
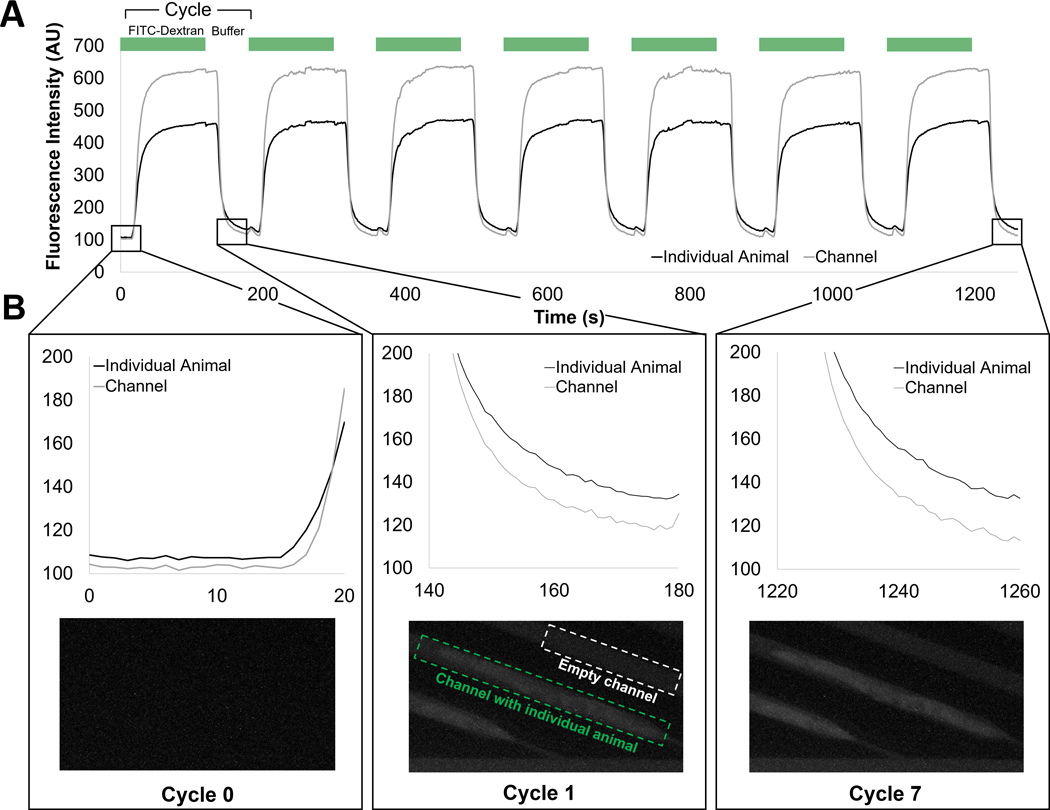
Multiple cycles of reagent delivery can be consistently delivered using the microfluidic platform. (A) Fluorescence intensity of the individual animal and the reference empty channel directly preceding the worm. Cycles of FITC-Dextran and non-fluorescent buffer are exchanged while tracking the same traps. (B) FITC-Dextran is accumulating in the fixed and permeabilized worms over prolonged exposure to the fluorescent macromolecules.
After the fixation and permeabilization steps of smFISH, the worm became a hydrated nanoporous matrix. The reagent transport in the traditional off-chip approach is dominated by diffusion.25 To enhance the transport into other biological systems, vortex shakers can be used; however, this treatment introduces harsh shear that may damage the fragile worms’ tissues. In contrast, gentle flow on the microfluidic chip introduced convection, enhancing the transport of reagents into the worm. We hypothesized that reagent delivery was more consistent and much faster than the off-chip approach (Fig. 3A). When quantifying the amount of FITC-Dextran delivered into the worm after 10 mins of exposure to the fluorescent FITC-Dextran solution, the on-chip worms were significantly brighter than when using the off-chip protocol (Fig. 3B). This indicated that more of the fluorescent reagent was transported into the worm in the same time frame. To characterize the kinetics of the reagent delivery, we quantified the dynamics of reagent delivery over time and fit a one phase association curve to each trial, allowing us to calculate the transport time constants, τ (Fig. 3C,D), to compare the speeds of delivery. We found not only that the on-chip approach had shorter time constants on-chip, which corresponded to faster delivery, but also, they were much more consistent and repeatable when compared to off-chip (Fig. 3C,D). This consistency was crucial for smFISH in order to minimize any experimental variability and capture true biological variability. Overall, we found the dynamics and delivery of reagents were more efficient on-chip than off-chip, supporting that our flow-enhanced reagent exchange was more effective than diffusion.
Fig. 3.
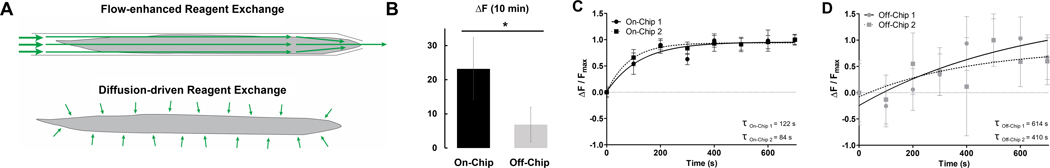
The microfluidic-based platform enhances reagent exchange by introducing convection. (A) On-chip reagent exchange is flow-enhanced. Off-chip reagent exchange is slower as it is diffusion-dominated. (B) After 10 minutes of exposure to FITC-Dextran, worms on-chip were significantly brighter than worms incubated off-chip. (C) On-chip dynamics of FITC-Dextran accumulation is much faster and more consistent than (D) off-chip dynamics. Error bars represent standard deviation. An unpaired T-test was performed. (*P < 0.05)
Adapting smFISH in whole organisms on-chip
We next aimed to demonstrate the utility of the device, using smFISH as an example of an end-point assay. smFISH in whole organisms requires multiple reagents to be delivered over multiple days. By using our microfluidic-based platform, we eliminated the need for difficult manual handling of the samples. Our device also allowed us to maintain individuality from loading the device to performing smFISH. Qualitatively, we were able to validate our process by visualizing individual smFISH puncta for the neuronally-expressed gene, gpa-3 (Fig. 4A). With DAPI staining, we were also able to localize the expression to individual cells in neurons and interneurons, with most of the expression concentrated in the nerve ring of the animals (Fig. 4A). Along with this visual verification, we validated the quantitative smFISH results (i.e. number of puncta or mRNA molecules).
Fig. 4.
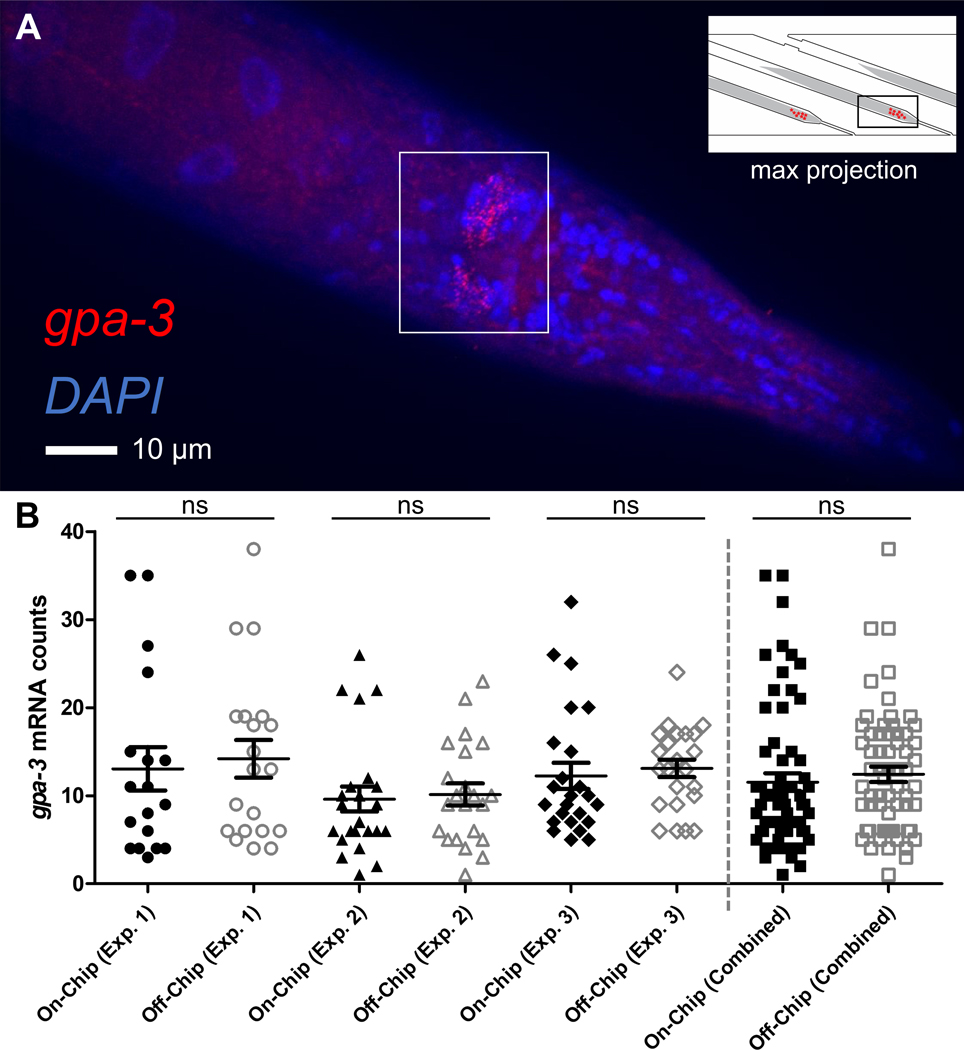
smFISH on whole animals using the microfluidic-based pipeline. (A) Representative maximum projection image of an individual animal after smFISH. Red puncta represent individual gpa-3 mRNA molecules; DAPI staining for nuclei is in blue. Puncta are concentrated on the nerve ring of the animal. (B) Quantification of the puncta is consistent between on- and off-chip controls, repeated in 3 separate experiments. Each experiment was a different cohort of animals while each on- and off-chip comparison was from the same population. Error bars represent standard error mean. The Mann-Whitney U test was performed. (ns, not significant)
To do so, groups of worms that were cultured together were separated for smFISH on- and off-chip over multiple experiments. Three independent biological and technical repeats were performed. We used FISH-quant, a standard MATLAB program developed by Mueller et. al, to quantify the mRNA counts.24 We found that the mRNA counts were consistent between the on- and off-chip conditions (Fig. 4B). We also found that the standard error of the mean was similar between our on- and off-chip conditions (Fig. S1), suggesting that the measured biological noise was similar between the two conditions, and our on-chip protocol did not introduce significant experimental variability. In this example, we investigated gpa-3, a gene expressed in the neurons and inter-neurons of the animal.10 Overall, these experiments validated that our smFISH protocol was reliable and compatible on-chip, did not bias the population, and was properly optimized for future investigations.
Measuring multiple gene expressions within individual organisms
We next sought to highlight the power of our microfluidic platform’s ability to maintain individuality by examining the relationship between two food-sensing genes, daf-7 and gpa-3. daf-7 expression has been characterized to be responsive to bacterial food levels. In comparison to well-fed worms, daf-7 expression in starved populations was reduced. DAF-7 functions in many diverse pathways, and its controlled expression is particularly important for organismal development, physiology, and longevity.26–28 gpa-3 encodes for a G protein α subunit and has olfactory receptor binding activity. Its function is primarily in chemosensation (e.g. chemoattraction and chemoaversion).29–31 There was evidence from mutant-based studies that gpa-3 acts upstream of daf-7, but the overall relationship is unclear: one study used qRT-PCR to find that gpa-3 negatively regulates daf-732, while another used a daf-7 fluorescent transcriptional reporter strain and smFISH for daf-7 to conclude that gpa-3 positively regulates daf-7 in the presence of pathogenic bacteria.10 These traditional approaches to study multiple genes used mutant strains (e.g. gain-of-function and knockout). This allowed researchers to study how expressions of one gene (regulated by environmental factors or otherwise) can influence the expression of another gene.
In our microfluidic-based approach, we were able to capture and quantify multiple pieces of information within the same animal, eliminating the reliance on population-based mutant studies. In this demonstration, we loaded a population of Pdaf-7::Venus transcriptional reporter strains and imaged them to measure the activity of the daf-7 promoter. Immediately after live imaging, we delivered fixation and smFISH reagents to the same samples within an hour of imaging. This fixation process crosslinked biomolecules, including the native mRNA, to its surrounding tissues. A second round of imaging these worms allowed us to quantify the gpa-3 mRNA counts using smFISH. Since the physiologically relevant time scale for significant gpa-3 mRNA change, Pdaf-7::Venus fluorescence intensity change, and the Venus fluorophore’s turnover rate (i.e. days)17, 33 was much longer than our live imaging and fixation time scale (1–2 hours), considered both measurements as a single time point.
Our microfluidic device retained the individuality at each step, enabling us to measure the daf-7 and gpa-3 expression in each animal (Fig. 5A). By comparing the gene expression levels in individuals of the whole population, we found a weak-to-moderate positive correlation (R = 0.39) (Fig. 5A). daf-7 plays important roles in many biological processes, and it receives inputs from additional genes including gpa-3. We also observed that the natural variability in daf-7 expression increases with higher gpa-3 expression, where worms with medium- and high-expression of gpa-3 tend to have more stochasticity in their daf-7 expression (Fig. S2). This was expected as an increase of gpa-3 expression led to a stronger, competing input to daf-7, which added complexity to its expression.
Fig. 5.
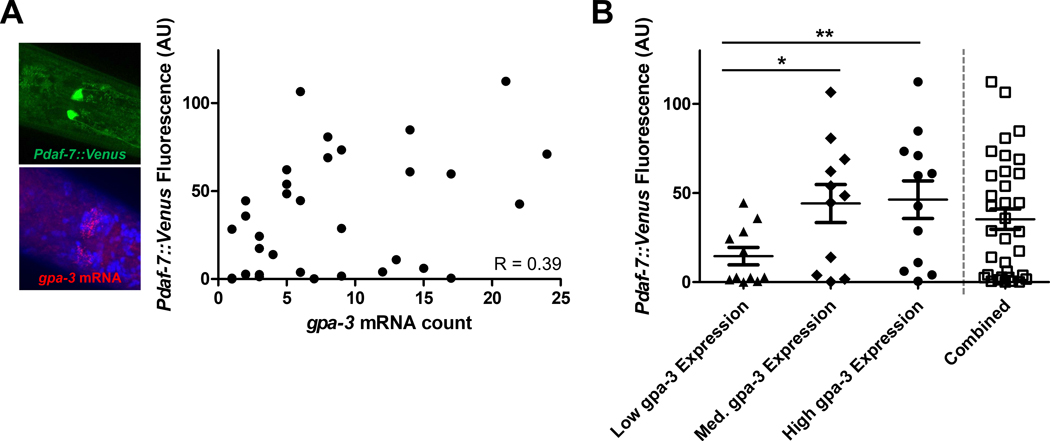
Multi-level gene expression quantification within individual animals. (A) Each point represents an individual animal with its daf-7 and gpa-3 expression, Pdaf-7::Venus fluorescence and gpa-3 mRNA count respectively. R = 0.39, indicating a low-to-moderate positive correlation. (B) Population binned by gpa-3 expression reveals a positive relationship. Higher gpa-3 expression leads to higher daf-7 expression levels. Error bars represent standard error mean. The Mann-Whitney U test was performed. (**P < 0.01; *P < 0.05)
Our approach allowed us to study the biological expression distributions of gpa-3 and daf-7 in detail with single-animal resolution. Since gpa-3 acts upstream of daf-7, our platform enabled us to quantify the resulting daf-7 expression of the natural stochasticity of gpa-3 expression. By separating and binning the individuals based on their gpa-3 expression, we found that the populations also had the same positive trend where higher gpa-3 expression led to significantly higher daf-7 expression (Fig. 5B), and this conclusion was robust to the exact binning (Fig. S3). This type of binning allowed us to recapitulate the traditional methods using mutants: a knockout or loss-of-function mutant (low gpa-3 expression), a wildtype condition (medium gpa-3 expression), and an overexpressing or gain-of-function mutant (high gpa-3 expression).
Using mutants, previous studies suggest that the relationship between gpa-3 and daf-7 is complex.10, 32 It was possible that the subtle nuances and natural variability of the relationship between this pair of genes were masked in the traditional off-chip population-based comparisons, while our method can preserve such information. Further, mutants may affect the expressions of related genes in unforeseen ways. Here, we presented a complementary approach to traditional mutant-based studies. Our methods measure gene expression and the gene-gene relationship in a single, native genetic background. When comparing the binned populations (Fig. 5B), we could see these trends emerge where higher gpa-3 expression correlated with higher daf-7 expression. These trends would have been less obvious when considering whole population averages, similar to our measurements in Figure 5A. Our gene-gene correlation result in single worms suggested that gpa-3 may play indirect roles in via daf-7 in processes, such as development and longevity.
Conclusions
In this study, we engineered a novel microfluidic-based pipeline and approach that enables us to discover gene expression relationships by retaining the inter-individuality of whole organisms throughout multiple bioassays. By automating reagent exchanges on-chip, we were able to perform complex biological assays robustly, repeatedly, and significantly faster than traditional, diffusion-dominated off-chip approaches. We demonstrated the biological utility of our platform by adapting and performing smFISH. Next, we applied our microfluidic-based approach and discovered the relationship between two food-sensing genes, gpa-3 and daf-7. By quantifying both gene distributions, we could examine how the natural variability of gpa-3 correlated with the stochastic daf-7 expression within individuals. While the traditional off-chip approach relied on the use of functional mutants, our microfluidic platform enabled us to preserve the native gene expression networks, presenting a new complementary technique to study gene expression relationships in a single genetic background.
In this study, we demonstrated an example of how coupling live imaging of a fluorescent transcriptional reporter with an endpoint assay, smFISH, can result in multi-dimensional data. This strategy is readily adaptable to other existing microfluidic devices, such as Drosophila embryo traps to study embryogenesis34 and arrays for high-throughput single-cell analysis35, to couple other dynamic live-imaging phenotypes to gene expression analysis. Additionally, live imaging does not have to be limited to fluorescent imaging; one possible example is first quantifying the behavior in a population of C. elegans using a microfluidic chamber array36 and then performing smFISH in the same array to identify genes related to the behavioral phenotype. Further, many genomic-based studies require non-transgenic animals, such as genome-wide association studies, which also makes smFISH an attractive method. This integrated method can also be applied to other popular small model organisms, such as tunicate, Hydra, Drosophila, and other roundworms, where one can investigate gene expression in a natural population and couple it to live phenotype.
In summary, we established a generalizable pipeline to investigate and quantify gene expression relationships. In this field alone, this pipeline could be adapted for future studies such as quantifying development- or age-related changes of gene expression, measuring the weights of expression relationships, or even studying how external perturbations impact gene expressions (e.g. toxicology screening). This tool presents a new opportunity to help uncover gene networks, pushing us one step closer to understanding how these networks arise and revealing any underlying mechanisms that govern living systems.
Supplementary Material
Acknowledgements
We would like to acknowledge the National Institutes of Health (R01AG056436 and R01GM088333) and the U.S. National Science Foundation (1707401). J.W. would like to acknowledge his support from the American Federation for Aging Research and the National Science Foundation Graduate Research Fellowships Program (DGE-1650044). J.D. would like to acknowledge her support from NSF REU Program (OCE-1559923).
Footnotes
Conflicts of interest
There are no conflicts to declare.
References
- 1.Tomancak P, Beaton A, Weiszmann R, Kwan E, Shu S, Lewis SE, Richards S, Ashburner M, Hartenstein V, Celniker SE and Rubin GM, Genome Biol., 2002, 3, research0088.0081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wang J. and Kim SK, Development, 2003, 130, 1621–1634. [DOI] [PubMed] [Google Scholar]
- 3.Lund J, Tedesco P, Duke K, Wang J, Kim SK and Johnson TE, Curr. Biol, 2002, 12, 1566–1573. [DOI] [PubMed] [Google Scholar]
- 4.Elowitz MB, Levine AJ, Siggia ED and Swain PS, Science, 2002, 297, 1183. [DOI] [PubMed] [Google Scholar]
- 5.Raj A. and van Oudenaarden A, Cell, 2008, 135, 216–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Herndon LA, Schmeissner PJ, Dudaronek JM, Brown PA, Listner KM, Sakano Y, Paupard MC, Hall DH and Driscoll M, Nature, 2002, 419, 808–814. [DOI] [PubMed] [Google Scholar]
- 7.Pincus Z, Smith-Vikos T. and Slack FJ, PLoS Genet., 2011, 7, e1002306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rea SL, Wu D, Cypser JR, Vaupel JW and Johnson TE, Nat. Genet, 2005, 37, 894–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Entchev EV, Patel DS, Zhan M, Steele AJ, Lu H. and Ch’ng Q, Elife, 2015, 4, e06259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Meisel JD, Panda O, Mahanti P, Schroeder FC and Kim DH, Cell, 2014, 159, 267–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cao J, Packer JS, Ramani V, Cusanovich DA, Huynh C, Daza R, Qiu X, Lee C, Furlan SN, Steemers FJ, Adey A, Waterston RH, Trapnell C. and Shendure J, Science, 2017, 357, 661–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shapiro E, Biezuner T. and Linnarsson S, Nat Rev Genet, 2013, 14, 618–630. [DOI] [PubMed] [Google Scholar]
- 13.Ly K, Reid SJ and Snell RG, MethodsX, 2015, 2, 59–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Boulin T, Etchberger JF, Hobert O, Wormbook, Reporter gene fusions, 2006, The C. elegans Research Community, 10.1895/wormbook.1.106.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hunt-Newbury R, Viveiros R, Johnsen R, Mah A, Anastas D, Fang L, Halfnight E, Lee D, Lin J, Lorch A, McKay S, Okada HM, Pan J, Schulz AK, Tu D, Wong K, Zhao Z, Alexeyenko A, Burglin T, Sonnhammer E, Schnabel R, Jones SJ, Marra MA, Baillie DL and Moerman DG, PLoS Biol., 2007, 5, e237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li X, Zhao X, Fang Y, Jiang X, Duong T, Fan C, Huang CC and Kain SR, J. Biol. Chem, 1998, 273, 34970–34975. [DOI] [PubMed] [Google Scholar]
- 17.Snapp EL, Trends Cell Biol., 2009, 19, 649–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Raj A, van den Bogaard P, Rifkin SA, van Oudenaarden A. and Tyagi S, Nat. Methods, 2008, 5, 877–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Harterink M, Kim D. h., Middelkoop TC, Doan TD, van Oudenaarden A. and Korswagen HC, Development, 2011, 138, 2915–2924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ji N. and van Oudenaarden A, Wormbook, Single molecule fluorescent in situ hybridization (smFISH) of C. elegans worms and embryos, 2012, The C. elegans Research Community, 10.1895/wormbook.1.153.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen KH, Boettiger AN, Moffitt JR, Wang S. and Zhuang X, Science, 2015, 348, aaa6090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Eng CL, Lawson M, Zhu Q, Dries R, Koulena N, Takei Y, Yun J, Cronin C, Karp C, Yuan GC and Cai L, Nature, 2019, 568, 235–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee H, Kim SA, Coakley S, Mugno P, Hammarlund M, Hilliard MA and Lu H, Lab Chip, 2014, 14, 4513–4522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mueller F, Senecal A, Tantale K, Marie-Nelly H, Ly N, Collin O, Basyuk E, Bertrand E, Darzacq X. and Zimmer C, Nat. Methods, 2013, 10, 277–278. [DOI] [PubMed] [Google Scholar]
- 25.Shaffer SM, Joshi RP, Chambers BS, Sterken D, Biaesch AG, Gabrieli DJ, Li Y, Feemster KA, Hensley SE, Issadore D. and Raj A, Lab Chip, 2015, 15, 3170–3182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.de Bono M, Tobin DM, Davis MW, Avery L. and Bargmann CI, Nature, 2002, 419, 899–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gallagher T, Kim J, Oldenbroek M, Kerr R. and You YJ, J Neurosci, 2013, 33, 9716–9724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shaw WM, Luo S, Landis J, Ashraf J. and Murphy CT, Curr. Biol, 2007, 17, 1635–1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hilliard MA, Bergamasco C, Arbucci S, Plasterk RHA and Bazzicalupo P, EMBO J, 2004, 23, 1101–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jansen G, Thijssen KL, Werner P, van derHorst M, Hazendonk E. and Plasterk RHA, Nat. Genet.s, 1999, 21, 414–419. [DOI] [PubMed] [Google Scholar]
- 31.Kim K, Sato K, Shibuya M, Zeiger DM, Butcher RA, Ragains JR, Clardy J, Touhara K. and Sengupta P, Science, 2009, 326, 994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hahm JH, Kim S. and Paik YK, Aging Cell, 2009, 8, 473–483. [DOI] [PubMed] [Google Scholar]
- 33.Rangaraju S, Solis GM, Thompson RC, Gomez-Amaro RL, Kurian L, Encalada SE, Niculescu AB 3rd, Salomon DR and Petrascheck M, Elife, 2015, 4, e08833-e08833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chung K, Kim Y, Kanodia JS, Gong E, Shvartsman SY and Lu H, Nat. Methods, 2011, 8, 171–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chung K, Rivet CA, Kemp ML and Lu H, Anal. Chem, 2011, 83, 7044–7052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chung K, Zhan M, Srinivasan J, Sternberg PW, Gong E, Schroeder FC and Lu H, Lab Chip, 2011, 11, 3689–3697. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.