

SUMMARY
Spermiogenesis is a highly orchestrated developmental process during which chromatin condensation decouples transcription from translation. Spermiogenic mRNAs are transcribed earlier and stored in a translationally inert state until needed for translation; however, it remains largely unclear how such repressed mRNAs become activated during spermiogenesis. We previously reported that the MIWI/piRNA machinery is responsible for mRNA elimination during late spermiogenesis in preparation for spermatozoa production. Here we unexpectedly discover that the same machinery is also responsible for activating translation of a subset of spermiogenic mRNAs to coordinate with morphological transformation into spermatozoa. Such action requires specific base-pairing interactions of piRNAs with target mRNAs in their 3′ UTRs, which activates translation through coupling with cis-acting AU-rich elements to nucleate the formation of a MIWI/piRNA/eIF3f/HuR super-complex in a developmental stage-specific manner. These findings reveal a critical role of the piRNA system in translation activation, which we show is functionally required for spermatid development.
In Brief
The piRNA pathway, through functional interplay with HuR and eIF3f, plays an important role in translational activation of a specific set of mRNAs during mouse spermiogenesis.
Graphical Abstract
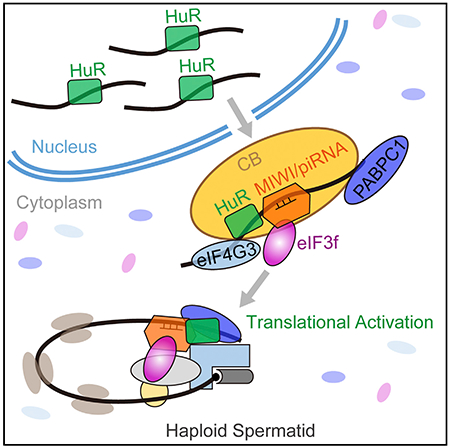
INTRODUCTION
The post-meiotic phase of male germ cell development in mammals undergoes a series of dramatic morphological changes, including acrosome and flagellum formation, nuclear condensation, and cytoplasmic exclusion, which are driven by a highly regulated sequence of biochemical reactions (Yan, 2009; Meistrich and Hess, 2013). This differentiation process, called spermiogenesis, consists of at least 16 sequential transition steps in mice, which occur from postpartum day 21 to 28 for round spermatid differentiation (steps 1–8), postpartum day 28–35 for morphological transformation into elongating spermatids (steps 9–11), elongated or condensed spermatids (steps 12–14), and finally spermatozoa (steps 15–16) (Yan, 2009; Meistrich and Hess, 2013). These sequential transitions are programmed by a series of highly orchestrated regulatory events in gene expression at each developmental step. Due to chromatin compaction during the course of spermatid elongation, transcription becomes gradually inhibited during spermiogenesis before ceasing completely later on (Steger, 1999; Sassone-Corsi, 2002). Thus, the program of spermiogenesis relies on gene products that are transcribed earlier and stored in translationally inert messenger ribonucleoproteins (mRNPs) until needed for translation (Steger, 1999; Sassone-Corsi, 2002). This phenomenon, known as uncoupling between transcription and translation, is a unique feature of gene regulation during spermiogenesis, which has been one of the major mysteries in understanding the germ cell development program.
The evolutionarily conserved PIWI family proteins have been well documented for their essential roles in germline development and gametogenesis on various animal models (Siomi et al., 2011; Iwasaki et al., 2015). Most recently, we showed a causative role of human Piwi (Hiwi) mutations in male infertility, establishing a definitive linkage of human Piwi genes to human infertility (Gou et al., 2017; Hasuwa et al., 2018). PIWI proteins are known to enlist germline-specific PIWI-interacting RNAs (piRNAs), whose primary function has been thought to silence transposable elements, thereby protecting the genome integrity in animal germ cells (Siomi et al., 2011; Iwasaki et al., 2015). Importantly, we and others demonstrated that the PIWI/piRNA machinery also mediates the degradation of mRNA transcripts via miRNA- and/or siRNA-like mechanisms in germ cells (Gou et al., 2014; Zhang et al., 2015; Goh et al., 2015; Watanabe et al., 2015; Vourekas et al., 2016; Shen et al., 2018; Zhang et al., 2018). These findings demonstrate that the piRNA system also regulates protein-coding genes beyond silencing transposable elements in germ cells.
To understand the mechanism for the PIWI/piRNA machinery to regulate spermiogenic mRNAs at post-transcriptional levels, we previously embarked on a systematic effort to identify mouse PIWI (MIWI)-associated mRNAs in mouse spermatids by RNA co-immunoprecipitation (RIP) in combination with UV crosslinking coupled with deep sequencing (CLIP-seq) and found that nearly all mRNAs in complex with MIWI could be matched to a specific set of piRNAs (Gou et al., 2014; Zhang et al., 2015). Using a mouse spermatocyte-derived GC-2spd(ts) cell line (Hofmann et al., 1995) as a cell-culture model, we determined the functional consequence of predicted piRNA:mRNA pairs on a 3′ UTR-based reporter system in response to individual synthetic piRNAs. While this cell line is unlikely to perfectly recapitulate PIWI/piRNA function during spermiogenesis, it is the only cell-culture model that we found to be functional for biochemical studies because piRNAs, MIWI, MILI, and Tudor proteins as well as MIWI/piRNA complexes are all present (Gou et al., 2014). Indeed, on this cellular model, we found that many tested piRNAs are able to induce downregulation of their cognate targets (Gou et al., 2014; Zhang et al., 2015).
During this analysis, we were surprised to note that among the predicted piRNA:mRNA pairs (~100) that we tested, five such targets (i.e., Plectin, Agfg1, Tbpl1, Cnot4, and Atg12) were instead activated by their cognate piRNAs. All of these piRNA-activated targets are characterized by AU-rich elements (AREs) in their 3′ UTRs, which we show are required for piRNA-mediated translational activation. Pursuing this unexpected regulatory paradigm, we uncover the involvement of the ARE-binding protein HuR and the translation initiation factor eIF3f in mediating translation activation of these MIWI/piRNA targets. Focusing on Agfg1 and Tbpl1, the two genes critical for acrosome formation during spermatid development (Kang-Decker et al., 2001; Zhang et al., 2001), we demonstrate that their activation by the piRNA system is indeed indispensable for acrosome formation in spermatozoa. These findings, coupled with the established role of MIWI/piRNA in massive mRNA degradation for inactivating vast cellular programs in preparation for spermatozoa production from spermatids, unveil a dual function of the MIWI/piRNA machinery in differential regulation of its target mRNAs in spermatids to instruct post-meiotic male germ cell development in mice.
RESULTS
piRNAs Activate Target mRNAs via Imperfect Base-Pairing Interactions
Our previous studies showed that the MIWI complex in mouse spermatids harbors both piRNAs and mRNAs and that most tested piRNAs repress mRNA targets via a base-pairing rule using a 3′ UTR-based reporter screen system in transfected GC-2spd(ts) cells (Gou et al., 2014). Surprisingly, we found that five specific piRNAs (i.e., piR_1029, piR_3146, piR_2617, piR_010111, and piR_3344) significantly upregulated their cognate target reporters Plectin, Agfg1, Tbpl1, Cnot4, and Atg12 (Figures 1A and 1B). None of these piRNAs caused any measurable change in reporter mRNA expression (Figure 1C), implying a function of piRNAs at the level of translation. We were particularly attracted to this possibility, as it would suggest a potential role of piRNAs in selective translation activation in mouse spermatids, which has been one of the major mysteries in understanding spermiogenesis.
Figure 1. piRNAs Activate the Translation of Target mRNAs via Imperfectly Base Pairing with the 3′ UTRs.
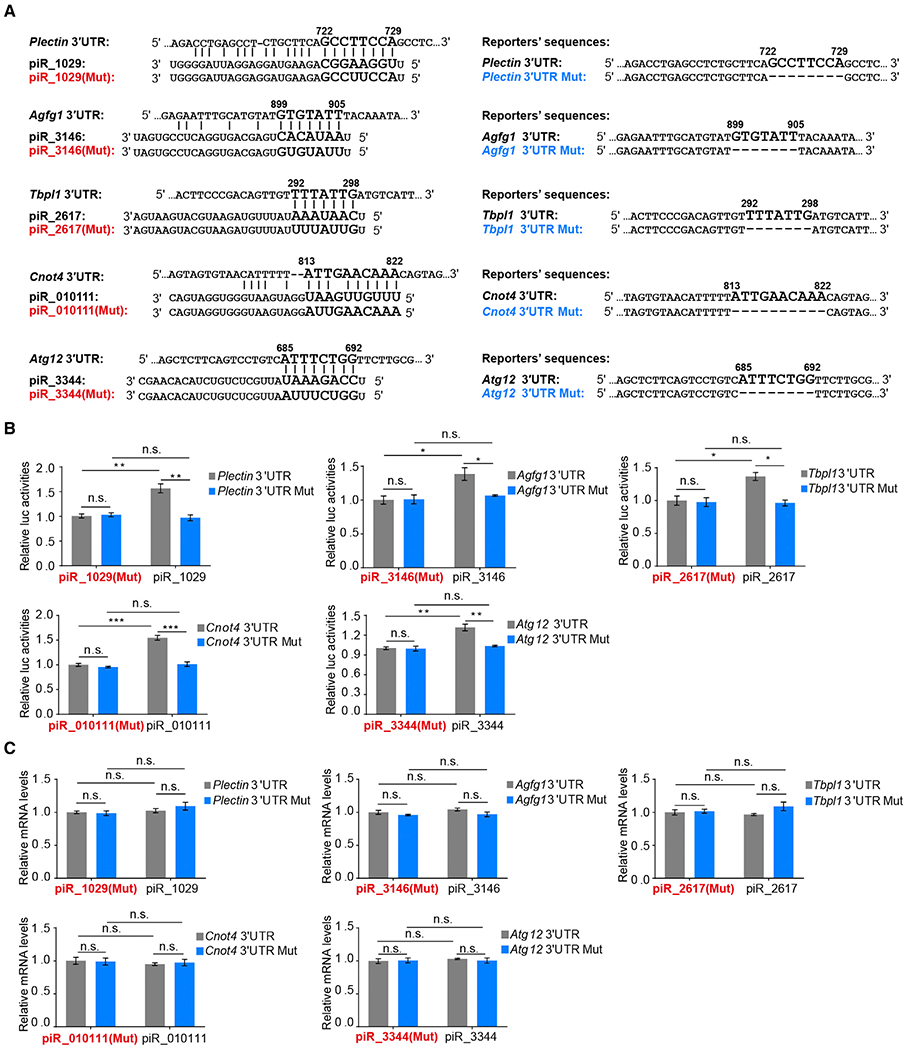
(A) Predicted piRNA regulatory elements at the 3′ UTRs of Plectin, Agfg1, Tbpl1, Cnot4, or Atg12 and the synthetic piRNAs, mutated piRNAs, and wild-type and mutated 3′ UTR Renilla luciferase reporters.
(B) Dual-luciferase reporter assay showing that piRNAs promoted the activities of 3′ UTR reporters in GC-2spd (ts) cells, with mutated piRNAs serving as negative controls.
(C) qPCR assay showing that piRNAs barely altered the levels of 3′ UTR reporter mRNAs.
The average values ± SD of three separate experiments were plotted. *p < 0.05, **p < 0.01, ***p < 0.001, n.s., not significant. See also Figure S1.
We next asked whether specific piRNA:mRNA pairing is required for this newly discovered piRNA function. To this end, we constructed mutant reporters in each with deletion of the 7~8-nt (nucleotide) sequence complementary to the 5′ part of the corresponding piRNAs and also synthesized mutant piRNAs to substitute the 7~8-nt sequence in each with complementary nucleotides (Figure 1A). We found that both deletion of piRNA-binding sites in the reporters and mutations in the corresponding piRNAs completely abolished the activation effect of piRNAs (Figure 1B), again without detectable impact on reporter mRNA levels (Figure 1C). This supports the requirement of piRNA:mRNA base pairing for piRNA-induced target activation. Moreover, in agreement with recent observations in C. elegans (Shen et al., 2018; Zhang et al., 2018), we found that one mismatch in the piR_1029:Plectin mRNA pairing region is sufficient to disrupt the piRNA function (Figure S1A), further demonstrating the indispensability of piRNA:mRNA base pairing for this new piRNA function.
Consistent with the possibility of translation activation, our sucrose gradient analysis showed that, relative to the piR_1029 (Mut) control, co-transfected piR_1029 significantly elevated the association of MIWI with the reporter mRNA in the polysome fractions, which was accompanied with a reduction in lighter RNP fractions (Figures S1B–S1D). These results confirm an active role of piRNA:mRNA base pairing in trafficking more target transcripts to the translation machinery, providing initial evidence for piRNA-dependent translation activation of target mRNAs through imperfect base pairing in the 3′ UTRs.
MIWI Is Required for piRNA-Mediated Translation Activation
To determine whether MIWI is required for piRNA-induced target activation, we took advantage of our previous MIWI CLIP data generated on enriched mouse spermatids (Gou et al., 2014; Zhang et al., 2015), showing clusters of specific MIWI-CLIP tags on cognate piRNA target sites on Plectin, Agfg1, Tbpl1, Cnot4, and Atg12 mRNAs (Figure 2A), thus pinpointing specific MIWI-binding sites in these mRNAs in vivo. To further substantiate these observations, we performed RIP and found that Flag-MIWI readily pulled down the Plectin reporter mRNA in piR_1029, but not piR_1029 (Mut) co-transfected GC-2spd(ts) (Figure S2A), indicating that specific piRNA:mRNA base pairing guides MIWI loading onto its targeted mRNA.
Figure 2. MIWI Is Essential for piRNA-Induced Target Activation.
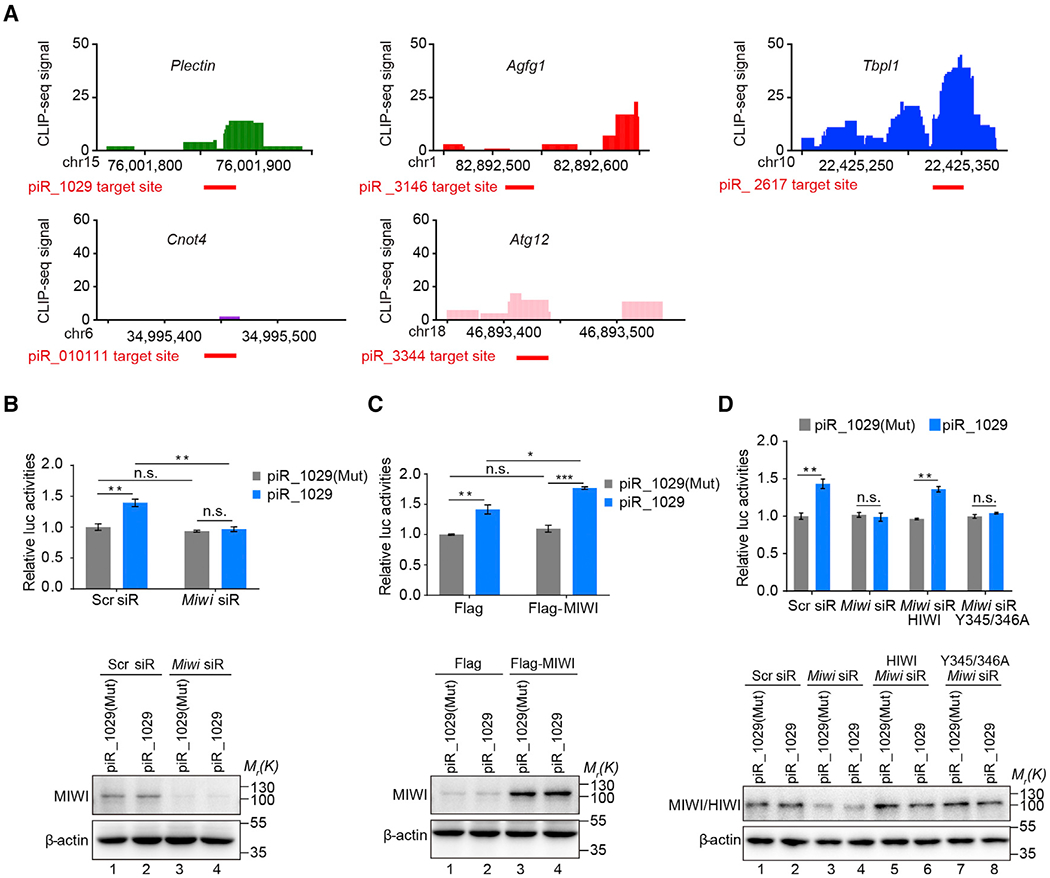
(A) Verification of piRNA target sites by MIWI crosslinking immunoprecipitation (CLIP) in mouse spermatids. Top: the cluster of MIWI CLIP tags on the five validated piRNA-activated genes, with the scales in genes numbered following the dimension of the chromosomes; bottom: the red thick lines indicate piRNA target sites.
(B and C) The effect of Miwi knockdown (B) or overexpression (C) on piR_1029-induced Plectin reporter activation in GC-2spd (ts) cells. Top: dual-luciferase reporter assay; bottom: western blotting assay of MIWI.
(D) Ectopic Flag-HIWI, but not its piRNA-loading-deficient Y345/346A mutant, rescued piR_1029-induced Plectin reporter activation in Miwi siR-treated GC-2spd(ts) cells. Top: dual-luciferase reporter assay; bottom: western blotting assay of MIWI or HIWI.
The average values ± SD of three separate experiments were plotted. *p < 0.05, **p < 0.01, ***p < 0.001, n.s., not significant. See also Figures S2A–S2D.
We next co-transfected Miwi siRNA (Miwi siR) to knock down endogenous Miwi in GC-2spd(ts) cells. We found that MIWI depletion abrogated the effect of piR_1029 on the Plectin reporter (Figure 2B). In contrast, depletion of either MILI (another mouse Piwi protein expressed in GC-2spd(ts) cells) or AGO2 (a key component of the microRNA/small interfering RNA [miRNA/siRNA] machinery) showed little impact on piRNA-induced target activation (Figure S2B). Conversely, ectopic expression of Flag-MIWI further enhanced the activation effect of piR_1029 on the Plectin reporter (Figure 2C). Neither Miwi knockdown nor overexpression altered the reporter mRNA expression (Figures S2C and S2D). These results indicate the functional requirement of MIWI for piRNA-induced target activation.
To further substantiate this piRNA-guided MIWI function in translation activation, we determined whether its piRNA loading capacity is required. We employed a Miwi siRNA to deplete endogenous MIWI in GC-2spd(ts) cells and then complemented the cells with either Flag-tagged wild-type human PIWI (HIWI) or its piRNA loading-deficient Y345/346A mutant (Figure 2D). As expected, wild-type HIWI was able to restore the piR_1029-dependent reporter activation in MIWI-depleted GC-2spd(ts) cells, but the Y345/346A mutant failed to rescue, demonstrating that the piRNA-loading ability of PIWI is indispensable for piRNA-mediated target activation. This further supports the functional requirement of MIWI in this newly discovered piRNA function.
Identification of eIF3f as a Key Partner of MIWI in Translation Activation
To understand the mechanism, we next aimed to identify a potential cofactor(s) with the MIWI/piRNA machinery to activate target mRNA translation. To this end, we carried out a yeast two-hybrid screen to search for candidate MIWI-interacting proteins, paying particular attention to hits with links to translation using mouse testis and human fetal brain cDNA libraries. The f-subunit of eukaryotic translation initiation factor 3 (eIF3f) stood out from the screen as a potent MIWI-interacting protein, as 37 out of 71 positive clones all contained eIF3f cDNA (Table S1). We confirmed the interaction between MIWI and eIF3f by co-immunoprecipitation (coIP) in both co-transfected 293T cells (Figure 3A) and mouse testes (Figure 3B). In addition, we found that MIWI was also able to pull down eIF3a and eIF3c, two other eIF3 core subunits, in mouse testes (Figure 3B). Furthermore, unbiased proteomic analysis of anti-MIWI IPed complexes from mouse testes identified eIF3a, eIF3b, eIF3c, eIF3e, eIF3f, eIF3h, eIF3l, and eIF3m (see Figure S3H). These findings suggest that MIWI may recruit the entire eIF3 complex via eIF3f in mouse testes.
Figure 3. eIF3f, a MIWI-Interacting Protein, Is Required for piRNA-Induced Target Activation.
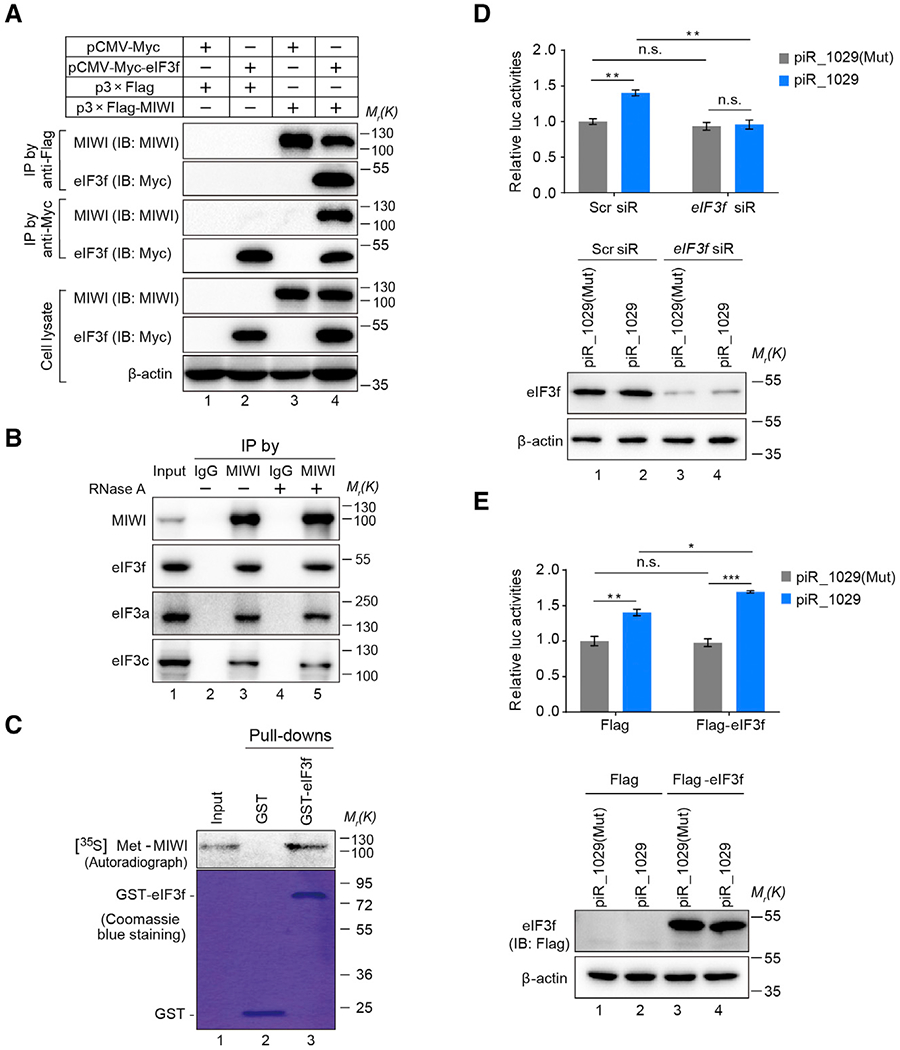
(A and B) CoIP assay of MIWI-eIF3f interaction in Myc-eIF3f and Flag-MIWI co-transfected 293T cells (A) or adult mouse testes (B).
(C) GST pulldown assay showing a direct interaction between eIF3f and MIWI. Samples were analyzed by autoradiography (top) or visualized by Coomassie blue staining (bottom).
(D and E) The effect of eIF3f knockdown (D) or overexpression (E) on piR_1029-induced Plectin reporter activation in GC-2spd (ts) cells. Top: dual-luciferase reporter assay; bottom: western blotting assay of eIF3f.
The average values ± SD of three separate experiments were plotted. *p < 0.05, **p < 0.01, ***p < 0.001, n.s., not significant. See also Figures S2E–S2G and Table S1.
We next performed the GST pull-down assay using purified recombinant GST-eIF3f and in vitro translated [35S]Met-MIWI, showing the direct interaction between them (Figure 3C). Such an interaction was barely affected in response to treatment with RNase A (Figure 3B) or in the presence of piRNA (Figure S2E), indicating that RNA is not required for the MIWI-eIF3f interaction. Importantly, knockdown of eIF3f in GC-2spd(ts) cells completely attenuated the effect of piR_1029 on the Plectin reporter (Figure 3D), whereas the ectopic expression of Flag-eIF3f further strengthened the impact of piR_1029 on the reporter activity (Figure 3E). Neither eIF3f knockdown nor overexpression altered the reporter mRNA level (Figure S2F). The data strongly suggested an essential role of eIF3f in piRNA-induced translation activation. Of note, eIF3f has been shown as a nonessential eukaryotic initiation subunit, which appears to function in a tissue-specific or intracellular and signaling context-dependent manner (Marchione et al., 2013). Indeed, our high-resolution sucrose gradient analysis revealed that eIF3f knockdown caused little, if any, alteration in the polysome-to-monosome (P/M) ratio in GC-2spd(ts) cells (Figure S2G), suggesting that eIF3f is not essential for global mRNA translation in these cells. These data point to a specific function of eIF3f in piRNA-activated translation.
AU-Rich Elements in 3′ UTR Are Responsible for piRNA-Induced Target Activation
We next investigated how the MIWI/piRNA complex induces mRNA decay in the vast majority of target transcripts while activating translation of a subset. Given the established roles of cis-elements in the 3′ UTRs in regulating mRNA metabolism in male germ cells (Kleene, 2013), we hypothesized that certain specific cis-elements in the 3′ UTRs might be responsible for differentiating translation activation and mRNA degradation by the piRNA system. Close inspection of the five validated piRNA-activated targets revealed AREs in their 3′ UTRs (Figure S3A), and indeed, mutations in the AREs abolished the activation effect of piRNAs on the reporters (Figure 4A). These observations suggest that AREs in the 3′ UTRs may be a critical sequence feature for piRNA-mediated translation activation.
Figure 4. AREs in the 3′ UTRs of Target mRNAs Are Required for piRNA-Induced Target Activation.
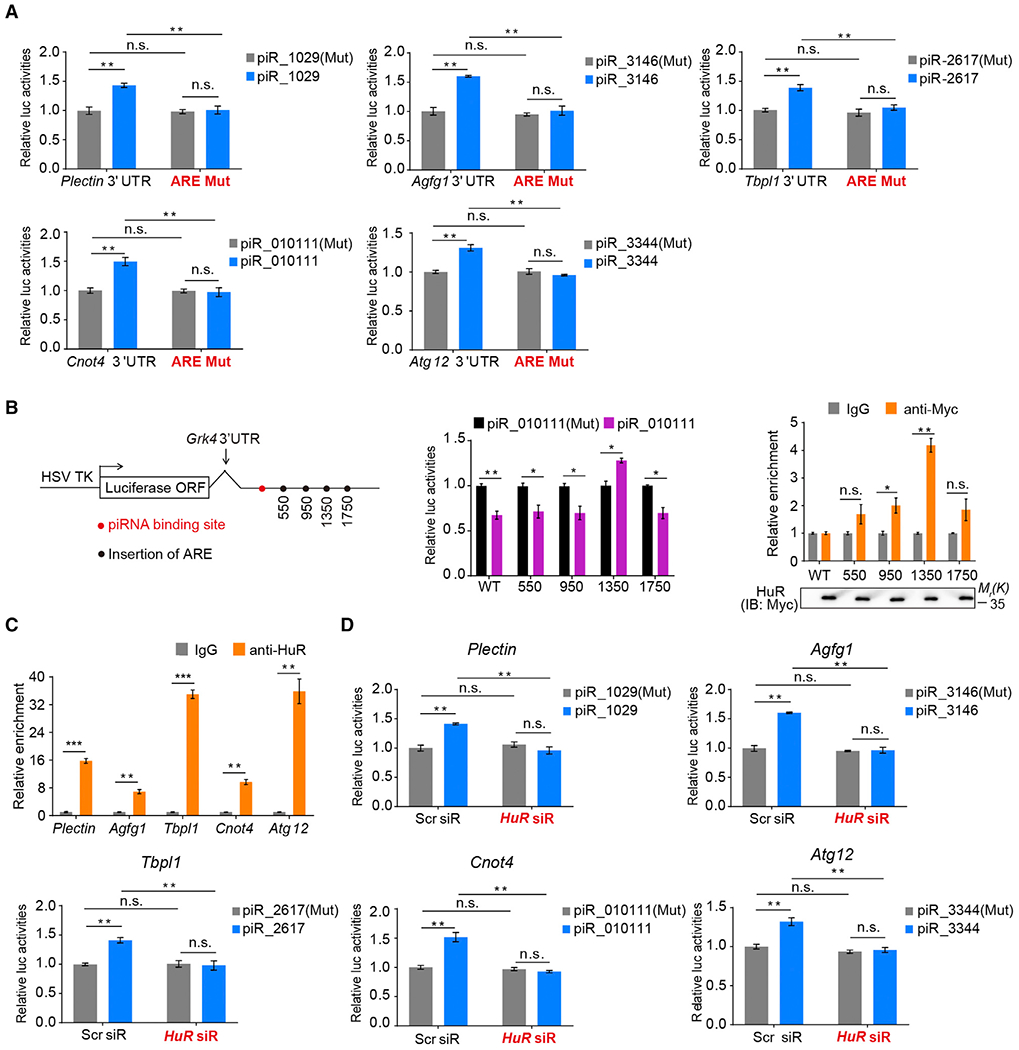
(A) Dual-luciferase reporter assay showing that deletion of AREs in target mRNAs impaired piRNA-induced target activation.
(B) Insertion of an ARE at position 1,350 nt downstream of piRNA-binding site reversed the repressive effect of piR_010111 to activation on the Grk4 reporter in GC-2spd (ts) cells. Left: schematic diagram showing the position of piRNA-binding site (red spot) and respective ARE insertion (black spot) in Grk4 reporter; middle: dual-luciferase reporter assay; right: RIP combined with qRT-PCR assays of HuR-binding to the ARE-containing Grk4 reporter mRNAs, with relative enrichment of reporter mRNA in anti-HuR IP compared with IgG IP (top), using anti-HuR IB as loading references (bottom).
(C) RIP combined with qRT-PCR assays of HuR-binding to the five validated piRNA-activated mRNAs in mouse testes. Relative enrichment of indicated mRNA in HuR IP was compared with IgG IP.
(D) HuR knockdown substantially impaired the effect of piRNAs on the target reporters.
The average values ± SD of three separate experiments were plotted. *p < 0.05, **p < 0.01, ***p < 0.001, n.s., not significant. See also Figure S3 and Table S2.
To further test whether an ARE is sufficient to trigger piRNA-induced translation activation, we inserted an ARE into the Grk4 3′ UTR reporter, a target to be repressed by piR_010111 (Gou et al., 2014), and to additionally test potential position-sensitive effects, we inserted the ARE at 550 nt, 950 nt, 1,350 nt, or 1,750 nt downstream of the piRNA-binding site, respectively (Figure 4B, left). An ARE at 1,350 nt, but not other locations, successfully reversed the repressive effect of piR_010111 to activation on the reporter (Figure 4B, middle). These results indicate that an ARE is sufficient to enable piRNA-mediated target activation in a position-sensitive manner.
HuR Plays an Essential Role in piRNA-Induced, ARE-Dependent Target Activation
HuR has been known to be essential for spermatogenesis in mice (Nguyen-Chi et al., 2011). A previous study reported that HuR, a canonical ARE-binding protein, is associated with the traffic of ARE-containing mRNAs from the chromatoid body, a germ granule in round spermatids that has been suggested as a center for mRNA storage (Meikar et al., 2011), to polysomes (Nguyen-Chi et al., 2009). These prior observations thus point to a positive role of HuR in regulating a switch from mRNA storage to translation during spermiogenesis.
To test the hypothesis that HuR might be involved in piRNA-mediated translation activation, we first performed RIP assay on the series of Grk4 3′ UTR reporters and found that the ARE inserted at 1,350 nt in the Grk4 3′ UTR reporter led to a more efficient HuR-binding compared to its insertion at other locations in the same reporter (Figure 4B, right), correlating HuR-binding to piRNA-dependent translation activation. We extended the analysis to all five validated piRNA target mRNAs in mouse testes, finding that Plectin, Agfg1, Tbpl1, Cnot4, and Atg12 mRNAs were all substantially enriched in anti-HuR immunoprecipitant compared to IgG control (Figure 4C), indicative of a strong association of these mRNAs with HuR in vivo. We further found that HuR knockdown completely abolished the effect of piRNAs on activating their target reporters (Figure 4D), which again barely altered the reporter mRNA levels (Figure S3B). These results strongly suggested HuR in target activation by the piRNA system.
We next examined whether HuR associates with MIWI and eIF3f. By coIP, we found that either Flag-tagged MIWI or eIF3f was unable to pull down Myc-HuR in co-transfected 293T cells (Figure S3C). However, co-transfection of the Plectin reporter and piR_1029 enabled the interaction between HuR and the MIWI/eIF3f complex (Figure S3D), suggesting that piRNA:mRNA pairing may bridge the interaction between HuR and the MIWI/eIF3f complex. Unlike 293T cells, mouse testes contain active piRNAs, and indeed, we detected significant association of endogenous HuR with both MIWI and eIF3f in mouse testes (Figure S3E). We further performed proteomic analyses in this biologically relevant system and detected eIF3f and HuR in anti-MIWI immunoprecipitant and, conversely, MIWI in anti-HuR immunoprecipitant from mouse testes (Table S2). As expected from RNA-bridged association, we confirmed that the association between HuR and MIWI was RNase A sensitive (Figure S3E). Together, these data provide both biochemical and functional evidence for a vital role of HuR in translation activation of ARE-containing MIWI/piRNA targets.
Both MIWI and HuR Associate with Multiple Translation-Related Proteins in Mouse Testes
Interestingly, we also identified several translation-related proteins in both anti-MIWI and anti-HuR IPed complexes from mouse testes (Table S2), including translation initiation factor eIF4G3 and poly(A) tail-binding proteins (PABPs). To further dissect the functional complexes, we immunopurified MIWI- or HuR-containing complexes from polysome fractions prepared from mouse testes for proteomic analysis and again confirmed the presence of these translation initiation factors in both anti-MIWI and anti-HuR IPed complexes (Figure S3H). These results suggest that MIWI and HuR associate with the competent translation initiation machinery in mouse testes.
eIF4G is essential for the assembly of the eIF4F complex and initiation of cap-dependent translation (Merrick, 2015). Interestingly, among three eIF4G isoforms, eIF4G3 is essential for male fertility in mice (Sun et al., 2010). We noted that a recent report also identified eIF4G3 as a HuR-interacting protein in mouse testes to facilitate dramatic translational reprogramming in post-meiotic spermatids together with additional associated translation factors (i.e., eIF4A2, eIF4E, and PABPC1; Hu et al., 2018). These findings thus point to a role of eIF4G3 in activating mRNA translation in post-meiotic spermatids. We confirmed the interaction of HuR with eIF4G3 by coIP in both mouse testes (Figure S3F) and co-transfected 293T cells (Figure S3G) and showed that their interaction was insensitive to RNase A treatment. These results thus suggest eIF4G3 as a component of the MIWI/piRNA/eIF3f/HuR super-complex in mouse testes.
PABP facilitates translation initiation via its association with eIF4G (Imataka et al., 1998), which has also been reported to associate with MIWI in mouse testes (Kimura et al., 2009). Two cytoplasmic PABP proteins, PABPC1 and PABPC2, have been characterized in mouse male germ cells, where PABPC1 appears to associate with actively translating polysomes whereas PABPC2 likely functions in translation repression during spermatogenesis (Kimura et al., 2009). Interestingly, we found that HuR effectively promoted the association of MIWI with PABPC1 (Figure S3I). Collectively, these biochemical data suggest that HuR may orchestrate translation activation of ARE-containing target mRNAs through recruiting multiple translation activators, particularly eIF4G3 and PABPC1.
Requirement of Both MIWI and HuR for Translation Activation in Mouse Spermatids
We next focused on examining the effect of the piRNA system on translation activation during spermatogenesis. To this end, we isolated spermatocytes (SCs) and haploid spermatids (STs) from mouse testes (Figure S4A) and examined the expression of three validated piRNA-activated targets, i.e., Plectin, Agfg1, and Tbpl1, in SCs and STs. We found similar levels of their mRNAs in both cell populations (Figure 5A, left), but their proteins were only readily detectable in STs (right). These developmental expression profiles are thus fully in line with the possibility that these piRNA target genes are transcribed in SCs but translationally activated in spermatids.
Figure 5. piRNA System Contributes to the Translation Activation Program in Mouse Spermatids.
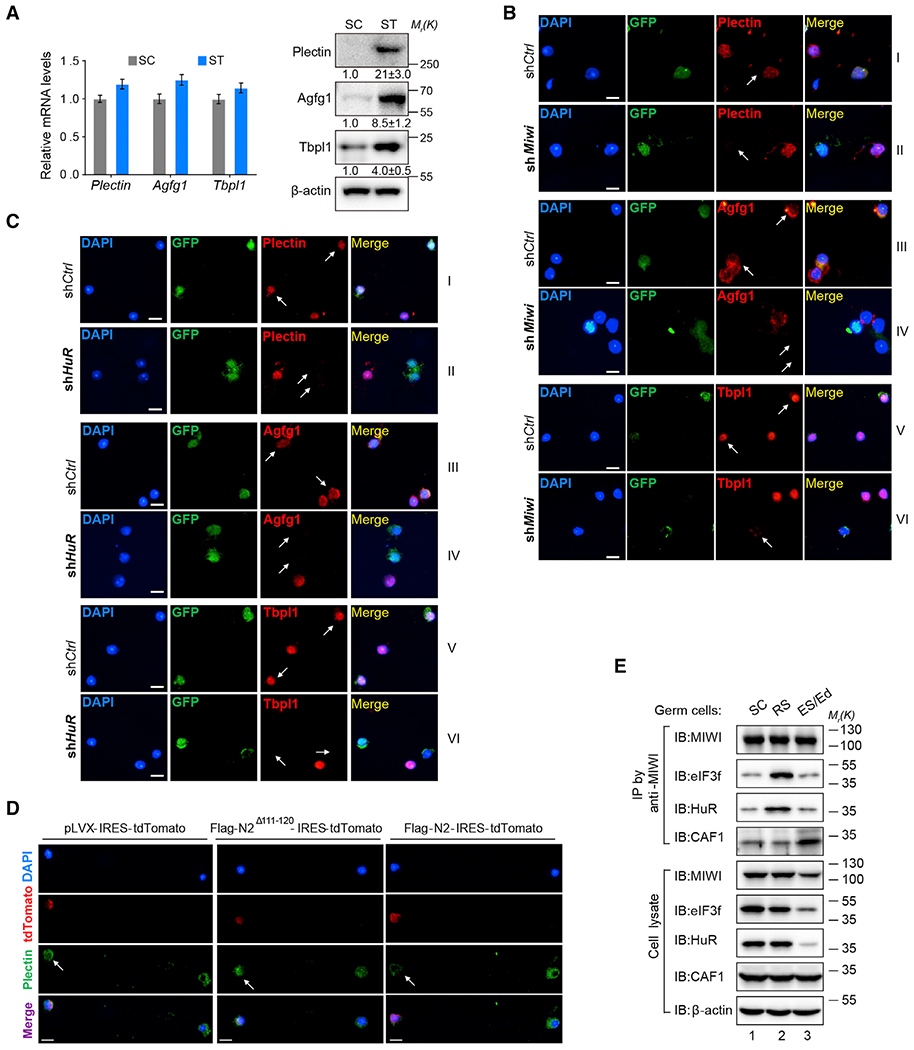
(A) qRT-PCR (left) and western blotting assays (right) of the expression of Plectin, Agfg1, and Tbpl1 in enriched SCs and STs. Quantification of blotting intensity for indicated proteins is shown in parentheses (the one in SC is set as 1.0 after normalization with β-actin blotting).
(B and C) Immunofluorescent assay of the effect of shMiwi:GFP (B) or shHuR:GFP transduction (C) on Plectin (II), Agfg1 (IV), and Tbpl1 expression (VII) in mouse spermatids. GFP (green) and arrows indicate the transduced cells. Scale bar, 10 μm.
(D) Immunofluorescent assay of the effect of MIWI-N2 or MIWI-N2Δ111–120 peptides on Plectin expression in control pLVX-IRES-tdTomato (left), Flag-MIWI-N2Δ111–120 (middle), or Flag-MIWI-N2-transduced spermatids (right). tdTomato (red) and arrows indicate the transduced cells.
(E) CoIP assay of the association of MIWI with eIF3f, HuR, and CAF1 in SCs (lane 1), RSs (lane 2), and later stages of spermatids (ESs/elongated spermatids [Eds], lane 3).
Results shown are representative of three independent experiments. See also Figures S4, S5, and S6.
To directly demonstrate these decoupled transcription and translation events, we performed sucrose gradient analysis to assess the translation activity of the three mRNAs during male germ cell development. Due to severe collapse of polysomes from highly purified SCs and STs (data not shown), we then compared the polysome profiles of testicular tissues at two developmental time points: 18 days postpartum (dpp), which contains SCs but lacks STs, versus 30-dpp, which contains both SCs and STs. In spermatid-containing 30-dpp testes, the levels of all three mRNAs (Figure S4B) and their cognate piRNAs (Figure S4C), as well as MIWI and HuR proteins (Figure S4D), were significantly elevated in the polysome fractions with a concomitant decrease in lighter RNP and 40S-80S fractions compared to 18-dpp testes. Importantly, all three mRNAs showed increased association with MIWI and HuR on polysomes in 30-dpp testes relative to 18-dpp testes from our RIP analysis (Figure S4E). These data support the functional roles of MIWI and HuR in translation activation of these piRNA targets in mouse spermatids.
To directly document the requirement of the piRNA system in target mRNA translation in mouse spermatids, we used high-titer short hairpin RNA (shRNA) lentivirus to knock down Miwi, HuR, or eIF3f in mouse spermatids through testis transduction on 3- to 4-week-young mice. We found little toxicity with control pSilencer:GFP or shMiwi lentivirus on mouse testes. However, the shRNA lentivirus against eIF3f caused severe damage and necrosis in transduced testes, and that shHuR lentivirus gave rise to moderately damaged but still viable testes. These phenotypes likely result from eIF3f or HuR depletion-induced destruction of testicular structures, given that both eIF3f and HuR, unlike MIWI, are also constitutively expressed in testicular somatic cells. In any case, we performed immunostaining on the remaining GFP+ spermatids and found that GFP+ spermatids from both shMiwi:GFP and shHuR:GFP-transduced mice, but not pSilencer:GFP controls, showed a marked reduction of Plectin, Agfg1, and Tbpl1 proteins compared to GFP− cells (Figures 5B, 5C, S4F, and S4G). We conclude from these data that both MIWI and HuR are required for translation activation of piRNA targets in mouse spermatids.
Blocking the MIWI-eIF3f Interaction Inhibits Plectin Expression in Mouse Spermatids
To begin to determine whether MIWI-binding to eIF3f is required for translation activation of piRNA targets in mouse spermatids, we mapped the domain(s) in MIWI responsible for eIF3f-binding by testing a series of MIWI truncation mutants (Figure S5A) for coIP with full-length eIF3f. We found that a 150-aa (amino acid) MIWI N-terminal peptide (MIWI-N2) efficiently interacted with eIF3f and deletion of 10 aa at position 111–120 (MIWI-N2Δ111–120) abolished its binding with eIF3f (Figure S5B). Importantly, MIWI-N2, but not MIWI-N2Δ111–120, effectively impaired the binding of full-length MIWI to eIF3f (Figure S5C). We further found that MIWI-N2, but not MIWI-N2Δ111–120, significantly attenuated the effect of piR_1029 on the Plectin reporter (Figure S5D). These results indicate that MIWI-N2 is able to compete with full-length MIWI for binding to eIF3f.
The identification of such an interfering peptide afforded us a critical tool to test the functional requirement of the MIWI-eIF3f interaction for piRNA target expression in mouse spermatids. We therefore cloned Flag-tagged MIWI-N2 or MIWI-N2Δ111–120 into pLVX-IRES-tdTomato lentiviral vector (Figure S5E) and expressed them in mouse spermatids through testis transduction on 3- to 4-week-young mice. Although this experiment could only yield a small number of tdTomato+ spermatids, we managed to examine the effect of wild-type and mutant peptides on the expression of Plectin, a representative target, in tdTomato+ spermatids by immunostaining. We found that transduction of the MIWI-N peptide, but not MIWI-N2Δ111–120, led to an average of ~40% reduction of Plectin protein in mouse spermatids compared to untransduced cells (Figures 5D, S5F, and S5G). These results provide critical evidence for the functional requirement of the MIWI-eIF3f interaction for translation activation of piRNA targets in mouse spermatids.
Stage-Specific Association of MIWI with eIF3f/HuR during Spermatogenesis
Having established the functional requirement of MIWI and HuR proteins and the MIWI-eIF3f interaction for translation activation of piRNA targets in mouse spermatids, we next investigated how piRNA target mRNAs are selectively activated in a defined developmental window. To address this, we examined the assembly of the MIWI/eIF3f/HuR complex using isolated mouse male germ cells (Figure S6, left). By double immunostaining of MIWI and eIF3f, we found that MIWI was largely separated from eIF3f in SCs, but the MIWI-containing granules came to partially colocalize with or in close proximity to eIF3f-containing granules in round spermatids (RSs), and finally, MIWI and eIF3f were separated from each other again in elongating spermatids (ESs; Figure S6, right). This indicates that the association of MIWI with eIF3f is temporally regulated during spermatogenesis.
To further corroborate this, we performed coIP experiments on three isolated cell populations, again confirming that MIWI was inefficient in association with either eIF3f or HuR in enriched SCs but showing significant interactions with both in enriched RSs, which were greatly attenuated in enriched ESs/Eds (elongating spermatids [ESs]/elongated spermatids [Eds]) (Figure 5E). In contrast, we found that MIWI was switched to interact with the deadenylase CAF1 in ES/Ed (Figure 5E), which is consistent with our previous observation for the assembly of the pi-RISC in late spermatids (Gou et al., 2014). This indicates that MIWI is switched from the translation-enhancing complex to the RNA decay complex during the progression of spermiogenesis. In fact, we further noted that both eIF3f and HuR proteins were sharply reduced in abundance in ES/Ed, while the levels of CAF1 protein were relatively consistent in all stages of spermatids. Collectively, our results indicate that the association of MIWI with eIF3f/HuR or CAF1 is temporally regulated during spermatogenesis and that the MIWI-containing translation activation complex is mainly assembled in round spermatids.
Global Evidence for a Large MIWI-Activated Translation Program in Mouse Spermatids
To generalize the findings based on a few piRNA targets characterized above, we next assessed how many genes might be subjected to activation by the piRNA system in mouse spermatids by performing ribosome profiling in parallel with RNA sequencing (RNA-seq). Because spermatogenesis is arrested at step 4 spermatids (corresponding to 24-dpp; Deng and Lin, 2002), we chose to compare between wild-type and Miwi null testes at this developmental stage. Having first shown typical 3-nt periodicity of ribosome protected fragments (RPFs) and established the reproducibility of our libraries (Figures S7A and S7B), we identified 3,959 mRNAs that showed significantly reduced translational efficiency based on RNA-seq-normalized RPFs in Miwi null testes relative to wild-type control (Figure 6A; Table S3). Gene Ontology (GO) analysis showed that many direct targets are involved in spermatogenesis, spermatid development including acrosome and flagellum formation, and sperm motility (Figure S7C). We noted that 3 out of the 5 validated piRNA targets (i.e., Agfg1, Tbpl1, and Cnot4) were identified with significantly reduced translational efficiency in Miwi null testes and the translational efficiency of Atg12 appeared to be also reduced, though without sufficient statistical significance (Figure 6A; Table S3). However, few RPFs on the Plectin transcript were detected in either 24-dpp Miwi null or wild-type testes, suggesting little translation in spermatids prior to step 4, consistent with a previous report showing that Plectin protein began to be detectable at steps 8–9 spermatids (Johnson et al., 2004). These data suggest that MIWI may be directly or indirectly involved in regulating the translation of a large group of mRNAs in mouse testes.
Figure 6. A Subset of Genes in Mouse Spermatids Is Subjected to Activation by the piRNA System.
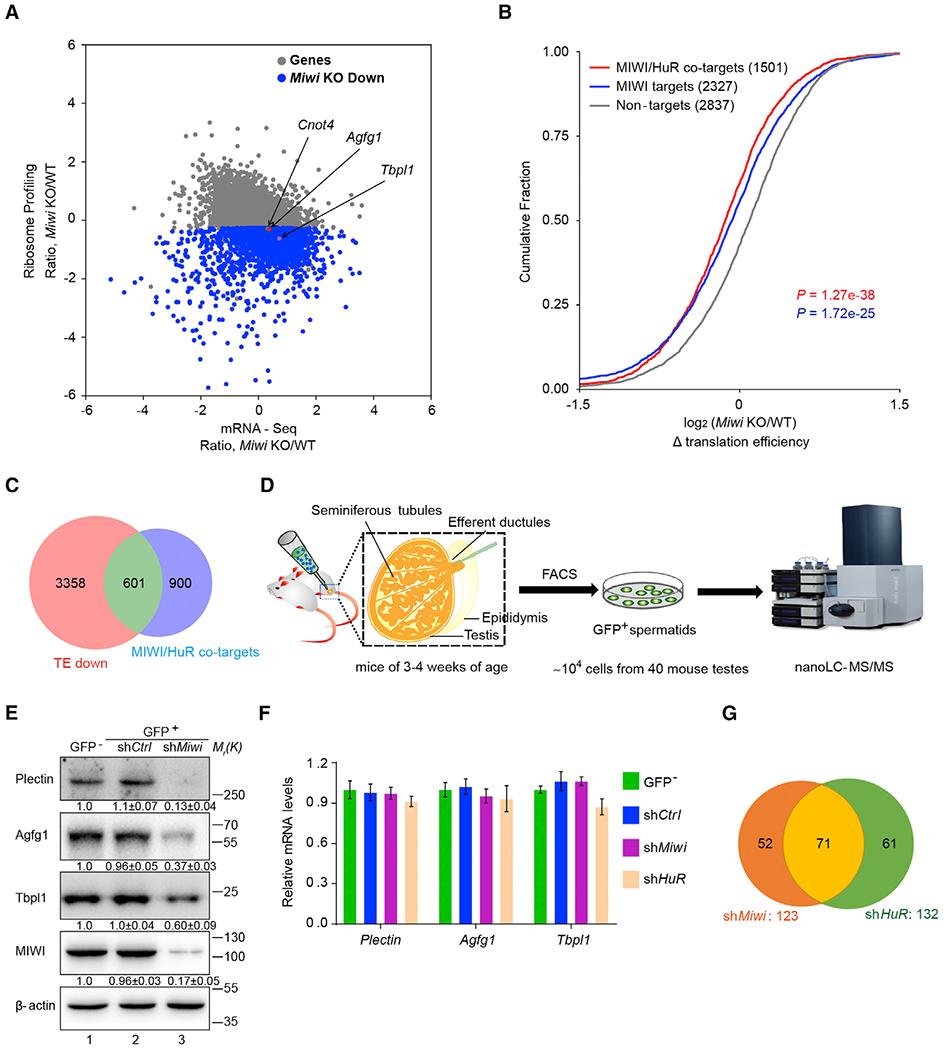
(A) Scatterplot showing mRNA level changes (x axis) against ribosome profiling changes (y axis) between Miwi null and wild-type testes. Arrows indicate the three validated piRNA-activated targets (i.e., Agfg1, Tbpl1, and Cnot4).
(B) Cumulative distribution of the log2-fold changes of translation efficiency (TE, ratio of ribosome-protected fragments [RPFs] and mRNA input) between the Miwi null and wild-type control samples for non-targets (no MIWI CLIP reads, gray line), MIWI targets (MIWI CLIP peaks in 3′ UTRs, blue line), and MIWI/HuR co-targets (containing both MIWI and HuR-binding sites in 3′ UTRs, red line)
(C) Venn diagram showing the cross of MIWI/HuR co-target mRNAs (light blue) and translational efficiency-reduced mRNAs (TE down, pink) in Miwi null testes.
(D) Schematic diagram illustrating the experimental design.
(E) Western blotting assay of Plectin, Agfg1, and Tbpl1 expression in sbMiwi:GFP-transduced spermatids (lane 3), with quantification of blotting intensity shown in parentheses (the one in control GFP− spermatids is set as 1.0 after normalization with β-actin blotting).
(F) qRT-PCR assay of the effect of Miwi or HuR knockdown on Plectin, Agfg1, and Tbpl1 mRNA levels in mouse spermatids, with β-actin served as an internal control.
(G) Venn diagram representing the number of proteins that were downregulated (>2-fold) in shMiwi:GFP and shHuR:GFP-transduced spermatids compared with pSilencer:GFP control.
Results shown are representative of three independent experiments. See also Figure S7 and Tables S3 and S4.
We next sorted out MIWI and HuR co-bound piRNA targets for further comparative analysis. Using our previous MIWI CLIP-seq data in mouse spermatids (Gou et al., 2014; Zhang et al., 2015) and reported HuR-binding motifs (Lebedeva et al., 2011), we categorized mRNA transcripts in mouse spermatids into three groups: (1) non-targets (no MIWI CLIP reads), (2) MIWI targets (MIWI CLIP peaks in 3′ UTRs), and (3) MIWI/HuR co-targets (containing both MIWI and HuR-binding sites in 3′ UTRs). Indeed, relative to non-targets, we observed significantly decreased translational efficiency for MIWI targets and more pronounced decrease for MIWI/HuR co-targets in Miwi null testes compared to wild-type control (p < 0.001, Mann-Whitney U test; Figure 6B). Cross analysis revealed that 601 out of the 1,501 MIWI/HuR co-targets showed significantly reduced translational efficiency in Miwi null testes (Figure 6C), implicating this subset of genes as direct targets. These results suggest that MIWI may promote translation of its target genes in testes via multiple mechanisms, one of which is through co-binding with HuR to achieve robust translation activation.
To further determine the role of the piRNA system in translation activation during spermiogenesis, we examined the global effect of MIWI or HuR knockdown on protein expression in mouse spermatids. To engineer MIWI or HuR depletion in round spermatids, we carried out testis transduction on young mice at 3–4 weeks of age, which allowed us to obtain ~104–105 GFP+ spermatids from 40 shHuR:GFP or shMiwi:GFP-transduced testes by unit gravity sedimentation coupled with fluorescence-activated cell sorting (FACS) procedures (Figure 6D). Western blotting verified a marked reduction of Plectin, Agfg1, and Tbpl1 proteins in shMiwi:GFP-transduced spermatids (Figure 6E), with few changes in their mRNA levels as quantified by qRT-PCR (Figure 6F). Using ~1 × 104 GFP+ spermatids, we performed protein profiling on a sensitive nanoLC-MS/MS (liquid chromatography-tandem mass specrometry) instrument and detected ~300–500 proteins in spermatids transduced with control pSilencer:GFP, shMiwi:GFP, or shHuR:GFP, respectively. We noted that Plectin showed a ~3- to 6-fold reduction in MIWI- or HuR-depleted spermatids relative to controls (Table S4). Importantly, we found 123 and 132 proteins that were significantly reduced (>2-fold) in shMiwi:GFP- or shHuR:GFP-transduced cells, respectively (Figure 6G; Table S4). Cross analysis revealed that 71 proteins were shared between MIWI- and HuR-regulated proteins (Figure 6G), among which 37 genes contain MIWI and HuR-binding sites in their 3′ UTRs. These results together strongly support that MIWI and HuR may act in the same pathway to activate the translation of a subset of mRNAs in mouse spermatids.
MIWI-Activated Genes Critical for Acrosome Formation
Finally, we wished to provide the biological significance for MIWI/piRNA-induced target activation during spermatid development. To this end, we constructed a pLKO.1-shRNA-Cyto IV-fused EGFP lentiviral vector that contains a mitochondrial targeting signal (Cyto IV) fused to EGFP (Cyto IV-EGFP; Figure 7A, top), thus allowing us to track the progression of sperm maturation from transduced spermatids. We detected GFP+ sperm in the epididymides of shMiwi-Cyto IV-EGFP-transduced mice, but not in shHuR-Cyto IV-EGFP or sheIF3f-Cyto IV-EGFP-transduced animals, indicating that both HuR and eIF3f have broader roles than MIWI in testes. Importantly, MIWI knockdown clearly impaired sperm maturation, because the counts of GFP+ sperm in shMiwi-Cyto IV-EGFP-transduced mice were dramatically reduced relative to the shRNA-Cyto IV-EGFP-transduced controls, supporting a functional requirement of MIWI for spermatozoa formation.
Figure 7. The piRNA System Is Required for Acrosome Formation during Spermiogenesis.
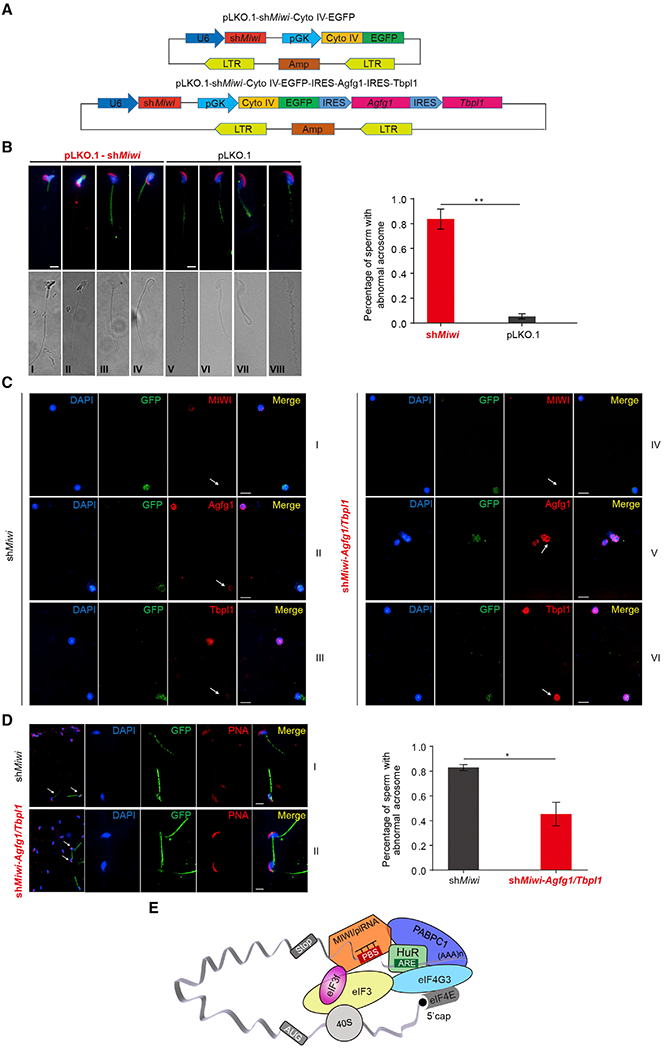
(A) Schematic diagram showing the design of lentiviral shRNA vector pLKO.1-shRNA-Cyto IV-EGFP for Miwi knockdown (top) as well as co-expression of shMiwi, Agfg1, and Tbpl1 (bottom). shRNA expression is driven by human U6 promoter, and Cyto IV (Cytochrome c oxidase subunit IV)-fused EGFP expression is driven by human phosphoglycerate kinase promoter (pGK). Cyto IV, a mitochondria-localized signal peptide; LTR, long terminal repeat; EGFP, enhanced green fluorescent protein; Amp, ampicillin resistance gene.
(B) Acrosome staining (Peanut agglutinin [PNA], red) of sperm progressed from shMiwi (left) or control shRNA-Cyto IV-EGFP-transduced spermatids (right), with nuclei counterstained by DAPI (blue). Left: representative PNA staining (top) and DIC micrograph images of GFP+ sperm; right: percentages of GFP+ sperm with abnormal acrosomes from indicated transduction (n = 100 per group). Scale bar, 10 μm.
(C) Immunofluorescent assay of MIWI (I and IV, red), Agfg1 (II or V, red) and Tbpl1 proteins (III and VI, red) in shMiwi (left) or shMiwi-Agfg/Tbpl1-transduced spermatids (right). GFP (green) indicates the transduced cells. Scale bar, 10 μm.
(D) Acrosome staining (red) of sperm progressed from shMiwi or shMiwi-Agfg1/Tbpl1-transduced spermatids (bottom), with nuclei counterstained by DAPI (blue). Left: representative PNA staining images of GFP+ sperm; right: percentages of GFP+ sperm with abnormal acrosomes from indicated transduction (n = 100 per group). Scale bar, 10 μm.
(E) Schematic model showing that MIWI/piRNA, eIF3f, and HuR cooperate to activate the translation of specific mRNAs in mouse spermatids. piRNAs guide MIWI to bind to the 3′ UTRs of ARE-containing target mRNAs and trigger mRNA looping through a direct interaction between MIWI and eIF3f; the mRNA looping is stabilized by ARE-guided HuR-binding to target mRNAs, thereby mediating the association of multiple other translation initiation factors, including eIF4G and PABPC1, to activate translation.
Results shown are representative of three independent experiments.
Given that Agfg1 and Tbpl1 have been shown to be essential for acrosome formation during spermatid development (Kang-Decker et al., 2001; Zhang et al., 2001), we inspected acrosomes in the sperm progressed from shMiwi-Cyto IV-EGFP-transduced spermatids by Peanut agglutinin (PNA) staining. Indeed, we observed severe defects at acrosomes in the shMiwi-Cyto IV-EGFP-transduced sperm, but not in the shRNA-Cyto IV-EGFP-transduced control (Figure 7B, top). These results demonstrated a functional requirement of MIWI for acrosome formation during spermiogenesis.
To further substantiate the functional role of MIWI/piRNA-induced target activation in acrosome formation, we asked whether the defective acrosome formation in shMiwi-Cyto IV-EGFP-transduced spermatids could be rescued by restoring Agfg1 or Tbpl1 protein in these cells. To this end, we constructed the pGK-driven Cyto IV-EGFP-IRES-Agfg1 or Tbpl1 into the pLKO.1-shMiwi lentiviral vector, in which shMiwi was co-expressed with the expression unit of Agfg1 or Tbpl1. We found little improvement of acrosomes in GFP+ sperm from either shMiwi-Agfg1 or shMiwi-Tbpl1-transduced mice (data not shown), suggesting that single restoration of either Agfg1 or Tbpl1 protein in MIWI-depleted spermatids was not enough to rescue acrosomes in resulting sperm. We thus attempted to insert the pGK-driven Cyto IV-EGFP-IRES-Agfg1-IRES-Tbpl1 into the pLKO.1-shMiwi vector (Figure 7A, bottom) to co-express shMiwi with Agfg1 and Tbpl1 proteins. Immunostaining of enriched spermatids showed a dramatic reduction of Agfg1 and Tbpl1 proteins in shMiwi-Cyto IV-EGFP-transduced cells relative to untransduced GFP− controls (Figure 7C, left) and successful restoration of both Agfg1 and Tbpl1 proteins in shMiwi-Agfg1/Tbpl1 vector-transduced spermatids (right). Remarkably, acrosomes were significantly recovered in >50% of GFP+ sperm from shMiwi-Agfg1/Tbpl1-transduced mice (Figure 7D). These results unequivocally demonstrated that restoration of Agfg1 and Tbpl1 expression is able to significantly rescue defective acrosome formation in MIWI-depleted spermatids. Together, these results demonstrated that translationally activated Agfg1 and Tbpl1 by the newly elucidated function of the piRNA system plays a central role in acrosome formation during spermiogenesis.
DISCUSSION
An Unprecedented Function of the piRNA Machinery in Translational Control
Post-transcriptional gene regulation plays a vital role in postmeiotic male germ cell development in mammals. Although previous studies have revealed specific mechanisms for translational control of several spermiogenic mRNAs (Kwon and Hecht, 1991, 1993; Zhong et al., 1999, 2001; Tsai-Morris et al., 2012; Kleene, 2013; Nguyen-Chi and Morello, 2011), how a large variety of repressed mRNAs are activated during spermiogenesis has remained an open question. We now provide evidence that the MIWI/piRNA system plays a key role in translation activation in mouse spermatids, thus uncovering a previously unknown function of the piRNA machinery in male germ cell development.
Our findings provide mechanistic insights into the reported association of mouse PIWI proteins with the translation machinery (Grivna et al., 2006; Unhavaithaya et al., 2009) and a putative role of MIWI in stabilizing certain spermiogenic mRNA in mouse testes (Vourekas et al., 2012). Interestingly, the Drosophila PIWI protein Aubergine was also reported to interact with several translation initiation factors to promote translation of self-renewal and differentiation factors in germline stem cells (Ma et al., 2017), and the same protein was suggested to help stabilize certain mRNAs in a piRNA-dependent manner (Dufourt et al., 2017). These observations suggest that piRNA-activated translation may be a conserved mechanism in animals.
Stage-Specific Recruitment of Translation Initiation Factors to MIWI/piRNA Targets
As schemed in Figure 7E, piRNA-programmed translation activation requires specific recruitment of eIF3f by MIWI. However, this alone seems to be insufficient to activate translation, which also depends on the presence of an ARE adjacent to a piRNA target site in 3′ UTR. Such AREs help recruit the RNA-binding protein HuR, which in turn attracts eIF4G3, a specific eIF4G isoform known to be required for male germ cell development (Hu et al., 2018). Interestingly, while the interaction between MIWI and eIF3f and the interaction between HuR and eIF4G3 are both independent of RNA, the formation of the MIWI/eIF3/HuR/eIF4G3 super-complex is RNA mediated. Therefore, although eIF3 has been reported to be able to recruit eIF4G but without specificity for different eIF4G isoforms in other cellular systems (Gingras et al., 1999), our data indicate that eIF4G3 might be recruited and then stabilized by both MIWI/eIF3 and HuR in a synergistic fashion.
Importantly, such a super-complex is formed specifically in round spermatids based on our coIP and co-colocalization data. This stage-specific complex assembly likely accounts for the switch of target mRNAs from the stored to the activated state. However, all of these factors appear to be constitutively expressed in multiple developmental stages from SCs to round spermatids to elongating spermatids with detectable decline only in later elongated spermatids. We currently do not know how the super-complex is assembled in such a stage-specific manner, which may involve a stage-specific factor(s) and/or post-translational modifications of an existing factor(s) to be investigated in future studies.
The MIWI/piRNA System Drives a Large Translation Activation Program in Testes
Our current study began with pursuing an unexpected activation of a few MIWI/piRNA targets. However, given the enormous repertoire of the piRNA targeting capacity and the prevalent distribution of HuR-binding sites in the 3′ UTRs of numerous mRNAs (Lebedeva et al., 2011), the initial characterized targets appear to be the tip of the iceberg. Indeed, by ribosome profiling using Miwi null testes, we identified a group of ~600 mRNAs as direct piRNA targets for such regulation in mouse spermatids, and our nanoLC-MS/MS analysis further confirmed a number of targets at the protein levels.
Importantly, we showed that this newly elucidated function of piRNA system is critical for acrosome formation during spermiogenesis. Moreover, besides abnormal acrosomes, we found several other defects in the shMiwi:Cyto IV-EGFP-transduced sperm, including abnormal morphology, curved back heads, etc. This implies the contribution of the MIWI/piRNA machinery to multiple developmental events during spermiogenesis via controlling various target genes. It will be interesting to determine whether and how the piRNA system plays such seemingly broad roles in facilitating the transformation of spermatids into spermatozoa.
A Dual Role of the MIWI/piRNA Machinery during Spermiogenesis
Our current findings extend in the function of the Piwi/piRNA machinery in regulating coding genes in animal germ cells. Previous studies from us and others demonstrate the function of piRNAs in inducing mRNA decay by using a miRNA/siRNA-like mechanism (Gou et al., 2014; Zhang et al., 2015; Goh et al., 2015; Watanabe et al., 2015; Vourekas et al., 2016; Shen et al., 2018; Zhang et al., 2018). Thus, the MIWI/piRNA machinery appears to have a dual function in regulating the fate of target mRNAs in mouse spermatids. In earlier stages of spermatids, the machinery is responsible for activating translation through the formation of the MIWI/piRNA/eIF3f/HuR super-complex. This complex appears to be switched to the MIWI/piRNA/CAF1 super-complex in later stages of spermatids to trigger massive mRNA elimination to inactivate the vast cellular programs in preparation for spermatozoa production from spermatids.
In conclusion, our present findings reveal an important role for the piRNA system in translation activation of spermiogenic mRNAs and demonstrate the critical importance of such regulation in acrosome formation during spermatid development in mice. Together with the elucidated function of the piRNA system in mRNA decay, our findings highlight a multifaceted function of the piRNA system in a highly orchestrated developmental program in male germ cells.
STAR★METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Mo-Fang Liu (mfliu@sibcb.ac.cn). All unique/stable reagents generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell Culture and Transfections
Two cell lines were used in this study: Female Human Embryonic Kidney 293T (293T) and Male mouse spermatocyte-derived GC-2spd (ts) cells have been authenticated. Cells were cultured in DMEM with 10% FBS. Transfections were performed with Lipofectamine 2000 (Invitrogen) for 293T cells or Lipofectamine 3000 (Invitrogen) for GC-2spd (ts) cells according to the manufacturer’s instructions. All DNA plasmids were cleared of endotoxin.
Mice
Male C57BL6 mice at 18-dpp, 30-dpp, 3-4 weeks (young) or 8 weeks (adult) of age were used in this study. Miwi knockout mice (Deng and Lin, 2002) were purchased from Mutant Mouse Resource Research Centers. Male Miwi null and control mice at 24-dpp of age were used in this study. All mice were housed in the SIBCB animal facility under SPF conditions in accordance with institutional guidelines and ethical regulations and fed with regular chow and water by the facility staff. All experimental animal procedures were approved by the Institutional Animal Care and Research Advisory Committee at SIBCB, CAS.
METHOD DETAILS
Plasmids and RNA oligonucleotides
For Renilla luciferase 3′ UTR reporters, the 3′ UTRs of mouse Plectin (~1.2 kb), Agfg1 (~1.2 kb), Tbpl1 (~1.9 kb), Cnot4 (~1.5 kb) and Atg12 (~2 kb) were cloned downstream of the Renilla luciferase gene in pRL-TK (Promega), respectively. To generate the mutant 3′ UTR reporters, seven or eight nucleotides in each 3′ UTR complementary to the 5′ portion of cognate piRNA were deleted using KOD-Plus-mutagenesis kit (Figure 1A). p3xFlag-MIWI, p3xFlag-HIWI, and Flag-Y345/346A HIWI were constructed as we recently reported. p3xFlag-eIF3f, p3xFlag-eIF4G3, pCMV-Myc-eIF3f, pCMV-Myc-HuR, pCMV-Myc-PABPC1 and pCMV-Myc-PABPC2 were constructed through insertion of the cDNAs without the 3′ UTRs of mouse eIF3f (NM_025344.2), eIF4g3 (NM_172703.3), HuR (NM_010485.3), Pabpc1 (NM_008774.3) and Pabpc2 (NM_011033.2) into p3 × Flag-CMV-10 (Sigma) and pCMV-Myc (Clontech), respectively. p3xFlag-MIWI was used to generate MIWI truncation mutants (Figure S5A) with KOD-Plus-mutagenesis kit. To construct GST-fused eIF3f for bacterial expression, mouse eIF3f cDNA was inserted into pGEX-KG (ATCC). The lentiviral shRNA vectors targeting mouse Miwi and HuR were constructed using pSilencer-H1-LV as we recently described (Gou et al., 2014; Zhao et al., 2013). The lentiviral vectors pLKO.1-shRNA-CytoIV-fused EGFP were generated using pLKO.1 (Addgene), and inserted with pGK-driven CytoIV-EGFP [a mitochondrial targeting signal (Cyto) fused EGFP] or pGK-driven CytoIV-EGFP-IRES-Agfg1-IRES-Tbpl1 (Figure 7A). The lentiviral vectors pLVX-Flag-MIWI-N2-IRES-tdTomato and pLVX-Flag-MIWI-N2Δ111–120-IRES-tdTomato were generated using pLVX-IRES-tdTomato (Clontech; Figure S5E). All constructs were confirmed by DNA sequencing. All RNA oligonucleotides, including piRNAs and siRNAs, were synthesized by Ribobio or Biotend (China), and their sequences were listed in the Key Resource Table and Tables S5 and S6. All chemically synthetic piRNAs are containing phosphate at the 5′ ends and 2′-O-methylation at the 3′ ends.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-MIWI | This paper | Gou et al., 2014 |
| Mouse monoclonal antibody anti-Flag | Sigma | Cat#F3165; RRID: AB_259529 |
| Mouse monoclonal antibody anti-β-actin | Sigma | Cat#A3854; RRID: AB_262011 |
| Rabbit polyclonal anti-c-Myc | Sigma | Cat#C3956; RRID: AB_439680 |
| Mouse monoclonal antibody anti-HuR | Invitrogen | Cat#39-0600; RRID: AB_2533394 |
| Mouse monoclonal antibody anti-GFP | Abways | Cat#AB0005 |
| Mouse monoclonal antibody anti-AGO2 | Abcam | Cat#ab57113; RRID: AB_2230916 |
| Goat polyclonal anti-Mili | Santa Cruz | Cat#sc-67504;RRID: AB_2283941 |
| Goat polyclonal anti-PIWIL1 | Santa Cruz | Cat#sc-22685;RRID: AB_2165430 |
| Goat polyclonal anti-Plectin | Santa Cruz | Cat#sc-7572; RRID: AB_654218 |
| Rabbit polyclonal anti-Agfg1 | Proteintech | Cat#12670-1-AP; RRID: AB_2258077 |
| Rabbit polyclonal anti-Tbpl1 | Proteintech | Cat#12258-1-AP; RRID: AB_10642841 |
| Rabbit polyclonal anti-eIF3f | Abgent | Cat#AP2900a; RRID: AB_1967585 |
| Rabbit polyclonal anti-eIF4G3 | Novus | Cat#NBP2-16309 |
| Rabbit polyclonal anti-GFP | MBL | Cat#598 |
| Rabbit polyclonal anti-eIF3a | Bioworld | Cat#BS70526 |
| Rabbit polyclonal anti-eIF3c | Bioworld | Cat#BS8653 |
| Bacterial and Virus Strains | ||
| TOP10 chemically competent E.coli | TIANGEN | CB104 |
| BL21 chemically competent E.coli | TIANGEN | CB105 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Cycloheximide | MCE | HY-12320 |
| Turbo DNase | Invitrogen | AM2238 |
| SUPERase·In | Invitrogen | AM2694 |
| GST-eIF3f | This paper | N/A |
| Critical Commercial Assays | ||
| Lipofectamine 2000 | ThermoFisher | 11668019 |
| Lipofectamine 3000 | ThermoFisher | L3000015 |
| RNAiso Plus | Takara | 9109 |
| PrimeScript RT reagent Kit with gDNA Eraser | Takara | RR047A |
| SYBR Premix Ex Taq II | Takara | RR820A |
| Deposited Data | ||
| Elongating spermatids CLIP-seq data | Gou et al., 2014 | SRP043638 |
| Round spermatids CLIP-seq data | Zhang et al., 2015 | GSE67683 |
| Ribosome profiling | This paper | GSE139399 |
| Experimental Models: Cell Lines | ||
| Human: 293T | ATCC | CRL-3216 |
| Mouse: GC-2spd (ts) | ATCC | CRL-2196 |
| Experimental Models: Organisms/Strains | ||
| C57BL6 male mice | SLAC | N/A |
| B6.129-Piwil11Hfl/Mmmh | MMRRC | 29995 |
| Oligonucleotides | ||
| Sequences for qPCR Primers, see Table S5 | This paper | N/A |
| Sequences for shRNA primers, see Table S5 | This paper | N/A |
| Sequences for piRNAs, see Table S6 | This paper | N/A |
| Sequences for siRNAs, see Table S6 | This paper | N/A |
| Recombinant DNA | ||
| pRL-TK | Promega | E2241 |
| pRL-TK-Plectin-3′ UTR | This paper | N/A |
| pRL-TK-Agfg1-3′ UTR | This paper | N/A |
| pRL-TK-Tbpl1-3′ UTR | This paper | N/A |
| pRL-TK-Cnot4-3′ UTR | This paper | N/A |
| pRL-TK-Atg12-3′ UTR | This paper | N/A |
| pGL3 | Promega | E1761 |
| p3xFLAG-CMV™-10 | Sigma | E7658 |
| p3xFLAG-CMV™-10-MIWI | This paper | N/A |
| p3xFLAG-CMV™-10-HIWI | This paper | N/A |
| p3xFLAG-CMV™-10-HIWI-Y345/346A | This paper | N/A |
| p3xFLAG-CMV™-10-eIF3f | This paper | N/A |
| pCMV-Myc | Clontech | 631604 |
| pCMV-Myc-eIF3f | This paper | N/A |
| pCMV-Myc-HuR | This paper | N/A |
| pCMV-Myc-PABPC1 | This paper | N/A |
| pCMV-Myc-PABPC2 | This paper | N/A |
| pGEX-KG | ATCC | 77103 |
| pGEX-KG-eIF3f | This paper | N/A |
| pSilencer-H1 | N/A | N/A |
| pSilencer-H1-shMiwi | This paper | N/A |
| pSilencer-H1-shHuR | This paper | N/A |
| pLKO.1 | Addgene | N/A |
| pLKO.1-Cyto IV-EGFP | This paper | N/A |
| pLKO.1-shMiwi-CytoIV-EGFP | This paper | N/A |
| pLKO.1-shHuR-CytoIV-EGFP | This paper | N/A |
| pLKO.1-shMiwi-CytoIV-EGFP-IRES-Agfg1-IRES-Tbpl1 | This paper | N/A |
| p3xFLAG-CMV-10-MIWI-N | This paper | N/A |
| p3xFLAG-CMV-10-MIWI-PAZ+MID | This paper | N/A |
| p3xFLAG-CMV-10-MIWI-PIWI | This paper | N/A |
| p3xFLAG-CMV-10-MIWI-N1 | This paper | N/A |
| p3xFLAG-CMV-10-MIWI-N2 | This paper | N/A |
| p3xFLAG-CMV-10-MIWI-N3 | This paper | N/A |
| p3xFLAG-CMV-10-MIWI-N4 | This paper | N/A |
| p3xFLAG-CMV-10-MIWI-N2Δ111–120 | This paper | N/A |
| p3xFLAG-CMV-10-eIF4G3 | This paper | N/A |
| pLVX-IRES-tdTomato | Clontech | 631238 |
| pLVX-FLAG-MIWI-N2-IRES-tdTomato | This paper | N/A |
| pLVX-FLAG-MIWI-N2Δ111–120-IREStdTomato | This paper | N/A |
| Software and Algorithms | ||
| GraphPad Prism | GraphPad | N/A |
| Photoshop CS3 | Adobe | N/A |
| ImageJ | NIH | https://imagej.nih.gov/ij/ |
Luciferase reporter assay
Luciferase reporter assays were carried out as we described previously (Gou et al., 2014). In brief, GC-2spd (ts) cells were co-transfected in 24-well plates with chemically synthetic piRNAs, Renilla luciferase 3′ UTR reporters, or/and siRNAs or protein expression vectors. The firefly luciferase plasmid pGL3 was used as an internal control. Dual luciferase reporter assays were performed 24 h after transfection using Dual-Glo Luciferase Assay Systems (Promega).
Immunoprecipitation, immunoblotting, and immunofluorescent microscopy (IF)
For immunoprecipitation, cells or testicular tissue were homogenized in iced lysis buffer [50 mM Tris-HCl (pH 7.4), 1% Triton X-100, 150 mM NaCl, 5 mM EDTA, and protease inhibitor cocktail]. Cell or tissue extracts were incubated with antibody-coupled Protein A beads or Protein G beads (Sigma) according to the manufacturer’s Instructions. For immunoblotting, IP pellets or cell/tissue extracts were diluted in SDS loading buffer and subjected to standard SDS-PAGE and western blotting procedures. Western blotting images were captured by Tanon-5200 Chemiluminescent Imaging System (Tanon). For IF, enriched spermatids or sperm from the cauda epididymis were fixed with 4% paraformaldehyde, permeabilized with 0.5% Triton X-100 in PBS, and incubated with primary antibodies and Alexa Fluor 488 or Cy3-conjugated secondary antibodies or with PNA conjugated with Alexa Fluor 594 (Molecular probes). Nuclei were counterstained with DAPI (Vector Laboratories). Images were captured using a Nikon DS-Ri2 microscope, and immunofluorescent densities were quantified using imageJ software.
RNA isolation, qRT-PCR, and RIP assays
The assays were performed as we described recently (Zhao et al., 2013; Gou et al., 2014). In brief, total RNAs in cells or tissues were extracted with RNAiso Plus (Takara) according to the manufacturer’s protocol. Reverse transcription and qRT-PCR were performed using PrimeScript RT and SYBR Green kits (Takara) on a QuantStudio 3 system (Applied Biosystems) according to the manufactures’ protocols. For RIP assays, cells or tissues were homogenized in lysis buffer [100 mM KCl, 5 mM MgCl2,10 mM HEPES, 0.5% NP-40 containing 10 U/mL RNase inhibitor (Takara) and a protease inhibitor cocktail (Roche)] and extracts were incubated with IgG, anti-MIWI, or anti-HuR antibody-coupled Protein A/G for 4 h at 4°C. RIP analyses of sucrose gradient fractions were performed as described previously (Vasudevan and Steitz, 2007). In brief, the indicated fractions derived from sucrose gradients were incubated with IgG, anti-MIWI, or anti-HuR antibody-coupled Protein A/G for 4 h at 4°C. After stringently washing the beads with washing buffer [50 mM Tris-HCl (pH 7.4), 0.1% Triton X-100, 500 mM NaCl, 5 mM EDTA, protease inhibitor cocktail and RNase inhibitor], total RNAs were extracted with TRIzol and subjected to qRT-PCR assays. The primer sequences for qRT-PCR were provided in the Key Resource Table and Table S5. The results were analyzed and shown as relative piRNA or mRNA levels of the CT (cycle threshold) values, which were then converted as fold change.
GST pull-down
GST pull-down was performed as we previously described (Zhao et al., 2013). In brief, MIWI protein was prepared in the presence of S35-labeled methionine using the transcription and translation (TNT) system (Promega). GST-fusion eIF3f was expressed in E.coli BL21, purified and immobilized on glutathione agarose beads. The beads were blocked with PBS containing 0.1% NP-40 (v/v), 5 mM EDTA, 5 mM EGTA, 20 mM leupeptin, 25 mg/mL ALLN, 5 mg/mL pepstatin A, and 2 mg/mL aprotinin plus 5% nonfat milk, and incubated with [35S]Met-MIWI for 2 h at 4°C. After five time stringently washing, the bound proteins were dissolved in SDS sample buffer, separated by SDS-PAGE, and subjected to autoradiography.
Sucrose gradient analysis
Sucrose gradient analysis was performed as described previously (Hawthorne et al., 2006). In brief, cells or testicular tissues from 18-dpp or 30-dpp mice were homogenized in extraction buffer (PEB) [100 mM NaCl, 50 mM Tris·HCl, pH 7.5, 5 mM MgCl2,1% Triton X-100,100 μg/mL cycloheximide (CHX)], and were subjected to centrifugation at 13,000 g for 2 min to remove nuclear pellets. The extracts were then layered over 10%–60% sucrose gradients and centrifuged at 39,000 rpm on the SW 41 rotor for 2 h at 4°C. Twelve fractions were collected, and each fraction was subjected to western blotting and RT-qPCR analyses of indicated proteins and RNAs, respectively. For Sucrose gradient analysis of GC-2spd (ts) cells, 24 h post-transfection, cells were pre-incubated with cycloheximide (Sigma; 100 μg/mL for 15 min) and then subjected to preparation of cytoplasmic lysates. The lysates were fractionated by ultra-centrifugation through 10%–60% sucrose gradients and then separated into ~1,200 fractions using a Piston Gradient Fractionator (Biocomp, Fredericton, Canada).
Isolation of mouse SCs, STs, and GFP+ spermatids
Isolation of SCs and STs from mouse testes was performed as we recently described (Zhao et al., 2013; Gou et al., 2014). In brief, total spermatogenic cells were extracted from the seminiferous tubules of indicated male mice, and SCs and STs were separated through the unit gravity sedimentation procedure. Then round spermatids and later stages of spermatids (consisting of ESs and Eds) were separated from haploid spermatid fractions through FACS. GFP+ spermatids were sorted through FACS from haploid spermatid fractions obtained from transduced testes. The isolated spermatogenic cells were all confirmed by their distinct nuclear morphology (DAPI staining of nuclei).
Lentivirus packaging and testis transduction
Lentivirus packaging and testis transduction were performed as we recently described (Zhao et al., 2013). In brief, the lentiviral vectors were packaged into pseudovirus with their cognate packaging plasmids following standard procedures. High-titer lentivirus (> 108 transduction units/mL) was prepared by ultra-centrifugation. About 10 μL of high-titer lentivirus were injected into seminiferous tubule through the efferent duct via a sharp glass capillary with a tip diameter of 50 μm. The testes were harvested a week post-transduction of shRNA expression vectors for examining transduced spermatids, while epididymides were harvested two weeks post-transduction for examining sperm progressed from transduced spermatids.
Proteomics and protein profiling analyses
Proteomics analysis of anti-MIWI or anti-HuR IPed complexes from mouse testicular lysates or polysome fractions from mouse testicular lysates prepared by 10%–60% sucrose gradients were analyzed by gel-free LC-MS/MS (Thermo Scientific) according to the manufacture’s protocol. In brief, Cells were lysed in a buffer (8 M urea, 4% CHAPS, 40 mM Tris-HCl, 65 mM DTT, 1 mM PMSF, 1 mM EDTA, 0.5 mM EGTA). After shaking for 1 min, the cells were subjected to ultrasonic treatment for 10 min (at an 80 W sonic power, working for 3 s and stop for 3 s). Lysates were then clarified at 20,000 g for 15 min. Total protein concentrations were determined using the bicinchoninic acid (BCA) protein assay kit. The proteins were alkylated with 300 mM iodoacetamide in the dark for 40 min, and digested with trypsin (1:25, Promega) overnight at 37°C via the FASP protocol with spin ultrafiltration units of nominal molecular weight cutoff 10,000 (Wiśniewski et al., 2009). Protein profiling of GFP+ spermatids (~1×104) were performed on the nanoLC-MS/MS system. The nanoLC-MS/MS analysis was performed using an EASY-nL 1200 system coupled online to a QExactive plus mass spectrometer (Thermo Scientific, Bremen, Germany). Briefly, aliquots containing 1-2 μg of peptides were injected and separated on a reversed-phase column (15cm μ 50 μm, 2 μm, Dionex, Thermofisher) using a 2 h acetonitrile gradient in 0.1% formic acid at a flow rate of 300 nL/min.
The MaxQuant (version 1.5.3.30) software was used to analyze the MS/MS raw data. For the database searching parameters, the precursor mass tolerance was set to 15 ppm. Trypsin/P was set as the protease, accounting for in-source fragmentation of lysine or arginine residues followed by proline. Two missed cleavages were allowed. All data were searching against with the UniProt Mouse database (sequences) including oxidation (+15.9949 Daltons) of methionine as dynamic modifications, and carbamidomethylation (+57.0215 Daltons) of cysteine as static modifications.
Ribosome profiling
Ribosome profiling was performed with a procedure modified from the method described previously (Ingolia et al., 2012). In brief, mouse testicular lysates were generated using 24-dpp Miwi−/− testes or wild-type controls in the presence of 100 μg/mL cycloheximide. The DNase-treated lysates were clarified by centrifugation and subjected to RNA extraction & sequencing or ribosome profiling analyses. For ribosome profiling, the lysates were further layered over 10%–60% sucrose gradients to prepare polysome fractions. The obtained polysomes were treated with RNase I and then subjected to purification of ~30 nt ribosome protected fragments using a 15% TBE-Urea gel. The fragments were subjected to RNA sequencing analysis using the Illumina sequencing technology, and sequences were processed and analyzed in the same manner as CLIP-seq data below.
QUANTIFICATION AND STATISTICAL ANALYSIS
CLIP-seq analysis
For CLIP-seq analysis, the anti-MIWI CLIP-seq data in enriched mouse spermatids were obtained from the Gene Expression Omnibus: SRP043638 and GSE67683 (Gou et al., 2014; Zhang et al., 2015). For quality control, the reads of CLIP-seq were subjected to removal of adapter sequences by cutadapt program (Martin, 2011) under parameters “—max-n 3 –e 0.1”. For the samples from GSE67683, the first 4 nt was extracted as barcode, and only one read from the reads group with the same sequence and barcode was kept to remove potential PCR bias. The reads that can be mapped to mouse rRNA sequences were removed using bowtie program (Langmead et al., 2009). The reads longer than 15-nt were mapped to GRCm38.p4 genome sequences by bowtie2 under parameters “--no-unal --end-to-end -k 10 --al” (Langmead and Salzberg, 2012). Only the primary alignments were retained for analysis. To call MIWI peaks, the transcript was scanned using a 10-nt sliding window with 1-nt step. The window was called as positive if the mean coverage was larger than or equal to sample-specific threshold from 3 to 6 by library size, and overlapping positive windows were merged as peaks. The genes with peak at their 3′ UTR are considered as MIWI-CLIP target genes, among which the genes containing more than two HuR motifs (UUUUUUU, UUUAUUU, UUUGUUU, UAUUUAU, AUUUUUA, AUUUAUU, AAUUUUA, and AAUAUUU) in their 3′ UTR are considered as common target genes.
RNA-seq and Ribo-seq analyses
For RNA-seq and Ribo-seq analyses, the adaptor sequences were first removed from Illumina sequencing reads. For RNA-seq reads, the adapters were trimmed by cutadapt program (Martin, 2011) with parameters “-m 18 -e 0.15,” while for Ribo-seq reads, the adapters were trimmed, and the random barcodes were extracted and appended to read names by custom script. To remove potential rRNA contamination, the reads were mapped to rRNA sequences with bowtie program (Langmead et al., 2009), and those not mapped were then mapped to the mouse genome with gene annotation (GRCm38.p4 and Gencode.vM20) by using STAR program (Dobin et al., 2013) in global mode. The primary and proper-paired alignments were extracted from mapping results by BAMTools (Barnett et al., 2011) for further downstream analysis. For Ribo-seq samples, only one fragment from those with the same genomic signature (chromosome, start, end, and gaps) and same barcode was kept. For fragments within range from 28 nt to 30 nt, the 13th base located at the 1st position of codons were treated as canonical ribosome-protected fragments (RPFs) for the translation efficiency analysis. The FPKMcds (FPKM of CDS region) of each transcript was calculated as C * 1e9 / (L * N), where C represents the count of fragments that overlap with CDS region; L is the length of CDS; and N represents the total fragment count of the library. FPKMcds_RPF and FPKMcds_rna for each transcript were calculated from Ribo-seq and matched RNA-seq data, respectively. The translation efficiency was calculated as FPKMcds_RPF/FPKMcds_rna, and only those transcripts with FPKMcds_rna > = 1 and Ccds > = 50 were used for downstream analysis.
Other analyses
Gene Ontology (GO) term analyses were performed by DAVID (https://david.ncifcrf.gov/summary.jsp).
Student’s t test was performed to compare the differences between treated groups relative to their paired controls. All results were presented as the mean ± SD P-values are indicated in the text or figures above the two groups compared with a value < 0.05 (denoted by asterisks) considered significant (***p < 0.001, **p < 0.01).
DATA AND CODE AVAILABILITY
All data presented are available in the main text and supplementary materials. The accession number for Ribo-seq reported in this paper is GSE139399.
Supplementary Material
Highlights.
MIWI/piRNA activates mRNA translation via imperfect base-pairing interactions
HuR and eIF3f are required for MIWI/piRNA-mediated target mRNA activation
piRNA system controls the translation of a subset of mRNAs in mouse spermatids
piRNA system plays a central role in acrosome formation during spermiogenesis
ACKNOWLEDGMENTS
We thank members of the M.-F.L. and Y.Z. labs and Dr. Hong Cheng from SIBCB, CAS, for helpful comments. We thank Drs. Jianhua Yang from Sun Yat-sen University; Hu Zhou and Hongwen Zhu from SIMM, CAS; Peng Zhang from the University of Texas MD Anderson Cancer Center; and Xiaorong Zhang and Yuanchao Xue from IBP, CAS, for experimental assistance. This work was supported by grants from the National Key R&D Program of China (2017YFA0504400), Chinese Academy of Sciences (“Strategic Priority Research Program” grant XDB19010203), National Natural Science Foundation of China (31830109, 31821004, 91940305, and 91640201), Shanghai Municipal Science and Technology Major Project (17JC1420100, 19JC1410200, and 2017SHZDZX01), and the Foundation of Key Laboratory of Gene Engineering of the Ministry of Education. P.D. was supported by the SA-SIBS Scholarship Program and China Postdoctoral Science Foundation (2016M600335). L.-T.G. and X.-D.F. were supported by the NIH (grants GM052872 and HG004659).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.cell.2019.11.022.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Barnett DW, Garrison EK, Quinlan AR, Strömberg MP, and Marth GT (2011). BamTools: a C++ API and toolkit for analyzing and managing BAM files. Bioinformatics 27, 1691–1692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng W, and Lin H (2002). miwi, a murine homolog of piwi, encodes a cytoplasmic protein essential for spermatogenesis. Dev. Cell 2, 819–830. [DOI] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dufourt J, Bontonou G, Chartier A, Jahan C, Meunier AC, Pierson S, Harrison PF, Papin C, Beilharz TH, and Simonelig M (2017). piRNAs and Aubergine cooperate with Wispy poly(A) polymerase to stabilize mRNAs in the germ plasm. Nat. Commun 8, 1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gingras AC, Raught B, and Sonenberg N (1999). eIF4 initiation factors: effectors of mRNA recruitment to ribosomes and regulators of translation. Annu. Rev. Biochem 68, 913–963. [DOI] [PubMed] [Google Scholar]
- Goh WS, Falciatori I, Tam OH, Burgess R, Meikar O, Kotaja N, Hammell M, and Hannon GJ (2015). piRNA-directed cleavage of meiotic transcripts regulates spermatogenesis. Genes Dev. 29, 1032–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gou LT, Dai P, Yang JH, Xue Y, Hu YP, Zhou Y, Kang JY, Wang X, Li H, Hua MM, et al. (2014). Pachytene piRNAs instruct massive mRNA elimination during late spermiogenesis. Cell Res. 24, 680–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gou LT, Kang JY, Dai P, Wang X, Li F, Zhao S, Zhang M, Hua MM, Lu Y, Zhu Y, et al. (2017). Ubiquitination-Deficient Mutations in Human Piwi Cause Male Infertility by Impairing Histone-to-Protamine Exchange during Spermiogenesis. Cell 169, 1090–1104.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grivna ST, Pyhtila B, and Lin H (2006). MIWI associates with translational machinery and PIWI-interacting RNAs (piRNAs) in regulating spermatogenesis. Proc. Natl. Acad. Sci. USA 103, 13415–13420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasuwa H, Ishino K, and Siomi H (2018). Human PIWI (HIWI) is an azoospermia factor. Sci. China Life Sci 61, 348–350. [DOI] [PubMed] [Google Scholar]
- Hawthorne SK, Busanelli RR, and Kleene KC (2006). The 5′ UTR and 3′ UTR of the sperm mitochondria-associated cysteine-rich protein mRNA regulate translation in spermatids by multiple mechanisms in transgenic mice. Dev. Biol 297, 118–126. [DOI] [PubMed] [Google Scholar]
- Hofmann MC, Abramian D, and Millán JL (1995). A haploid and a diploid cell coexist in an in vitro immortalized spermatogenic cell line. Dev. Genet 16, 119–127. [DOI] [PubMed] [Google Scholar]
- Hu J, Sun F, and Handel MA (2018). Nuclear localization of EIF4G3 suggests a role for the XY body in translational regulation during spermatogenesis in mice. Biol. Reprod 98, 102–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imataka H, Gradi A, and Sonenberg N (1998). A newly identified N-terminal amino acid sequence of human eIF4G binds poly(A)-binding protein and functions in poly(A)-dependent translation. EMBO J. 17, 7480–7489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingolia NT, Brar GA, Rouskin S, McGeachy AM, and Weissman JS (2012). The ribosome profiling strategy for monitoring translation in vivo by deep sequencing of ribosome-protected mRNA fragments. Nat. Protoc 7, 1534–1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwasaki YW, Siomi MC, and Siomi H (2015). PIWI-Interacting RNA: Its Biogenesis and Functions. Annu. Rev. Biochem 84, 405–433. [DOI] [PubMed] [Google Scholar]
- Johnson KJ, Zecevic A, and Kwon EJ (2004). Protocadherin alpha3 acts at sites distinct from classic cadherins in rat testis and sperm. Biol. Reprod 70, 303–312. [DOI] [PubMed] [Google Scholar]
- Kang-Decker N, Mantchev GT, Juneja SC, McNiven MA, and van Deursen JM (2001). Lack of acrosome formation in Hrb-deficient mice. Science 294, 1531–1533. [DOI] [PubMed] [Google Scholar]
- Kimura M, Ishida K, Kashiwabara S, and Baba T (2009). Characterization of two cytoplasmic poly(A)-binding proteins, PABPC1 and PABPC2, in mouse spermatogenic cells. Biol. Reprod 80, 545–554. [DOI] [PubMed] [Google Scholar]
- Kleene KC (2013). Connecting cis-elements and trans-factors with mechanisms of developmental regulation of mRNA translation in meiotic and haploid mammalian spermatogenic cells. Reproduction 146, R1–R19. [DOI] [PubMed] [Google Scholar]
- Kwon YK, and Hecht NB (1991). Cytoplasmic protein binding to highly conserved sequences in the 3′ untranslated region of mouse protamine 2 mRNA, a translationally regulated transcript of male germ cells. Proc. Natl. Acad. Sci. USA 88, 3584–3588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon YK, and Hecht NB (1993). Binding of a phosphoprotein to the 3′ untranslated region of the mouse protamine 2 mRNA temporally represses its translation. Mol. Cell. Biol 13, 6547–6557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, and Salzberg SL (2009). Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10, R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebedeva S, Jens M, Theil K, Schwanhäusser B, Selbach M, Landthaler M, and Rajewsky N (2011). Transcriptome-wide analysis of regulatory interactions of the RNA-binding protein HuR. Mol. Cell 43, 340–352. [DOI] [PubMed] [Google Scholar]
- Ma X, Zhu X, Han Y, Story B, Do T, Song X, Wang S, Zhang Y, Blanchette M, Gogol M, et al. (2017). Aubergine Controls Germline Stem Cell Self-Renewal and Progeny Differentiation via Distinct Mechanisms. Dev. Cell 41, 157–169.e5. [DOI] [PubMed] [Google Scholar]
- Marchione R, Leibovitch SA, and Lenormand JL (2013). The translational factor eIF3f: the ambivalent eIF3 subunit. Cell. Mol. Life Sci 70, 3603–3616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet 17. 10.14806/ej.17.1.200. [DOI] [Google Scholar]
- Meikar O, Da Ros M, Korhonen H, and Kotaja N (2011). Chromatoid body and small RNAs in male germ cells. Reproduction 142, 195–209. [DOI] [PubMed] [Google Scholar]
- Meistrich ML, and Hess RA (2013). Assessment of spermatogenesis through staging of seminiferous tubules. Methods Mol. Biol 927, 299–307. [DOI] [PubMed] [Google Scholar]
- Merrick WC (2015). eIF4F: a retrospective. J. Biol. Chem 290, 24091–24099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen-Chi M, Auriol J, Jégou B, Kontoyiannis DL, Turner JM, de Rooij DG, and Morello D (2011). The RNA-binding protein ELAVL1/HuR is essential for mouse spermatogenesis, acting both at meiotic and postmeiotic stages. Mol. Biol. Cell 22, 2875–2885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen-Chi M, Chalmel F, Agius E, Vanzo N, Khabar KS, Jégou B, and Morello D (2009). Temporally regulated traffic of HuR and its associated ARE-containing mRNAs from the chromatoid body to polysomes during mouse spermatogenesis. PLoS ONE 4, e4900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen-Chi M, and Morello D (2011). RNA-binding proteins, RNA granules, and gametes: is unity strength? Reproduction 142, 803–817. [DOI] [PubMed] [Google Scholar]
- Sassone-Corsi P (2002). Unique chromatin remodeling and transcriptional regulation in spermatogenesis. Science 296, 2176–2178. [DOI] [PubMed] [Google Scholar]
- Shen EZ, Chen H, Ozturk AR, Tu S, Shirayama M, Tang W, Ding YH, Dai SY, Weng Z, and Mello CC (2018). Identification of piRNA Binding Sites Reveals the Argonaute Regulatory Landscape of the C. elegans Germline. Cell 172, 937–951.e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siomi MC, Sato K, Pezic D, and Aravin AA (2011). PIWI-interacting small RNAs: the vanguard of genome defence. Nat. Rev. Mol. Cell Biol 12, 246–258. [DOI] [PubMed] [Google Scholar]
- Steger K (1999). Transcriptional and translational regulation of gene expression in haploid spermatids. Anat. Embryol. (Berl.) 199, 471–487. [DOI] [PubMed] [Google Scholar]
- Sun F, Palmer K, and Handel MA (2010). Mutation of Eif4g3, encoding a eukaryotic translation initiation factor, causes male infertility and meiotic arrest of mouse spermatocytes. Development 137, 1699–1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai-Morris CH, Sato H, Gutti R, and Dufau ML (2012). Role of gonadotropin regulated testicular RNA helicase (GRTH/Ddx25) on polysomal associated mRNAs in mouse testis. PLoS ONE 7, e32470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unhavaithaya Y, Hao Y, Beyret E, Yin H, Kuramochi-Miyagawa S, Nakano T, and Lin H (2009). MILI, a PIWI-interacting RNA-binding protein, is required for germ line stem cell self-renewal and appears to positively regulate translation. J. Biol. Chem 284, 6507–6519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasudevan S, and Steitz JA (2007). AU-rich-element-mediated upregulation of translation by FXR1 and Argonaute 2. Cell 128, 1105–1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vourekas A, Zheng Q, Alexiou P, Maragkakis M, Kirino Y, Gregory BD, and Mourelatos Z (2012). Mili and Miwi target RNA repertoire reveals piRNA biogenesis and function of Miwi in spermiogenesis. Nat. Struct. Mol. Biol 19, 773–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vourekas A, Alexiou P, Vrettos N, Maragkakis M, and Mourelatos Z (2016). Sequence-dependent but not sequence-specific piRNA adhesion traps mRNAs to the germ plasm. Nature 531, 390–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe T, Cheng EC, Zhong M, and Lin H (2015). Retrotransposons and pseudogenes regulate mRNAs and lncRNAs via the piRNA pathway in the germline. Genome Res. 25, 368–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiśniewski JR, Zougman A, Nagaraj N, and Mann M (2009). Universal sample preparation method for proteome analysis. Nat. Methods 6, 359–362. [DOI] [PubMed] [Google Scholar]
- Yan W (2009). Male infertility caused by spermiogenic defects: lessons from gene knockouts. Mol. Cell. Endocrinol 306, 24–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang D, Penttila TL, Morris PL, Teichmann M, and Roeder RG (2001). Spermiogenesis deficiency in mice lacking the Trf2 gene. Science 292, 1153–1155. [DOI] [PubMed] [Google Scholar]
- Zhang P, Kang JY, Gou LT, Wang J, Xue Y, Skogerboe G, Dai P, Huang DW, Chen R, Fu XD, et al. (2015). MIWI and piRNA-mediated cleavage of messenger RNAs in mouse testes. Cell Res. 25, 193–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang D, Tu S, Stubna M, Wu WS, Huang WC, Weng Z, and Lee HC (2018). The piRNA targeting rules and the resistance to piRNA silencing in endogenous genes. Science 359, 587–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao S, Gou LT, Zhang M, Zu LD, Hua MM, Hua Y, Shi HJ, Li Y, Li J, Li D, et al. (2013). piRNA-triggered MIWI ubiquitination and removal by APC/C in late spermatogenesis. Dev. Cell 24, 13–25. [DOI] [PubMed] [Google Scholar]
- Zhong J, Peters AH, Lee K, and Braun RE (1999). A double-stranded RNA binding protein required for activation of repressed messages in mammalian germ cells. Nat. Genet 22, 171–174. [DOI] [PubMed] [Google Scholar]
- Zhong J, Peters AH, Kafer K, and Braun RE (2001). A highly conserved sequence essential for translational repression of the protamine 1 messenger rna in murine spermatids. Biol. Reprod 64, 1784–1789. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data presented are available in the main text and supplementary materials. The accession number for Ribo-seq reported in this paper is GSE139399.