

Abstract
Intracellular Ca2+ level is under strict regulation through calcium channels and storage pools including the endoplasmic reticulum (ER). Mutations in certain ion channel subunits, which cause mis-regulated Ca2+ influx, induce the excitotoxic necrosis of neurons. In the nematode Caenorhabditis elegans, dominant mutations in the DEG/ENaC sodium channel subunit MEC-4 induce six mechanosensory (touch) neurons to undergo excitotoxic necrosis. These necrotic neurons are subsequently engulfed and digested by neighboring hypodermal cells. We previously reported that necrotic touch neurons actively expose phosphatidylserine (PS), an “eat-me” signal, to attract engulfing cells. However, the upstream signal that triggers PS externalization remained elusive. Here we report that a robust and transient increase of cytoplasmic Ca2+ level occurs prior to the exposure of PS on necrotic touch neurons. Inhibiting the release of Ca2+ from the ER, either pharmacologically or genetically, specifically impairs PS exposure on necrotic but not apoptotic cells. On the contrary, inhibiting the reuptake of cytoplasmic Ca2+ into the ER induces ectopic necrosis and PS exposure. Remarkably, PS exposure occurs independently of other necrosis events. Furthermore, unlike in mutants of DEG/ENaC channels, in dominant mutants of deg-3 and trp-4, which encode Ca2+ channels, PS exposure on necrotic neurons does not rely on the ER Ca2+ pool. Our findings indicate that high levels of cytoplasmic Ca2+ are necessary and sufficient for PS exposure. They further reveal two Ca2+-dependent, necrosis-specific pathways that promote PS exposure, a “two-step” pathway initiated by a modest influx of Ca2+ and further boosted by the release of Ca2+ from the ER, and another, ER-independent, pathway. Moreover, we found that ANOH-1, the worm homolog of mammalian phospholipid scramblase TMEM16F, is necessary for efficient PS exposure in thapsgargin-treated worms and trp-4 mutants, like in mec-4 mutants. We propose that both the ER-mediated and ER-independent Ca2+ pathways promote PS externalization through activating ANOH-1.
Author summary
Necrosis is a type of cell death that exhibits distinct morphological features such as cell and organelle swelling. Necrotic cells expose phosphatidylserine (PS)–a type of phospholipid—on their outer surfaces. Receptor molecules on phagocytes detect PS on necrotic cells and subsequently initiate the engulfment process. As necrosis is associated with stroke, cancer, neurodegenerative diseases, and heart diseases, studying necrotic cell clearance has important medical relevance. In the model organism the nematode C. elegans, we previously identified membrane proteins that promote the exposure of PS on necrotic cell surfaces by studying neurons that are induced to undergo necrosis by dominant mutations in ion channels. Here, in C. elegans, we have discovered that the necrotic insults trigger an increase of the cytoplasmic calcium ion (Ca2+), which in turn promotes PS externalization on necrotic cell surfaces. Furthermore, we have identified two different mechanisms that increase cytoplasmic Ca2+ levels, one dependent on the Ca2+ contribution from the endoplasmic reticulum (ER), the other independent of the ER. The Ca2+ signal targets ANOH-1, a worm homolog of mammalian proteins capable of externalizing PS, for promoting PS exposure on necrotic cells. Our findings reveal novel upstream regulatory mechanisms that promote necrotic cell clearance in animals.
Introduction
Cells undergoing necrosis—a type of cell death morphologically distinct from apoptosis—display cell and organelle swelling, excessive intracellular membranes, and the eventual rupture of the intracellular and plasma membranes [1,2]. Necrosis is frequently observed during cell injury, and is closely associated with stroke, heart diseases, inflammatory diseases, diabetes, cancer, and neurodegeneration [3–8]. Although necrosis has historically been considered an uncontrolled cell death event, recent discoveries from multiple organisms demonstrated that cells possess genetic programs that trigger necrosis in response to extracellular stimuli [9–12]. In the nematode Caenorhabditis elegans, dominant (d) mutations in certain ion channel subunits of the DEG/ENaC (degenerin/epithelial sodium channel) superfamily, in the nicotinic acetylcholine receptor, in trimeric GTPases, and in a few other proteins induce neurons of specific identities to undergo a type of necrosis that resembles the excitotoxic necrosis of mammalian neurons [10]. One such gene is mec-4, which encodes a core subunit of a multimeric, mechanically gated sodium channel that belongs to the DEG/ENaC family and functions in the mechanosensory (touch) neurons for sensing gentle touch along the worm body [13]. Dominant mutations in mec-4 trigger the necrosis of all six touch neurons (AVM, PVM, ALM(L/R) and PLM(L/R)) [13,14]. Unlike apoptosis, the necrosis triggered by the mec-4(d) mutations does not require the function of CED-3, the C. elegans caspase [15]. Furthermore, many of the cellular events occurring during necrosis are different from those occurring during apoptosis [1,2].
Cells undergoing necrosis, like those undergoing apoptosis, are recognized, engulfed, and degraded by neighboring engulfing cells [16,17]. Apoptotic cells are known to present phosphatidylserine (PS), a membrane phospholipid, on their outer surfaces to attract the phagocytic receptors on engulfing cells [18]. PS is thus referred to as an “eat me” signal recognized by engulfing cells and triggering subsequent engulfment [18]. We previously discovered that the outer surfaces of necrotic touch neurons in mec-4(d) mutant worms expose PS, like apoptotic cells [19,20]. As a consequence, PS interacts with the phagocytic receptor CED-1 residing on the surfaces of neighboring engulfing cells, allowing necrotic cells to be recognized by engulfing cells [19]. We found that PS was actively exposed on the surface of necrotic neurons while the plasma membrane remained intact [19]. In addition, we identified two proteins that act in parallel to promote the externalization of PS from the inner leaflet of the plasma membrane to the outer leaflet during necrosis [19]. One of these proteins is CED-7, the C. elegans homologs of the mammalian ABCA transporter [19,21]. Mammalian ABCA possesses the PS-externalization activity [22], and CED-7 is also essential for promoting PS exposure on the outer surfaces of apoptotic cells [20,23]. The other protein is ANOH-1, the C. elegans homolog of mammalian phospholipid scramblase TMEM16F, which catalyzes the random, bi-directional “flip-flop” of phospholipids across the membrane bilayer [19,24]. Whereas CED-7 is expressed broadly and its function needed in both apoptotic and necrotic cells for efficient PS exposure, ANOH-1 is specifically expressed in neurons and acts specifically in necrotic neurons, but not apoptotic cells, to facilitate PS exposure [19–21]. Mammalian TMEM16F was reported to promote PS exposure on the surfaces of platelets during the blood clotting process [24–26]. To our knowledge, C. elegans ANOH-1 is the first phospholipid scramblase reported to promote PS exposure on necrotic cells.
The proteins responsible for PS externalization on C. elegans touch neurons during necrosis are presumably activated by upstream signal(s). However, the identity of such upstream signal(s) remains unknown. In comparison, during apoptosis, caspases are known to be the critical upstream molecules that trigger the exposure of PS and other cellular events. In living cells, PS is almost exclusively localized to the inner leaflet of the plasma membrane [18]. In mammalian cells undergoing apoptosis, caspase 3 cleaves and inactivates the aminophospholipid translocase ATP11C, which selectively flips PS from the outer to the inner leaflet in living cells [27]. The mammalian caspase 3 and C. elegans CED-3 cleave and activate Xk-related protein 8 (Xk8), also a phospholipid scramblase, and Xk8’s worm homolog CED-8, respectively [28,29].
Caspase activity is not involved in excitotoxic necrosis, including the necrosis of the touch neurons triggered by mec-4(d) mutations [15,30,31]. Rather, intracellular calcium ions were considered important signaling molecules that induce the necrosis of multiple types of cells [32–34], including the C. elegans touch neurons [35]. The mec-4(d) mutations alter the conformation of the mechanosensory Na+ channel (of which MEC-4 is a subunit) and make it permeable to extracellular Ca2+ [36]. Remarkably, mec-4(d) induced necrosis is partially suppressed by loss-of-function (lf) mutations in crt-1 –which encodes calreticulin, the ER residential Ca2+ chaperone that facilitates the accumulation of Ca2+ in the ER—and by inactivating genes encoding the ER Ca2+-release channels [35]. Additionally, mec-4(d)-induced necrosis is suppressed by chemical treatment that impairs the release of Ca2+ from the ER to the cytoplasm [35]. Based on the knowledge that (1) the cytoplasmic Ca2+ level is generally much lower than that in the extracellular space or intracellular Ca2+ pools [37], (2) the ER is one of the primary storage pools for intracellular Ca2+ [37], and (3) the Ca2+ release channels in the ER are activated by the increase of cytoplasmic Ca2+ [38], Xu et al [35] proposed that the small amount of extracellular Ca2+ that entered the touch neurons through the MEC-4(d)-containing Na+ channel further induced a robust Ca2+ release from the ER to the cytoplasm. Moreover, they propose that necrosis is induced once the level of cytoplasmic Ca2+ reaches a certain threshold level [35]. Closely related to this discovery and to our investigation of PS exposure on necrotic cells, the scramblase activity of mammalian TMEM16F is known to be Ca2+-dependent [24].
We set out to investigate whether cytoplasmic Ca2+ triggers PS exposure on necrotic cells and whether the ER, as an intracellular Ca2+ pool, takes part in the externalization of PS. Using a fluorescently tagged-MFG-E8, a secreted reporter that detects PS on the outer surfaces of cells [19,20], we quantitatively monitored PS exposure on the cell surface. Previously, the level of Ca2+ in touch neurons has not been monitored over the course of necrosis. To study the relationship between cytoplasmic Ca2+ and PS exposure, we developed a touch neuron-specific Ca2+ indicator. By monitoring the Ca2+ indicator and the PS reporter, we observed a close relationship between a surge of cytoplasmic Ca2+ and the subsequent exposure of PS on the cell surface of necrotic cells. We further determined that the exposure of PS was regulated by Ca2+. Moreover, we found that the release of Ca2+ from the ER was essential for the exposure of PS in mutants of the DEC/ENaC sodium channel subunits, yet not necessary for that in mutants of two different Ca2+ channels. In addition, PS exposure is independent of cell swelling, another necrosis event. Lastly, the efficient Ca2+-triggered PS exposure depends on the function of ANOH-1. Our findings reveal necrotic cell-specific, Ca2+-mediated regulatory mechanisms that act to trigger the exposure of the “eat me” signal and the clearance of necrotic cells.
Results
Multiple kinds of necrotic neurons expose phosphatidylserine (PS) on their surfaces
The six C. elegans touch neurons, when undergoing necrosis in the mec-4(e1611d) mutant strain, expose PS on their surfaces [19]. This PS signal is detected by an mCherry-tagged MFG-E8, a PS-binding protein that is expressed in neighboring hypodermal cells and subsequently secreted to the extracellular space (Fig 1C and 1D) [19]. Conversely, MFG-E8::mCherry does not detect PS on the surfaces of live touch neurons (marked by Pmec-7GFP) in wild-type larvae (Fig 1A and 1B) [19]. To determine whether PS exposure is a common feature for necrotic neurons of different kinds and induced to die by different insults, we examined a number of necrotic neurons in deg-1(u38), unc-8(n491sd), trp-4(ot337), and deg-3(u662) dominant mutant L1 larvae. deg-1 encodes a DEG/ENaC family sodium channel subunit involved in chemotaxis [39]. unc-8 encodes another DEG/ENaC family sodium channel subunit that functions as a critical regulator of locomotion [40,41]. trp-4 encodes a transient receptor potential (TRP) channel expressed in dopaminergic neurons and a number of other neurons [42]. deg-3 encodes a ligand-gated calcium channel belonging to the nicotinic acetylcholine receptor family [43]. Dominant mutations in these genes induces necrosis of neurons: the deg-1(u38) mutation causes necrosis of the IL1 sensory neurons and the AVD, AVG, and PVC interneurons [44]; the unc-8(n491sd) mutation induces the necrosis of a number of motor neurons at L1 stage [45]; the trp-4(ot337) mutation results in the necrosis of dopaminergic neurons and a few non-dopaminergic neurons, including DVA and DVC, two mechanosensory neurons in the tail [31]; and the deg-3(u662) mutation triggers the necrosis of a number of sensory neurons including the six touch neurons, IL1, and PVD, and interneurons AVG and PVC [43]. deg-1(u38), unc-8(n491sd), trp-4(ot337), and deg-3(u662) mutants all exhibited strong PS signal on the surfaces of necrotic cells (Fig 1), indicating that PS exposure is a common event occurring on necrotic sensory neurons, interneurons, and motor neurons of different identities and induced to undergo necrosis by mutations of different genes.
Fig 1. PS exposure is a common phenomenon observed on various necrotic neurons.
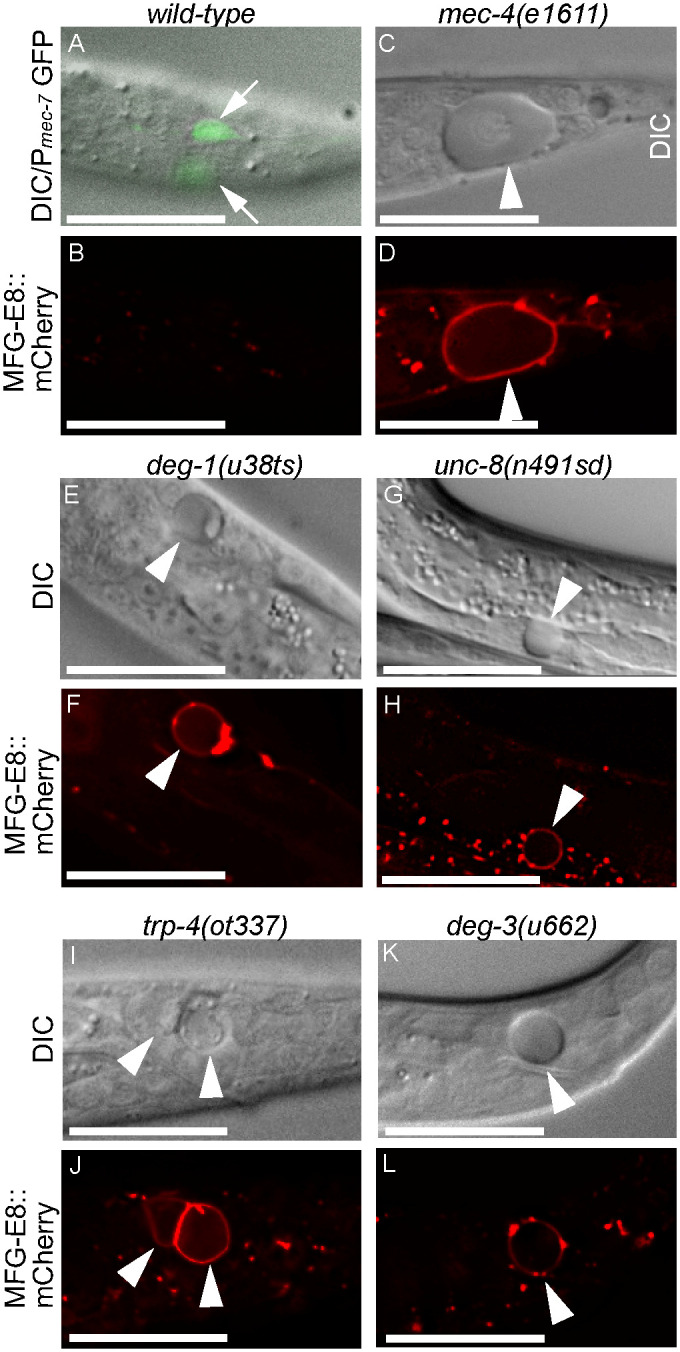
DIC (A, C, E, G, I, K) and epifluorescence (B, D, F, H, J, L) images of two live touch neurons (white arrows) and six necrotic neurons (white arrowheads) in the L1 larvae of wild-type and different mutant strains expressing Pdyn-1mfg-e8::mCherry, including the PLM touch neuron in the tail (A-D), the AVG interneuron in the head (E-F), a ventral motor neuron (G-H), the DVA and DVC neurons in the tail (I-J), and a putative PVD neuron in the tail (K-L). In (A), Pmec-7GFP labels two PLM neurons (white arrows). In (E-F), AVG interneurons in the head are observed at an incubation temperature of 25°C, as deg-1(u38ts) is a temperature sensitive mutant. The bright puncta seen in (B, D, F, H, J, L) are non-specific aggregates of MFG-E8::mCherry. Scale bars are 15μm.
A robust and transient increase in the cytoplasmic Ca2+ level is detected preceding PS exposure on necrotic neurons
To determine whether cytoplasmic Ca2+ level changes as predicted during the necrosis of touch neurons [35], we constructed a Ca2+ indicator (GCaMP5G) that is expressed in touch neurons under the control of the Pmec-7 promoter [46] and introduced Pmec-7 GCaMP5G into the mec-4(e1611) mutant strain (Materials and methods). In the absence of Ca2+, GCaMP5G only emits a very low background level of fluorescence; in the presence of Ca2+, due to the conformation change of this fusion protein induced by its interaction with Ca2+, GCaMP5G generates a detectable GFP signal [47]. The Pmec-7 GCaMP5G reporter is designed to detect cytoplasmic Ca2+ but not the Ca2+ inside the ER lumen because it does not have any ER signal sequence. We co-expressed GCaMP5G with the PS reporter MFG-E8::mCherry and established a time-lapse recording protocol that allowed us to simultaneously monitor Ca2+ and PS signals throughout embryonic development (Materials and methods). In mec-4(e1611) mutants, touch neurons PLML and PLMR (Fig 2A) undergo necrosis, evident by cell swelling (Figs 2B and S1E), during the late embryonic developmental stage [19]. We monitored throughout the necrosis process of PLML and PLMR and detected robust and transient increases of GCaMP5G intensity in the cytoplasm of the PLM neurons in the form of peaks that gradually reduced to the basal level (Figs 2B, 2D, 4B, S1A–S1C, and S2C). In all cases (Figs 2, 4B, S1A–S1C, and S2C), multiple peaks were observed, and at least one peak in each cell occurs prior to cell swelling. PS exposure occurs after cell swelling; and immediately prior to the detection of PS, there are additional transient Ca2+ peaks occurring (Figs 2B–2D and S1A–S1C). These observations demonstrate that the rise in cytoplasmic Ca2+ level precedes cell swelling and PS exposure. Together, the recording of multiple samples revealed that although there were broad ranges of variation of both the peak Ca2+ signal value and the width of the peak, a general pattern of cytoplasmic Ca2+ peaks preceding cell swelling and PS exposure remained consistent among necrotic neurons (Figs 2C, 4, S1, and S2C).
Fig 2. The cytoplasmic Ca2+ signal increases prior to the appearance of necrosis morphology and the exposure of PS, and displays a linear correlation with the PS level.

Reported here are results of time-lapse recording experiments monitoring cytoplasmic Ca2+ levels, cell swelling, and PS signal in or on the surfaces of PLML and PLMR neurons. Embryos are mec-4(e1611) homozygotes carrying the enIs92 transgenic array co-expressing Pmec-7GCaMP5G and Pdyn-1mfg-e8::mCherry. (A) Diagram of a hermaphrodite indicating the positions and names of the six touch neurons. (B) Time-lapse images monitoring the necrosis of one PLML neuron (white arrowheads). Time points are marked as min post-first embryonic cell division. Recording started at 100 min after the embryo reached 2-fold stage, at 560 min, and ended at 900 min. The GCaMP5G signal first appears in (b) and reaches its peak value in (e). PLML starts swelling in (g) and continues swelling in (j, m, p, s). The MFG-E8::mCherry signal on the surface of PLML becomes visible in (o) and increases over time (r, u). The scale bar is 10μm. Additional time-lapse recording for three more PLM neurons are shown in S1 Fig. (C) Relative signal levels (in comparison to the background levels) of GCaMP5G in the cytoplasm, and of MFG-E8::GFP on the membrane surface, of the PLML shown in (A). The green, blue, and red arrows underneath the X-axis mark the time points when the rise of Ca2+ signal, the distinct cell swelling morphology becomes obvious, and PS is first seen on the surface of the neuron, respectively, by eye observation. (D) Summary of the time points when each of the three cellular events in PLM neurons during the onset of necrosis: the appearance of Ca2+ in the cytoplasm (GCaMP5G), the distinct cell swelling morphology observed under DIC microscope, and the appearance of PS on the surface (MFG-E8::mCherry). Data represent mean ± standard deviation. n, the numbers of PLM neurons analyzed. (E) Linear regression analysis between the log-transformed relative cytoplasmic Ca2+ levels inside the PLM neurons and relative PS levels on the surfaces of the same neurons. The integral values of the Ca2+ and PS signals throughout the observed time period (560 min to 900 min post-1st embryonic division) were calculated. Linear regression and Pearson’s correlation coefficient analysis were performed by the R Software. The plot was derived from time points from 4 independent time-lapse recording series (Fig 2B and S1A–S1C Fig). The data of different series are labeled in different colors. The data include 60 time points (18 from embryo 1, 14 each from embryos 2–4). X: the integral value of the relative Ca2+ intensity; Y: the integral value of the relative PS intensity; R2: coefficient of determination; r: correlation coefficient.
Fig 4. Dantrolene reduces the level of Ca2+ in the cytoplasm of touch neurons.

Shown here are results of time-lapse recording experiments measuring the intensity of cytoplasmic Ca2+ and cell swelling of PLM neurons over time. The strain is homozygous for mec-4(e1611) allele and carries a transgenic array expressing Pmec-7 GCaMP5G::gfp. (A) Time-lapse recording images of two necrotic PLM neurons in one untreated embryo (a-n) and two living PLM neurons in one embryo from the 4μM dantrolene treated adult (o-bb). (a-n) Because PLML and PLMR do not appear in the same focal planes, at every time point only one of the two (identity labeled) is visible. (o-bb) No PLM swelling was observed in this embryo. Scale bars are 10μM. (B) The relative fluorescence intensities of cytoplasmic GCaMP5 in the PLM neurons shown in (A) are plotted over time. Arrowheads mark the time points when cell swelling became distinct in the 0μM dantrolene samples. The two 4μM dantrolene samples did not display cell swelling morphology.
On the contrary, in live PLM neurons in the mec-4(+) embryos, only weak, background levels of GCaMP5G signal were observed (S2A and S2B Fig). Although the GCaMP5G signal also fluctuates, both the peak and basal values are much lower than in the necrotic PLM neurons in the mec-4(e1611) embryos, so are the duration period of the increased signal. We further calculated the integral Ca2+ signal levels over two periods: the first period covers from 560 min to 695 min post-1st embryonic division, the mean time point that cell swelling is visible; the 2nd period covers 696–800 min post-1st embryonic division, during which PS signal starts to be detectable on the surfaces of necrotic cells and its intensity continues to increase (S1F Fig). We found that in mec-4(+) embryos, the mean Ca2+ integral values in periods 1 and 2 were 52.6% and 30.7% of that observed from necrotic PLM neurons in mec-4(e1611) mutants (S1D Fig). The low peak value, short duration period of the peak, and the overall low integral value (40.4% of that of mec-4(e1611) mutants) (S1D Fig) of the cytoplasmic Ca2+ signal together correlate with the lack of necrosis and PS exposure in live PLM neurons in wild-type embryos.
The above observations demonstrate that the cytoplasmic Ca2+ exhibits a unique and dynamic pattern of fluctuation in necrotic neurons that is not observed in their living counterparts. To examine the potential correlation between the signal levels of cytoplasmic Ca2+ and PS on the cell surface, in addtion to the integral values of Ca2+, we quantified the integral values of the relative signal intensities of PS over time. We subsequently conducted a linear-regression analysis between the log-transformed integral values of the cytoplasmic Ca2+ and cell-surface PS levels. Remarkably, we identified a very strong positive correlation between these two values (the correlation coefficient (r) is about 0.96) (Fig 2E). Together, the above observations and analyses suggest that the cytoplasmic Ca2+ makes a strong contribution to the level of PS exposure on the surfaces of necrotic neurons. Both the fold of increase of the Ca2+ intensity and the duration of the increased intensity are likely important factors for the activation of PS exposure machinery.
Suppression of the release of Ca2+ from the ER by dantrolene represses the exposure of PS on necrotic neurons
To determine whether the exposure of PS on necrotic touch neurons requires the release of Ca2+ from the ER, we examined whether blocking the release of Ca2+ from the ER in the mec-4(e1611) mutants would affect PS exposure on necrotic neurons. The ryanodine receptor (RyR) and InsP3 receptor (InsP3R) are two major kinds of Ca2+ release channels on the ER membrane [38]. Dantrolene, a small molecule antagonist of ryanodine receptors [48], was reported to suppress mec-4(d) induced necrosis in touch neurons [35]. We placed hermaphrodites that carried homozygous mec-4(e1611) alleles on NGM plates containing different doses of dantrolene (Materials and methods), and measured the cell surface PS intensity on the PLML and PLMR touch neurons in their progeny as newly hatched L1 larvae using MFG-E8::mCherry (Fig 3).
Fig 3. Dantrolene inhibits the exposure of PS on the surface of necrotic neurons.

(A) The normalized relative PS intensity (normalized PSR) of necrotic PLMs in the tails of newly hatched ced-1(e1735); mec-4(e1611) L1 larvae. Adult worms were placed on NGM plates with different doses of dantrolene and allowed to lay eggs; shortly after hatching, larvae were scored. The “0 μM” samples were given DMSO with no dissolved dantrolene. Data are presented as mean ± standard error of the mean (s.e.m.). Numbers of necrotic PLM neurons scored for each dantrolene concentration are in paratheses. ***, p < 0.001, Student t-test. (B) DIC (a, d, g, j, m), Pmec-7GFP (b, e, h, k, n), and MFG-E8::mCherry (c, f, i, l, o) images of the necrotic PLM neurons (arrows) in the newly hatched L1 larvae. The mother worms were incubated with different doses of dantrolene as labeled. PLM neurons are labeled with Pmec-7GFP. Scale bars are 15 μm. (C) The mean numbers of necrotic PLM neurons in the tails of newly hatched L1 larvae generated by adult worms incubated with different doses of dantrolene. Data are presented as mean ± s.e.m. The numbers of L1 larvae scored after dantrolene treatments are in parentheses. **, 0.001 < p < 0.01; ns, no significant difference, Student t-test.
In every mec-4(e1611) mutant embryo, both the PLML and PLMR neurons undergo necrosis [19]. In worms that exhibit normal clearance of dying cells, these necrotic neurons are rapidly engulfed and degraded; as a consequence, some of them disappear before the embryo hatches [19,49]. To allow necrotic cells to persist during the larval stages, we included a ced-1(e1735) null mutation, which perturbs necrotic cell clearance [19], in the strains to be tested. In each ced-1(e1735); mec-4(e1611) double mutant L1 larva hatched within 1 hr, on average 1.7 instead of 2 necrotic PLMs were observed (Fig 3C), due to the residual cell corpse clearance activity that is still present in ced-1 mutants [50,51]. We found that low doses of dantrolene (e.g. 1.0 and 2.0 μM) did not significantly affect the number of PLM neurons that undergo necrosis (Fig 3C), yet resulted in a ~40% reduction of PS level on the surface of necrotic PLMs (Fig 3A and 3B (d-i)). These results suggest that a modest level of inhibition of the release of Ca2+ from the ER affects the exposure of PS on necrotic touch neurons without significantly inhibiting necrosis. At higher doses (e.g. 3.0 and 4.0 μM), dantrolene resulted in 50% reduction of the PS intensity (Fig 3A and 3B) and significant reduction of necrosis events (Fig 3C). Collectively, these results suggest that while the Ca2+ released from the ER facilitates both necrosis and PS exposure, and PS exposure requires the support of a higher level of cytoplasmic Ca2+ than cell swelling does. Moreover, they imply that cytoplasmic Ca2+ might directly induce PS exposure independently of cell swelling.
To examine whether the dantrolene treatment indeed reduces the concentration of cytoplasmic Ca2+ in the PLM neurons in mec-4(e1611) mutant embryos, we monitored the GCaMP5G reporter over time. We first measured the Ca2+ levels in PLMs whose necrosis was suppressed by 4 μM dantrolene and thus remained living and compared them to their untreated counterparts, and found that dantrolene treatment greatly reduced the Ca2+ levels in the cytoplasm of PLM neurons throughout embryonic development (Figs 4 and S2C). Remarkably, dantrolene treatment disrupted the transient cytoplasmic Ca2+ peak characteristic of necrosis (Fig 4A (o-bb)). Secondly, we compared the cytoplasmic Ca2+ levels in necrotic PLM neurons in worms either through 3 μM dantrolene treatment or not treated with dantrolene. After 3 μM dantrolene treatment, the GCaMP5G signal intensity in necrotic PLM neurons display peaks that are lower and more ephemeral than their untreated counterparts (S2C and S2D Fig). Together, the heavily suppressed cytoplasmic Ca2+ signal patterns demonstrate that dantrolene treatments indeed cause the reduction of cytoplasmic Ca2+. in the mec-4(d) background. These patterns explain the reduced PS level on the surface of necrotic PLM neurons and the partial suppression of necrosis, which is represented by cell swelling (Fig 3).
Impairing the establishment of the Ca2+ pool in the ER inhibits PS exposure on necrotic neurons
To further dertermine the role of the ER Ca2+ pool in inducing PS exposure, we analyzed the crt-1(lf) mutants in which the establishment of Ca2+ pool in the ER is defective. crt-1 encodes the only C. elegans homolog of mammalian calreticulin, an ER-residential calcium chaperon that binds free Ca2+ and enables the accumulation of Ca2+ in the ER [52]. Calreticulin-deficient cells are defective in the storage of Ca2+ in ER [53]. In C. elegans, the crt-1(bz29) null mutation ([35] and Materials and methods) fully suppressed the necrosis of touch neurons induced by mec-4(e1611) mutation (0 PLM neuron underwent necrosis among 1000 L1 larvae scored), supporting the notion that in crt-1(bz29) mutants, the ER fails to release sufficient amount of Ca2+ to the cytoplasm [35]. We measured the level of cytoplasmic Ca2+ in crt-1(bz29); mec-4(e1611) double mutant embryos carrying the GCaMP5G reporter and found that in four PLM neurons whose cell-swelling morphology was suppressed, the cytoplasmic Ca2+ level remained low throughout the recording period from mid-embryogenesis to hatching (S3 Fig). Even when a small and transient peak of signal was observed, the peak value never exceeded 1.2-fold of the background signal value (S3A and S3B Fig). In comparison, in crt-1(+); mec-4(e1611) strain, the peak Ca2+ signal levels vary from 1.4- to 5-fold of the background level (Figs 2B–2C and S1A–S1C). The integral Ca2+ intensity values in crt-1(bz29); mec-4(e1611) embryos for the entire time-lapse recording period (560–800 min post 1st embryonic division) and for the early (560–695 min) and late (696–800 min) periods, respectively, are less than 25%, 35%, and 30% of that in mec-4(e1611) embryos, respectively (S3C Fig). The low level of cytoplasmic Ca2+ suggests a failure of the ER to release Ca2+ to the cytoplasm and further implies that the ER Ca2+ pool is not properly established in crt-1 null mutant background.
Two crt-1 mutations, the bz29 null mutation and bz50, another loss-of-function mutation [35], partially suppress necrosis in deg-1(u38ts) mutants and reduce the number of necrotic cells (Fig 5A) [35]. In crt-1(lf); deg-1(u38ts) double mutants and deg-1(u38ts) single mutant worms, we examined PS exposure on necrotic neurons using Pced-1mfg-e8::mKate2 (Materials and methods). In deg-1(u38ts) mutants, a small number of sensory and interneurons undergo necrosis at 25°C, the restrictive temperature [44] (Fig 5A), and expose PS on their surfaces (Fig 5B (b)). In both crt-1 alleles, the levels of PS signal on the existing necrotic cells is reduced to <10% of that in the crt-1(+) animals (Fig 5B (c-f) and 5C). The crt-1 mutation also partially suppresses necrosis in the unc-8(n491) background (Fig 5D). In the unc-8(n491); crt-1(bz29) double mutants (Materials and methods), PS signal intensity is reduced to on average 29% of that of the unc-8 single mutants (Fig 5E and 5F). Together, the above results strongly suggest that a normal ER pool of Ca2+ and the release of Ca2+ into the cytoplasm are essential for proper PS exposure on cells induced to undergo necrosis by hyperactive mutations in subunits of the DEG/ENaC channels.
Fig 5. Mutations in crt-1 inhibit the exposure of PS on the surface of necrotic cells in deg-1 and unc-8 dominant mutant animals.

Newly hatched L1 larvae of labeled genotypes expressing the PS reporter Pced-1mfg-e8::mKate2 or Pced-1mfg-e8::mCherry are scored for the the number of necrotic cells (A and D) and the relative PS intensity (C and F). All strains carry the deg-1(u38ts) allele were incubated at 25°C. (A and D) The mean numbers of necrotic cells in the head of each newly hatched larva (A) or in the entire body of a late L1/early L2 stage larva (D) are represented as bars. Error bars represent s.e.m.. Numbers in parentheses indicate the number of animals analyzed for each strain. “**”, <0.001<p<0.01, Student t-test. (B) DIC (a, c, e) and mKate2 (b, d, f) images of necrotic cells (arrowheads) in the heads of L1 larvae. Scale bars are 15μm. (E) DIC (a, c) and mCherry (b, d) images of necrotic cells (arrowheads) in the tails of ealy L2 larvae. Scale bars are 10μm. (C and F) The normalized relative signal intensities of PS on the surfaces of necrotic cells in the heads (C) or tails (F) are presented as the mean ratios relative to crt-1(+) worms. Bars represent the mean values of each strain. Error bars represent s.e.m.. Numbers in parentheses represent the number of necrotic cells analyzed. “***”, p<0.001, Student t-test.
Ectopically increasing cytoplasmic Ca2+ induces necrosis and the exposure of PS
If the level of cytoplasmic Ca2+ determines whether a cell exposes PS and undergoes necrosis or not, presumably an ectopic increase of cytoplasmic Ca2+ level should induce cells to expose PS. To test this possibility, we treated wild-type worms with thapsigargin (Materials and methods). Thapsigargin inhibits Sarco-Endoplasmic Reticulum Ca2+ ATPase (SERCA), a Ca2+ re-uptake pump located on the ER and sarcoplasmic reticulum (SR) that pumps Ca2+ from the cytoplasm to the ER and SR [54,55]. Inhibiting SERCA activity increases the level of cytoplasmic Ca2+ [55]. Thapsigargin treatment was reported to induce PS exposure on the surfaces of platelets [56]. We observed that, consistent with the previously report [35], thapsigargin treatment induced the onset of necrosis of cells that are otherwise destined to live in the head (Fig 6A and 6B), whereas untreated worms exhibited nearly no necrotic cells (only one necrotic cell observed in a popuation of 83 larvae) (Fig 6A). In addition, thapsigargin-induced necrotic cells expose PS on their surfaces; furthermore, a higher concentration of thapsigargin correlates with stronger PS exposure (6 μg/ml vs 3 μg/ml thapsigargin) (Fig 6B and 6C). These results support a model proposing that the PS exposure on cell surfaces is dependent on the concentration of cytoplasmic Ca2+.
Fig 6. Thapsigargin induces living cells to undergo necrosis and expose PS.

Adult worms carrying the integrated PS reporter Pced-1 mfg-e8::mKate2 were placed on NGM plates with different doses of thapsigargin (TG) and incubated at 25°C; their progeny were allowed to hatch into L1 larvae and promptly scored for the number of necrotic corpses in the head (A) and the intensity of PS on the surface of necrotic cells (B-D). (A) The mean numbers of necrotic cells in each wild-type L1 larva are represented in the bar graph. Error bars represent s.e.m.. “**”, 0.001<p<0.01, Student t-test. (B and D) MFG-E8::mKate2 (a, b) fluorescence and DIC (c, d) images of necrotic cells (white arrowheads) observed in the heads of wild-type (B) and anoh-1 mutant (D) larvae treated with differet doses of TG. Small yellow arrows in (a, c) mark two small, necrotic-like cells (possibly intermediate necrotic cells) that expose PS. Because of their small sizes, they were not counted as necrotic cells. Scale bars are 10μm. (C) The normalized relative MFG-E8::mKate2 signal intensities on the surfaces of necrotic cells in the heads of wild-type and anoh-1(tm4762) mutant L1 larvae are presented as the mean ratios relative that measured from worms subject to 3 μM thapsigargin treatment. Genotypes are indicated underneath the X-axis. Error bars represent s.e.m.. Numbers in parentheses represent the number of animals analyzed for each strain. “***”, p<0.001, “*”, 0.01<p<0.05, “ns”, non-sigificant, Student t-test.
Induction of PS exposure is independent of other necrosis features
Our observation reported in Fig 3 implies that cytoplasmic Ca2+ might induce cell swelling and PS exposure by targeting independent effectors. We reason that if PS exposure is a consequence of the execution of necrosis, blocking the intermediate steps of necrosis execution would inhibit PS exposure even when cytoplasmic Ca2+ level is increased via ER-mediated Ca2+ release; otherwise, PS exposure will not be blocked but other necrotic cell events such as cell swelling will. We applied two different methods to impair the intermediate necrosis events, and examined whether PS exposure is affected in each case.
We first explored the effect of an unc-51(e369) mutation on PS exposure. unc-51 encodes a homolog of yeast ATG1 and mammalian ULK1 (UNC-51-like autophagy activating kinase), an important component of the autophagy pathway [57]. Autophagy genes actively participate in the lysosomal-dependent necrosis, which causes cell swelling and other cellular events [58,59]. Specifically, a loss-of-function mutation of unc-51 was reported to partially block the execution of necrosis induced by a mec-4(d) allele [58,59]. We introduced the unc-51(e369) lf allele into the ced-1(e1735); mec-4(e1611) mutant strain that carries the MFG-E8::mCherry and Pmec-7gfp reporters. We specifically monitored the PLML and PLMR neurons in the tail of young L1 larvae. Consistent with previous reports [58,59], we observed a significant and sizable reduction (25%) in the number of necrotic PLM neurons in newly hatched ced-1(e1735); unc-51(e369); mec-4(e1611) L1 larvae comparing to that observed in ced-1(e1735); mec-4(e1611) L1 larvae (Fig 7A). Remarkably, in ced-1(e1735); unc-51(e369); mec-4(e1611) L1 larvae, PS was detected on 27.4% of the PLM neurons (labeled with Pmec-7 GFP) that appeared live and displayed a relative normal and non-swelling morphology under the DIC microscope (Fig 7B and 7C), whereas in ced-1(e1735); unc-51(+); mec-4(+) L1 larvae in which all PLM neurons were alive, 0% of living PLM neurons exposed PS (Fig 7B and 7C). Fig 7B (d-g) displayed an example of an apparently living PLM neuron that exposed PS. This phenomenon reveals that PS exposure could be induced by the mec-4(d) mutation in a manner independent of necrotic cell swelling.
Fig 7. Impairing the execution of necrosis does not block PS exposure.

(A) The mean number of PLM neurons (labeled with the Pmec-7 GFP reporter) in ced-1(e1735); mec-4(e1611) and ced-1(e1735); unc-51(e369); mec-4(e1611) L1 larvae that displayed the necrotic swelling phenotype were presented in the graph. L1 larvae were scored within 1 hr of hatching. Bars represent the mean values of each sample. Error bars represent s.e.m.. The numbers in the parentheses represent the numbers of animals scored. “***”, p<0.001, Student t-test. (B) DIC, GFP, and mCherry images of four living PLM neurons, two in a wild-type L1 larva (a-c, white arrowheads), and two in a ced-1(e1735); unc-51(e369); mec-4(e1611) L1 larva (d-f, white arrows and arrowheads). PLM neurons are labeled with the Pmec-7GFP reporter (c, f). DIC images (a, d) show that none of the four PLM neurons display the necrotic swelling morphology. PS presentation (MFG-E8::mCherry) on the surface of one PLM neuron (e) is marked by a white arrow (e). White arrowheads in (a-f) mark living PLM neurons that do not expose PS. Scale bars are 5μm. (g) 3.6-fold enlarged image of the region where the PS+ living cell (e, arrow) is. (C) A bar graph representing the percentage of live PLM neurons that expose PS on their surfaces among all living PLM neurons in early L1 larvae with the indicated genotypes. Error bars represent s.e.m.. The numbers in the parentheses represent the total numbers of living PLMs. “***”, p<0.001, Student t-test. (D) Relative PS intensity on the surfaces of PLM neurons in newly hatched L1 larvae. In mec-4(e1611) mutants, necrotic PLM neurons were measured. In unc-51(e369); mec-4(e1611) mutants, living PS+ PLM neurons were measured. Data are presented by mean ± sem. ns, non-significant, Student t-test.
We next attempted to impair the execution of necrosis through disrupting lysosomal function. During excitotoxic necrosis, increased cytoplasmic concentration of Ca2+ is known to activate a cascade of events that lead to the breakage of lysosomes, the acidification of the entire cell, and the release of lysosomal hydrolytic enzymes into the cytoplasm [60,61]. Cell swelling is a primary consequence of these events [60,61]. To disrupt lysosomal function, we applied NH4Cl, a reagent that impairs lysosomal acidification and partially blocks the execution of necrosis of touch neurons in the mec-4(d) mutants [60], to a liquid worm culture (Materials & methods). In the ced-1(e1735); mec-4(e1611) mutant animals, 5mM NH4Cl caused a 40% reduction in the necrosis of the PLM neurons in L1 larvae (S4A Fig), consistent with a previous report [60]. In an average of 5.0% of 78 live (non-swelling) PLM neurons analyzed, we observed PS exposure on their cell surfaces (S4B(d-f) and S4C Fig, white arrows). Together, the results obtained from the unc-51 mutants and the NH4Cl experiment indicate that the exposure of PS is not dependent on the full execution of necrosis.
In deg-3 and trp-4 mutants, the exposure of PS does not rely on the ER Ca2+ pool
deg-3 encodes a ligand-gated calcium channel belonging to the nicotinic acetylcholine receptor family [43]. trp-4 encodes a transient receptor potential (TRP) channel belonging to the TRPN subfamily [42]. TRP channels are non-voltage gated Ca2+ channels that are also permeable to Na+, and K+ [62,63]. In deg-3(u662) and trp-4(ot337) dominant mutants, PS is observed on the surfaces of necrotic cells (Fig 1I–1L). We constructed crt-1(bz29); deg-3(u662) and crt-1(bz29); trp-4(ot337) double mutants that carry the MFG-E8::mCherry reporter by introducing the crt-1(bz29) mutation into the crt-1 gene in the genome via CRISPR/Cas9 mediated gene editing (Materials and methods). Unlike in crt-1(bz29); deg-1(u38) or crt-1(bz29); unc-8(n491) double mutants (Fig 5), in crt-1(bz29) deg-3(u662) and trp-4(ot337); crt-1(bz29) double mutants, the relative PS intensities on necrotic cells are the same as in the deg-3 and trp-4 single mutants, respectively (Fig 8A, 8B, 8D, and 8E). These results indicate that in the cells induced to undergo necrosis by hyperactive Ca2+ channels, a normal Ca2+ pool in the ER appears not essential for PS exposure. Previously it was reported that the crt-1(bz29) mutation does not suppress the necrosis induced by deg-3(u662) [35]. We confirmed this result (Fig 8C). Xu et al [35] proposed that the deg-3 dominant mutation results in the influx of ample amount of Ca2+ into the neurons so that the contribution of Ca2+ from the ER is not needed for the induction of necrosis. Similarly, the crt-1(bz29) mutation does not suppress the necrosis of neurons in the tail in trp-4 mutant larvae (Fig 8F). Together, our results indicate that the hyperactive Ca2+ channel mutations induce PS exposure through an ER-independent mechanism.
Fig 8. The crt-1 null mutation does not affect the exposure of PS on necrotic cells in deg-3 or trp-4 mutants.

(A) and (D) DIC (b, d) and MFG-E8::mCherry (a, c) images of necrotic cells (arrows) in the heads (A) and tails (D) of L1 larvae, respectively. Genotypes are labeled on the top. Scale bars are 10μm. (B) and (E) The normalized relative signal intensities of MFG-E8::mCherry on the surfaces of necrotic cells in the heads (B) or tails (E) of L1 larvae are presented as the mean ratios relative to crt-1(+) worms. Bars represent the mean values of each strain. Error bars represent s.e.m.. Numbers in parentheses represent the number of necrotic cells analyzed. Mutant alleles are listed underneath the X-axis. ns, no significant difference, Student t-test. (C) and (F) The mean numbers of necrotic cells in the heads (C) and tails (F) of each L1 larva are represented as bars. Error bars represent s.e.m.. Numbers in parentheses indicate the number of animals analyzed for each strain. Mutant alleles are listed underneath the X-axis. ns, no significant difference, Student t-test.
The Ca2+-induced PS exposure on necrotic cells is dependent on ANOH-1
The anoh-1(tm4762) lf mutation reduces the PS signal intensity on the surfaces of necrotic cells in the mec-4(e1611) mutants ([19] and see Introduction). To obtain direct evidence that the increase of cytoplasmic Ca2+ induces PS exposure through targeting ANOH-1, we tested whether the PS exposure induced by thapsigargin requires the function of ANOH-1. We found that in anoh-1(tm4762) mutant L1 larvae, the PS intensity on necrotic cells were reduced (Fig 6C and 6D). For example, after the treatment of 6μg/ml thapsigargin, the mean PS signal intensity displayed by anoh-1 mutants is only 32.7% of that measured from the wild-type background (Fig 6C). These results indicate that ANOH-1 plays an important role in Ca2+-induced PS exposure on necrotic cells.
The trp-4(ot337)-induced PS exposure does not appear to require the ER release of Ca2+ (Fig 8D and 8E). We further tested whether ANOH-1 was needed for the exposure of PS on the two necrotic cells located in the tails of L1 larvae. We found that, in trp-4(ot337); anoh-1(tm4762) double mutants, the PS signal on the necrotic cells was reduced to 49% of that observed in trp-4(ot337) single mutants (Fig 9). This observation indicates that ANOH-1 function is also required for the ER-independent PS exposure pathway.
Fig 9. The trp-4(ot337)-induced PS exposure is dependent on the function of ANOH-1.

(A) DIC (b, d) and MFG-E8::mKate2 (a, c) images of necrotic cells (arrows) in the tails of L1 larvae. Genotypes are labeled on the top. Scale bars are 10μm. (B) Bar graph of the normalized relative PS signal intensities on the surfaces of two necrotic cells in the tail of each trp-4(ot337) single mutant and trp-4(ot337); anoh-1(tm4762) double mutant L1 larva. Bars represent the mean values of each strain. Error bars represent s.e.m.. Numbers in parentheses represent the number of necrotic cells analyzed. Mutant alleles are listed underneath the X-axis. ***, p<0.001, Student t-test.
Impairing the release of ER Ca2+ into the cytoplasm does not affect PS exposure on the surfaces of apoptotic cells
During apoptosis in C. elegans, whether cytoplasmic Ca2+ contributes to the PS exposure on apoptotic cells was not known. To address this question, we examined whether a crt-1(bz29) null mutation, which disrupts the Ca2+ pool in the ER, would affect PS exposure on apoptotic cells. In the ced-1(e1735) mutants, the clearance of apoptotic cells is largely inhibited and many apoptotic cells persist in the head of newly hatched L1 larvae (Fig 10A and 10B). Loss of ced-1 function does not affect PS dynamics on the surfaces of dying cells [19,20], and thus all of these apoptotic cells expose PS on their cell surfaces (Fig 10B and 10C). In the ced-1(e1735); crt-1(bz29) double mutant L1 larvae, the crt-1(bz29) null mutation does not affect either the number of apoptotic cells or the level of PS exposure on apoptotic cells (Fig 10A–10C).
Fig 10. A null mutation in crt-1 does not affect the exposure of PS on the surface of apoptotic cells.

(A) In ced-1(e1735) and ced-1(e1735); crt-1(bz29) mutant strains, the number of apoptotic cells in the head of L1 larvae hatched within 1 hr were scored. Bars represent the mean values of each sample. Error bars represent s.e.m.. Numbers in parentheses represent the the numbers of L1 larvae scored. ns, statistically not significant (p>0.05, Student t-test). (B) DIC and fluorescence images of the heads of two larvae displaying numerous apoptotic cells (a and c, arrows). The PS signal exposed on their surfaces are detected by MFG-E8::mCherry (b and d, arrows). Scale bars are 10 μm. (C) Apopotic cells in the heads of larvae were scored for the normalized MFG-E8::mCherry signal intensity on their cell surfaces. The signal intensity was compared between two different mutant strains. Bars represent the mean values of each sample. Error bars represent s.e.m.. Numbers in parentheses are the the numbers of apoptotic cells analyzed. ns, statistically not significant (p>0.05, Student t-test). (D-F) The crt-1 promoter is expressed in apoptotic cells (D), a necrotic touch neurons (E), and two living touch neurons (F). Transgenes were expressed in ced-1(e1735); mec-4(e1611) double (D and E) and ced-1(e1735) single (F) mutant strains. Scale bars are 15 μm. (D) DIC (a) and fluorescence (b) images of the head of an L2/L3-stage larva expressing Pcrt-1NLS-mNeonGreen. Arrows mark apoptotic cells. (E) DIC (a) and fluorescence (b and c) images of the tail of an L1-stage larva co-expressing Pcrt-1NLS-GFP (b) and the touch neuron marker Pmec-7mCherry (c). Arrowheads mark one necrotic PLM neurons. (F) DIC (a) and fluorescence (b and c) images of the tail of an L1-stage larva co-expressing Pcrt-1NLS-GFP (b) and Pmec-7mCherry (c). Arrowheads mark living PLM neurons.
crt-1 is broadly expressed in C. elegans [35,64]. We next determined whether crt-1 was expressed in cells destined to undergo apoptosis and/or necrosis. We constructed Pcrt-1NLS-gfp and Pcrt-1NLS-mNeonGreen reporters, in which the GFP or mNeonGreen reporters were tagged with a nuclear localization signal (NLS) and expressed under the direct control of the crt-1 promoter (Pcrt-1) [64] (Materials & methods). The crt-1 open reading frame is not present in these two reporters. The NLS sequence facilitates the enrichment of GFP or mNeonGreen in the nucleus. In the ced-1(e1735); mec-4(e1611) double mutant animals that expressed the Pcrt-1 NLS-gfp or Pcrt-1 NLS-mNeonGreen constructs, mNeonGreen and GFP signals were observed broadly, including in many apoptotic cells retained in the head (Fig 10D) as well as the necrotic touch neurons in the tail of L1 larvae (Fig 10E). In addition, in ced-1(e1735) single mutant larvae, Pcrt-1 NLS-mNeonGreen expression was observed in live PLM neurons in the tail (Fig 10F). These results indicate that crt-1 is expressed in cells destined to die of apoptosis and necrosis. Therefore, collectively, our observations support the hypothesis that the ER Ca2+ pool does not regulate either the initiation of apoptosis or the exposure of PS on apoptotic cells, despite that crt-1 is expressed and presumably functional in cells distined to undergo apoptosis. Our findings thus demonstrate that necrotic and apoptotic cells employ different molecular mechanisms to trigger PS exposure.
Discussion
Previously, we discovered that necrotic touch neurons in C. elegans larvae remain intact and actively expose PS on their outer surfaces in order to attract engulfing cells [19]. Our continuing investigation reported here reveals a previously unknown signaling mechanism that triggers the exposure of PS on the surface of necrotic cells. Despite its essential roles in many cellular events, Ca2+ was not known to act as a trigger for the clearance of necrotic cells. We developed a time-lapse recording approach and monitored the dynamic change of the cytoplasmic Ca2+ concentration both prior to and throughout the necrosis of touch neurons in developing C. elegans embryos. The Ca2+ signal in touch neurons has previously been measured in adults [65] but has not been monitored in real time or during embryonic development. Our newly-developed real time-recording protocol has revealed a rapid and transient increase of the cytoplasmic Ca2+ in the touch neurons prior to cell swelling, a feature of necrotic cells, and the subsequent presentation of PS on necrotic cell surfaces. Further quantitative analysis demonstrated a close correlation between the levels of cytoplasmic Ca2+ and PS exposure. We further discovered that the ER Ca2+ pool was necessary for the the robust increase in cytoplasmic Ca2+ and the consequential PS exposure during the excitotoxic necrosis of neurons induced by mutations of DEG/ENaC family of sodium channels. On the other hand, the ER contribution of Ca2+ appears to be dispensable for PS exposure induced by constitutively active mutations of two different Ca2+ channels. Based on these findings and on the known mechanisms that regulate the intracellular Ca2+, we propose a model describing two different Ca2+-triggered PS exposure-mechanisms occuring in neurons undergoing necrosis, one ER-dependent, the other ER-independent. Our results further suggest that both mechanisms target ANOH-1, a putative Ca2+-dependent phospholipid scramblase, for PS externalization (Fig 11).
Fig 11. Two models illustrating how the increase of cytoplamic Ca2+ induces the exposure of PS on the surface of necrotic cells.

See Discussion for a more detailed explanation of the models. The cylinders embedded in the plasma membrane represent various types of ion channels. Blue arrows indicate the Ca2+-induced Ca2+ release from the ER. Burgundy arrows indicate the ER-dependent Ca2+ accumulation pathway. Yellow arrows indicate the ER-independent Ca2+ increase pathway. The establishment of the calcium pool in the ER requires the calcium chaperons calreticulin (CRT-1) and calnexin (CNX-1). The release of Ca2+ from the ER requires the ryanodine receptor, whose activity is inhibited by dantrolene, and the InsP3 receptor (InsP3R). SERCA is an ER-surface Ca2+ reuptake pump whose function is inhibited by thapsigargin. Dominant mutations of DEC/ENaC channel subunits allow a small amount of Ca2+ to enter neurons. This, in turn, triggers a “Ca2+-induced” Ca2+ release from the ER, results in a further increase of cytoplasmic Ca2+ concentration. The resulting surge in the level of the cytoplasmic Ca2+ activates multiple downstream targets to induce parallel necrosis events. Dominant mutations in Ca2+ channels DEG-3 or TRP-4 induce PS exposure independent of the ER Ca2+ pool, presumably due to the large amount of Ca2+ let in the cells by these constitutively open Ca2+ channels. ANOH-1 function is necessary for the efficient PS exposure triggered by both ER-dependent and ER-independent Ca2+ increase mechanisms and we propose ANOH-1 is a prime candidate for a Ca2+-target that facilitates PS exposure. In addition, question marks indicate that the increased cytoplasmic Ca2+ might also activate CED-7 or other unknown PS-externalization enzymes. Besides Ca2+, there might be other upstream signaling molecules that induce PS exposure.
A Ca2+-triggered, ER-assisted, “two-step” mechanism that promotes the exposure of PS on the surface of necrotic neurons
The DEG/ENaC sodium channel in touch neurons, of which MEC-4 is a subunit, plays an essential role in the mechanosensation [13]. Biochemical studies have revealed that the MEC-4(d) mutant protein alters the property of this sodium channel, making it permeable to Ca2+ [36]. This altered permeability was detected when overexpressed in Xenopus oocytes or in C. elegans touch neurons, although only a modest permeability of Ca2+ was detected [36]. In mec-4(d) mutants, multiple lines of evidence indicate that a further increase of the level of cytoplasmic Ca2+, provided by the ER through the calcium-induced Ca2+ release mechanism, triggers necrosis [35,38].
The exposure of PS is a common feature observed on multiple types of necrotic neurons, including necrosis induced by hyperactive mutations in three DEG/ENaC channel subunits (MEC-4, DEG-1 and UNC-8) and two Ca2+ channels (DEG-3 and TRP-4) (Fig 1 and [19]). Our time-lapse observation of Ca2+ signal intensity in mec-4(d) mutants has provided direct evidence of a transient increase in the cytoplasmic Ca2+ level prior to necrotic cell swelling and PS exposure. We further provide multiple lines of evidence to indicate that, similar to cell swelling, the exposure of PS on the surface of necrotic neurons in mec-4(d), deg-1(d), and unc-8(d) mutants is also regulated by the ER Ca2+ release. First, dantrolene, which inhibits the release of Ca2+ from the ER, attenuates PS exposure. Secondly, the crt-1 mutations that block the establishment of ER Ca2+ pool result in the great reduction of PS exposure. Calreticulin (CRT-1) is an ER-localized calcium chaperon that plays a critical role in establishing the Ca2+ pool in the ER by binding to Ca2+ with high capacity [52]. Upon upstream signals, such as the binding of the InsP3 receptor (InsP3R) by cytoplasmic Ca2+, more Ca2+ is released from ER chaperons such as calreticulin and calnexin and enter the cytoplasm [37]. By monitoring the Ca2+ signal intensity over time in crt-1(null); mec-4(d) double mutant embryos, we observed very low cytoplasmic Ca2+ intensity in the PLM neurons. This evidence verified the lack of Ca2+ release from the ER despite the presence of the necrosis signal, presumably due to the lack of a normal Ca2+ pool in the ER. The crt-1 null mutation severely reduced PS exposure in deg-1 and unc-8 mutants, indicating the essential role of a high level of cytoplasmic Ca2+ in inducing PS exposure. Thirdly, on the contrary, the thapsigargin treatment that inhibits reuptake of Ca2+ into ER not only induces necrosis of cells in otherwise wild-type animals, but also triggers the exposure of PS on the surfaces of these necrotic cells. Together, these results lead us to propose a “two-step” model for extracellular Ca2+ to induce PS exposure in response to hyperactive mutations of DEG/ENaC sodium channel subunits: step 1, a modest level of Ca2+ leaks into neurons through the mutated DEG/ENaC sodium channels, and step 2, these Ca2+ molecules activate the “Ca2+-triggered Ca2+ release” from the ER, which further increases the cytoplasmic Ca2+ level; once the cytoplasmic Ca2+ level reaches a threshold, necrosis and PS exposure are induced (Fig 11).
Is the exposure of PS merely a consequence of necrosis? Our further experiments demonstrate that this is not the case.
First of all, while low doses of dantrolene reduce the intensity of PS on the necrotic cell surfaces without suppressing cell swelling, higher doses both partially suppress cell swelling and further reduce PS intensity on the surfaces of the remaining necrotic cells (Fig 3). This phenomenon suggests that the threshold of cytoplasmic Ca2+ required for PS exposure is higher than that needed for inducing cell swelling, suggesting that PS exposure and cell swelling might be regulated by different calcium effectors. Secondly, we directly examined whether PS exposure is a downstream consequence of necrosis by blocking mec-4(d)-induced necrosis, through either inhibiting autophagy or impairing proper lysosomal function. As the execution of necrosis requires both lysosomes and the autophagy pathway [58–60], inhibiting autophagy or impairing proper lysosomal function each only suppresses cell swelling in a certain percentage of touch neurons. We observed that even when cell swelling was blocked, PS was still frequently detected on the plasma membrane of these normal-looking neurons. These observations indicate that blocking cell swelling does not necessarily block PS exposure, and further strongly suggest that the cytoplasmic Ca2+ targets multiple effectors in parallel, one of which leads to PS exposure. Similarly, thapsigargin-treated platelets expose PS on their surfaces but do not undergo cell death, supporting the notion that PS exposure is not dependent on cell death [56].
We have also discovered a Ca2+-dependent, ER-independent mechanism that triggers PS exposure
It would stand to reason that if the Ca2+ influx from the extracellular space is in a large enough amount, the ER-dependent Ca2+-release could be dispensible for PS exposure. Among the insults that are known to trigger the necrosis of neurons, a dominant mutation in deg-3, which encodes a subunit of a ligand-gated calcium channel belonging to the nicotinic acetylcholine receptor family [43], induces neuronal necrosis in an ER-independent manner [35]. The deg-3(u662) dominant mutation results in a calcium channel with increased conductivity [43,66]. In addition to deg-3, trp-4 encodes a protein belonging to the TRPN subfamily of TRP channels, which are Ca2+ channels [62]. We found that blocking the establishment of an ER Ca2+ pool by a crt-1 null mutation did not affect the exposure of PS on necrotic cells resulting from the deg-3 or trp-4 mutations, in contrast to that observed in the deg-1 and unc-8 mutant backgrounds. These results indicate that when there is a large enough amount of Ca2+ entering into the neurons through the hyperactive Ca2+ channels, the Ca2+-triggered PS exposure might be independent of the ER Ca2+ release (Fig 11).
The putative phospholipid scramblase ANOH-1 is likely a direct target of the cytoplasmic Ca2+
What protein(s) externalizes PS in response to the increased levels of cytoplasmic Ca2+? One potential candidate is C. elegans ANOH-1. TMEM16F, the mammalian homolog of ANOH-1, is both a Ca2+-activated cation channel and a Ca2+-dependent phospholipid scramblase [24,25]. Despite that its activity has not associated with necrotic cells in mammals, TMEM16F is essential for the PS exposure on platelets in response to Ca2+ signaling without compromising plasma membrane integrity [24,26]. Although its biochemical activity has not been determined, C. elegans ANOH-1 might act as a phospholipid scramblase whose activity is dependent on its association with Ca2+, as the Ca2+-binding motif identified in TMEM16F is conserved in ANOH-1 [19,67–70]. We found that, like in mec-4(d) mutants, in worms treated with thapsigargin, a null mutation of anoh-1 severely reduced the intensity of PS on the surfaces of necrotic cells. This observation provides strong evidence to suggest that the high level of cytoplasmic Ca2+ activates ANOH-1’s phospholipid scramblase activity. This activation might be through direct interaction between Ca2+ and ANOH-1 (Fig 11). Similarly, in trp-4(d); crt-1(null) mutants, PS exposure is also substantially reduced comparing to trp-4(d) single mutants, indicating that both of the two mechanisms that increase cytoplamsic Ca2+ levels target ANOH-1 to promote PS exposure (Fig 11).
Conversely, CED-7, the other protein important for PS exposure, is not known to bind Ca2+, neither is CED-7’s mammalian homolog ABCA1. It remains to be investigated whether CED-7 is also regulated by Ca2+ or by a different upstream signaling molecule (Fig 11). Given that CED-7 is important for PS exposure on the surface of both apoptotic and necrotic cells [19,20], it is possible that CED-7 can be activated by multiple upstream signals.
As to how Ca2+ mediates cell swelling, the Ca2+-activated protease calpain is a known target of necrosis signals, whose stimulated activity is associated with lysosomal rupture and the activation of autophagy, two major events that lead to many cell morphological changes and the degradation of cellular proteins during necrosis [71–74]. We propose that during the initiation of necrosis, the elevated cytoplasmic Ca2+ concentration activates, in parallel, a number of downstream targets that include the PS-exposure enzymes, calpain, and possibly other proteases, and in this manner induces cellular changes of multiple aspects, including PS exposure and cell swelling (Fig 11).
Does intracellular Ca2+ facilitate PS exposure on the surface of apoptotic cells?
Previously, extracellular Ca2+ was reported to stimulate the exposure of PS on the surface of a T lymphocyte hybridoma cell line undergoing apoptosis [75]. A Ca2+-dependent PS exposure mechanism was suggested to be a general mechanism utilized by apoptotic cells of different identities [76]; however, lines of evidence that do not support this hypothesis also exist [77,78]. Nonethless, it was unknown whether Ca2+ is involved in the developmentally programmed apoptosis and/or the subsequent exposure of the “eat me” signal in C. elegans. We determined that neither the number of apoptosis events nor the level of PS exposure is compromised during embryogenesis in crt-1 mutants. These results indicate that the fluctuation of the cytoplasmic Ca2+ level does not trigger either apoptosis or PS exposure in developing C. elegans embryos. This is consistent with our previous finding that the proposed Ca2+-dependent phospholipid scramblase ANOH-1 is not involved in the PS exposure on apoptotic cells [19]. Together, our findings demonstrate that apoptotic and necrotic cells regulate the exposure of “eat me” signals and their subsequent clearance through distinct mechanisms; apoptotic cells utilize Ca2+-independent mechanisms, while necrotic cells utilize Ca2+-dependent mechanisms.
The Ca2+-dependent regulatory mechanism for PS exposure is likely conserved throughout evolution
The regulatory mechanisms we have discovered in C. elegans might be mechanisms that are also employed in other organisms including mammals, particularly since the mammalian Ca2+-dependent scramblase TMEM16F is a close homolog of C. elegans ANOH-1. Whether TMEM16F plays a role similar to C. elegans ANOH-1 in facilitating the clearance of necrotic cells remains to be tested, although it is clear that TMEM16F is not involved in the PS exposure on apoptotic cells [79]. Insults that increase the cytoplasmic Ca2+ trigger the necrosis of neurons, glial cells, cardiomyocytes, cancer cells, and other cells [6,34,80–82], and it is possible that some or all types of these necrotic cells all utilize the Ca2+-dependent mechanism to activate the PS-exposure activity. We predict that the Ca2+-triggered PS exposure might be an evolutionarily conserved mechanism that facilitates the clearance of different types of necrotic cells. As calcium overload and the consequential excitotoxic necrosis play critical roles in the pathology of many diseases including neurodegenerative disorders, stroke, heart failure, and cancer [72,80–82], investigating the clearance of necrotic cells using C. elegans as a model organism will shed light on the pathology and therapeutics of these human diseases. For example, there are pre-clinical therapeutic attempts to use chemical inhibitors of the Ca2+-dependent protease calpain to hamper the necrosis occurring during pathological conditions such as brain ischemia [83]. What we have found suggests that this approach might not be able to efficiently inhibit the exposure of PS on cells whose necrosis is blocked and thus might not necessarily prevent these cells from being engulfed.
Materials and methods
Mutations, plasmids, strains, and transgenic arrays
C. elegans was grown at 20°C as previously described [84] unless indicated otherwise. The N2 Bristol strain was used as the wild-type strain. Mutations are described in [85] and in the Wormbase (http://www.wormbase.org) unless noted otherwise: LGI, ced-1(e1735), trp-4(ot337). LGII, enIs74[Pdyn-1mfg-e8::mCherry, Pmec-7 gfp, and punc-76(+)] (this study). LGIII, anoh-1(tm4762), crt-1 (bz29 and bz50). LGIV, unc-8(n491sd); enIs77[Pced-1mfg-e8::mKate2 and punc-76(+)] (this study). LGV, unc-76(e911), deg-3(u662), unc-51(e369), enIs92[Pmec-7GCaMP5G, Pdyn-1mfg-e8::mCherry, and punc-76(+)] (this study); LGX, deg-1(u38ts), mec-4(e1611).
Plasmids Pdyn-1mfg-e8::mCherry and Pmec-7gfp (pPD117.01, a gift from Andrew Fire) have been reported in [19]. Pced-1mfg-e8::mKate2 was generated by first replacing the dyn-1 promoter (Pdyn-1) [86] in Pdyn-1mfg-e8::mCherry with Pced-1 [23] and then replacing mCherry cDNA in Pced-1mfg-e8::mCherry with mKate2 cDNA [87]. Pmec-7GCaMP5G was generated by cloning the GCaMP5G cassette obtained from pCMV-GCaMP5G [47] to the BamH1 and EcoR1 sites of pPD117.01. Plasmid Pcrt-1NLS-gfp was generated by amplifying the genomic DNA that covers the first 6 amino acids including the start codon and a 1.5kb upstream region representing the crt-1 promoter (Pcrt-1) [88] and cloning this fragment into the Sph1 and BamH1 sites of pPD95.69 (a gift from Andrew Fire), in frame with the nuclear localization signal (NLS) tagged GFP coding sequence, generating a NLS-GFP reporter expressed under the control of Pcrt-1. Plasmid Pcrt-1NLS-mNeonGreen was constructed by replacing the coding sequence for GFP with that for mNeonGreen [89].
Extrachromosomal arrays were generated by microinjection [90] of plasmids with co-injection marker punc-76(+) [91] into strains carrying the unc-76(e911) mutation. Transgenic animals were isolated as non-Unc animals. enIs74, enIs77, and enIs92 are integrated transgenic arrays [90] generated from the corresponding transgenic arrays in this study. trp-4(ot337), crt-1(bz29), crt-1(bz50), unc-8(n491sd), and deg-1(u38) were provided by the C. elegans Genetic Center (CGC).
Standard genetic crosses [84] were used for generating double and triple mutant strains except for the following three double mutants that carry enIs74: (1) unc-8(n491sd); crt-1(bz29); (2) deg-3(u662) crt-1(bz29); and (3) trp-4(ot337); crt-1(bz29). These three strains were generated by CRISPR/Cas9-mediated gene editing of the bz29 mutation into the corresponding unc-8, deg-3, and trp-4 single mutants carrying enIs74. The crt-1(bz29) mutation is a change of nucleotide 83 (the 1st nucleotide of the start codon is designated as nucleotide 1) from G to A, resulting in a Trp28-to-stop codon nonsense mutation [35]. We used the Paix protocol [92] to introduce the G83A mutation into each of the three above single mutant strain. The tracer RNA and crRNAs, and the Cas9 protein, were from Integrated DNA technologies (IDT), Inc. The repair template oligo and other oligos were ordered from Sigma-Aldrich, Inc. Sequencing results confirmed the expected changes as homozygous in the three corresponding double mutant strains.
Chemical treatments of C. elegans
Dantrolene and thapsigargin treatments
Worms were treated with various doses of dantrolene (Tocris Bioscience, Inc.) as described previously [35]. Dantrolene was dissolved in DMSO and was both spread on unseeded NGM plates and added to the suspension of OP50 (the E. coli strain seeded onto the NGM plates). One day after seeding the NGM plates with the drugged OP50, L4 hermaphrodites were placed onto the dantrolene plates and raised at room temperature (20–22°C). Twenty-four to 36 hrs later, L1 larvae (F1 progeny) hatched within 1 hr were examined by both DIC and fluorescence imaging. Dantrolene is likely affecting embryos through entry into the germline of adult hermaphrodites and subsequently being incorporated in the embryos. The 0 μM dantrolene control sample represent worms from NGM plates and seeded OP50 that were treated with DMSO, the solvant for dantrolene. The thapsigargin (Sigma-Aldrich, Inc.) treatment protocol is similar to that of dantrolene treatment, except that the worms were raised at 25°C instead of room temperature (20–22°C).
NH4Cl treatment
As described previously [35], 1M NH4Cl (Sigma-Aldrich, Inc.) solution in water was added to 10 mL S medium containing 50 young adult hermaphrodites and appropriate amount of concentrated bacteria OP50 (food for worms) to reach a final concentration of 5 mM. This suspension was grown at room temperature and with constant shaking. Fifteen hours later, young L1 larvae were collected and analyzed by DIC and fluorescence microscopy. Note that because the progeny of the young adults were grown in the liquid culture and could not be distinctly timed, the L1 larvae that we collected and analyzed are between 1–3 hrs after hatching. As a consequence, the mean number of necrotic PLMs observed in the tail is slightly lower than that of the L1 larvae scored within 1 hr of hatching due to the extra time after hatching.
DIC microscopy and scoring the number of necrotic cells and apoptotic cells
DIC microscopy was performed using an Axioplan 2 compound microscope (Carl Zeiss) equipped with Nomarski DIC imaging apparatus, a digital camera (AxioCam MRm; Carl Zeiss), and imaging software (AxioVision; Carl Zeiss), or with an Olympus IX70-Applied precision DeltaVision microscope equipped with a DIC imaging apparatus, a Photometrics Coolsnap 2 digital camera, and the SoftWoRx imaging software (GE Healthcare, Inc.). In ced-1(e1735); mec-4(e1611) mutant and ced-1(e1735); mec-4(+) control backgrounds, the presence of two necrotic PLM neurons (PLML and PLMR) in the tail was scored by their swelling morphology in L1 larvae as previously described [93]; these larvae were collected within 1 hr after hatching, or within 1-3hrs after hatching, depending on what is annotated in the figure legends. Between 30–60 L1 larvae were scored for each sample. These larvae were grouped as 10 animals per group. The mean values of the number of necrotic PLM neurons of each group were calculated, and the mean values and standard errors of the means of all groups were presented in bar graphs. To examine deg-1(u38ts)-induced necrosis, adults were grown at 25°C for one day, at which point we collected the embryos that they laid. These embryos were incubated at 25°C and allowed to hatch, and we examined the L1 larvae within 1hr after they hatched. Between 50–129 larvae were scored for the number of necrotic cells in the head region. These larvae were grouped as 10, 15, and 25 animals per group for the deg-1(u38ts), deg-1(u38ts); crt-1(bz50), and deg-1(u38ts); crt-1(bz29) strains, respectively. The mean values of the number of necrotic cells of each group were calculated, and the mean values and standard errors of the means of all groups were presented in bar graphs.
The number of apoptotic cells was scored in the head of newly hatched L1 larvae as described in [93].
The numerical values of the graphs can be found in S1 Data.
Fluorescence microscopy, time-lapse recording, and quantification of image intensity
An Olympus IX70-Applied Precision DeltaVision microscope equipped with a DIC imaging apparatus and a Photometrics Coolsnap 2 digital camera was used to acquire serial Z -stacks of fluorescence and DIC images, whereas the SoftWoRx software was utilized for image deconvolution and processing as described in [19,93]. To quantify the intensity of PS signal on the surface of a necrotic PLM neuron, the cell surface area containing the MFG-E8::mCherry signal was outlined by two closed polygons. The total signal inetensity as well as the area of each polygon were recorded. The unit PS signal intensity (PSPLM) of the “donut shape” area between the two polygons was calculated as follows: UPSPLM = (PSexternalpolygon−PSinternalpolygon)/(Areaexternalpolygon−Areainternalpolygon). The background mCherry signal (PSBackground) of a “circlular” area nearby and the area size (Areabackground) were obtained and the unit intensity UPSbackground = PSbackground/Areabackground. The relative PS signal intensity (RPSPLM) = UPSPLM / UPSbackground. According to this formular, the value 1 indicates no enrichment of signal comparing to the non-specific background signal. The PS intensity on the surfaces of other necrotic neurons, living PLM neurons, and apoptotic cells were also measured using the above method.
To quantify the Ca2+ intensity in the cytoplasm of live or necrotic PLM neurons, the cell body of a PLM neuron was outlined by a polygon and the GCaMP5G signal value within the polygon was obtained and referred to as Cax. CaBackground of an area in equivalent size in a neighboring region was similarly obtained. The relative Ca2+ intensity of a particular cell was defined as CaR = Cax / CaBackground.
To monitor the dynamics of Ca2+ release into the cytoplasm of PLM neurons, cell swelling during necrosis, and PS presentation during embryogenesis, we modified a previously established protocol [19]. We mounted embryos on an agar pad on a glass slide with M9 buffer. The recording period covered 560 min (100 min after worms reached the 2-fold stage (460min)) to 900 min post-1st embryonic cleavage. Serial Z stacks were captured in 10–15 sections at 0.5μm per section. Recording interval was 2.5 min from 560–680 min post-1st embryonic cleavage and 5 min from 685–900 min post 1st cleavage. Unfortunately, since embryos being recording are constantly moving inside the eggshell, images recorded in most of the time points do not have the PLML or PLMR neurons in focus. Thus images useful for signal quantification are far fewer than those captured. We set the minimum time points required for constructing a curve to 10 per embryo. Each curve used in our integral analysis includes between 10 and 20 time points. The numerical values of the graphs and plots can be found in S1 Data.
Supporting information
(Related to Fig 2) (A-C) Reproted here are results of three time-lapse series recording cytoplasmic Ca2+ levels and PS signal in or on the surface of PLML and PLMR neurons, respectively. Embryos are mec-4(e1611) homozygotes carrying the enIs92 transgenic array. Relative signal levels (in comparison to the background levels) of GCaMP5G in the cytoplasm, and of MFG-E8::GFP on the membrane surface, of the PLM neurons are ploted over time. The green, blue, and red arrows underneath the X-axis mark the time points when the rise of Ca2+ signal, the distinct cell swelling morphology becomes obvious, and PS is first seen on the surface of the neuron, respectively, by eye observation. (D) The mean integral values of relative cytoplasmic Ca2+ intensities in each cell measured from 4 necrotic PLM neurons in mec-4(e1611) embryos and 4 live PLM neurons in mec-4(+) embryos were obtained by calculating the integral value of GCaMP5G intensities within three time periods, 560–800 min, the entire time-lapse recording period, 560–695 min, the period prior to the mean time point when cell swelling is obvious, and 696–800 min post-1st embryonic division, respectively. Error bars represent s.e.m. (E) Time-lapse DIC images following the necrosis of one PLML neuron (white arrows). Yellow arrows mark the PLMR neuron which went out of focus in later time points. Time points are marked as min post-first embryonic cell division. Recording started at 560 min and ended at 900 min. The PLML neuron starts swelling at 675 min and continues swelling in the later time points until reaching its maximal size at 870 min. The scale bar is 10μm.
(TIF)
(Related to Figs 2 and 4) Presented here are results of time-lapse recording experiments that measure the intensity of cytoplasmic Ca2+ and cell swelling of PLM neurons during embryonic development. All embryos carry the transgenic array expressing Pmec-7GCaMP5G. (A) DIC and fluorescence time-lapse images of two live PLM neurons in one mec-4(+) embryo. Time points are marked as min post-1st embryonic division. White and yellow arrowheads mark the PLML and PLMR neurons, respectively. Scale bars are 10μM. (B) The relative signal levels of GCaMP5G were measured in 4 live PLM neurons and plotted over time. Graph (a) displays the plots of the PLML and PLMR neurons shown in (A), whereas graph (b) displays the plots of the PLML and PLMR neurons in an additional embryo. (C) The relative signal levels of GCaMP5G were measured in two necrotic PLM neurons in a mec-4(e1611) mutant embryo from a plate treated with DMSO but no dantrolene. Two other examples are shown in Fig 4. (D) The relative GCaMP5G signal levels measured in 2 necrotic PLM neurons in mec-4(e1611) mutant embryos from a 3μM dantrolene treated plate are plotted over time.
(TIF)
(Related to Fig 5) (A-B) Presented here are results of time-lapse recording experiments that measure the intensity of cytoplasmic Ca2+ and cell swelling of 4 PLM neurons during the development of two crt-1(bz29); mec-4(e1611) mutant embryos carrying the transgenic array expressing Pmec-7GCaMP5G. The relative GCaMP5 intensity in living PLM neurons are plotted over time. (C) The mean integral values of relative cytoplasmic Ca2+ intensities in each cell measured from 4 necrotic PLM neurons in mec-4(e1611) embryos and 4 PLM neurons whose necrosis were suppressed in crt-1(bz29); mec-4(e1611) embryos were obtained by calculating the integral value of GCaMP5G intensities within three time periods, 560–800 min, the entire time-lapse recording period, 560–695 min, the period prior to the mean time point when cell swelling is obvious, and 696–800 min, the period after cell swelling, respectively. Error bars indicate s.e.m.
(TIF)
(Related to Fig 7) (A) The NH4Cl treatment partially suppresses necrosis induced by the mec-4(e1611) mutation. The mean numbers of PLM neurons (labeled with Pmec-7GFP) that display the swelling necrosis phenotype in the tails of young ced-1(e1735); mec-4(e1611) L1 larvae from liquid cultures treated or not treated with 5mM NH4Cl are presented in the graph. Bars represent the mean values of each sample. Error bars indicate s.e.m. The numbers in the parentheses represent the numbers of L1 larvae scored. “**”, 0.001<p<0.01, Student t-test. (B) Images of necrotic and live PLM neurons that present PS on their outer surfaces in mec-4(e1611) L1 larvae. L1 larvae were from liquid cultures either treated with 5mM NH4Cl (d-f, white arrowheads) or left untreated (a-c). PLM neurons (c (white arrow), f (white and yellow arrowheads)) are labeled with the Pmec-7GFP reporter. DIC images identify one necrotic PLM neuron (white arrow) and one apoptotic cell (yellow arrow) in the tail of an L1 larva not treated with NH4Cl (a), and one necrotic PLM (yellow arrowhead) and one live PLM (white arrowhead) in an L1 larva from the 5mM NH4Cl treated culture (d-f). PS presentation on the cell surfaces (b, e) is detected by MFG-E8::mCherry. Scale bars are 5μm. (C) A bar graph representing the percentage of live PLM neurons that expose PS on their surfaces among all living PLM neurons in early L1 larvae after NH4Cl treatment. Error bars represent s.e.m.. The numbers in the parentheses represent the numbers of living cells scored.
(TIF)
(XLSX)
Acknowledgments
We thank R. Haley for helpful comments. We thank A. Fire for the gift of plasmids pPD95.69 and pPD117.01. We thank CGC for providing strains.
Data Availability
All relevant data are within the manuscript and its Supporting information files.
Funding Statement
This project was supported by NIH R01GM104279 (URL: https://www.nih.gov). ZZ was supported by NIH R01GM104279 and R01GM067848. (URL: https://www.nih.gov). YF was supported by the “Tobitate Ryugaku JAPAN Nihondaihyou Program” Scholarship from the Independent Administrative Corporation Japan Student Service Organization. (URL: https://tobitate.jasso.go.jp/document/). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Golstein P, Kroemer G. Cell death by necrosis: towards a molecular definition. Trends Biochem Sci. 2007;32:37–43. 10.1016/j.tibs.2006.11.001 [DOI] [PubMed] [Google Scholar]
- 2.Kroemer G, Galluzzi L, Vandenabeele P, Abrams J, Alnemri ES, Baehrecke EH, et al. Classification of cell death: recommendations of the Nomenclature Committee on Cell Death. Cell Death Differ. 2009;16:3–11. 10.1038/cdd.2008.150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tymianski M Emerging mechanisms of disrupted cellular signaling in brain ischemia. Nat Neurosci. 2011;14:1369–1373. 10.1038/nn.2951 [DOI] [PubMed] [Google Scholar]
- 4.Lai TW, Zhang S, Wang YT Excitotoxicity and stroke: identifying novel targets for neuroprotection. Prog Neurobiol. 2014;115:157–188. 10.1016/j.pneurobio.2013.11.006 [DOI] [PubMed] [Google Scholar]
- 5.Jacobson MD, Bergeron L. Cell death in the nervous system In: Jacobson MD, McCarthy N, editors. Apoptosis, the molecular biology of programmed cell death: Oxford University Press; 2002;pp. 278–301. [Google Scholar]
- 6.Noch E, Khalili K. Molecular mechanisms of necrosis in glioblastoma: the role of glutamate excitotoxicity. Cancer Biol Ther. 2009;8:1791–1797. 10.4161/cbt.8.19.9762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Challa S, Chan FK. Going up in flames: necrotic cell injury and inflammatory diseases. Cell Mol Life Sci. 2010;67:3241–3253. 10.1007/s00018-010-0413-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Whelan RS, Kaplinskiy V, Kitsis RN. Cell death in the pathogenesis of heart disease: mechanisms and significance. Annu Rev Physiol. 2010;72:19–44. 10.1146/annurev.physiol.010908.163111 [DOI] [PubMed] [Google Scholar]
- 9.McCall K. Genetic control of necrosis—another type of programmed cell death. Curr Opin Cell Biol. 2010;22:882–888. 10.1016/j.ceb.2010.09.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vlachos M, Tavernarakis N. Non-apoptotic cell death in Caenorhabditis elegans. Dev Dyn. 2010;239:1337–1351. 10.1002/dvdy.22230 [DOI] [PubMed] [Google Scholar]
- 11.Moquin D, Chan FK. The molecular regulation of programmed necrotic cell injury. Trends Biochem Sci. 2010;35:434–441. 10.1016/j.tibs.2010.03.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhou W, Yuan J. Necroptosis in health and diseases. Semin Cell Dev Biol. 2014. 10.1016/j.semcdb.2014.07.013 [DOI] [PubMed] [Google Scholar]
- 13.Driscoll M, Chalfie M. The mec-4 gene is a member of a family of Caenorhabditis elegans genes that can mutate to induce neuronal degeneration. Nature. 1991;349:588–593. 10.1038/349588a0 [DOI] [PubMed] [Google Scholar]
- 14.Chalfie M, Sulston J. Developmental genetics of the mechanosensory neurons of Caenorhabditis elegans. Dev Biol. 1981;82:358–370. 10.1016/0012-1606(81)90459-0 [DOI] [PubMed] [Google Scholar]
- 15.Ellis HM, Horvitz HR. Genetic control of programmed cell death in the nematode C. elegans. Cell. 1986;44:817–829. 10.1016/0092-8674(86)90004-8 [DOI] [PubMed] [Google Scholar]
- 16.Krysko DV, D’Herde K, Vandenabeele P. Clearance of apoptotic and necrotic cells and its immunological consequences. Apoptosis. 2006;11:1709–1726. 10.1007/s10495-006-9527-8 [DOI] [PubMed] [Google Scholar]
- 17.Li Z, Zhou Z. How are necrotic cells recognized by their predators? Worm. 2016;5:e1120400 10.1080/21624054.2015.1120400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Segawa K, Nagata S. An Apoptotic ’Eat Me’ Signal: Phosphatidylserine Exposure. Trends Cell Biol. 2015;25:639–650. 10.1016/j.tcb.2015.08.003 [DOI] [PubMed] [Google Scholar]
- 19.Li Z, Venegas V, Nagaoka Y, Morino E, Raghavan P, Audhya A, et al. Necrotic Cells Actively Attract Phagocytes through the Collaborative Action of Two Distinct PS-Exposure Mechanisms. PLoS Genet. 2015;11:e1005285 10.1371/journal.pgen.1005285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Venegas V, Zhou Z. Two alternative mechanisms that regulate the presentation of apoptotic cell engulfment signal in Caenorhabditis elegans. Mol Biol Cell. 2007;18:3180–3192. 10.1091/mbc.e07-02-0138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wu Y, Horvitz HR. The C. elegans cell corpse engulfment gene ced-7 encodes a protein similar to ABC transporters. Cell. 1998a;93:951–960. [DOI] [PubMed] [Google Scholar]
- 22.Hamon Y, Broccardo C, Chambenoit O, Luciani MF, Toti F, Chaslin S, et al. ABC1 promotes engulfment of apoptotic cells and transbilayer redistribution of phosphatidylserine. Nat Cell Biol. 2000;2:399–406. 10.1038/35017029 [DOI] [PubMed] [Google Scholar]
- 23.Zhou Z, Hartwieg E, Horvitz HR. CED-1 is a transmembrane receptor that mediates cell corpse engulfment in C. elegans. Cell. 2001b;104:43–56. 10.1016/s0092-8674(01)00190-8 [DOI] [PubMed] [Google Scholar]
- 24.Suzuki J, Umeda M, Sims PJ, Nagata S. Calcium-dependent phospholipid scrambling by TMEM16F. Nature. 2010;468:834–838. 10.1038/nature09583 [DOI] [PubMed] [Google Scholar]
- 25.Yang H, Kim A, David T, Palmer D, Jin T, Tien J, et al. TMEM16F forms a Ca2+-activated cation channel required for lipid scrambling in platelets during blood coagulation. Cell. 2012;151:111–122. 10.1016/j.cell.2012.07.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fujii T, Sakata A, Nishimura S, Eto K, Nagata S. TMEM16F is required for phosphatidylserine exposure and microparticle release in activated mouse platelets. Proc Natl Acad Sci U S A. 2015;112:12800–12805. 10.1073/pnas.1516594112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Segawa K, Kurata S, Yanagihashi Y, Brummelkamp TR, Matsuda F, Nagata S. Caspase-mediated cleavage of phospholipid flippase for apoptotic phosphatidylserine exposure. Science. 2014;344:1164–1168. 10.1126/science.1252809 [DOI] [PubMed] [Google Scholar]
- 28.Suzuki J, Denning DP, Imanishi E, Horvitz HR, Nagata S. Xk-related protein 8 and CED-8 promote phosphatidylserine exposure in apoptotic cells. Science. 2013;341:403–406. 10.1126/science.1236758 [DOI] [PubMed] [Google Scholar]
- 29.Chen YZ, Mapes J, Lee ES, Skeen-Gaar RR, Xue D. Caspase-mediated activation of Caenorhabditis elegans CED-8 promotes apoptosis and phosphatidylserine externalization. Nat Commun. 2013;4:2726 10.1038/ncomms3726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kraemer BC, Zhang B, Leverenz JB, Thomas JH, Trojanowski JQ, Schellenberg G D. Neurodegeneration and defective neurotransmission in a Caenorhabditis elegans model of tauopathy. Proc Natl Acad Sci U S A. 2003;100:9980–9985. 10.1073/pnas.1533448100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nagarajan A, Ning Y, Reisner K, Buraei Z, Larsen JP, Hobert O, et al. Progressive degeneration of dopaminergic neurons through TRP channel-induced cell death. J Neurosci. 2014;34:5738–5746. 10.1523/JNEUROSCI.4540-13.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Berliocchi L, Bano D, Nicotera P. Ca2+ signals and death programmes in neurons. Philos Trans R Soc Lond B Biol Sci. 2005;360:2255–2258. 10.1098/rstb.2005.1765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Celsi F, Pizzo P, Brini M, Leo S, Fotino C, Pinton P, et al. Mitochondria, calcium and cell death: a deadly triad in neurodegeneration. Biochim Biophys Acta. 2009;1787:335–344. 10.1016/j.bbabio.2009.02.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bano D, Ankarcrona M. Beyond the critical point: An overview of excitotoxicity, calcium overload and the downstream consequences. Neurosci Lett. 2018;663:79–85. 10.1016/j.neulet.2017.08.048 [DOI] [PubMed] [Google Scholar]
- 35.Xu K, Tavernarakis N, Driscoll M. Necrotic cell death in C. elegans requires the function of calreticulin and regulators of Ca(2+) release from the endoplasmic reticulum. Neuron. 2001;31:957–971. 10.1016/s0896-6273(01)00432-9 [DOI] [PubMed] [Google Scholar]
- 36.Bianchi L, Gerstbrein B, Frokjaer-Jensen C, Royal DC, Mukherjee G, Royal MA, et al. The neurotoxic MEC-4(d) DEG/ENaC sodium channel conducts calcium: implications for necrosis initiation. Nat Neurosci. 2004;7:1337–1344. 10.1038/nn1347 [DOI] [PubMed] [Google Scholar]
- 37.Sammels E, Parys JB, Missiaen L, De Smedt H, Bultynck G. Intracellular Ca2+ storage in health and disease: a dynamic equilibrium. Cell Calcium. 2010;47:297–314. 10.1016/j.ceca.2010.02.001 [DOI] [PubMed] [Google Scholar]
- 38.Santulli G, Nakashima R, Yuan Q, Marks AR. Intracellular calcium release channels: an update. J Physiol. 2017;595:3041–3051. 10.1113/JP272781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Garcia-Anoveros J, Ma C, Chalfie M. Regulation of Caenorhabditis elegans degenerin proteins by a putative extracellular domain. Curr Biol. 1995;5:441–448. 10.1016/s0960-9822(95)00085-6 [DOI] [PubMed] [Google Scholar]
- 40.Tao L, Porto D, Li Z, Fechner S, Lee SA, Goodman MB, et al. Parallel Processing of Two Mechanosensory Modalities by a Single Neuron in C. elegans. Dev Cell. 2019;51:617–631 e613. 10.1016/j.devcel.2019.10.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tavernarakis N, Shreffler W, Wang S, Driscoll M. unc-8, a DEG/ENaC family member, encodes a subunit of a candidate mechanically gated channel that modulates C. elegans locomotion. Neuron. 1997;18:107–119. 10.1016/s0896-6273(01)80050-7 [DOI] [PubMed] [Google Scholar]
- 42.Li W, Feng Z, Sternberg PW, Xu XZ. A C. elegans stretch receptor neuron revealed by a mechanosensitive TRP channel homologue. Nature. 2006;440:684–687. 10.1038/nature04538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Treinin M, Chalfie M. A mutated acetylcholine receptor subunit causes neuronal degeneration in C. elegans. Neuron. 1995;14:871–877. 10.1016/0896-6273(95)90231-7 [DOI] [PubMed] [Google Scholar]
- 44.Chalfie M, Wolinsky E. The identification and suppression of inherited neurodegeneration in Caenorhabditis elegans. Nature. 1990;345:410–416. 10.1038/345410a0 [DOI] [PubMed] [Google Scholar]
- 45.Shreffler W, Magardino T, Shekdar K, Wolinsky E. The unc-8 and sup-40 genes regulate ion channel function in Caenorhabditis elegans motorneurons. Genetics. 1995;139:1261–1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hamelin M, Scott IM, Way JC, Culotti JG. The mec-7 beta-tubulin gene of Caenorhabditis elegans is expressed primarily in the touch receptor neurons. EMBO J. 1992;11:2885–2893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Akerboom J, Chen TW, Wardill TJ, Tian L, Marvin JS, Mutlu S, et al. Optimization of a GCaMP calcium indicator for neural activity imaging. J Neurosci. 2012;32:13819–13840. 10.1523/JNEUROSCI.2601-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Inan S, Wei H. The cytoprotective effects of dantrolene: a ryanodine receptor antagonist. Anesth Analg. 2010;111:1400–1410. 10.1213/ANE.0b013e3181f7181c [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chung S, Gumienny TL, Hengartner MO, Driscoll M. A common set of engulfment genes mediates removal of both apoptotic and necrotic cell corpses in C. elegans. Nat Cell Biol. 2000;2:931–937. 10.1038/35046585 [DOI] [PubMed] [Google Scholar]
- 50.Ellis RE, Jacobson DM, Horvitz HR. Genes required for the engulfment of cell corpses during programmed cell death in Caenorhabditis elegans. Genetics. 1991a;129:79–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mangahas PM, Zhou Z. Clearance of apoptotic cells in Caenorhabditis elegans. Semin Cell Dev Biol. 2005;16:295–306. 10.1016/j.semcdb.2004.12.005 [DOI] [PubMed] [Google Scholar]
- 52.Gelebart P, Opas M, Michalak M. Calreticulin, a Ca2+-binding chaperone of the endoplasmic reticulum. Int J Biochem Cell Biol. 2005;37:260–266. 10.1016/j.biocel.2004.02.030 [DOI] [PubMed] [Google Scholar]
- 53.Nakamura K, Zuppini A, Arnaudeau S, Lynch J, Ahsan I, Krause R, et al. Functional specialization of calreticulin domains. J Cell Biol. 2001;154:961–972. 10.1083/jcb.200102073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Doan NT, Paulsen ES, Sehgal P, Moller JV, Nissen P, Denmeade SR, et al. Targeting thapsigargin towards tumors. Steroids. 2015;97:2–7. 10.1016/j.steroids.2014.07.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Treiman M, Caspersen C, Christensen SB. A tool coming of age: thapsigargin as an inhibitor of sarco-endoplasmic reticulum Ca(2+)-ATPases. Trends Pharmacol Sci. 1998;19:131–135. 10.1016/s0165-6147(98)01184-5 [DOI] [PubMed] [Google Scholar]
- 56.Dachary-Prigent J, Pasquet JM, Freyssinet JM, Nurden AT. Calcium involvement in aminophospholipid exposure and microparticle formation during platelet activation: a study using Ca2+-ATPase inhibitors. Biochemistry. 1995;34:11625–11634. 10.1021/bi00036a039 [DOI] [PubMed] [Google Scholar]
- 57.Zhang H, Baehrecke EH. Eaten alive: novel insights into autophagy from multicellular model systems. Trends Cell Biol. 2015. 10.1016/j.tcb.2015.03.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Toth ML, Simon P, Kovacs AL, Vellai T. Influence of autophagy genes on ion-channel-dependent neuronal degeneration in Caenorhabditis elegans. J Cell Sci. 2007;120:1134–1141. 10.1242/jcs.03401 [DOI] [PubMed] [Google Scholar]
- 59.Samara C, Syntichaki P, Tavernarakis N. Autophagy is required for necrotic cell death in Caenorhabditis elegans. Cell Death Differ. 2008;15:105–112. 10.1038/sj.cdd.4402231 [DOI] [PubMed] [Google Scholar]
- 60.Artal-Sanz M, Samara C, Syntichaki P, Tavernarakis N. Lysosomal biogenesis and function is critical for necrotic cell death in Caenorhabditis elegans. J Cell Biol. 2006;173:231–239. 10.1083/jcb.200511103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Syntichaki P, Samara C, Tavernarakis N. The vacuolar H+ -ATPase mediates intracellular acidification required for neurodegeneration in C. elegans. Curr Biol. 2005;15:1249–1254. 10.1016/j.cub.2005.05.057 [DOI] [PubMed] [Google Scholar]
- 62.Venkatachalam K, Montell C. TRP channels. Annu Rev Biochem. 2007;76:387–417. 10.1146/annurev.biochem.75.103004.142819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Guéguinou M, Chantôme A, Fromont G, Bougnoux P, Vandier C, Potier-Cartereau M. KCa and Ca(2+) channels: the complex thought. Biochim Biophys Acta. 2014;1843:2322–2333. 10.1016/j.bbamcr.2014.02.019 [DOI] [PubMed] [Google Scholar]
- 64.Park BJ, Lee DG, Yu JR, Jung SK, Choi K, Lee J, et al. Calreticulin, a calcium-binding molecular chaperone, is required for stress response and fertility in Caenorhabditis elegans. Mol Biol Cell. 2001;12:2835–2845. 10.1091/mbc.12.9.2835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Suzuki H, Kerr R, Bianchi L, Frokjaer-Jensen C, Slone D, Xue J, et al. In vivo imaging of C. elegans mechanosensory neurons demonstrates a specific role for the MEC-4 channel in the process of gentle touch sensation. Neuron. 2003;39:1005–1017. 10.1016/j.neuron.2003.08.015 [DOI] [PubMed] [Google Scholar]
- 66.Treinin M, Gillo B, Liebman L, Chalfie M. Two functionally dependent acetylcholine subunits are encoded in a single Caenorhabditis elegans operon. Proc Natl Acad Sci U S A. 1998;95:15492–15495. 10.1073/pnas.95.26.15492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Brunner JD, Lim NK, Schenck S, Duerst A, Dutzler R. X-ray structure of a calcium-activated TMEM16 lipid scramblase. Nature. 2014;516:207–212. 10.1038/nature13984 [DOI] [PubMed] [Google Scholar]
- 68.Falzone ME, Rheinberger J, Lee BC, Peyear T, Sasset L, Raczkowski AM, et al. Structural basis of Ca(2+)-dependent activation and lipid transport by a TMEM16 scramblase. Elife. 2019;8 10.7554/eLife.43229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Alvadia C, Lim NK, Clerico Mosina V, Oostergetel GT, Dutzler R, Paulino C. Cryo-EM structures and functional characterization of the murine lipid scramblase TMEM16F. Elife. 2019;8 10.7554/eLife.44365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kalienkova V, Clerico Mosina V, Bryner L, Oostergetel GT, Dutzler R, Paulino C. Stepwise activation mechanism of the scramblase nhTMEM16 revealed by cryo-EM. Elife. 2019;8 10.7554/eLife.44364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Syntichaki P, Xu K, Driscoll M, Tavernarakis N. Specific aspartyl and calpain proteases are required for neurodegeneration in C. elegans. Nature. 2002;419:939–944. 10.1038/nature01108 [DOI] [PubMed] [Google Scholar]
- 72.Yamashima T. Ca2+-dependent proteases in ischemic neuronal death: a conserved ’calpain-cathepsin cascade’ from nematodes to primates. Cell Calcium. 2004;36:285–293. 10.1016/j.ceca.2004.03.001 [DOI] [PubMed] [Google Scholar]
- 73.Yamashima T, Oikawa S. The role of lysosomal rupture in neuronal death. Prog Neurobiol. 2009;89:343–358. 10.1016/j.pneurobio.2009.09.003 [DOI] [PubMed] [Google Scholar]
- 74.Kourtis N, Tavernarakis N. Autophagy and cell death in model organisms. Cell Death Differ. 2009;16:21–30. 10.1038/cdd.2008.120 [DOI] [PubMed] [Google Scholar]
- 75.Verhoven B, Krahling S, Schlegel RA, Williamson P. Regulation of phosphatidylserine exposure and phagocytosis of apoptotic T lymphocytes. Cell Death Differ. 1999;6:262–270. 10.1038/sj.cdd.4400491 [DOI] [PubMed] [Google Scholar]
- 76.Zhivotovsky B, Orrenius S. Calcium and cell death mechanisms: a perspective from the cell death community. Cell Calcium. 2011;50:211–221. 10.1016/j.ceca.2011.03.003 [DOI] [PubMed] [Google Scholar]
- 77.Schoenwaelder SM, Yuan Y, Josefsson EC, White MJ, Yao Y, Mason KD. et al. Two distinct pathways regulate platelet phosphatidylserine exposure and procoagulant function. Blood. 2009;114:663–666. 10.1182/blood-2009-01-200345 [DOI] [PubMed] [Google Scholar]
- 78.Hampton MB, Vanags DM, Porn-Ares MI, Orrenius S. Involvement of extracellular calcium in phosphatidylserine exposure during apoptosis. FEBS Lett. 1996;399:277–282. 10.1016/s0014-5793(96)01341-5 [DOI] [PubMed] [Google Scholar]
- 79.Suzuki J, Fujii T, Imao T, Ishihara K, Kuba H, Nagata S, et al. Calcium-dependent phospholipid scramblase activity of TMEM16 protein family members. J Biol Chem. 2013;288:13305–13316. 10.1074/jbc.M113.457937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Nakayama H, Chen X, Baines CP, Klevitsky R, Zhang X, Zhang H, et al. Ca2+- and mitochondrial-dependent cardiomyocyte necrosis as a primary mediator of heart failure. J Clin Invest. 2007;117:2431–2444. 10.1172/JCI31060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wojda U, Salinska E, Kuznicki J. Calcium ions in neuronal degeneration. IUBMB Life. 2008;60:575–590. 10.1002/iub.91 [DOI] [PubMed] [Google Scholar]
- 82.Danese A, Patergnani S, Bonora M, Wieckowski MR, Previati M, Giorgi C, et al. Calcium regulates cell death in cancer: Roles of the mitochondria and mitochondria-associated membranes (MAMs). Biochim Biophys Acta Bioenerg. 2017;1858:615–627. 10.1016/j.bbabio.2017.01.003 [DOI] [PubMed] [Google Scholar]
- 83.Ono Y, Saido TC, Sorimachi H. Calpain research for drug discovery: challenges and potential. Nat Rev Drug Discov. 2016;15:854–876. 10.1038/nrd.2016.212 [DOI] [PubMed] [Google Scholar]
- 84.Wood WB, the Community of C. elegans Researchers. The Nematode Caenorhabditis elegans. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press;1988. [Google Scholar]
- 85.Riddle DL, Blumenthal T, Meyer BJ, Priess JR, editors. C. elegans II. Plainview, NY: Cold Spring harbor Laboratory Press;1997. [PubMed] [Google Scholar]
- 86.Yu X, Odera S, Chuang CH, Lu N, Zhou Z. C. elegans Dynamin mediates the signaling of phagocytic receptor CED-1 for the engulfment and degradation of apoptotic cells. Dev Cell. 2006;10:743–757. 10.1016/j.devcel.2006.04.007 [DOI] [PubMed] [Google Scholar]
- 87.Shcherbo D, Murphy CS, Ermakova GV, Solovieva EA, Chepurnykh TV, Shcheglov AS, et al. Far-red fluorescent tags for protein imaging in living tissues. Biochem J. 2009;418:567–574. 10.1042/BJ20081949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Lee D, Singaravelu G, Park BJ, Ahnn J. Differential requirement of unfolded protein response pathway for calreticulin expression in Caenorhabditis elegans. J Mol Biol. 2007;372:331–340. 10.1016/j.jmb.2007.06.071 [DOI] [PubMed] [Google Scholar]
- 89.Shaner NC, Lambert GG, Chammas A, Ni Y, Cranfill PJ, Tsien RY, et al. A bright monomeric green fluorescent protein derived from Branchiostoma lanceolatum. Nat Methods. 2013;10:407–409. 10.1038/nmeth.2413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Jin Y. Transformation In: Hope IA, editor. C elegans, a practical approach. Oxford: Oxford University Press;1999. pp. 69–96. [Google Scholar]
- 91.Bloom L, Horvitz HR. The Caenorhabditis elegans gene unc-76 and its human homologs define a new gene family involved in axonal outgrowth and fasciculation. Proc Natl Acad Sci U S A. 1997;94:3414–3419. 10.1073/pnas.94.7.3414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Paix A, Folkmann A, Seydoux G. Precision genome editing using CRISPR-Cas9 and linear repair templates in C. elegans. Methods. 2017;121–122:86–93. 10.1016/j.ymeth.2017.03.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Li Z, Lu N, He X, Zhou Z. Monitoring the clearance of apoptotic and necrotic cells in the nematode Caenorhabditis elegans. Methods Mol Biol. 2013;1004:183–202. 10.1007/978-1-62703-383-1_14 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(Related to Fig 2) (A-C) Reproted here are results of three time-lapse series recording cytoplasmic Ca2+ levels and PS signal in or on the surface of PLML and PLMR neurons, respectively. Embryos are mec-4(e1611) homozygotes carrying the enIs92 transgenic array. Relative signal levels (in comparison to the background levels) of GCaMP5G in the cytoplasm, and of MFG-E8::GFP on the membrane surface, of the PLM neurons are ploted over time. The green, blue, and red arrows underneath the X-axis mark the time points when the rise of Ca2+ signal, the distinct cell swelling morphology becomes obvious, and PS is first seen on the surface of the neuron, respectively, by eye observation. (D) The mean integral values of relative cytoplasmic Ca2+ intensities in each cell measured from 4 necrotic PLM neurons in mec-4(e1611) embryos and 4 live PLM neurons in mec-4(+) embryos were obtained by calculating the integral value of GCaMP5G intensities within three time periods, 560–800 min, the entire time-lapse recording period, 560–695 min, the period prior to the mean time point when cell swelling is obvious, and 696–800 min post-1st embryonic division, respectively. Error bars represent s.e.m. (E) Time-lapse DIC images following the necrosis of one PLML neuron (white arrows). Yellow arrows mark the PLMR neuron which went out of focus in later time points. Time points are marked as min post-first embryonic cell division. Recording started at 560 min and ended at 900 min. The PLML neuron starts swelling at 675 min and continues swelling in the later time points until reaching its maximal size at 870 min. The scale bar is 10μm.
(TIF)
(Related to Figs 2 and 4) Presented here are results of time-lapse recording experiments that measure the intensity of cytoplasmic Ca2+ and cell swelling of PLM neurons during embryonic development. All embryos carry the transgenic array expressing Pmec-7GCaMP5G. (A) DIC and fluorescence time-lapse images of two live PLM neurons in one mec-4(+) embryo. Time points are marked as min post-1st embryonic division. White and yellow arrowheads mark the PLML and PLMR neurons, respectively. Scale bars are 10μM. (B) The relative signal levels of GCaMP5G were measured in 4 live PLM neurons and plotted over time. Graph (a) displays the plots of the PLML and PLMR neurons shown in (A), whereas graph (b) displays the plots of the PLML and PLMR neurons in an additional embryo. (C) The relative signal levels of GCaMP5G were measured in two necrotic PLM neurons in a mec-4(e1611) mutant embryo from a plate treated with DMSO but no dantrolene. Two other examples are shown in Fig 4. (D) The relative GCaMP5G signal levels measured in 2 necrotic PLM neurons in mec-4(e1611) mutant embryos from a 3μM dantrolene treated plate are plotted over time.
(TIF)
(Related to Fig 5) (A-B) Presented here are results of time-lapse recording experiments that measure the intensity of cytoplasmic Ca2+ and cell swelling of 4 PLM neurons during the development of two crt-1(bz29); mec-4(e1611) mutant embryos carrying the transgenic array expressing Pmec-7GCaMP5G. The relative GCaMP5 intensity in living PLM neurons are plotted over time. (C) The mean integral values of relative cytoplasmic Ca2+ intensities in each cell measured from 4 necrotic PLM neurons in mec-4(e1611) embryos and 4 PLM neurons whose necrosis were suppressed in crt-1(bz29); mec-4(e1611) embryos were obtained by calculating the integral value of GCaMP5G intensities within three time periods, 560–800 min, the entire time-lapse recording period, 560–695 min, the period prior to the mean time point when cell swelling is obvious, and 696–800 min, the period after cell swelling, respectively. Error bars indicate s.e.m.
(TIF)
(Related to Fig 7) (A) The NH4Cl treatment partially suppresses necrosis induced by the mec-4(e1611) mutation. The mean numbers of PLM neurons (labeled with Pmec-7GFP) that display the swelling necrosis phenotype in the tails of young ced-1(e1735); mec-4(e1611) L1 larvae from liquid cultures treated or not treated with 5mM NH4Cl are presented in the graph. Bars represent the mean values of each sample. Error bars indicate s.e.m. The numbers in the parentheses represent the numbers of L1 larvae scored. “**”, 0.001<p<0.01, Student t-test. (B) Images of necrotic and live PLM neurons that present PS on their outer surfaces in mec-4(e1611) L1 larvae. L1 larvae were from liquid cultures either treated with 5mM NH4Cl (d-f, white arrowheads) or left untreated (a-c). PLM neurons (c (white arrow), f (white and yellow arrowheads)) are labeled with the Pmec-7GFP reporter. DIC images identify one necrotic PLM neuron (white arrow) and one apoptotic cell (yellow arrow) in the tail of an L1 larva not treated with NH4Cl (a), and one necrotic PLM (yellow arrowhead) and one live PLM (white arrowhead) in an L1 larva from the 5mM NH4Cl treated culture (d-f). PS presentation on the cell surfaces (b, e) is detected by MFG-E8::mCherry. Scale bars are 5μm. (C) A bar graph representing the percentage of live PLM neurons that expose PS on their surfaces among all living PLM neurons in early L1 larvae after NH4Cl treatment. Error bars represent s.e.m.. The numbers in the parentheses represent the numbers of living cells scored.
(TIF)
(XLSX)
Data Availability Statement
All relevant data are within the manuscript and its Supporting information files.