

Summary
Cysteine is required for maintaining cellular redox homeostasis in both normal and transformed cells. Deprivation of cysteine induces the iron-dependent form of cell death known as ferroptosis; however, the metabolic consequences of cysteine starvation beyond impairment of glutathione synthesis are poorly characterized. Here, we find that cystine starvation of non-small cell lung cancer cell lines induces an unexpected accumulation of γ-glutamyl-peptides, which are produced due to a non-canonical activity of glutamate-cysteine ligase catalytic subunit (GCLC). This activity is enriched in cell lines with high levels of NRF2, a key transcriptional regulator of GCLC, but is also inducible in healthy murine tissues following cysteine limitation. γ-glutamyl-peptide synthesis limits the accumulation of glutamate, thereby protecting against ferroptosis. These results indicate that GCLC has a glutathione-independent, non-canonical role in the protection against ferroptosis by maintaining glutamate homeostasis under cystine starvation.
Keywords: cystine, cysteine, ferroptosis, GCLC, glutamate, γ-glutamyl, NRF2
Graphical Abstract
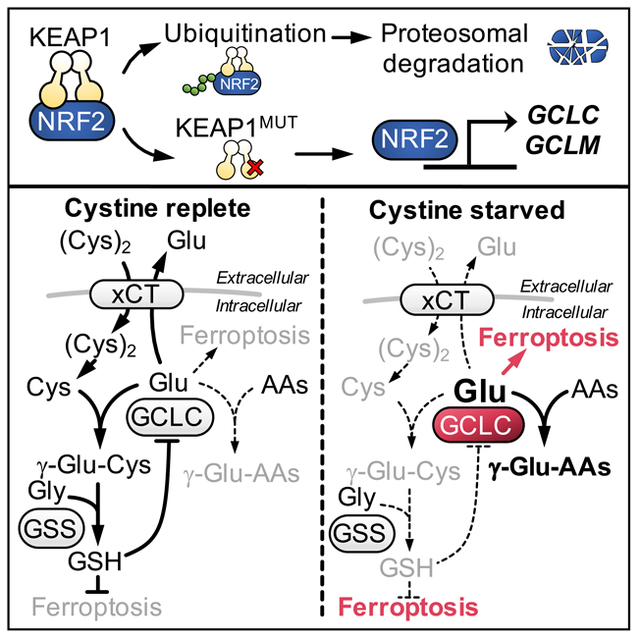
eTOC blurb
GCLC catalyzes the first step in glutathione synthesis via the ligation of cysteine with glutamate. In this issue of Cell Metabolism, Kang et al. demonstrate that under cysteine limiting conditions, GCLC instead ligates glutamate with alternative amino acids, thereby scavenging glutamate to protect against ferroptosis.
Introduction
Amino multiple cellular processes as a consequence of their sulfur moiety, which facilitates diverse functions, including enzyme catalysis, energy transfer, and redox metabolism (Furuyama and Sassa, 2000; Martinez-Reyes et al., 2016; Rouault, 2012; Solmonson and DeBerardinis, 2018; Vyas et al., 2016). Cysteine is a rate-limiting substrate for the synthesis of glutathione (GSH) (Stipanuk et al., 2006), the most abundant intracellular antioxidant (Winterbourn and Hampton, 2008). GSH is a tripeptide consisting of the amino acids cysteine, glutamate and glycine. The synthesis of GSH occurs in two steps (Anderson, 1998). First, glutamate and cysteine are ligated by GCLC, producing the dipeptide γ-glutamyl-cysteine (γ-Glu-Cys). Next, glycine is added to γ-Glu-Cys, producing the tripeptide GSH (γ-Glu-Cys-Gly). The antioxidant activity of GSH is a consequence of its function as a cofactor to multiple antioxidant proteins, including glutaredoxins (GRXs), GSH peroxidases (GPXs), and GSH S-transferases, thereby removing reactive oxygen species (ROS) (Harris and DeNicola, 2020).
Because of both its reactive thiol moiety and its essential function in redox homeostasis, cysteine levels are tightly regulated. While cysteine excess is prevented by overflow into the taurine pathway (Stipanuk et al., 2009), cysteine demand is met by inducible regulation of cystine import. Following oxidative stress, the expression of the cystine/glutamate exchange transporter xCT is induced, (Habib et al., 2015) thereby facilitating the uptake of cystine and its reduction to cysteine. In some tissues, most notably the liver, cysteine is also synthesized from homocysteine and serine via the transsulfuration pathway (Beatty and Reed, 1980; Rao et al., 1990; Reed and Orrenius, 1977). Given the important roles of cysteine, many cancers overexpress xCT (Ji et al., 2018; Takeuchi et al., 2013; Timmerman et al., 2013), which is positively regulated by oncogenic RAS (Lim et al., 2019) and NRF2 (Sasaki et al., 2002), and negatively regulated by the tumor suppressor p53 (Jiang et al., 2015). Pharmacological targeting of cystine uptake can effectively induce cancer cell death (Cramer et al., 2017; Dixon et al., 2012; Zhang et al., 2019), and cystine starvation can impair growth in multiple in vivo cancer models (Cramer et al., 2017; Zhang et al., 2019).
Cysteine inadequacy can induce an iron-dependent form of cell death known as ferroptosis (Dixon et al., 2012). Ferroptosis is triggered by the reaction of polyunsaturated fatty acids (PUFA) in membrane lipids with peroxyl radicals produced from iron (Fe2+) and ROS (Cao and Dixon, 2016; Yang et al., 2014), thereby inducing lipid peroxidation. Consistently, processes that promote ferroptosis include increased ferritin uptake (Gao et al., 2015), ferritin degradation (Mancias et al., 2014), synthesis of PUFA containing lipids (Dixon et al., 2015; Doll et al., 2017), polyamine synthesis-derived ROS (Zhang et al., 2020), and ROS production via other mechanisms (Gao et al., 2015; Gao et al., 2019). Ferroptosis protection is conferred by parallel pathways that protect cells against membrane lipid peroxidation in and GSH-dependent (Yang et al., 2014) and GSH-independent (Bersuker et al., 2019; Doll et al., 2019; Soula et al., 2020) manners. However, while cysteine is directly linked to GSH synthesis, which can influence both the levels of ROS and the activity of the lipid peroxidase GPX4, cysteine availability can also influence the levels of cofactors and metabolites associated with ferroptosis beyond its use for GSH synthesis, including the production of coenzyme A (Badgley et al., 2020; Leu et al., 2019) and iron-sulfur clusters (Alvarez et al., 2017). Importantly, the metabolic consequences of cystine starvation are poorly understood.
To understand the metabolic consequences of cystine starvation, we analyzed cysteine metabolism and cystine starvation-induced metabolic changes in non-small cell lung cancer (NSCLC) cells, which we previously found are sensitive to cystine starvation (Kang et al., 2019). Surprisingly, we found that under cysteine-deprived conditions, GCLC substituted other small, non-charged amino acids for cysteine in the ligation with glutamate to generate γ-glutamyl-peptides. γ-glutamyl-peptide synthesis by GCLC was also evident in mouse tissues. This promiscuous activity prevented glutamate accumulation to protect against ferroptosis.
Results
Transsulfuration cannot support NSCLC cysteine pools
To evaluate the consequence of cystine starvation in NSCLC cells, we starved a panel of cell lines of extracellular cystine and first monitored viability using fluorescent dyes that stain the nuclei of dead cells (Figure S1A), which were monitored over time using the Incucyte system. Cumulative cell death was calculated from the area under the curve (AUC), thereby facilitating comparisons between cell lines or treatments as previously described by others (Bersuker et al., 2019; Cao et al., 2019). Consistent with prior reports in lung and other cancer cell types (Badgley et al., 2020; Poursaitidis et al., 2017; Zhang et al., 2020), NSCLC cell death in response to cystine starvation exhibited features of the iron-dependent form of cell death known as ferroptosis. First, dead cells exhibited morphological features consistent with ferroptosis, rather than apoptosis, which could be rescued by both the lipid ROS scavenger and ferroptosis inhibitor Ferrostatin-1 (Fer-1) and the iron chelator deferoxamine (DFO) (Dixon et al., 2012) (Figure S1B). Moreover, lipid peroxide accumulation was observed prior to the onset of cell death, which was rescued by Fer-1 treatment (Figure S1C). Response of the NSCLC cell line panel to cystine starvation was heterogeneous, from the complete resistance of cell death out to 72 hours to the immediate onset of cell death within hours, which was lipid peroxide- and iron-dependent in all lines assayed (Figures 1A, S1D).
Figure 1. Transsulfuration cannot support NSCLC cysteine pools.
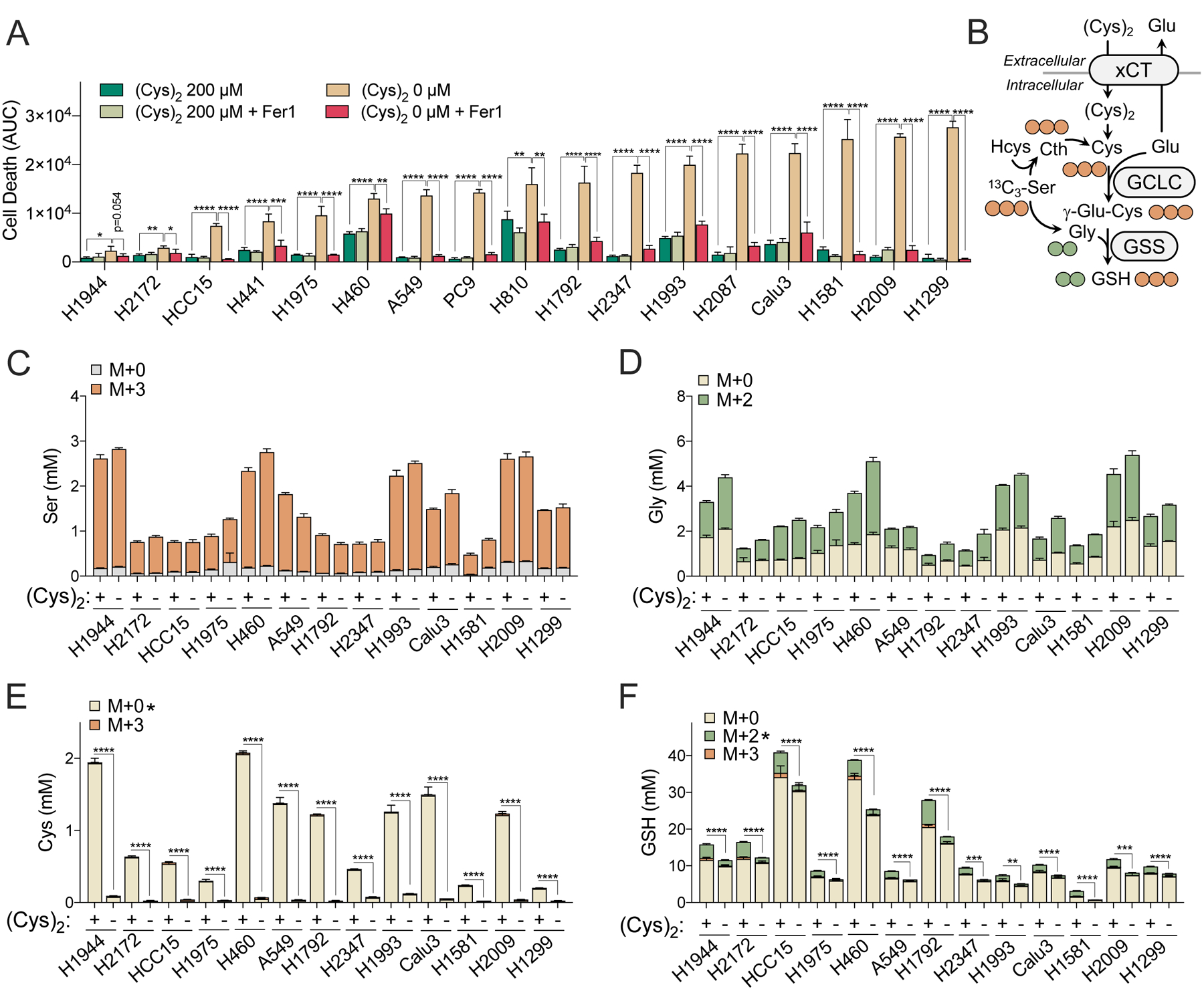
(A) Measurement of NSCLC cell death under cystine starved (0 mM) or replete (200 mM) conditions treated with Vehicle (0.1% DMSO) or Ferrostatin-1 (Fer-1, 10 μM) (N=4). Cell death was determined by Incucyte analysis of Sytox Green staining over 73 hrs, followed by normalization to cell density. Area under the curve (AUC) calculations are presented here. Full curves can be found in Figure S1D. (B) Schematic depiction of [13C3]-serine tracing into cysteine (M+3, 3 carbons labeled) and glutathione (M+2, 2 carbons labeled from glycine; M+3, 3 carbons labeled from cysteine). (C-F) Quantitation of [13C3]-serine tracing into (C) serine, (D) glycine, (E) cysteine, and (F) glutathione (GSH) following culture under cystine starved (−) or replete (+) conditions for 4 hrs (N=3). For A and C-F, data are shown as mean ± SD. N is number of biological replicates. *P<0.05, **P<0.01, ***P<0.001, and ****P<0.0001. For A, a one-way ANOVA with Bonferroni’s multiple comparison test was used for statistical analyses. An unpaired two-tailed t test was used for the statistical comparisons between non-labeled M+0 fraction in E and M+2 labeling fractions in F.
NSCLC cell lines have a diverse spectrum of mutations associated with diverse metabolic phenotypes and metabolic requirements (Chen et al., 2019; DeNicola et al., 2015). To examine whether the heterogeneous onset of ferroptosis under cystine starvation was associated with maintenance of the cysteine pool via the transsulfuration pathway, which synthesizes cysteine de novo from methionine-derived homocysteine and serine, we performed a quantitative analysis of 13C3-serine tracing into both cysteine and GSH. Cysteine is a rate limiting metabolite of GSH synthesis (Stipanuk et al., 2006) and GSH also plays an important role in ferroptosis via ROS metabolism (Dixon et al., 2012) and as a substrate of GPX4 (Conrad and Friedmann Angeli, 2015). 13C3-serine is metabolized to 13C2-glycine (M+2) and 13C3-cysteine (M+3), which are subsequently incorporated into GSH (M+2 and M+3, respectively, Figure 1B). Cells were labeled under cystine replete and starved conditions for 4 hrs, which was sufficient to label most of the serine fraction and half of the glycine fraction irrespective of the presence of cystine (Figure 1C and 1D). In contrast, minimal to no M+3 labeling of cysteine was detected under both fed and starved conditions, demonstrating little to no transsulfuration capacity (Figure 1E). Consequently, cystine starvation for 4 hours resulted in robust depletion of intracellular cysteine (Figure 1E). Moreover, both the amount of M+2 glycine incorporated into GSH and total GSH levels were lower across all cell lines following starvation (Figure 1F), demonstrating that GSH synthesis was impaired. These results indicate that de novo cysteine synthesis via transsulfuration cannot support intracellular cysteine levels in NSCLC cell lines, leading to impaired GSH synthesis as a consequence of cysteine depletion under cystine starved conditions.
Cystine starvation induces glutamate-derived γ-glutamyl-peptide accumulation
Despite a uniform depletion in intracellular cysteine, NSCLC cell lines had a heterogeneous induction of ferroptosis following cystine starvation. To identify additional cellular responses to starvation, we conducted non-targeted metabolomics in A549 cells. While most metabolites were depleted, we discovered an interesting cluster of unknown metabolites that were highly accumulated in cystine starved cells (Figure 2A). Using authentic standards, these unknown metabolites were identified as γ-glutamyl-di- or tripeptides, which all contain a glutamate moiety (Figure 2A). Authentic standards for γ-glutamyl-threonine (γ-Glu-Thr) and γ-glutamyl-alanyl-glycine (γ-Glu-Ala-Gly) were not available, and thus we further validated their identity via stable isotope metabolite tracing. 13C5, 15N2-glutamine tracing validated that both γ-Glu-Thr and γ-Glu-Ala-Gly were derived from glutamate (Figure 2B and 2C), and 2, 3, 3-2H3-serine tracing validated that γ-Glu-Ala-Gly was derived from glycine (Figure S2A). Moreover, we found that the xCT inhibitor erastin, which could deplete intracellular cysteine in a manner similar to cystine starvation (Figure S2B), also induced the accumulation of γ-glutamyl-di- or tripeptides to similar levels in A549 cells (Figure S2C). In addition, although γ-glutamyl-dipeptides are typically thought to be derived from GSH by γ-glutamyl transferase (GGT) via the transfer of the GSH γ-glutamyl group to recipient amino acids extracellularly (Hanigan and Pitot, 1985), 13C5, 15N2-glutamine tracing demonstrated that while the newly labeled GSH fraction was very small, as expected, glutamate and γ-glutamyl-dipeptides were approximately 50% labeled in cystine starved A549 cells (Figure 2B and 2C), suggesting that the γ-glutamyl-dipeptides were derived from glutamate but not from GSH. Finally, the levels of γ-glutamyl-peptides were inversely correlated with cystine starvation-induced ferroptosis sensitivity across the panel of NSCLC cell lines (Figure 2D). These results demonstrate that cystine starvation promotes the accumulation of glutamate-derived γ-glutamyl-peptides, which may play a role in the cellular response to cystine starvation.
Figure 2. Cystine starvation induces glutamate-derived γ-glutamyl-peptide accumulation.
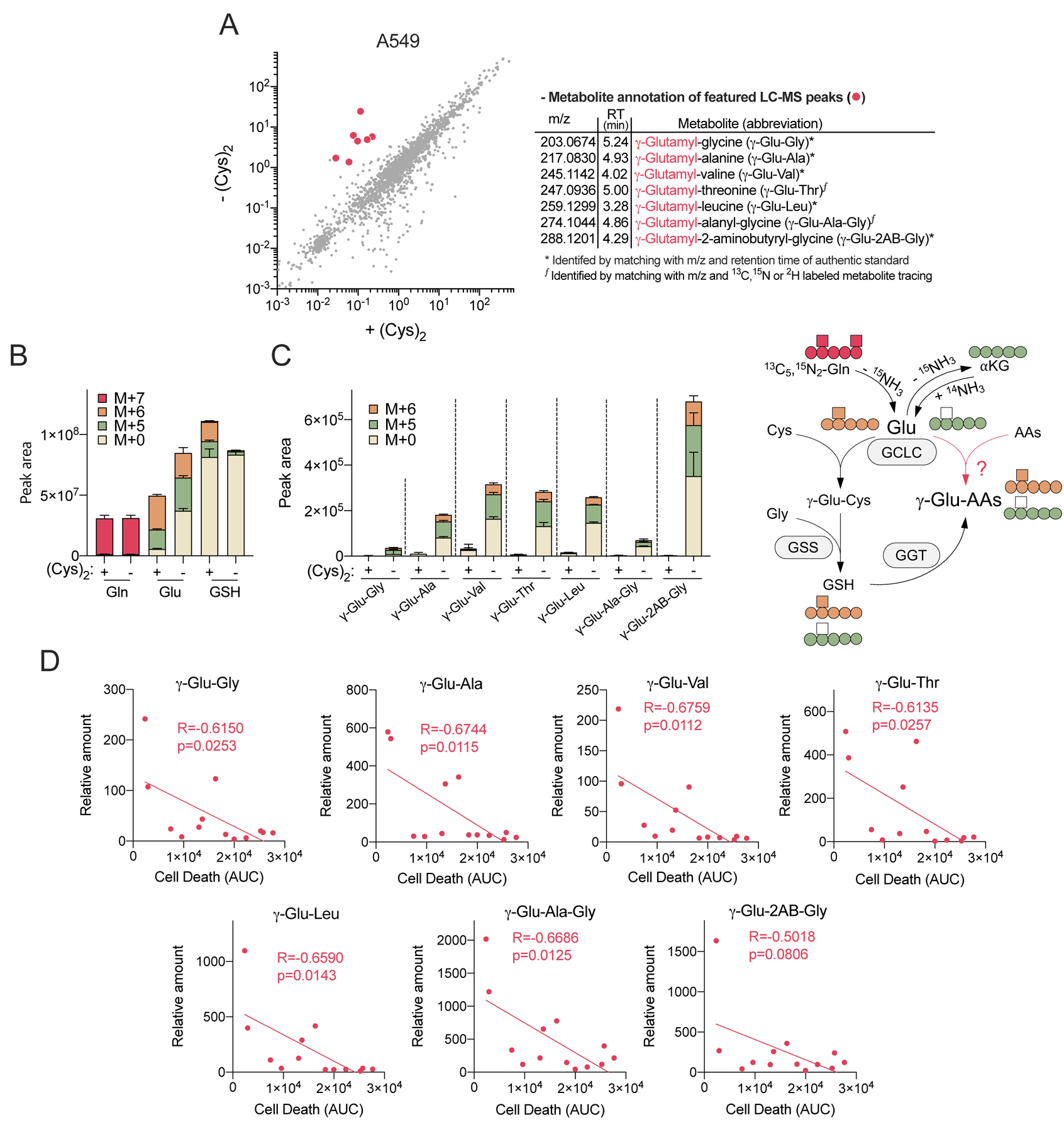
(A) Scatter plot comparison of non-targeted metabolomics features in A549 cells cultured under cystine replete (+Cys2) and starved conditions (-Cys2) for 4 hrs (left). The mean intensity of median-normalized LC-MS peaks of each group (N=3) are plotted on the axes, and each dot represents an individual LC-MS peak. The LC-MS peaks that highly accumulated under cystine starvation (red dots) were further identified and annotated (right). (B-C) A549 cell 13C5, 15N2-Gln tracing into (B) Gln, Glu, GSH, and (C) γ-Glu-peptides following culture in cystine replete or starved conditions for 4 hrs (N=3). (D) Correlation between ferroptotic cell death (AUC, from Figure 1A) and the levels of γ-Glu-peptides across 13 NSCLC cell lines. The γ-Glu-peptides were analyzed following cystine starvation for 12 hrs and normalized to the mean value of H1581 cells under cystine replete conditions (N=13). For B-D, data are shown as mean ± SD. N is number of biological replicates. For D, Pearson correlation test was used for statistical analysis.
GCLC mediates γ-glutamyl-peptide synthesis
We next wanted to understand how cells were producing γ-glutamyl-peptides. Among our accumulated γ-glutamyl-peptides was the GSH-similar tripeptide γ-glutamyl-2-aminobutyryl-glycine (γ-Glu-2AB-Gly), also known as ophthalmic acid, which is synthesized by GCLC and GSS under cysteine-limiting conditions. In this process, 2-aminobutyrate is substituted for cysteine (Huang et al., 1988; Oppenheimer et al., 1979), suggesting GCLC may directly synthesize γ-glutamyl-peptides. Moreover, γ-glutamyl-valine is synthesized by the Saccharomyces cerevisiae glutamate-cysteine ligase (Sofyanovich et al., 2019), and gglutamyl-dipeptide synthesis by mouse liver extracts was recently shown to be GCLC-dependent (Kobayashi et al., 2020). Thus, we hypothesized that the γ-glutamyl-dipeptides were directly generated by GCLC rather than by GGT. Consistent with this hypothesis, we observed a strong correlation between the levels of γ-glutamyl-dipeptides with GCLC expression under cysteine starved, but not cysteine replete, conditions across the NSCLC cell lines (Figure 3A).
Figure 3. GCLC mediates γ-glutamyl-peptide synthesis in cell culture.
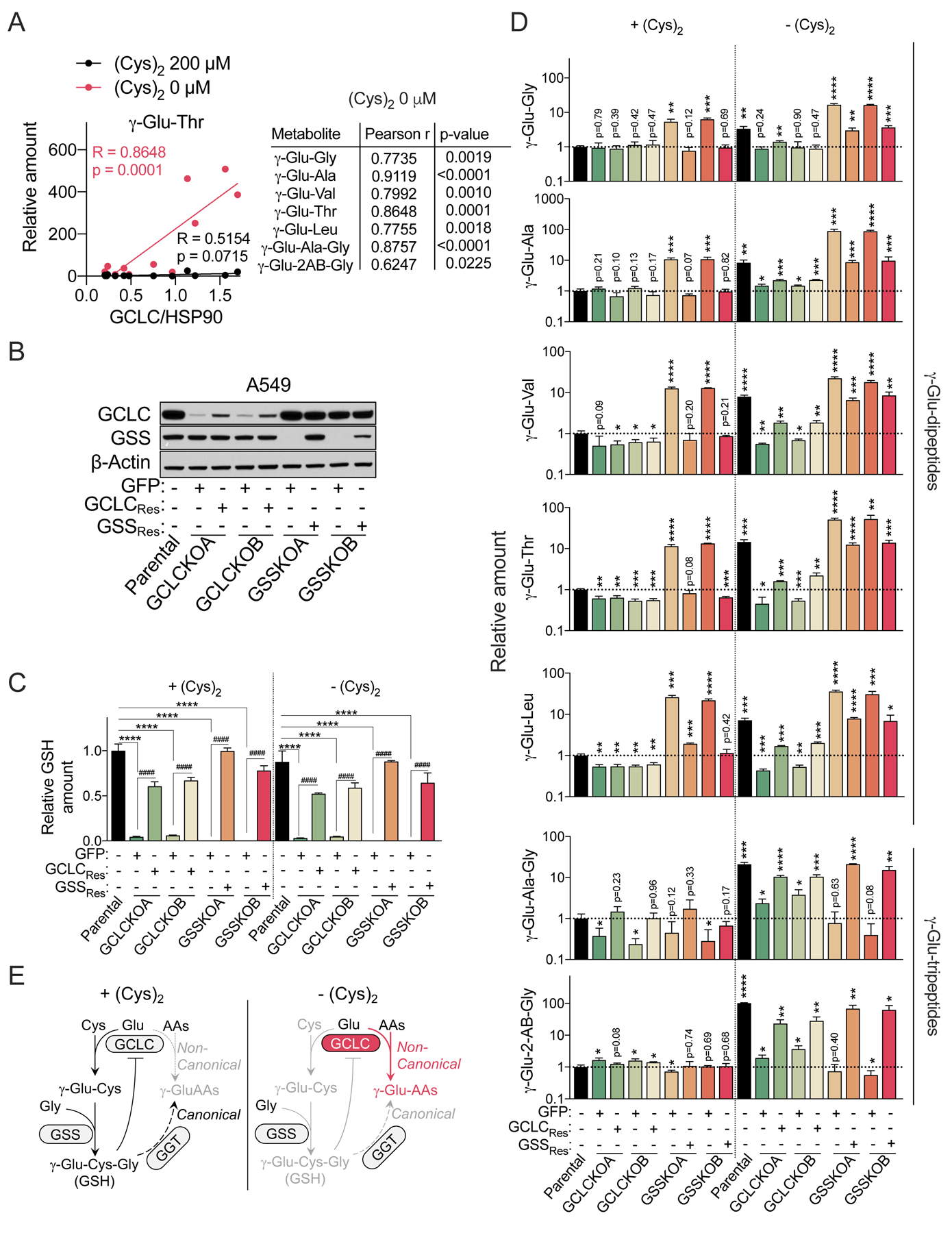
(A) Correlation of γ-Glu-dipeptides (data from Figure 2D) with GCLC expression in NSCLC cell lines. The GCLC protein expression of each cell line was normalized to the amount of HSP90 protein. The western blot can be found in Figure 5A. (B) Representative immunoblots of parental A549 cells, GCLC KO clones reconstituted with GFP (+GFP) or sgRNA-resistant GCLC (+GCLCRes), and GSS KO clones reconstituted with GFP (+GFP) or sgRNA-resistant GSS (+GSSRes). β-actin was used for the loading control. (C-D) Intracellular GSH levels (C) and γ-Glu-peptides levels (D) in the cells from (B) under cystine replete or starved conditions for 2.5 hrs (N=3). The data were normalized to the mean value of parental A549 cells under cystine replete conditions. (E) Schematic depicting the non-canonical, γ-Glu-peptide synthesis activity of GCLC. For C and D, data are presented as mean ± SD. n.d., not detected. N is number of biological replicates. *P<0.05, **P<0.01, ***P<0.001, and ****P<0.0001; ##P<0.01, ###P<0.001, ####P<0.0001. For C, a one-way ANOVA with Bonferroni’s multiple comparison test was used for statistical analyses for the comparison of Parental, GCLCKOA/B + GFP, and GSSKO A/B + GFP. For the comparison between GCLC KO or GSS KO group (GCLCKO A/B + GFP or GSSKO A/B + GFP) and their GCLC or GSS reconstituted group (GCLC KOA/B + GCLCRes or GSSKO A/B + GSSKOA/B + GSSRes), an unpaired two-tailed t test was used. For D, an unpaired two-tailed t test was used for the comparison with parental cells under cystine replete conditions [+(Cys)2].
To directly evaluate the production of γ-glutamyl-dipeptides by GCLC, we generated both GCLC and GSS KO A549 clones using CRISPR/Cas. In both cases, the clones should be deficient in GSH synthesis, but only GCLC KO cells should lack direct γ-glutamyl-dipeptide synthesis capacity. Two clones were generated for both GCLC and GSS KO cells and reconstituted with sgRNA resistant cDNAs (GCLCRes and GSSRes) to control for any clonal effects during selection (Figure 3B). Importantly, both clones were defective in GSH synthesis as evidenced by significantly reduced intracellular GSH levels effectively compared to parental cells (Figure 3C). However, while the GSS KO clones lacked detectable GSS protein and GSH, the GCLC KO clones were hypomorphic, with a small amount of residual GCLC expression and GSH. Despite repeated attempts, clones completely deficient for GCLC could not be obtained. However, despite this limitation, GCLC KO clones were dramatically impaired in γ-glutamyl-dipeptide accumulation following cystine starvation (Figure 3D). In contrast, GSS KO clones instead had increased γ-glutamyl-dipeptide levels under both fed and starved conditions, although their levels were enhanced by cystine starvation. This is likely explained by the feedback inhibition of GCLC by GSH, and more weakly by γ-Glu-2AB-Gly (Richman and Meister, 1975), which cannot be synthesized in GSS KO cells, leading to hyperactivation of GCLC. The production of γ-Glu-2AB-Gly and γ-Glu-Ala-Gly tripeptides required both GCLC and GSS, because GSS activity is required for the ligation of glycine. These phenotypes were all rescued by the reconstitution of KO cells with sgRNA-resistant cDNAs, confirming the specificity of the KO. Consistent alterations of GSH and γ-glutamyl-peptides were also observed in polyclonal GCLC and GSS KO H1299 cells, which were also rescued by GCLC or GSS restoration (Figure S3A–C). The accumulation of γ-glutamyl-peptides following erastin treatment or cystine starvation was also blocked by co-treatment with the GCLC inhibitor buthionine sulfoximine (BSO, Figure S3D–F), demonstrating that the enzyme activity of GCLC was necessary for their accumulation. These results indicate that γ-glutamyl-dipeptides are directly generated by GCLC under cystine starved conditions (Figure 3E).
GCLC mediates γ-glutamyl-peptide synthesis in vivo
Next, we examined whether GCLC mediates the synthesis of γ-glutamyl-peptides in vivo under normal physiological conditions. To this end, mice were treated with a single dose of PBS or cyst(e)inase to deplete serum cystine for 24 hours, followed by with saline or BSO to inhibit Gclc for 4 hours (Figure 4A). In a separate experiment, systemic Gclc deletion was induced in an adult mouse to achieve chronic Gclc inhibition for 2 weeks, which was verified by loss of mRNA expression in the lung, liver and kidney (Figure 4B). The consequences of these two independent perturbations were examined in these three tissues and the serum. While glutathione and cystine were present in their reduced forms (GSH, Cys) in tissues, the serum had predominantly the oxidized form (GSSG, Cys2), which may either be due to the oxidizing extracellular conditions or oxidation during sample preparation. Cyst(e)inase effectively depleted cyst(e)ine in the serum (Figure 4C), liver (Figure 4F), and lung (Figure S4A), but unexpected elevated cysteine in the kidney (Figure S4D). Cyst(e)ine depletion was accompanied by a modest decrease in liver, kidney and lung GSH (Figures 4G, S4B, S4E), but elevated serum GSSG (Figure 4D). As expected, BSO robustly depleted tissue GSH (Figures 4G, S4B, S4E) and serum GSSG (Figure 4D). Interestingly, BSO did not significantly influence cysteine levels in most tissues (Figures 4F, S4A, S4D), but did significantly elevate serum (Cys)2 (Figure 4C), possibly as a consequence of impaired GSH synthesis. In addition, the efficacy of Gclc deletion was evident by the depletion of glutathione by 75–90% in the serum and tissues (Figures 4I, 4K, S4G, S4i). As observed in cell culture, cysteine depletion elevated the levels of γ-glutamyl-peptides, particularly in the serum and liver, which was reversed by BSO (Figures 4E, 4H, S4C, S4F). Furthermore, we found that inhibition of Gclc with BSO or enzyme deletion depleted the basal levels of γ-glutamyl-peptides, including both the dipeptides and tripeptides, in all tissues (Figures 4J, 4L, S4H, S4J). Overall, these results indicate that GCLC mediates the in vivo synthesis of γ-glutamyl-peptides, which is responsive to cysteine availability.
Figure 4. GCLC mediates γ-glutamyl-peptide synthesis in vivo.
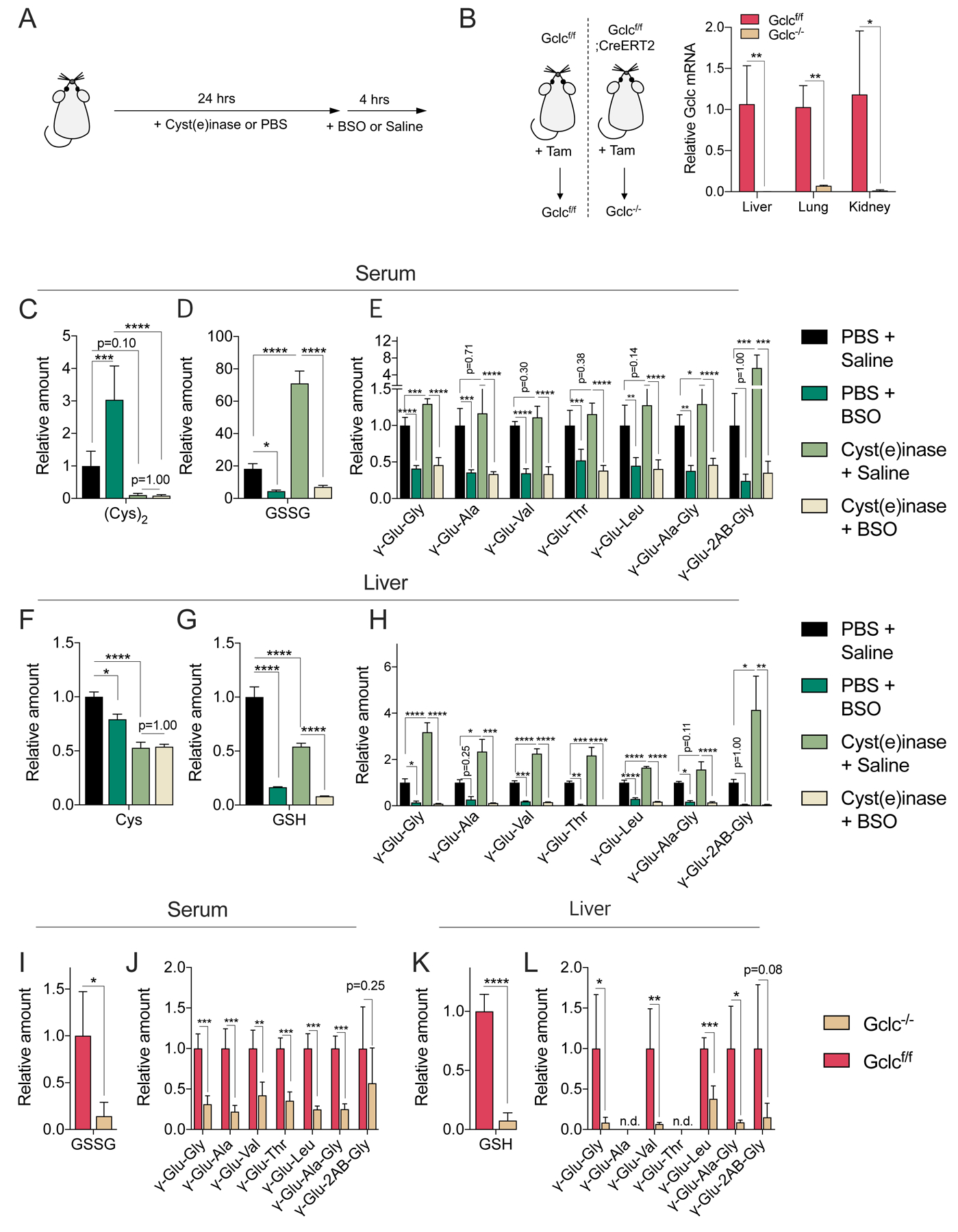
(A) Schematic depicting the Cyst(e)inase and BSO treatment schedule for the depletion of extracellular cyst(e)ine and inhibition of Gclc. Cyst(e)inase (75 mg/kg) or vehicle (PSB) were administered, followed by treatment with BSO (100 mmol/kg) or vehicle (saline) 24 hours later. Tissues were collected after 4 hrs. (B) Evaluation of Gclc mRNA expression after tamoxifen (Tam)-inducible Gclc deletion in the adult mouse. Gclcf/f (control, Gclc functional) and Gclc−/− (Gclc knockout). The mRNA levels are normalized to the mean value of Gclcf/f mouse tissues (N=4). (C-E) Analysis of serum cystine (C), GSSG (D), and γ-Glu-peptide levels (E) in mice treated with Cyst(e)inase/BSO. (F-H) Analysis of liver cysteine (F), GSH (G), and γ-Glu-peptide levels (H) in the mice from (C-E). The metabolite levels are normalized to the mean value of PBS/saline treated mice (N=5). (I-J) Analysis of serum GSSG (I) and γ-Glu-peptide levels (J) in Gclcf/f and Gclc−/− mice. (K-L) Analysis of and liver GSH (K) and γ-Glu-peptide levels (L) in the mice from (I-J). The metabolite levels are normalized to the mean value of Gclcf/f mice (N=4). For B-L, data are presented as mean ± SD. N is number of biological replicates. n.d., not detected. *P<0.05, **P<0.01, ***P<0.001, and ****P<0.0001. For B and I-L, an unpaired two-tailed t test was used for the statistical comparisons. For C-H, a one-way ANOVA with Bonferroni’s multiple comparison test was used for statistical analyses.
NRF2 promotes γ-glutamyl-peptide synthesis via GCLC
NSCLC cell lines demonstrated a wide range in the degree of γ-glutamyl-peptide accumulation (Figure 2D), despite similar depletion of intracellular cysteine following cystine starvation (Figure 1E), suggesting additional regulatory mechanisms may explain their levels. We examined the expression of GCLC protein across the NSCLC panel and found the expression of GCLC, and its modifier subunit GCLM, but not GSS, were associated with high activity of NRF2 (Figure 5A), a known regulator of both GCLC and GCLM expression. Consistently, KEAP1 mutant NSCLC cell lines had significantly higher levels of γ-glutamyl-peptides following cystine starvation compared to KEAP1 wild-type cell lines (Figure 5B, Figure S5A). To directly assay the effect of NRF2 on GCLC expression and γ-glutamyl-peptide synthesis, we used NRF2 KO A549 cells reconstituted with empty vector or NRF2, which we previously used to study cysteine metabolism (Kang et al., 2019). NRF2 reconstitution in this system led to elevated expression of both GCLC and GCLM (Figure 5C), and significantly increased levels of γ-glutamyl-dipeptides and γ-Glu-2AB-Gly, but not γ-Glu-Ala-Gly, under cystine starved conditions (Figure 5D). which was inhibited by BSO. In contrast, NRF2 promoted an increase in GSH only under cystine replete conditions (Figure 5E). These results were recapitulated by NRF2 activation in the KEAP1 wild type cell line Calu3 with the KEAP1 inhibitor KI-696 (Figures 5F–H) (Davies et al., 2016). Collectively, these results demonstrate that the regulation of GCLC by NRF2 promotes GSH synthesis under cystine replete conditions, but γ-glutamyl-peptide synthesis under cystine starved conditions.
Figure 5. NRF2 promotes γ-glutamyl-peptide synthesis via GCLC.
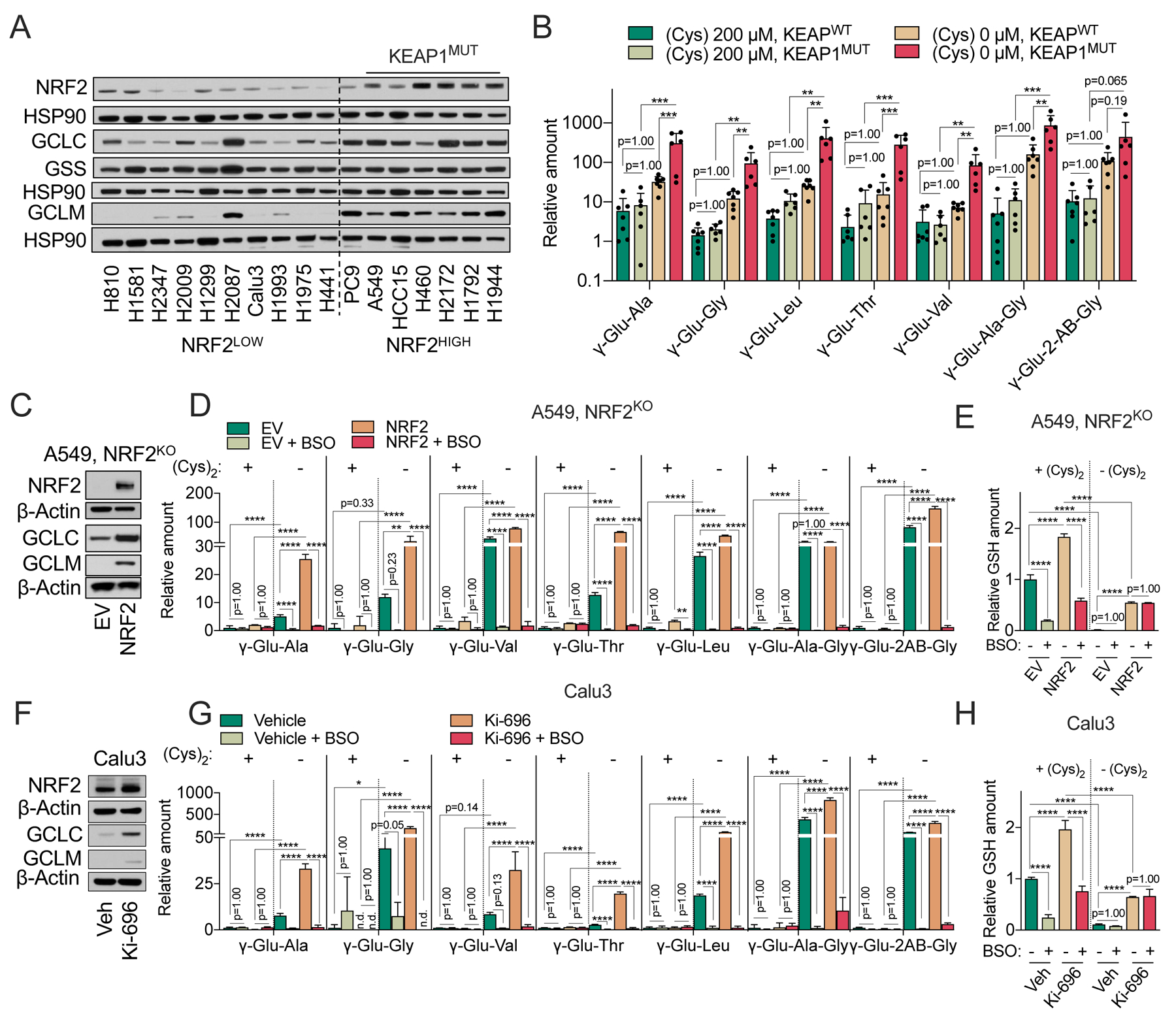
(A) Representative immunoblot of NRF2, GCLC, GSS, and GCLM expression in NRF2LOW and NRF2HIGH (KEAP1 mutant: A549, HCC15, H460, H2172, H1792, and H1944) NSCLC cell lines. HSP90 is used as loading control. (B) Comparison of γ-Glu-dipeptide levels in KEAP1WT and KEAPMUT NSCLC cell lines. Individual cell line data are found in Figure 5SA, and also correspond to the data in Figure 2D. The γ-Glu-peptides were analyzed following culture in cysteine replete and starved conditions for 12 hrs and normalized to the mean value of H1581 cells under cystine replete conditions. (C) Representative immunoblot of NRF2, GCLC, and GCLM expression in NRF2 KO A549 cells transduced with empty vector (EV) or NRF2. β-actin is used as loading control. (D-E) Analysis of intracellular γ-Glu-dipeptides (D) and GSH levels (E) in the cells from (C) under cystine replete or starved conditions in the presence and absence of 100 μM BSO for 12 hrs. (F) Representative immunoblot of NRF2, GCLC, and GCLM expression in Calu3 cells pre-treated with 100 nM of KI-696 or vehicle (Veh, 0.1% DMSO) for 48 hrs. b-actin is used as loading control. (G-H) Analysis of intracellular γ-Glu-dipeptides (G) and GSH levels (H) in the cells from (F) under cystine replete or starved conditions in the presence and absence of 100 μM BSO for 12 hrs. For B, D, E, G, and H, data are presented as mean ± SD. N is number of biological replicates. n.d., not detected. **P<0.01, ***P<0.001, and ****P<0.0001. For B, D, E, G, and H, a one-way ANOVA with Bonferroni’s multiple comparison test was used for statistical analyses.
Dipeptide synthesis protects KEAP1 mutant cells from ferroptosis
We next examined whether γ-glutamyl-peptides play a causal role in protection against ferroptosis. While we found that GCLC expression was anticorrelated with ferroptosis (Figure 6A), exogenous γ-Glu-Ala, γ-Glu-Leu, γ-Glu-Val, or γ-Glu-Gly did not protect GCLC KO A549 cells against cystine starvation-induced ferroptosis (Figure 6B), despite robust uptake of these dipeptides into the cells (Figure S6A). Multiple studies have demonstrated a potent, synergistic effect of the GCLC inhibitor BSO with limitation of cystine uptake or availability (Badgley et al., 2020; Cramer et al., 2017; Harris et al., 2015), leading us to hypothesis it was the synthesis of the γ-glutamyl-peptides, rather than the peptides themselves, that was protective. Indeed, we observed that BSO treatment consistently promoted ferroptosis of the KEAP1 mutant cell lines under cystine starvation but did not significantly affect most KEAP1 wild-type lines (Figure 6C, Figure S6B). We directly tested the effect of NRF2 on ferroptosis by reintroducing NRF2 into NRF2 KO A549 cells, which dramatically suppressed cystine-starvation induced ferroptosis; this effect was partially rescued by GCLC inhibition with BSO (Figure 6D). Similarly, KEAP1 inhibition in KEAP1 wild type Caul3 cells suppressed cystine starvation-induced ferroptosis, which was completely rescued by BSO (Figure 6E). Because we found that GSH synthesis is dramatically impaired under cystine starvation (Figure 1F, Figure 2B), these results suggest that BSO promotes ferroptosis by inhibiting γ-glutamyl-peptide synthesis. To directly interrogate the role of the γ-glutamyl-peptide synthesis function of GCLC in ferroptosis protection, we used the GCLC and GSS KO A549 clones (Figure 3B), which are both defective in GSH synthesis. Importantly, the GCLC KO clones demonstrated accelerated ferroptosis induction under cystine starvation compared to parental cells, which could be rescued by GCLC cDNA, while the GSS KO clones did not (Figure 6F). Similar results were observed following treatment with the xCT inhibitor erastin (Figure 6G). Furthermore, BSO treatment induced ferroptosis in the GSS KO clones (Figure 6H), but did not further sensitize the GCLC KO clones, further confirming the GSH-independent role of GCLC in ferroptosis protection and the specificity of BSO for GCLC. Finally, both GCLC KO and GSS KO accelerated ferroptosis induction under cystine starvation following acute deletion in KEAP1 WT H1299 cells, although GCLC KO was more potent (Figure 6I), suggesting NRF2 activation state may decrease cellular reliance on GSH for ferroptosis protection. Interestingly, we observed KEAP1 WT cell lines had higher GPX4 expression, suggesting they may be more reliant on GSH for lipid peroxide detoxification (Figure S6C). Together, these data indicate that GCLC has an additional, GSH-independent function to prevent cystine starvation-induced ferroptosis of NSCLC cells.
Figure 6. Dipeptide synthesis protects KEAP1 mutant cells from ferroptosis.
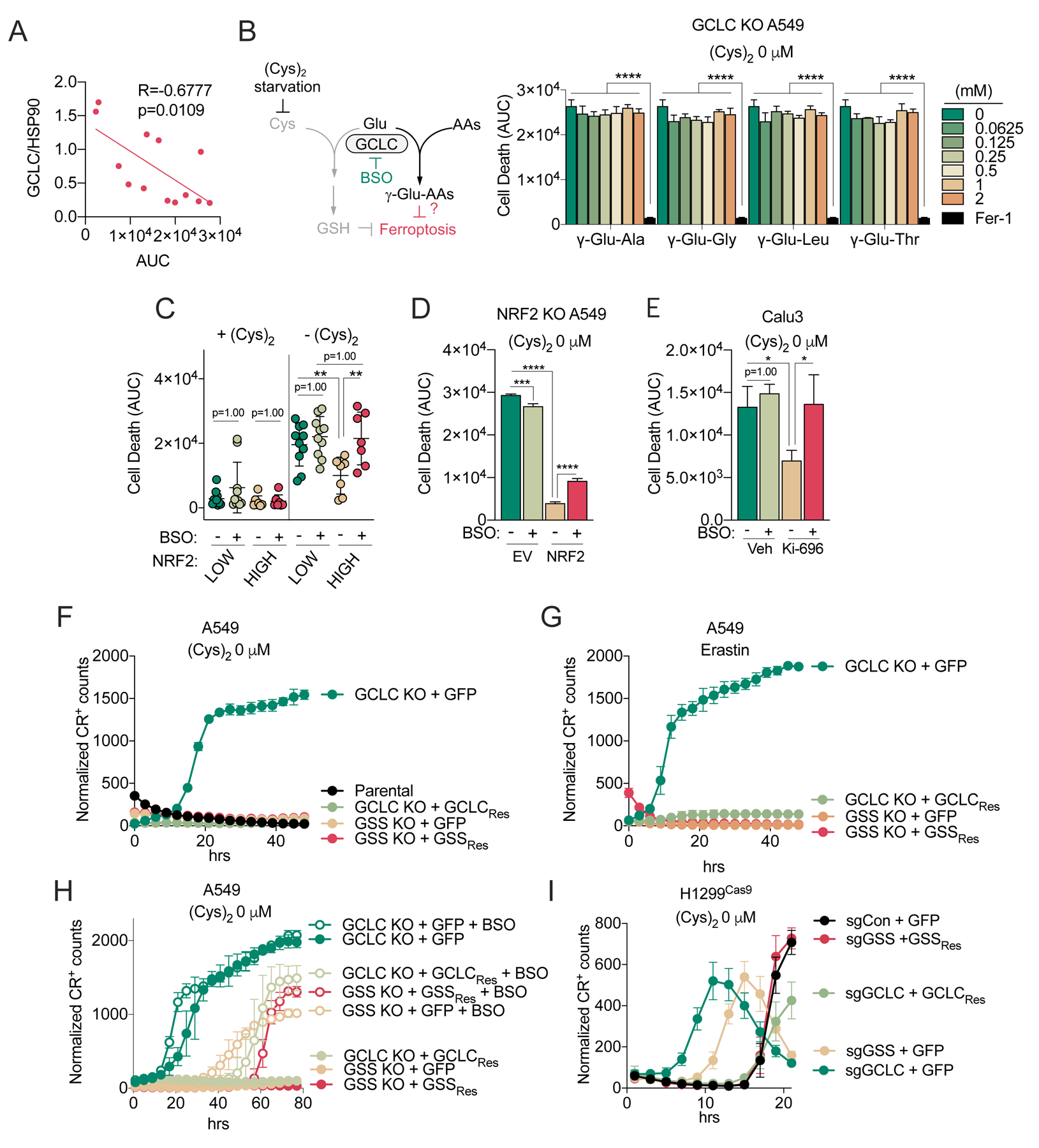
(A) Correlation between cell death under cystine starvation (AUC, from Figure 1A) and GCLC expression in NSCLC cells (N=13). GCLC expression is normalized to HSP90 expression and can be found in Figure 5A. (B) Evaluation of the influence γ-Glu-dipeptide treatment on the death of GCLC KO A549 cells under cystine starvation. Cells were treated with γ-Glud-ipeptides at the indicated concentration or Fer-1 (10 μM) as a positive control. Cell death was monitored with Sytox Green every 2 hrs for 49 hrs, normalized to cell density, and then AUCs were calculated (N=3). (C) Evaluation of the death of NRF2HIGH (N=7) and NRF2LOW (N=10) cell lines under cysteine replete and starved conditions in the presence and absence of 100 μM BSO. Each dot represents the mean AUC of each NSCLC cell line. Cell death was monitored with Sytox Green and individual cell line data is found in Figure S6B. (D) Evaluation of the death of NRF2 KO A549 cells transduced with empty vector (EV) or NRF2 under cystine replete and starved conditions in the presence and absence of 100 μM BSO (N=3). Cell death was monitored by Sytox Green every 2 hours for 65 hours, followed by AUC calculation. (E) Evaluation of the death of Calu3 cells pre-treated with 100 nM of KI-696 or vehicle (Veh, 0.1% DMSO) for 48 hrs, followed by culture under cystine replete and starved conditions in the presence and absence of 100 μM BSO (N=3). Cell death was monitored with Sytox Green every 2 hours for 47 hours, followed by AUC calculation. (F) Evaluation of the death of parental A549 cells, GCLC KO clones reconstituted with GFP (+GFP) or sgRNA-resistant GCLC (+GCLCRes), and GSS KO clones reconstituted with GFP (+GFP) or sgRNA-resistant GSS (+GSSRes) following cystine starvation for the indicated time points (N=3). (G) Evaluation of the death of GCLC KO clones reconstituted with GFP (+GFP) or sgRNA-resistant GCLC (+GCLCRes), and GSS KO clones reconstituted with GFP (+GFP) or sgRNA-resistant GSS (+GSSRes) treated with of Erastin (5 μM) for the indicated time points (N=3). (H) Evaluation of the death of the cells from (G) under the cystine starved conditions in the presence and absence of 100 μM BSO. (N=3). (I) Evaluation of the death of H1299Cas9 cells infected with sgRNAs (sgCon, sgGCLC or sgGSS), followed by reconstitution with GFP (+GFP), sgRNA-resistant GCLC (+GCLCRes), or sgRNA-resistant GSS (+GSSRes) under cystine starved conditions (N=4). For F-H, results are representative of 2 independent GCLC and GSS KO clones. For F-I, cell death was monitored by Cytotox Red (CR) every 2 hrs (I), 3 hrs (F, G) or 4 hrs (H). For B-I, data are presented as mean ± SD. N is number of biological replicates. *P<0.05, ***P<0.001, and ****P<0.0001. For A, a Pearson correlation analysis was used. For B-E, a one-way ANOVA with Bonferroni’s multiple comparison test was used for statistical analyses.
Dipeptide synthesis scavenges glutamate
The γ-glutamyl-peptides all have glutamate in common but contain other amino acids that may play a causal role in ferroptosis. To examine this possibility, we examined the association of their levels under cystine starvation with ferroptosis sensitivity. We found that glutamate levels, but not the levels of the other amino acids within γ-glutamyl-peptides, were strongly correlated with ferroptosis (Figure 7A). Importantly, prior work from Papagiannakopoulos and colleagues demonstrated that activation of NRF2 increases the consumption of glutamate for both glutathione synthesis and glutamate secretion by xc− antiporter system, thereby limiting glutamate availability for biosynthetic reactions (Sayin et al., 2017). As expected, while there was substantial heterogeneity in the expression of the glutamine transporter SLC1A5 and the glutamate transaminase GOT1 across NSCLC cell lines, we observed that cell lines with high NRF2 levels had high xCT expression and elevated glutamate export under cystine replete, but not starved conditions (Figures S7A and S7B). Furthermore, we observed a positive correlation between xCT expression and glutamate export only under cystine replete conditions (Figure 7B). In contrast, GCLC protein levels and intracellular glutamate were anticorrelated under both cystine replete and starved conditions (Figure 7C), suggesting that GCLC was still capable of limiting intracellular glutamate in the absence of cysteine. Furthermore, we observed that KEAP1 mutant cells had lower intracellular levels of glutamate, but not glycine (Figure S7C), which was preserved under cystine starved conditions (Figure 7D).
Figure 7. Dipeptide synthesis scavenges glutamate.
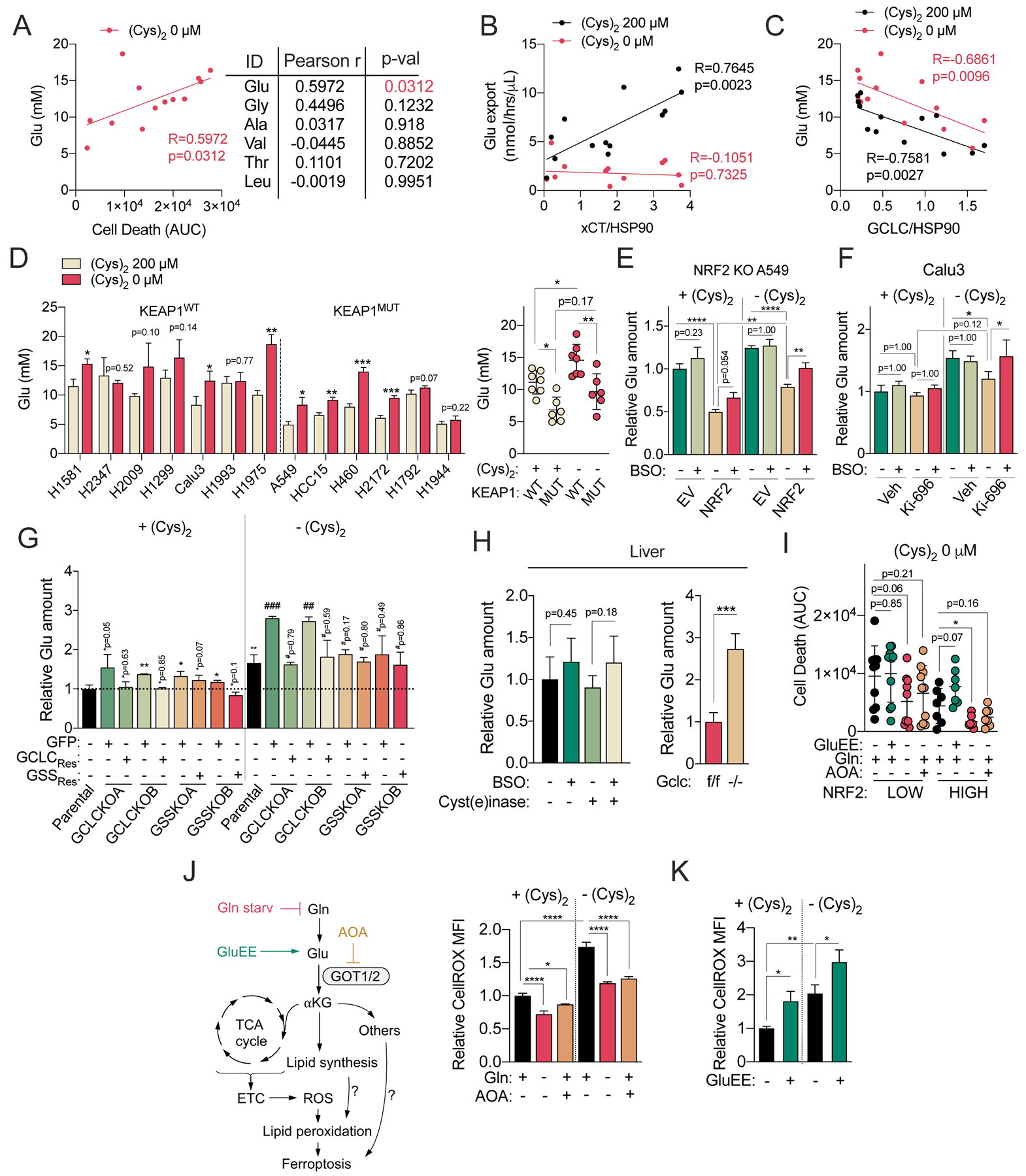
(A) Correlation between the intracellular concentrations of γ-Glu-peptide substrate amino acids (Glu, Gly, Ala, Val, Thr, Leu) following cystine starvation for 12 hours and cystine starvation AUCs (from Figure 1A) (N=13). Individual cell line Glu concentrations can be found in Figure 7D. (B) Correlation between xCT protein expression and the Glu export rate under cystine replete and starved conditions for 12 hrs in NSCLC cells. xCT expression is normalized to HSP90 expression and can be found in Figure S7A (N=13). (C) Correlation between GCLC protein expression and intracellular Glu levels following NSCLC culture in cysteine replete and starved conditions for 12 hours. GCLC expression is normalized to HSP90 expression and can be found in Figure 5A (N=13). (D) (Left) Analysis of intracellular Glu concentrations in NSCLC cell lines cultured under cystine replete or starved conditions for 12 hrs (N=3). (Right) Comparison of Glu concentrations between KEAP1 WT (N=7) and KEAP1 MUT NSCLC (N=6) cells. (E) Analysis of relative intracellular Glu levels in the NRF2 KO A549 cells transduced with empty vector (EV) or NRF2 cultured under cystine replete or starved conditions in the presence and absence of 100 μM BSO for 12 hrs (N=3). (F) Analysis of relative intracellular Glu levels in the Calu3 cells pre-treated with 100 nM of KI-696 or vehicle (Veh, 0.1% DMSO) for 48 hrs, followed by culture in cystine replete or starved conditions in the presence and absence of 100 μM BSO for 12 hrs (N=3). (G) Analysis of relative intracellular Glu levels of the parental A549 cells, GCLC KO clones reconstituted with GFP (+GFP) or sgRNA-resistant GCLC (+GCLCRes), and GSS KO clones reconstituted with GFP (+GFP) or sgRNA-resistant GSS (+GSSRes) cultured under cystine replete or starved conditions for 2.5 hrs (N=3). (H) (Left) Relative Glu levels in mouse liver treated with Cyst(e)inase or vehicle (PBS) for 24 hrs followed by treatment with BSO or vehicle (saline) for 4 hrs (for all exp group, N=5), and (Right) Relative Glu levels in the liver of Gclcf/f and Gclc−/− mice (for all exp group, N=4). (I) Analysis of the death of NRF2LOW (N=10) and NRF2HIGH (N=7) cells under cystine starved conditions treated with GluEE (5 mM) or AOA (0.5 mM), or Gln starvation for 49 hrs. Cell death was analyzed with Sytox Green and the mean AUC of each cell lines can be found in Figure S7I. (J) Analysis of intracellular A549 ROS levels with CellROX green following culture under cystine replete and starved conditions in the presence and absence of AOA (0.5 mM) or Gln starvation for 14 hrs (N=3). (K) Analysis of intracellular A549 ROS levels with CellROX green following culture under cystine replete and starved conditions in the presence and absence of GluEE (5 mM) for 8 hrs (N=3). (L) Schematic depiction of the GSH-independent, non-canonical role of GCLC in the protection against ferroptosis by maintaining glutamate homeostasis under cystine starvation. For D-K, data are presented as mean ± SD. N is number of biological replicates. *P<0.05, **P<0.01, ***P<0.001, and ****P<0.0001. For A-C, a Pearson correlation analysis was used. For D (left), G, H (right), and I, Unpaired two-tailed t tests were used for the statistical comparisons. For G, * is for the comparison with parental cells under cystine replete conditions [+(Cys)2] and # is for the comparison of parental group under cystine starved conditions [−(Cys)2]. For D (right), E, F, H (left), J, and K, one-way ANOVA with Bonferroni’s multiple comparison tests were used for statistical analyses.
To directly evaluate whether NRF2 influences glutamate levels under cystine starvation, we used the NRF2 KO A549 cells system, which exhibited increased GCLC-dependent γ-glutamyl-dipeptide synthesis upon NRF2 reconstitution (Figure 5D). NRF2 expression reduced intracellular glutamate under both cystine replete and starved conditions, which was partially reversed by GCLC inhibition with BSO (Figure 7E). Similar results were observed in the KEAP1 wild-type cell line Calu3 (Figure 7F). Moreover, treatment of A549 cells with BSO promoted glutamate accumulation following erastin-mediated xCT inhibition (Figure S7D). Next, we evaluated the ability of γ-glutamyl-dipeptides to serve as a glutamate sink. GCLC KO A549s demonstrated increased intracellular glutamate levels under cystine starvation, while GSS KO A549s did not (Figure 7G), which was also observed in H1299s to a lesser degree (Figure S7E). Finally, GCLC KO liver dramatically accumulated glutamate, with a similar trend observed in wild-type livers following a single treatment of BSO, even under cysteine-limiting conditions (Figure 7H). Together, these results demonstrate that dipeptide synthesis via GCLC serves as a glutamate sink to limit glutamate accumulation under cystine starvation.
Because glutamate was previously shown to contribute ferroptosis (Dixon et al., 2012; Gao et al., 2015), we examined whether glutamate plays a causal role in cystine-starvation induced ferroptosis in NSCLC cells. We depleted intracellular glutamate by starving cells of glutamine, or inhibited glutamate metabolism to α-ketoglutarate by treatment with the pan transaminase inhibitor AOA. While glutamine starvation depleted intracellular glutamate, α-ketoglutarate, and downstream metabolites like succinate, AOA treatment elevated glutamate while depleting succinate (Figure S7F). To elevate intracellular glutamate, cells were treated with membrane permeable glutamate diethyl ester (GluEE) at 5mM, a concentration we confirmed could increase intracellular glutamate (Figure S7G). GluEE did not inhibit xCT activity, unlike glutamate itself, which was a potent xCT inhibitor (Figure S7H). Glutamine starvation and AOA treatment could both mitigate ferroptosis independent of NRF2 expression status, while GluEE treatment promoted ferroptosis in most NRF2HIGH cell lines, but not NRF2LOW cell lines (Figures 7I and S7I). Moreover, cystine starvation-induced ferroptosis of GCLC KO cells was rescued by glutamine starvation, which was further enhanced by co-starvation of glutamate (Figure S7J). Glutamate was previously shown to promote ROS accumulation following cystine starvation in multiple studies. Using the general oxidative stress indicator CellROX green, we found that cystine starvation promoted ROS accumulation, which rescued by Gln starvation and AOA treatment (Figure 7J) and exacerbated by GluEE treatment (Figure 7K). However, while some studies suggest that glutamate promotes mitochondrial ROS via the electron transport chain (Gao et al., 2015; Gao et al., 2019), others have demonstrated that the mitochondria are dispensable for glutamate-induced ferroptosis (Dixon et al., 2012; Gaschler et al., 2018). Using MitoSOX, we found that cystine starvation did not increase mitochondrial superoxide, consistent with the observations of Dixon et al. (Figure S7K), suggesting mitochondria-derived superoxide is likely not the source of glutamate-induced ROS in our system. Collectively, these results suggest that glutamate promotes ROS accumulation, but the precise mechanism remains to be determined and requires future study.
Discussion
The findings reported herein demonstrate that γ-glutamyl-peptide synthesis by GCLC provides GSH-independent protection from cystine starvation-induced ferroptosis in cells with NRF2 activation. NRF2 has previously been linked to ferroptosis protection (Fan et al., 2017; Sun et al., 2016), and many of its targets have canonical functions that are protective against cellular oxidation, including those promoting glutathione synthesis, ROS metabolism, iron metabolism, and others. The ability of NRF2 to protect against ferroptosis has important implications for cancer, particularly lung cancers that commonly have NRF2 activation via mutations in KEAP1 and NRF2 (Hayes and McMahon, 2009). Further work is needed to determine whether NRF2 protects against specific classes of ferroptosis inducers in vivo. Moreover, the contribution of the non-canonical function of GCLC vs. other NRF2-regulated processes to protection against these specific classes of ferroptosis inducers remains to be explored.
While cystine starvation-induced ferroptosis has commonly been attributed to the depletion of cellular GSH, we show that cystine starvation induces complex metabolic changes within cells. Our work does not exclude the importance of GCLC-derived GSH in the protection against ferroptosis. GSH is a major intracellular antioxidant and substrate for GPX4 for lipid hydroperoxide detoxification (Dixon and Stockwell, 2019; Yang et al., 2014). However, our work demonstrates that cystine starvation causes a metabolic imbalance and accumulation of glutamate, which plays a causal role in ferroptosis induction. Our findings are consistent with prior reports demonstrating that glutamate contributes to ferroptosis via ROS (Dixon et al., 2012; Gao et al., 2015; Gao et al., 2019), although more work is needed to understand the mechanism. Glutamate-derived α-ketoglutarate can contribute to fatty acid metabolism via citrate, and lipid metabolism plays a key role in ferroptosis susceptibility (reviewed in Zou and Schreiber, 2020). Moreover, recent work has established an important role for amino acid levels in ferroptosis sensitivity through mTOR-dependent and independent mechanisms (Conlon et al., 2019). More work is needed to understand the signaling effects of glutamate on ferroptosis sensitivity.
Previous studies have found that combined inhibition of cystine uptake with glutathione synthesis can synergistically inhibit the viability of cells and tumors (Cramer et al., 2017; Harris et al., 2015). Our work has important implications for the interpretation of studies using BSO to inhibit GCLC. While many of those results may be attributed to GSH depletion, the contribution of GCLC to γ-glutamyl-peptide synthesis and glutamate scavenging may also play a very important role, particularly in the context of xCT inhibition, where cells cannot export glutamate. Our findings warrant the development of potent GSS inhibitors for the study of ferroptosis to distinguish these mechanisms. These inhibitors would also be valuable for therapeutic combinations with ferroptosis inducers, although they may increase glutamate scavenging, which may affect cellular responses.
Our in vivo results provide direct genetic evidence to support the GCLC-mediated γ-glutamyl-peptide production that was recently been reported in mouse liver extracts (Kobayashi et al., 2020), where glutamate could be ligated with other amino acids in a reaction inhibited by BSO. The promiscuity of GCLC toward amino acids other than cysteine is not a unique feature of this enzyme and has been shown for many other metabolic enzymes. For example, serine palmitoyl transferase will also metabolize alanine or glycine when serine is limiting (Penno et al., 2010) and glutamate-aspartate aminotransferase will also metabolize cysteine sulfinic acid (Weinstein et al., 1988). In the case of GCLC, this feature may have been selected for during evolution, as the S. cerevisiae homolog (Gsh1p) also has the ability to at least use valine (Sofyanovich et al., 2019). Additional work is needed to determine which other amino acids are accepted by S. cerevisiae Gsh1p. For the human enzyme, small, non-charged amino acids that are structurally similar to cysteine can be used based on their appearance in γ-glutamyl-peptides, although the full spectrum of amino acids has not been tested in a direct enzymatic assay.
Our findings may extend beyond conditions of cysteine deficiency. Systemic deletion of mouse Gclc revealed that Gclc plays a role in the regulation of glutamate and γ-glutamyl-peptides levels in normal tissue. Notably, glutamate accumulation was only observed in the liver but not other tissues (not shown). Liver plays a critical role in GSH synthesis to supply the rest of the organism, which may consume significantly more glutamate in liver than lung or kidney (Ookhtens and Kaplowitz, 1998). Similar to the liver, cancer cells synthesize a significant amount of GSH (Balendiran et al., 2004; Huang et al., 2001; Soini et al., 2001; Sun et al., 2019; Tatebe et al., 2002) and use glutamate for cystine export (Ji et al., 2018; Shin et al., 2017; Takeuchi et al., 2013; Timmerman et al., 2013), which may explain the robust accumulation of glutamate following cystine starvation in our NSCLC cells. It is important to note that, in contrast to cell culture, depletion of γ-glutamyl-peptides in Gclc KO tissue may be a consequence of both canonical, extracellular GGT mediated γ-glutamyl-dipeptide production and the intracellular GCLC-mediated pathway. However, the accumulation of glutamate and the depletion of γ-glutamyl-tripeptides, which require the activity of GSS, strongly suggests that these peptides are being produced intracellularly. Supportively, the activity of GGT is negligible in the mouse liver compared to kidney (Kobayashi et al., 2020). It is not known whether γ-glutamyl-peptides have additional functions in tissues beyond serving as a reservoir for glutamate, and potentially other amino acids. γ-glutamyl-peptides levels have been shown to be increased under conditions of liver injury, including drug-induced injury, hepatitis infection, liver cirrhosis, and hepatocellular carcinoma (Soga et al., 2011). Additional work is needed to understand the role of γ-glutamyl-peptide synthesis in these diseases.
We also find that GSS may regulate glycine homeostasis by producing γ-glutamyl-tripeptides, including γ-Glu-2AB-Gly and γ-Glu-Ala-Gly. While we found that glycine also accumulated following cystine starvation (Figure S7C), this was not associated with ferroptosis (Figure 7A). Future work is needed to both understand whether GSS can use other amino acids besides glycine and determine the full spectrum of γ-glutamyl-tripeptides produced by GSS. Further, we find that GSS deficiency actually enhances γ-glutamyl-dipeptide synthesis, which can be explained by loss of feedback inhibition of GCLC by GSH. These findings raise interesting implications for the metabolic phenotypes of patients with inborn errors of glutathione metabolism. Although extremely rare, mutations in GCLC and GSS result in hemolytic anemia. Interestingly, GSS mutant patients also present with 5-oxo-prolinuria, which is not observed in GCLC deficiency (Ristoff and Larsson, 2007). While this 5-oxo-prolinuria has been attributed to the accumulation of γ-glutamyl-cysteine and its metabolism to 5-oxo-proline by γ-glutamylcyclotransferase (GGCT) (Ristoff and Larsson, 2007), our work suggests that other γ-glutamyl-amino acids are likely produced in this situation to contribute to 5-oxo-prolinuria. This is likely to depend on the availability of cysteine, which would likely become limiting if the feedback inhibition of GCLC is lost due to an inability to synthesize GSH.
Limitations of the study
Although this study demonstrates a non-canonical role for GCLC in ferroptosis protection via glutamate scavenging, the study is limited to NSCLC cells and cystine-starvation induced ferroptosis. The role of glutamate scavenging by GCLC in other cancer types and in response to other ferroptosis inducers remains to be determined. While we provide in vivo evidence for γ-glutamyl-peptide synthesis by GCLC and its role in controlling glutamate levels, it will be important to examine this pathway in various tumor models. Finally, as discussed above, more work is needed to understand how glutamate influences ferroptosis.
STAR Methods
Resource Availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Dr. Gina M. DeNicola (Gina.DeNicola@moffitt.org)
Material Availability
All unique reagents generated in this study are available from the Lead Contact without restriction.
Data and Code Availability
This study did not generate any unique datasets or code.
Experimental Model and Subject Details
Animal Experiments
All animal studies with Gclcf/f mice were performed according to protocols approved by the Institutional Animal Care and Use Committee, the Standing Committee on Animals at Harvard University. Mice were housed under standard housing conditions. Gclcf/f (Chen et al., 2007) and R26-CreERT2 (Ventura et al., 2007) mice were bred to generate Gclcf/f; R26-CreERT2 mice (C57BL/6 background). Gclcf/f (Gclc WT) and Gclcf/f; R26-CreERT2 (Gclc KO) adult male and female mice (aged 11–17 weeks old) were treated with tamoxifen (20 mg/ml; dissolved in corn oil) via daily intraperitoneal injection for 5 consecutive days at 160 mg/kg (tamoxifen/mouse body mass). At 14–16 days following the last tamoxifen injection, blood was collected via the retro-orbital venous sinus into BD Microtainer serum separator tubes (catalog #365967) and placed on ice. Mice were euthanized (isoflurane inhalation followed by cervical dislocation) and the liver, kidney, and lung were immediately harvested and small tissue pieces were snap frozen in dry ice. Serum was isolated by centrifugation (16 min at 3000g) and serum aliquots were snap frozen on dry ice. Tissues and serum were stored at −80℃ prior to analysis. All studies with cyst(e)inase/BSO experiments were approved by and conducted in accordance to the ethical standards established by the University of South Florida IACUC (protocol # R IS00007922). Mice were housed under standard housing conditions. For cyst(e)inase/BSO experiments, C57BL/6J female mice (aged 7 weeks old) were treated with Cyst(e)inase (75 mg/kg) or vehicle (PBS) via intraperitoneal injection. After 24 hrs, the mice were further treated with BSO (10 mmol/kg) prepared as previously described (Monroe and Eaton, 1988) or vehicle (saline) via intraperitoneal injection. After 4 hrs, blood was collected from the submandibular vein into BD Microtainer serum separator tubes and placed on ice. Mice were then euthanized by cervical dislocation and the liver, kidney, and lung were immediately harvested and small pieces snap frozen in liquid nitrogen. Serum was isolated by centrifugation (16 min at 3000g) and serum aliquots were snap frozen on dry ice. Tissues and serum were stored at −80℃ prior to analysis.
Lentivirus generation
Lentiviruses were generated by overnight PEI transfection of 90% confluent Lenti-X 293T cells (Clontech) with target lentiviral plasmid, and packaging plasmids pCMV-dR8.2 dvpr and pCMV-VSV-G in DMEM (10% FBS). The next day, the medium was changed to fresh DMEM (10% FBS). After 24 and 48 hrs, the virus containing medium was collected and filtered with a 0.45 μm PES filter. The two collections were combined and stored at −80°C until virus infection.
Lentiviral infection of NSCLC cells.
To increase gene deletion efficiency in a polyclonal population, H1299 cells with stable Streptococcus pyogenes Cas9 expression (H1299Cas9) were first established by lentiviral transduction with lentiCas9-Blast (Greenfeld et al., 2015) followed by blasticidin selection (3 μg/mL) for 5 days. To generate GCLC or GSS KO cells, H1299Cas9 and A549 cells were transduced with empty pLenti-CRISPR-V2 or pLenti-CRISPR-V2 encoding sgGCLC or sgGSS with 2 μg/mL of polybrene, followed by puromycin selection (1 μg/mL) for 4 days. To select single clones for GCLC or GSS KO, transduced A549 cells were diluted to 0.5 cells/well in 96 well dishes in RPMI supplemented with 1 μM of Fer-1. Cells were grown for 2 weeks, followed by expansion of clones. GCLC or GSS KO was verified by loss of GCLC or GSS protein via immunoblotting. Subsequently, cells were reconstituted with sgRNA-resistant cDNAs by transduction with pLenti-hygro-GFP, pLenti-hygro-sgRNA-resistant GCLC (pLGH-GCLCRes), or pLenti-hygro-sgRNA-resistant GSS (pLGH-GSSRes) followed by hygromycin selection (300 μg/mL) for 5 days. NRF2 KO A549 cells were reconstituted with NRF2 or empty vector as described previously (Kang et al., 2019).
Dead cell measurement with IncuCyte
Cells were plated in black walled 96 well pates at a density of 10,000 cells/well in 100 μL final volume. The next day, the medium was changed to 100 μL of experimental medium containing 20 nM of Sytox Green or 250 nM Cytotox Red as follows: In cystine starvation experiments, cysteine/cystine, methionine and glutamine free RPMI (MP Biomedicals) was supplemented with 100 μM methionine and 10% dialyzed FBS (dFBS) containing 2 mM glutamine with or without 200 μM cystine as indicated. In experiments additionally lacking glutamine and/or glutamate, RPMI 1640 Medium without glucose and amino acids (US Biological) was prepared from powder according the manufacturer’s instructions and supplemented with glucose and amino acids to meet the RPMI formulation with the exception of cystine, glutamine, and glutamate. This media was further supplemented with 200 μM of cystine, 2 mM of glutamine, and/or 137 μM of glutamate as indicated. In addition, 10 μM Ferrostatin-1 (Fer-1), 100 μM DFO, 5 mM of GluEE, 0.5 mM AOA, 5 μM Erastin, 0.0625–2 mM gamma-glutamyl-dipeptides, and/or 100 μM BSO were added as indicated. The number of dead cells and cell confluence were measured by the IncuCyte Zoom or S3 live-cell analysis system (Essen BioScience, Ann Arbor, MI, USA) in a humidified tissue culture incubator at 37°C with 5% CO2. Data were acquired with a 4X or 10X objective lens in phase contrast and green fluorescence (Ex/Em: 460/524 nm, acquisition time: 400 ms) or red fluorescence (Ex/Em: 585/635 nm, acquisition time: 300 ms) channels. Images were acquired from each well at 2–4 hr intervals. Image and data processing were performed with IncuCyte Zoom 2018A, S3 2018B, or 2020A software (Essen BioScience, Ann Arbor, MI, USA). Dead cell number was normalized to cell confluence [Number of Sytox Green or Cytotox Red positive cells/mm2/cell confluence (% of total image)]. The area under the curve (AUC) was calculated as the sum of the dead cell numbers at each time point.
Metabolomics sample preparation
For the tissue targeted metabolomics, the frozen liver, lung, and kidney tissue samples were pulverized using a pre-chilled BioPulverizer (59012MS, BioSpec), weighed frozen, and then placed on dry ice. The tissue metabolites were extracted in 80% MeOH at a final tissue concentration of 50 mg/mL for 24 hrs at −80°C. After centrifugation (17000 g, 20 min, 4°C), the supernatant was analyzed by LC-HRMS.
For the quantitative intracellular 13C3-serine tracing and cysteine quantification, NSCLC cells were plated in 6 well dishes and pre-conditioned in RPMI medium containing dFBS (10%) overnight. For serine tracing, RPMI 1640 Medium without glucose and amino acids (US Biological) was prepared from powder according the manufacturer’s instructions and supplemented with glucose and amino acids to meet the RPMI formulation with the exception of serine and cystine. The following day, the cells were quickly washed with 1 mL of warm serine/cystine-free RPMI medium, followed by feeding with 13C3-serine containing medium (serine/cystine-free RPMI + 10% dFBS + 1% Pen/Strep + 300 μM 13C3-Serine) lacking or supplemented with 200 μM cystine. After 4 hrs, the medium was aspirated and the cells were quickly washed with ice cold PBS. As described in our previous study (Kang et al., 2019), the cellular metabolites were extracted and derivatized with 0.5 mL of ice-cold extraction solvent (80% MeOH:20% H2O containing 25 mM NEM and 10 mM ammonium formate, pH 7.0). The concentration of internal standards in the extraction solvent were as follows: 4.18 μM [2H5]-GSH-NEM, 2.49 μM [13C3, 15N]-serine, 2.48 μM [13C2, 15N]-glycine, and 2.49 mM [13C5, 15N]-glutamate from METABOLOMICS AMINO ACID MIX STANDARD (Cambridge Isotope Laboratories). For cysteine quantification, the extraction solvent contained 10 μM [13C2, 15N]-cysteine-NEM. [2H5]-GSH-NEM and [13C2, 15N]-cysteine-NEM were pre-prepared by reaction of 50 mM NEM (10 mM ammonium formate, pH 7.0) for 30 min as previously described (Kang et al., 2019). After incubation on ice for 30 min, the NEM-derivatized metabolite extracts were cleared by centrifugation and the supernatant was analyzed by LC-HRMS at the positive mode. For metabolite concentration calculations, the total volume of the cell pellet from a duplicate well was used to calculate the intracellular metabolite concentrations. Cell volume was determined with a Scepter 2.0 cell counter (Millipore), and cell numbers were determined using either the Scepter 2.0 cell counter or a Beckman coulter cell counter (Z1 S). Total volume of the cell pellet was used to calculate the intracellular metabolite concentrations.
For the intracellular 2, 3, 3- 2H3-serine tracing, A549 cells were prepared as described for 13C3-serine tracing but medium containing 300 μM [2, 3, 3-2H3-serine]-was used. Further, 0.5 μM of SHIN-1 (Ducker et al., 2017) was included in the cystine starved conditions. After 12 hrs, the medium was aspirated, and cells were quickly washed with ice cold PBS, and cellular metabolites were extracted with 1 mL of 80% MeOH (−80°C, 15 min). After scraping, the metabolite extract was transferred into an Eppendorf tube and cleared by centrifugation (17000 g, 20 min, 4°C), followed by LC-HRMS analysis in negative mode.
For the intracellular 13C5, 15N2-glutamine tracing, A549 cells were plated in 6 well dishes and pre-conditioned in RPMI medium containing dFBS (10%) overnight. Cysteine/cystine, methionine and glutamine free RPMI (MP Biomedicals) was supplemented with 100 μM methionine and 10% dialyzed FBS (dFBS) containing 2 mM 13C5, 15N2-glutamine with or without 200 μM cystine as indicated. The following day, the cells were quickly washed with 1 mL of warm glutamine/cystine-free RPMI medium, followed by feeding with 13C5, 15N2-glutamine containing medium lacking or supplemented with 200 μM cystine. After 4 hrs, the medium was aspirated and the cells were quickly washed with ice cold PBS, and cellular metabolites were extracted with 1 mL of 80% MeOH (−80°C, 15 min). After scraping, the metabolite extract was transferred into an Eppendorf tube and cleared by centrifugation (17000 g, 20 min, 4°C), followed by LC-HRMS analysis in negative mode.
For the intracellular non-targeted metabolomics, and Glycine and Glutamate quantification, NSCLC cells were plated in 6 well dishes so they were 70% confluent at the time of extraction and pre-conditioned in RPMI medium containing dFBS (10%) overnight. The following day, the medium was aspirated and the cells were quickly washed with 1 mL of cystine free RPMI (10% dFBS, 1% P/S), followed by feeding with conditioning medium as indicated. 1 mL of the medium supernatant was collected to assay glutamate as indicated. For the non-targeted metabolomics approach, medium was aspirated and cells were quickly washed with ice cold PBS, followed by extraction of cellular metabolites with 0.5 mL of 80% MeOH (−80°C, 15 min). For intracellular glutamate, glycine, alanine, valine, threonine, and leucine quantification, the extraction solvent also contained 2.49 μM of [13C5, 15N]-glutamate, 2.48 μM of [13C2, 15N]-glycine, 2.48 μM of [13C3, 15N]-alanine, 2.49 μM of [13C5, 15N]-valine, 2.45 μM of [13C4, 154 N]-threonine, and 2.48 μM of [13C6, 15N]-leucine from METABOLOMICS AMINO ACID MIX STANDARD (Cambridge Isotope Laboratories). After scraping, the metabolite extract was transferred into an Eppendorf tube and cleared by centrifugation (17000 g, 20 min, 4°C), followed by LC-HRMS analysis in the negative and/or positive mode. For metabolite concentration calculations, the total volume of the cell pellet from a duplicate well was used to calculate the intracellular metabolite concentrations. Cell volume was determined with a Scepter 2.0 cell counter (Millipore), and cell numbers were determined using either the Scepter 2.0 cell counter or a Beckman coulter cell counter (Z1 S).
Quantitation of glutamate exportation
The extracellular medium collected above was transferred to the 96 well plates and glutamate concentrations were measured with an YSI 2900 (Yellow springs, OH, USA) using the 2755 glutamate standard (5 mM). The extracellular glutamate secretion rate (nmol/μL of cell volume/hrs) was determined from cell volume measurements as described above.
LC-MS analysis
The LC-MS conditions were identical to previously established methods (Kang et al., 2019). For the chromatographic metabolite separation, the Vanquish UPLC systems were coupled to a Q Exactive HF (QE-HF) mass spectrometer equipped with HESI (Thermo Fisher Scientific, Waltham, MA). The column was a SeQuant ZIC-pHILIC LC column, 5 μm, 150 × 4.6 mm (MilliporeSigma, Burlington, MA) with a SeQuant ZIC-pHILIC guard column, 20 × 4.6 mm (MilliporeSigma, Burlington, MA). Mobile phase A was 10 mM (NH4)2CO3 and 0.05% NH4OH in H2O while mobile phase B was 100% ACN. The column chamber temperature was set to 30°C. The mobile phase condition was set according to the following gradient: 0–13min: 80% to 20% of mobile phase B, 13–15min: 20% of mobile phase B. The ESI ionization mode was positive or negative. The MS scan range (m/z) was set to 60–900. The mass resolution was 120,000 and the AGC target was 3 × 106. The capillary voltage and capillary temperature were set to 3.5 KV and 320°C, respectively. 5 μL of sample was loaded. For targeted metabolomics, the LC-MS peaks were manually identified and integrated by EL-Maven (Version 0.6.1, 0.10.0, or 0.11.0) by matching with a previously established in-house library (Kang et al., 2019). The peak areas of target metabolites were further normalized by the median value of identified metabolite peak areas or the peak area of stable isotope labeled internal standards for further quantification as previously described (Bennett et al., 2008). For the non-targeted metabolomics approach, the LC-MS peaks were automatically extracted and aligned using the Automated Feature Detection function of EL-Maven. After the normalization with the median value of the intensities of LC-MS peaks, the statistical analysis was conducted. The γ-glutamyl-peptide peaks were putatively identified by matching their m/z value with the online HMDB database (http://www.hmdb.ca), and further confirmed by matching the m/z value and retention time of authentic standards. The standards for γ-glutamyl-threonine and γ-glutamyl-alanyl-glycine were not available and were instead validated by stable isotope labeled metabolite tracing (Figure 2C and S2A), as described in the results. The γ-glutamyl-peptide levels relative to the average of γ-glutamyl-peptide levels in cystine starved A549 cells were used to compare γ-glutamyl-peptide levels across panel of NSCLC cell lines.
ROS and lipid peroxidation measurements
NSCLC cells were plated in 24 well dishes at 70,000 cells/well and pre-conditioned in RPMI medium containing dFBS (10%) overnight. The following day, the medium was aspirated, and the cells were quickly washed with warm PBS followed by feeding with the indicated medium conditions. For intracellular ROS measurements, CellROX green was added to the cells at a final concentration of 5 μM for the final 30 min of the experiment. Cells were washed with PBS, detached with trypsin, and transferred to Eppendorf tubes. Following centrifugation (10sec, 17,000g), the supernatant was aspirated, and the pellet was washed with FACS buffer (PBS containing 0.5% BSA, 2 mM EDTA, 1% P/S, and 25 mM HEPES), and fixed with 4% Paraformaldehyde (PFA) in PBS for 10 min. Following centrifugation (10 sec, 17,000g), the pellet was further washed with FACS buffer, re-suspended to 500 μL of FACS buffer, and filtered into FACS tubes. For mitochondrial superoxide measurements, MitoSOX Red was added to the cells at a final centration of 5 μM for the final 10 min of the experiment. For lipid peroxidation measurements, C11-BODIPY was added to the cells at a final concentration of 10 μM for the final 30 min of the experiment. For both MitoSOX and C11-BODIPY, cells were processed as described for CellROX green, except no fixation with PFA was performed. The samples were analyzed by Accuri™ C6 flow cytometer (BD biosciences) or BD LSR II flow cytometer (BD Biosciences) using the FITC (CellROX, C11-BODIPY) or PE (MitoSOX) filters. The mean fluorescence intensity (MFI) or median fluorescence intensity (Median FI) were analyzed with the Accuri™ C6 (BD biosciences) or FlowJo (Ver 10.7.1) software, and further normalized by the control of experimental set as indicated.
Immunoblotting
Cell lysates were prepared in RIPA buffer (20 mM Tris-HCl, pH7.5; 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% NP-40, 1% sodium deoxycholate) containing protease inhibitors. After protein quantification with the Bio-Rad DC assay, the samples were mixed with reducing buffer (v/v, 5:1) containing 2-mercaptoethanol. The proteins were separated by SDS-PAGE using NuPAGE 4–12% Bis-Tris gels (Invitrogen) and transferred to 0.45 um Nitrocellulose membrane (GE Healthcare). The membrane was blocked in blocking buffer (5% non-fat milk in TBST) for 15 min, and the membrane was incubated with primary antibodies overnight at the following dilutions: GCLC (1:1000), GCLM (1:1,000), GSS (1:2,000), xCT (1:1,000), GPX4 (1:1,000) β-Actin (1:100,000), HSP90 (1:5,000). After washing the membrane 3 times with TBST for 10 min, the membrane was incubated with secondary antibody (goat anti-rabbit or goat anti-mouse, 1:10,000 dilution, Jackson ImmunoResearch) in blocking buffer for 1 hr. After wash the membrane in TBST 3 times for 10 min, the enhanced chemo-luminescence signal was measured by exposing to x-ray film.
Statistical analysis
Statistical analyses were conducted with Graph Pad Prism 8. For the comparison of two groups, two-tailed Student’s t-test was used. For the comparison of more than 3 experimental groups, one-way ANOVA was used with Bonferroni’s multiple comparison test.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| GCLC (Mouse polyclonal Ab) | Santa Cruz Biotechnology | Cat#: sc-390811 Lot#: 1917 RRID: AB-2736837 |
| GSS (Mouse polyclonal Ab) | Novus Biologicals | Cat#: NBP2–03351 Lot#: A01 RRID: N/A |
| GCLM (Rabbit polyclonal Ab) | GeneTex | Cat#: GTX114075 Lot#: 40156 RRID: AB_10619535 |
| GPX4 (Mouse monoclonal Ab) | R&D Systems | Cat#: MAB5457 Lot#: CCXW0218061 RRID: AB_2232542 |
| β-Actin (Mouse monoclonal Ab) | Thermo Fisher Scientific | Cat#: AM4302 Lot#: 00867595 RRID: AB_2536382 |
| xCT (Rabbit polyclonal Ab) | Abcam | Cat#: ab37185 Lot#: GR3275067–8 RRID: AB_778944 |
| HSP90 (Rabbit polyclonal Ab) | Cell Signaling Technology | Cat#: 4874S Lot# 5 RRID: 2121214 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Sytox Green | Thermo Fisher Scientific | Cat#: S7020 |
| Cytotox Red | Thermo Fisher Scientific | Cat#: NC1015259 |
| DMSO | VWR Scientific Inc | Cat#: 97063–136 |
| 0.4% PFA in PBS | Thermo Fisher Scientific | Cat#: J19943-K2 |
| Tamoxifen | Sigma-Aldrich | Cat#: T5648–5G |
| Arginine | Sigma-Aldrich | Cat#:A6969–25G |
| Aspartate | MP Biomedicals | Cat#:219463380 |
| Asparagine | Sigma-Aldrich | Cat#:A4159–25G |
| Glutamate | Sigma-Aldrich | Cat#:G8415–100G |
| Glutamine | VWR | Cat#:VWRL0131–0100 |
| Glycine | VWR | Cat#:BP381–1 |
| Histidine | Sigma-Aldrich | Cat#:H5659–25G |
| Hydroxy-L-proline | VWR | Cat#:TCH0296–5G |
| Isoleucine | VWR | Cat#:AAJ63045–14 |
| Leucine | Sigma-Aldrich | Cat#:L8912–25G |
| Lysine | Sigma-Aldrich | Cat#:L8662–25G |
| Methionine | Sigma-Aldrich | Cat#:M5308–25G |
| Phenylalanine | Sigma-Aldrich | Cat#:P5482–25G |
| Proline | Sigma-Aldrich | Cat#:P5607–25G |
| Threonine | VWR | Cat#:97064–026 |
| Tryptophan | Sigma-Aldrich | Cat#:T8941–25G |
| Tyrosine | Sigma-Aldrich | Cat#:T1145–25G |
| Valine | Sigma-Aldrich | Cat#:V0513–25G |
| Glucose | Sigma-Aldrich | Cat#:G7021–100G |
| [2H5]-GSH | Santa Cruz Biotechnology | Cat#: sc-489493 |
| [13C3, 15N]-cysteine | Cambridge Isotope Laboratories | Cat#: CNLM-3871-H-0.25 |
| [2, 3, 3-2H3]-serine | Cambridge Isotope Laboratories | Cat#: DLM-582–0.1 |
| [13C3]-serine | Cambridge Isotope Laboratories | Cat#: CLM-1574-H-0.1 |
| [13C5, 15N2]-glutamine | Cambridge Isotope Laboratories | Cat#: CNLM-1275-H-0.1 |
| METABOLOMICS AMINO ACID MIX STANDARD | Cambridge Isotope Laboratories | Cat#: MSK-A2–1.2 |
| γ-glutamyl-alanine | Santa Cruz Biotechnology | Cat#: sc-300878 |
| γ-glutamyl-glycine | Bachem | Cat#: 4003498.0001 |
| γ-glutamyl-leucine | Bachem | Cat#: 4005004.0001 |
| γ-glutamyl-valine | Bachem | Cat#: 4003707.0250 |
| MeOH (HPLC grade) | Sigma Aldrich | Cat#: 34860–1 L-R |
| H2O (HPLC grade) | Fisher Chemical | Cat#: W5–1 |
| Acetonitrile (HPLC grade) | Honeywell | Cat#: 34967 |
| N-ethylmaleimide (NEM) | Alfa Aesar | Cat#: 40526–06 |
| Blasticidin | Invivogen | Cat#: ant-bl-1 |
| Puromycin | Invivogen | Cat#: ant-pr-1 |
| Hygromycin | Invivogen | Cat#: ant-hg-1 |
| Glutamate diethyl ester (GluEE) | TCI Chemicals | Cat#: G0179–5G |
| Cystine | Sigma Aldrich | Cat#: C6727–25G |
| Erastin | Cayman Chemical | Cat#: 17754 |
| Ferrostatin-1 (Fer-1) | Cayman Chemical | Cat#: 17729 |
| Deferoxamine (DFO) | Sigma Aldrich | Cat#: D9533–1G |
| AOA | Santa Cruz Biotechnology | Cat#: sc-207410 |
| SHIN-1 | Dr. Joshua Rabinowitz Department of Chemistry and Lewis-Sigler Institute for Integrative Genomics (Princeton University) |
(Ducker et al., 2017) |
| L-Buthionine-(S,R)-Sulfoximine (BSO) | Cayman Chemical or Sigma Aldrich | Cat#: 14484 or Cat#: B2515–500MG |
| Cyst(e)inase | Dr. Everett Stone Department of Molecular Biosciences (University of Texas Austin) |
(Cramer et al., 2017) |
| KI-696 | MedChem Express | Cat#: HY-101140 |
| 2755 Glutamate Standard | YSI | Cat#: 027055 |
| Critical Commercial Assays | ||
| CellROX green | Fisher Scientific | Cat#: C10444 |
| BODIPY-C11 | Invitrogen | Cat#: C10445 |
| MitoSOX Red | Invitrogen | Cat#: M36008 |
| Experimental Models: Cell Lines | ||
| PC9 | Dr John Minna, Hamon Cancer Center Collection (University of Texas-Southwestern Medical Center) | RRID: CVCL_B260 |
| H810 | Dr John Minna, Hamon Cancer Center Collection (University of Texas-Southwestern Medical Center) | RRID: CVCL_1590 |
| H2172 | Dr John Minna, Hamon Cancer Center Collection (University of Texas-Southwestern Medical Center) | RRID: CVCL_1537 |
| Calu3 | Dr John Minna, Hamon Cancer Center Collection (University of Texas-Southwestern Medical Center) | RRID: CVCL_0609 |
| H1581 | Dr John Minna, Hamon Cancer Center Collection (University of Texas-Southwestern Medical Center) | RRID: CVCL_1479 |
| H1975 | Dr John Minna, Hamon Cancer Center Collection (University of Texas-Southwestern Medical Center) | RRID: CVCL_1511 |
| H2087 | Dr John Minna, Hamon Cancer Center Collection (University of Texas-Southwestern Medical Center) | RRID: CVCL_1524 |
| H2347 | Dr John Minna, Hamon Cancer Center Collection (University of Texas-Southwestern Medical Center) | RRID: CVCL_1550 |
| H1792 | Dr John Minna, Hamon Cancer Center Collection (University of Texas-Southwestern Medical Center) | RRID: CVCL_1495 |
| H1944 | Dr John Minna, Hamon Cancer Center Collection (University of Texas-Southwestern Medical Center) | RRID: CVCL_1508 |
| H460 | Dr John Minna, Hamon Cancer Center Collection (University of Texas-Southwestern Medical Center) | RRID: CVCL_0459 |
| HCC15 | Dr John Minna, Hamon Cancer Center Collection (University of Texas-Southwestern Medical Center) | RRID: CVCL_2057 |
| H2009 | ATCC | Cat#: CRL-5911 RRID: CVCL_1514 |
| H1299 | ATCC | Cat#: CRL-5803 RRID: CVCL_0060 |
| H1993 | ATCC | Cat#: CRL-5909 RRID: CVCL_1512 |
| H441 | ATCC | Cat#: HTB-174 RRID: CVCL_1512 |
| A549 | ATCC | Cat#: CCL-185 RRID:CVCL_1561 |
| NRF2 KO A549 | Dr. Laureano de la Vega | (Torrente et al., 2017) |
| Lenti-X 293T | Clontech | Cat#: 632180 RRID: N/A |
| Experimental Models: Organisms/Strains | ||
| Gclcf/f | (Chen et al., 2007) | N/A |
| R26-CreERT2 | (Ventura et al., 2007) | N/A |
| Oligonucleotides | ||
| Guide RNA for lentiCRISPR-V2 GCLC. Forward: 5’-caccgTAGATGTGCAGGAACTGG-3’ Reverse: 5’-aaacCCAGTTCCTGCACATCTAc-3 |
(Harris et al., 2019) | N/A |
| Guide RNA for lentiCRISPR-V2 GSS. Forward: 5’-caccgGGTCTCTGGACCAAGACCGA-3’ Reverse: 5’-aaacTCGGTCTTGGTCCAGAGACc-3’ |
This study | N/A |
| PCR primer for pLenti-hygromycin-GCLC. Forward: 5’-cgactctagaggatccatggggctgctgtcc-3’ Reverse: 5’-gaggttgattgtcgacctagttggatgagtcagttttacttcc-3’ |
This study | N/A |
| PCR primer for pLenti-hygromycin-GSS. Forward: 5’-cgactctagaggatccatggccaccaactgg-3’ Reverse: 5’-gaggttgattgtcgactcacacagggtatgggttgtc-3’ |
This study | N/A |
| Site directed mutagenesis primer for pLenti-hygromycin-GCLCRes. Forward: 5’- CCTGCACATCTACCACG −3’ Reverse: 5’- AACTGGAAGATCCCGTGCCG −3’ |
This study | N/A |
| Site directed mutagenesis primer for pLenti-hygromycin-GSSRes. Forward: 5’- AAGACCGAAGACTGTTTGTGG −3’, Reverse: 5’- GGTCCAGAGACCCCTTTT-3’ |
This study | N/A |
| Recombinant DNA | ||
| lentiCas9-Blast | Addgene | Cat#: 52962 |
| lentiCRISPR-V2 | Addgene | Cat#: 52961 |
| lentiCRISPR-V2 GCLC; Using BsmBI restriction site, primers were annealed and cloned to progenitor of lentiCRISPR-V2. |
This study | N/A |
| lentiCRISPR-V2 GSS; Using BsmBI restriction site, primers were annealed and cloned to progenitor of lentiCRISPR-V2 |
This study | N/A |
| MGC Human GCLC Sequence-Verified cDNA (pCMV-SPORT6-GCLC) | Dharmacon | Cat#: MHS6278–202759380 |
| MGC Human GSS Sequence-Verified cDNA (pOTB7-GSS) | Dharmacon | Cat#: MHS6278–202830404 |
| pLenti-hygro-GFP | Addgene | Cat#: 17446 |
| pLenti-hygro-GCLC resistant to sgGCLC (pLGH-GCLCRes); The GFP of pLenti-hygro-GFP was excised and replaced with human GCLC cDNA using MGC Human GCLC Sequence-Verified cDNA (pCMV-SPORT6-GCLC) as a PCR template. The pLenti-hygro-GCLC resistant to sgGCLC was further generated by the site-directed mutagenesis. | This study | N/A |
| pLenti-hygro-GSS resistant to sgGSS (pLGH-GSSRes); The GFP of pLenti-hygro-GFP was excised and replaced with human GSS cDNA using MGC Human GSS Sequence-Verified cDNA (pOTB7-GSS) as a PCR template. The pLenti-hygro-GSS resistant to sgGSS was further generated by the site-directed mutagenesis. |
This study | N/A |
| pCMV-dR8.2 dvpr | Addgene | Cat#: 8455 |
| pCMV-VSV-G | Addgene | Cat#: 8454 |
| Software and Algorithms | ||
| EL-Maven | https://resources.elucidata.io/elmaven | Version 0.6.1, 0.10.0, or 0.11.0 |
| GraphPad Prism | https://www.graphpad.com/scientific-software/prism/ | Version 8 |
| IncuCyte Zoom | Essen BioScience | IncuCyte Zoom 2018A |
| IncuCyte S3 | Essen BioScience | IncuCyte S3 2018B or 2020A |
| Accuri™ | BD Biosciences | Version C6 |
| FlowJo | BD Biosciences | Version 10.7.1 |
| Other | ||
| RPMI 1640 Medium Modified w/o L-Glutamine, w/o Amino acids, Glucose (Powder) | US Biological | Cat# R9010–01 |
| cysteine/cystine/glutamine/methionine free RPMI | MP Biomedicals | Cat# 091646454 |
| Dialyzed FBS (dFBS) | Sigma Aldrich | Cat# F0392 |
Highlights.
Cystine starvation induces γ-glutamyl peptide accumulation in NSCLC cells.
GCLC catalyzes γ-glutamyl-peptide synthesis via a GSH-independent mechanism.
NRF2 protects against ferroptosis via γ-glutamyl peptide synthesis.
γ-glutamyl peptide synthesis prevents ferroptosis by reducing glutamate stress.
Acknowledgement
We would like to thank Dr. Joshua D. Rabinowitz for SHIN-1; Dr. Vince Luca, Dr. David Gonzalez-Perez, Elliot Medina, and Sae Bom Lee for flow cytometry assistance; Dr. Min Liu and Jayden K. Cline for LC-MS assistance; Dr. Joseph O. Johnson, Tingan Chen, Dr. Anna P. Gomes, and Dr. Didem Ilter for Incucyte assistance; and all members of the DeNicola laboratory for their very helpful discussions. This work was supported by grants from the NIH/NCI (R37-CA230042) and NIH/NIDDK (R01-DK123738) to G.M.D, NIH/NCI (CA189623) to E.S., the Ludwig Center at Harvard to I.S.H., the AACR-Takeda Oncology Lung Cancer Research Fellowship (19-40-38-KANG) to Y.P.K., and a National Pancreas Foundation grant to C.J. This work was also supported by Miles for Moffitt funds awarded to the Lung Cancer Metabolism Working Group, the Analytic Microscopy and the Proteomics/Metabolomics Cores, which are funded in part by Moffitt’s Cancer Center Support Grant (NCI, P30-CA076292), and grants from the Moffitt Foundation, and a Florida Bankhead-Coley grant (06BS-02-9614) to the Proteomics/Metabolomics core.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
I.S.H. is a consultant for Ono Pharma USA. E.S. is an inventor of intellectual property related cyst(e)inase, and has an equity interest in Aeglea Biotherapeutics, a company pursuing the commercial development of cyst(e)inase. These companies had no role in funding or the design of this study.
References
- Alvarez SW, Sviderskiy VO, Terzi EM, Papagiannakopoulos T, Moreira AL, Adams S, Sabatini DM, Birsoy K, and Possemato R (2017). NFS1 undergoes positive selection in lung tumours and protects cells from ferroptosis. Nature 551, 639–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson ME (1998). Glutathione: an overview of biosynthesis and modulation. Chem Biol Interact 111, 1–14. [DOI] [PubMed] [Google Scholar]
- Badgley MA, Kremer DM, Maurer HC, DelGiorno KE, Lee HJ, Purohit V, Sagalovskiy IR, Ma A, Kapilian J, Firl CEM, et al. (2020). Cysteine depletion induces pancreatic tumor ferroptosis in mice. Science 368, 85–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balendiran GK, Dabur R, and Fraser D (2004). The role of glutathione in cancer. Cell Biochemistry and Function: Cellular biochemistry and its modulation by active agents or disease 22, 343–352. [DOI] [PubMed] [Google Scholar]
- Beatty PW, and Reed DJ (1980). Involvement of the cystathionine pathway in the biosynthesis of glutathione by isolated rat hepatocytes. Arch Biochem and Biophys 204, 80–87. [DOI] [PubMed] [Google Scholar]
- Bennett BD, Yuan J, Kimball EH, and Rabinowitz JD (2008). Absolute quantitation of intracellular metabolite concentrations by an isotope ratio-based approach. Nat protoc 3, 1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bersuker K, Hendricks JM, Li Z, Magtanong L, Ford B, Tang PH, Roberts MA, Tong B, Maimone TJ, Zoncu R, et al. (2019). The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature 575, 688–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao JY, and Dixon SJ (2016). Mechanisms of ferroptosis. Cell and Mol Life Sci 73, 2195–2209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao JY, Poddar A, Magtanong L, Lumb JH, Mileur TR, Reid MA, Dovey CM, Wang J, Locasale JW, Stone E, et al. (2019). A Genome-wide Haploid Genetic Screen Identifies Regulators of Glutathione Abundance and Ferroptosis Sensitivity. Cell Rep 26, 1544–1556 e1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen PH, Cai L, Huffman K, Yang C, Kim J, Faubert B, Boroughs L, Ko B, Sudderth J, McMillan EA, et al. (2019). Metabolic Diversity in Human Non-Small Cell Lung Cancer Cells. Mol Cell 76, 838–851 e835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Yang Y, Miller ML, Shen D, Shertzer HG, Stringer KF, Wang B, Schneider SN, Nebert DW, and Dalton TP (2007). Hepatocyte-specific Gclc deletion leads to rapid onset of steatosis with mitochondrial injury and liver failure. Hepatology 45, 1118–1128. [DOI] [PubMed] [Google Scholar]
- Conlon M, Poltorack C, Forcina GC, Wells A, Mallais M, Kahanu A, Magtanong L, Pratt DA, and Dixon SJ (2019). A Compendium of Kinetic Cell Death Modulatory Profiles Identifies Ferroptosis Regulators. bioRxiv, 826925. doi: 10.1101/826925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad M, and Friedmann Angeli JP (2015). Glutathione peroxidase 4 (Gpx4) and ferroptosis: what’s so special about it? Mol Cell Oncol 2, e995047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cramer SL, Saha A, Liu J, Tadi S, Tiziani S, Yan W, Triplett K, Lamb C, Alters SE, and Rowlinson S (2017). Systemic depletion of L-cyst(e)ine with cyst(e)inase increases reactive oxygen species and suppresses tumor growth. Nat Med 23, 120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies TG, Wixted WE, Coyle JE, Griffiths-Jones C, Hearn K, McMenamin R, Norton D, Rich SJ, Richardson C, Saxty G, et al. (2016). Monoacidic Inhibitors of the Kelch-like ECH-Associated Protein 1: Nuclear Factor Erythroid 2-Related Factor 2 (KEAP1:NRF2) Protein-Protein Interaction with High Cell Potency Identified by Fragment-Based Discovery. J Med Chem 59, 3991–4006. [DOI] [PubMed] [Google Scholar]
- DeNicola GM, Chen PH, Mullarky E, Sudderth JA, Hu Z, Wu D, Tang H, Xie Y, Asara JM, Huffman KE, et al. (2015). NRF2 regulates serine biosynthesis in non-small cell lung cancer. Nat Genet 47, 1475–1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS, et al. (2012). Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 149, 1060–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon SJ, and Stockwell BR (2019). The hallmarks of ferroptosis. Annu Rev Cancer Biol 3, 35–54. [Google Scholar]
- Dixon SJ, Winter GE, Musavi LS, Lee ED, Snijder B, Rebsamen M, Superti-Furga G, and Stockwell BR (2015). Human haploid cell genetics reveals roles for lipid metabolism genes in nonapoptotic cell death. ACS Chem Biol 10, 1604–1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doll S, Freitas FP, Shah R, Aldrovandi M, da Silva MC, Ingold I, Goya Grocin A, Xavier da Silva TN, Panzilius E, Scheel CH, et al. (2019). FSP1 is a glutathione-independent ferroptosis suppressor. Nature 575, 693–698. [DOI] [PubMed] [Google Scholar]
- Doll S, Proneth B, Tyurina YY, Panzilius E, Kobayashi S, Ingold I, Irmler M, Beckers J, Aichler M, and Walch A (2017). ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat Chem Biol 13, 91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ducker GS, Ghergurovich JM, Mainolfi N, Suri V, Jeong SK, Hsin-Jung Li S, Friedman A, Manfredi MG, Gitai Z, Kim H, et al. (2017). Human SHMT inhibitors reveal defective glycine import as a targetable metabolic vulnerability of diffuse large B-cell lymphoma. Proc Natl Acad Sci U S A 114, 11404–11409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan Z, Wirth AK, Chen D, Wruck CJ, Rauh M, Buchfelder M, and Savaskan N (2017). Nrf2-Keap1 pathway promotes cell proliferation and diminishes ferroptosis. Oncogenesis 6, e371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuyama K, and Sassa S (2000). Interaction between succinyl CoA synthetase and the heme-biosynthetic enzyme ALAS-E is disrupted in sideroblastic anemia. J Clin Invest 105, 757–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao M, Monian P, Quadri N, Ramasamy R, and Jiang X (2015). Glutaminolysis and transferrin regulate ferroptosis. Mol Cell 59, 298–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao M, Yi J, Zhu J, Minikes AM, Monian P, Thompson CB, and Jiang X (2019). Role of mitochondria in ferroptosis. Mol Cell 73, 354–363. e353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaschler MM, Hu F, Feng H, Linkermann A, Min W, and Stockwell BR (2018). Determination of the Subcellular Localization and Mechanism of Action of Ferrostatins in Suppressing Ferroptosis. ACS Chem Biol 13, 1013–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenfeld H, Takasaki K, Walsh MJ, Ersing I, Bernhardt K, Ma Y, Fu B, Ashbaugh CW, Cabo J, and Mollo SB (2015). TRAF1 coordinates polyubiquitin signaling to enhance Epstein-Barr virus LMP1-mediated growth and survival pathway activation. PLoS pathog 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habib E, Linher-Melville K, Lin H-X, and Singh G (2015). Expression of xCT and activity of system xc− are regulated by NRF2 in human breast cancer cells in response to oxidative stress. Redox Biol 5, 33–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanigan MH, and Pitot HC (1985). Gamma-glutamyl transpeptidase–its role in hepatocarcinogenesis. Carcinogenesis 6, 165–172. [DOI] [PubMed] [Google Scholar]
- Harris IS, and DeNicola GM (2020). The Complex Interplay between Antioxidants and ROS in Cancer. Trends Cell Biol. 30, 440–451 [DOI] [PubMed] [Google Scholar]
- Harris IS, Endress JE, Coloff JL, Selfors LM, McBrayer SK, Rosenbluth JM, Takahashi N, Dhakal S, Koduri V, and Oser MG (2019). Deubiquitinases maintain protein homeostasis and survival of cancer cells upon glutathione depletion. Cell Metab 29, 1166–1181. e1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris IS, Treloar AE, Inoue S, Sasaki M, Gorrini C, Lee KC, Yung KY, Brenner D, Knobbe-Thomsen CB, and Cox MA (2015). Glutathione and thioredoxin antioxidant pathways synergize to drive cancer initiation and progression. Cancer Cell 27, 211–222. [DOI] [PubMed] [Google Scholar]
- Hayes JD, and McMahon M (2009). NRF2 and KEAP1 mutations: permanent activation of an adaptive response in cancer. Trends Biochem Sci 34, 176–188. [DOI] [PubMed] [Google Scholar]
- Huang C-S, Moore WR, and Meister A (1988). On the active site thiol of gamma-glutamylcysteine synthetase: relationships to catalysis, inhibition, and regulation. Proc Natl Acad Sci U S A 85, 2464–2468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Z-Z, Chen C, Zeng Z, Yang H, Oh J, Chen L, and Lu SC (2001). Mechanism and significance of increased glutathione level in human hepatocellular carcinoma and liver regeneration. The FASEB J 15, 19–21. [DOI] [PubMed] [Google Scholar]
- Ji X, Qian J, Rahman SJ, Siska PJ, Zou Y, Harris BK, Hoeksema MD, Trenary IA, Heidi C, and Eisenberg R (2018). xCT (SLC7A11)-mediated metabolic reprogramming promotes non-small cell lung cancer progression. Oncogene 37, 5007–5019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang L, Kon N, Li T, Wang S-J, Su T, Hibshoosh H, Baer R, and Gu W (2015). Ferroptosis as a p53-mediated activity during tumour suppression. Nature 520, 57–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang YP, Torrente L, Falzone A, Elkins CM, Liu M, Asara JM, Dibble CC, and DeNicola G (2019). Cysteine dioxygenase 1 is a metabolic liability for non-small cell lung cancer. Elife 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi S, Ikeda Y, Shigeno Y, Konno H, and Fujii J (2020). γ-Glutamylcysteine synthetase and γ-glutamyl transferase as differential enzymatic sources of γ-glutamylpeptides in mice. Amino Acids, 1–12. [DOI] [PubMed] [Google Scholar]
- Leu JI, Murphy ME, and George DL (2019). Mechanistic basis for impaired ferroptosis in cells expressing the African-centric S47 variant of p53. Proc Natl Acad Sci U S A 116, 8390–8396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim JK, Delaidelli A, Minaker SW, Zhang H-F, Colovic M, Yang H, Negri GL, von Karstedt S, Lockwood WW, and Schaffer P (2019). Cystine/glutamate antiporter xCT (SLC7A11) facilitates oncogenic RAS transformation by preserving intracellular redox balance. Proc Natl Acad Sci U S A 116, 9433–9442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mancias JD, Wang X, Gygi SP, Harper JW, and Kimmelman AC (2014). Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy. Nature 509, 105–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Reyes I, Diebold LP, Kong H, Schieber M, Huang H, Hensley CT, Mehta MM, Wang T, Santos JH, Woychik R, et al. (2016). TCA Cycle and Mitochondrial Membrane Potential Are Necessary for Diverse Biological Functions. Mol Cell 61, 199–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monroe DH, and Eaton DL (1988). Effects of modulation of hepatic glutathione on biotransformation and covalent binding of aflatoxin B1 to DNA in the mouse. Toxicol Appl Pharmacol 94, 118–127. [DOI] [PubMed] [Google Scholar]
- Ookhtens M, and Kaplowitz N (1998). Role of the liver in interorgan homeostasis of glutathione and cyst(e)ine In Seminars in liver disease (© 1998 by Thieme Medical Publishers, Inc.), pp. 313–329. [DOI] [PubMed] [Google Scholar]
- Oppenheimer L, Wellner VP, Griffith OW, and Meister A (1979). Glutathione synthetase. Purification from rat kidney and mapping of the substrate binding sites. J Biol Chem 254, 5184–5190. [PubMed] [Google Scholar]
- Penno A, Reilly MM, Houlden H, Laurá M, Rentsch K, Niederkofler V, Stoeckli ET, Nicholson G, Eichler F, and Brown RH (2010). Hereditary sensory neuropathy type 1 is caused by the accumulation of two neurotoxic sphingolipids. J Biol Chem 285, 11178–11187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poursaitidis I, Wang X, Crighton T, Labuschagne C, Mason D, Cramer SL, Triplett K, Roy R, Pardo OE, Seckl MJ, et al. (2017). Oncogene-Selective Sensitivity to Synchronous Cell Death following Modulation of the Amino Acid Nutrient Cystine. Cell Rep 18, 2547–2556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao AM, Drake MR, and Stipanuk MH (1990). Role of the transsulfuration pathway and of γ-cystathionase activity in the formation of cysteine and sulfate from methionine in rat hepatocytes. J Nutr 120, 837–845. [DOI] [PubMed] [Google Scholar]
- Reed DJ, and Orrenius S (1977). The role of methionine in glutathione biosynthesis by isolated hepatocytes. Biochem Biophys Res Commun 77, 1257–1264. [DOI] [PubMed] [Google Scholar]
- Richman P, and Meister A (1975). Regulation of gamma-glutamyl-cysteine synthetase by nonallosteric feedback inhibition by glutathione. J Biol Chem 250, 1422–1426. [PubMed] [Google Scholar]
- Ristoff E, and Larsson A (2007). Inborn errors in the metabolism of glutathione. Orphanet J Rare Dis 2, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouault TA (2012). Biogenesis of iron-sulfur clusters in mammalian cells: new insights and relevance to human disease. Dis Model Mech 5, 155–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki H, Sato H, Kuriyama-Matsumura K, Sato K, Maebara K, Wang H, Tamba M, Itoh K, Yamamoto M, and Bannai S (2002). Electrophile response element-mediated induction of the cystine/glutamate exchange transporter gene expression. J Biol Chem 277, 44765–44771. [DOI] [PubMed] [Google Scholar]
- Sayin VI, LeBoeuf SE, Singh SX, Davidson SM, Biancur D, Guzelhan BS, Alvarez SW, Wu WL, Karakousi TR, Zavitsanou AM, et al. (2017). Activation of the NRF2 antioxidant program generates an imbalance in central carbon metabolism in cancer. Elife 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin C-S, Mishra P, Watrous JD, Carelli V, D’Aurelio M, Jain M, and Chan DC (2017). The glutamate/cystine xCT antiporter antagonizes glutamine metabolism and reduces nutrient flexibility. Nat Commun 8, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sofyanovich OA, Nishiuchi H, Yamagishi K, Matrosova EV, and Serebrianyi VA (2019). Multiple pathways for the formation of the γ-glutamyl peptides γ-glutamyl-valine and γ-glutamyl-valyl-glycine in Saccharomyces cerevisiae. PloS one 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soga T, Sugimoto M, Honma M, Mori M, Igarashi K, Kashikura K, Ikeda S, Hirayama A, Yamamoto T, and Yoshida H (2011). Serum metabolomics reveals γ-glutamyl dipeptides as biomarkers for discrimination among different forms of liver disease. J Hepatol 55, 896–905. [DOI] [PubMed] [Google Scholar]
- Soini Y, Näpänkangas U, Järvinen K, Kaarteenaho‐Wiik R, Pääkkö P, and Kinnula VL (2001). Expression of γ-glutamyl cysteine synthetase in nonsmall cell lung carcinoma. Cancer: Interdisciplinary International Journal of the American Cancer Society 92, 2911–2919. [DOI] [PubMed] [Google Scholar]
- Solmonson A, and DeBerardinis RJ (2018). Lipoic acid metabolism and mitochondrial redox regulation. J Biol Chem 293, 7522–7530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soula M, Weber RA, Zilka O, Alwaseem H, La K, Yen F, Molina H, Garcia-Bermudez J, Pratt DA, and Birsoy K (2020). Metabolic determinants of cancer cell sensitivity to canonical ferroptosis inducers. Nat Chem Biol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stipanuk M, Ueki I, Dominy J, Simmons C, and Hirschberger L (2009). Cysteine dioxygenase: a robust system for regulation of cellular cysteine levels. Amino acids 37, 55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stipanuk MH, Dominy JE Jr, Lee J-I, and Coloso RM (2006). Mammalian cysteine metabolism: new insights into regulation of cysteine metabolism. J Nutr 136, 1652S–1659S. [DOI] [PubMed] [Google Scholar]
- Sun J, Zhou C, Ma Q, Chen W, Atyah M, Yin Y, Fu P, Liu S, Hu B, and Ren N (2019). High GCLC level in tumor tissues is associated with poor prognosis of hepatocellular carcinoma after curative resection. J Cancer 10, 3333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun X, Ou Z, Chen R, Niu X, Chen D, Kang R, and Tang D (2016). Activation of the p62-Keap1-NRF2 pathway protects against ferroptosis in hepatocellular carcinoma cells. Hepatology 63, 173–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi S, Wada K, Toyooka T, Shinomiya N, Shimazaki H, Nakanishi K, Nagatani K, Otani N, Osada H, and Uozumi Y (2013). Increased xCT expression correlates with tumor invasion and outcome in patients with glioblastomas. Neurosurgery 72, 33–41. [DOI] [PubMed] [Google Scholar]
- Tatebe S, Unate H, Sinicrope FA, Sakatani T, Sugamura K, Makino M, Ito H, Savaraj N, Kaibara N, and Kuo MT (2002). Expression of heavy subunit of γ-glutamylcysteine synthetase (γ-GCSh) in human colorectal carcinoma. Int J Cancer 97, 21–27. [DOI] [PubMed] [Google Scholar]
- Timmerman LA, Holton T, Yuneva M, Louie RJ, Padró M, Daemen A, Hu M, Chan DA, Ethier SP, and van’t Veer LJ (2013). Glutamine sensitivity analysis identifies the xCT antiporter as a common triple-negative breast tumor therapeutic target. Cancer Cell 24, 450–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torrente L, Sanchez C, Moreno R, Chowdhry S, Cabello P, Isono K, Koseki H, Honda T, Hayes JD, Dinkova-Kostova AT, et al. (2017). Crosstalk between NRF2 and HIPK2 shapes cytoprotective responses. Oncogene 36, 6204–6212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ventura A, Kirsch DG, McLaughlin ME, Tuveson DA, Grimm J, Lintault L, Newman J, Reczek EE, Weissleder R, and Jacks T (2007). Restoration of p53 function leads to tumour regression in vivo. Nature 445, 661–665. [DOI] [PubMed] [Google Scholar]
- Vyas S, Zaganjor E, and Haigis MC (2016). Mitochondria and Cancer. Cell 166, 555–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinstein C, Haschemeyer R, and Griffith O (1988). In vivo studies of cysteine metabolism. Use of D-cysteinesulfinate, a novel cysteinesulfinate decarboxylase inhibitor, to probe taurine and pyruvate synthesis. J Biol Chem 263, 16568–16579. [PubMed] [Google Scholar]
- Winterbourn CC, and Hampton MB (2008). Thiol chemistry and specificity in redox signaling. Free Radic Biol Med 45, 549–561. [DOI] [PubMed] [Google Scholar]
- Yang WS, SriRamaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS, Cheah JH, Clemons PA, Shamji AF, and Clish CB (2014). Regulation of ferroptotic cancer cell death by GPX4. Cell 156, 317–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang T, Bauer C, Newman AC, Uribe AH, Athineos D, Blyth K, and Maddocks ODK (2020). Polyamine pathway activity promotes cysteine essentiality in cancer cells. Nat Metab. 2, 1062–1076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Tan H, Daniels JD, Zandkarimi F, Liu H, Brown LM, Uchida K, O’Connor OA, and Stockwell BR (2019). Imidazole ketone erastin induces ferroptosis and slows tumor growth in a mouse lymphoma model. Cell Chem Biol 26, 623–633. e629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou Y, and Schreiber SL (2020). Progress in Understanding Ferroptosis and Challenges in Its Targeting for Therapeutic Benefit. Cell Chem Biol 27, 463–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate any unique datasets or code.