

Abstract
The localization of proteins at a tissue- or cell-type-specific level is tightly linked to the protein function. To better understand each protein’s role in cellular systems, spatial information constitutes an important complement to quantitative data. The standard methods for determining the spatial distribution of proteins in single cells of complex tissue samples make use of antibodies. For a stringent analysis of the human proteome, we used orthogonal methods and independent antibodies to validate 5981 antibodies that show the expression of 3775 human proteins across all major human tissues. This enhanced validation uncovered 56 proteins corresponding to the group of “missing proteins” and 171 proteins of unknown function. The presented strategy will facilitate further discussions around criteria for evidence of protein existence based on immunohistochemistry and serves as a useful guide to identify candidate proteins for integrative studies with quantitative proteomics methods.
Keywords: antibody-based proteomics, missing proteins, protein evidence, immunohistochemistry, transcriptomics, antibody validation, human proteome
Introduction
Human physiology is dependent on the complex interplay between intercellular interactions and cell-type-specific functions in organs and tissues. For a full understanding of the diseases disrupting these processes, it is necessary to study the tissue architecture and the molecular constituents with a single-cell resolution. Proteomics constitutes the functional representation of the genome, and the standard approach for spatial localization of proteins in tissues is immunohistochemistry (IHC).1 There are, however, several hurdles to overcome to validate the specificity and selectivity of antibodies,2−5 and there is a widely acknowledged need for improved reproducibility of IHC data. To provide a best estimate of protein expression across different tissues, it is therefore of utmost importance that antibodies undergo careful validation.5 The International Working Group for Antibody Validation (IWGAV) has suggested five different “pillars” to use for antibody validation, drawing increased attention to the implementation of standardized validation pipelines for antibody assays.6−8 A demand to adequately present validation strategies for antibodies used in publications has also been requested by multiple journals,9 which has led to an increase in the proportion of validated antibodies. Because samples are treated differently in different applications, which affects which epitopes of the target protein are exposed to the antibody, it is necessary that the validation is performed in an application-specific manner.10 Two main antibody validation strategies are suggested for IHC in human tissues: (i) orthogonal validation, comparing protein expression levels using an antibody-independent method, or (ii) independent antibody validation, comparing protein expression levels using two different antibodies targeting nonoverlapping regions of the same protein.
The largest initiative for the discovery of the entire human proteome using antibody-based proteomics is the Human Protein Atlas (HPA), with IHC data covering 15 308 proteins corresponding to 78% of the protein-coding genome. The HPA has spent a considerable effort establishing stringent pipelines for antibody validation and has implemented the five strategies for application-specific antibody validation, as suggested by the IWGAV. Recently, a streamlined pipeline for the validation of antibodies for Western blot applications was described,11 where more than 6000 antibodies could be confidently validated by at least one of the strategies. There is, however, no previous large-scale study outlining the exact criteria for the implementation of antibody validation strategies for IHC.
Another method for the detection of proteins in a tissue is mass spectrometry. A systematic initiative focusing on mapping the entire human proteome is the Human Proteome Project (HPP),12,13 a worldwide effort that together with its reference knowledgebase neXtProt14 has set up criteria for ranking proteins into categories according to evidence of their existence (PE). This coordinated effort that has adopted stringent interpretation guidelines of mass spectrometry data has resulted in experimental validation (PE1) of almost 90% of all proteins predicted by the human genome. Approximately 1900 proteins, however, still lack evidence of existence at the protein level and are defined as “missing proteins”. These proteins, scored as PE2, PE3, or PE4, constitute important targets for further investigation. In addition, despite evidence of their existence, many PE1 proteins lack information on known function,15,16 or data on cell-type-specific localization within tissues. Querying the UniProt database for reviewed PE1 proteins with experimental evidence of tissue specificity or subcellular location shows that ∼30% of PE1 proteins lack data for both tissue specificity and subcellular location. These proteins that lack a functional annotation together with the “missing proteins” may require alternative methods due to expression at low levels or in rare cell types. A small proportion of PE1 proteins have been validated using methods other than mass spectrometry, but only a handful of proteins scored as PE1 rely on antibody-based proteomics. For further discussions on criteria for how antibody-based data can be taken into consideration for evidence of protein existence, it is crucial to first define proper strategies for antibody validation using IHC. This is particularly important when studying missing proteins, as they are challenging to validate due to the lack of information on the expected staining pattern or well-characterized positive controls.
Here we present an approach for the enhanced validation of antibodies for IHC, confidently applied to 5981 antibodies covering 3775 human proteins. Among these were 56 proteins that correspond to missing proteins and an additional 171 proteins that do not have any assigned function. The presented strategies hold promise for the streamlined validation of antibodies for IHC that is suitable for both antibody providers and users and will facilitate discussions around the criteria for the potential integration of antibody-based data for the characterization of missing proteins. The data are also likely to aid in identifying targets that are relevant for integrated efforts using both mass spectrometry and IHC for further characterization of missing proteins.
Experimental Procedures
Target Gene Set and Antibodies Used
The HPA uses a whole-proteome approach in the effort to determine the expression and distribution of the human proteins across a wide variety of human tissues using spatial proteomics. Our gene set is based on the protein-coding genes of the Ensembl database, which, in version 92, corresponds to 19 670 genes with more than 82 000 protein-coding splice variants and almost 72 000 unique protein sequences. The aim is to target at least one splice variant of each gene with at least one antibody, and currently, there are antibodies validated by IHC for 78% (n = 15 308) of the genes. To decrease the risk of antibody cross-reactivity, the antigen sequences are selected on regions of the target protein with the lowest possible identity to proteins from other genes. A sliding window BLAST with three different window sizes is used to evaluate both the global antigen size identity (50 amino acid window) and the closer to epitope size identity (10 amino acid and 20 amino acid windows). The maximum identity for each window and window size is determined, and the resulting identity profiles are used to select the antigen sequence with the lowest identity to other proteins. All HPA antibodies are further affinity-purified, and only antibodies that in the protein array analysis selectively bind their target protein epitope signature tag (PrEST) and do not show any cross-reactivity to 383 randomly selected PrESTs are approved for use. The majority of the proteins (n = 14 111) have been analyzed with single-targeting antibodies for which the antigen sequence is known or with commercial antibodies for which no additional information on multiple recognition is provided. These antibodies are expected to target a single protein based on having low sequence identity (maximum 60%, with the vast majority having <40%) to all human transcripts, except for those corresponding to the gene of interest. However, for 1197 genes, it was not possible to generate single-targeting antibodies due to the high sequence identity among proteins belonging to different genes. These genes are, in many cases, closely related and belong to known gene families, and in these cases, a multitargeting antibody was produced that has >80% sequence identity to transcripts of the genes belonging to the family and low sequence identity to the transcripts of all other human genes.
Human Tissue Samples
Human tissues samples for the analysis of mRNA and protein expression in the HPA data sets were collected and handled in accordance with Swedish laws and regulation. Tissues were obtained from the Clinical Pathology department, Uppsala University Hospital, Sweden and collected within the Uppsala Biobank organization. All samples were anonymized for personal identity by following the approval and advisory report from the Uppsala Ethical Review Board (ref nos. 2002-577, 2005-388, 2007-159, 2011-473). Informed consent was obtained from all subjects in the study.
Transcript Profiling
The protocol for RNA sequencing of tissue types has been previously described.17,18 In this study, RNA expression data were used for the classification of genes into different RNA categories as well as for correlation with protein expression data. The classification of genes according to tissue specificity and tissue distribution is based on normalized expression (NX) values and includes data from 37 different tissues and tissue groups based on three different data sets (HPA, GTEx, and FANTOM5), as previously described.19 For correlation with protein expression levels, NX expression values corresponding to TMM-normalized TPMs for 37 normal tissues analyzed within the HPA were used.
Protein Profiling
The generation of tissue microarrays (TMAs), IHC staining, and the digitization of stained TMA slides were performed as previously described.20 In brief, formalin-fixed, paraffin-embedded (FFPE) tissue blocks were assembled into tissue microarrays (TMAs) based on 1 mm cores from 44 different normal tissue types, with 3 individuals per tissue. TMA blocks were cut in 4 μm sections, dried overnight at room temperature (RT), and baked at 50 °C for at least 12 h. Automated IHC was performed by using a Lab Vision Autostainer 480S module (ThermoFisher Scientific, Freemont, CA), as previously described in detail. Primary antibodies were optimized on a test TMA containing 20 different normal tissues. Antibody IDs and details of the antigen retrieval and dilutions for all antibodies used in IHC figures are available for each gene at https://v19.proteinatlas.org. The stained slides were digitized with ScanScope AT2 (Leica Aperio, Vista, CA) using a 20× objective. The annotation parameters included the staining intensity, defined as the saturation level of brown staining (negative, weak, moderate, or strong), and the quantity, defined as the ratio of stained cells versus the total number of cells within each analyzed tissue divided into the following groups: 0, 1–24, 25–75, and >75%. Both the intensity and the quantity were annotated separately for each cell type, and all individual samples (up to three samples per tissue type) were taken into consideration. All tissue samples were manually annotated by one observer and quality-controlled by a second observer, an experienced expert. The second observer double-checked all 44 normal tissues for tissue quality, cell-type identification, intensity, quantity, and subcellular localization. Both the first and second observer were blinded to previous literature and the corresponding RNA expression levels to avoid biased decisions. All annotations not agreed upon by the first and second observer were discussed with a third independent observer, an experienced histologist or certified pathologist, until a consensus decision was made.
Analysis of Data
Correlation of Protein Expression with RNA Expression
Data analysis and visualization were performed using R (version 3.6.1, Action of the Toes).21 A correlation matrix of Kendall’s tau for the genes with IHC data was calculated based on the HPA RNA NX values (values below 1 were set to 0) and the protein expression levels estimated from the protein staining and manual annotation of IHC images across all 37 tissues. The protein expression value for each tissue was calculated by multiplying the staining intensity (negative = 0, weak = 1, moderate = 2, or strong = 3) by the quantity (0% = 0, 1–24% = 1, 25–75% = 2, or >75% = 3), which yielded a semiquantitative protein expression value ranging from 0 to 9, and then choosing the maximum value obtained for each tissue. The obtained p values for the correlation were adjusted according to Benjamini–Hochberg, and both tau values and adjusted p values are included in Supplementary Table S1.
Comparison of Protein Expression Patterns for Independent Antibodies
Independent antibodies are antibodies with antigen sequences originating from nonoverlapping regions of the same gene. For the 4039 genes with at least two independent HPA antibodies, a correlation matrix of Kendall’s tau was calculated using the protein expression levels across 44 HPA normal tissues for two antibodies for each gene. The protein expression values were calculated by multiplying the staining intensity by the quantity, as previously described.
Data Availability
High-resolution images corresponding to immunohistochemically stained TMA cores of 44 different tissue types corresponding to all antibodies analyzed in the present investigation are available in the latest version 19.3 of the HPA (https://v19.proteinatlas.org). The normalized consensus transcript expression levels based on transcriptomics data from the HPA, GTEx, and FANTOM5 as well as the annotated protein expression levels based on IHC can be accessed under the download page (https://v19.proteinatlas.org/about/download).
Results
Antibody Reliability and Enhanced Validation of Antibodies for Immunohistochemistry
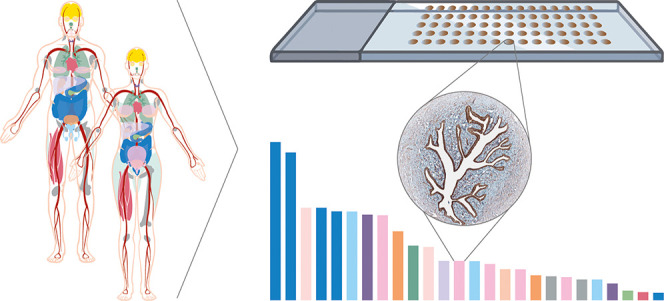
With the aim to characterize the entire human proteome, antibodies targeting 15 308 unique proteins were used for IHC on TMAs comprising 44 different normal tissues and organs, corresponding to all of the major parts in the human body. This large data set covering 78% of the human protein-coding genome formed the basis for a stringent antibody validation workflow (Figure 1).
Figure 1.

Validation of antibodies for the immunohistochemical analysis of the human protein-coding genes. Overview of the antibody validation workflow, where antibody-based proteomics data using IHC on TMAs is compared with mRNA levels from three sources and available gene/mRNA/protein characterization data from various databases and literature to determine a reliability score for the antibody data corresponding to each protein. Proteins with “Enhanced” validation have at least one antibody meeting the criteria for either (i) the orthogonal strategy, showing a high consistency between mRNA and protein levels, or (ii) the independent antibody strategy, where a similar spatial localization is observed between two independent antibodies.
IHC staining patterns were graded into the staining intensity and the quantity of stained cells for each analyzed cell type constituting the primary data of presumed protein expression levels, and by grading the performance of the antibodies in IHC, each characterized protein was assigned a reliability score (Table 1). The highest level of reliability, “Enhanced”, was assigned for 3775 proteins and corresponds to antibodies that meet the stringent criteria for enhanced validation based on the strategies adapted from the IWGAV, including orthogonal validation or independent antibody validation. The other three levels, “Supported” (1608 proteins), “Approved” (5514 proteins), and “Uncertain” (4411 proteins), rely on the comparison of the IHC staining pattern with the RNA expression levels (as defined in Table 2), available literature, and independent antibodies without meeting the criteria for enhanced validation. Literature was considered “valid” if UniProt had data on the protein level with information on both the tissue specificity and the subcellular localization and the tissue specificity was determined using human samples.
Table 1. Reliability Scorea.
| reliability score | description | number of proteins |
|---|---|---|
| Enhanced | At least one antibody meets the criteria for Enhanced validation using either Orthogonal validation or Independent antibody validation | 3775 |
| Supported | ONE OF THE FOLLOWING | 1608 |
| (i) At least one antibody has an RNA similarity score of high or medium consistency, but the antibody does not qualify for Orthogonal validation | ||
| AND | ||
| Staining pattern is consistent with valid literature, or there is no valid literature available | ||
| (ii) At least one antibody has an RNA similarity scored defined as “Cannot be evaluated” | ||
| AND | ||
| Staining pattern is consistent with valid literature | ||
| (iii) Paired antibodies show similar spatial expression patterns, but the antibodies do not qualify for Independent antibody validation, e.g., due to unknown target sequence | ||
| AND | ||
| Staining pattern is consistent with valid literature, or there is no valid literature available | ||
| Approved | ONE OF THE FOLLOWING | 5514 |
| (i) At least one antibody has an RNA similarity score of high or medium consistency | ||
| AND | ||
| Staining pattern is inconsistent with valid literature | ||
| (ii) At least one antibody has an RNA similarity score of low consistency | ||
| AND | ||
| Staining pattern is consistent with valid literature | ||
| (iii) At least one antibody has an RNA similarity scored defined as “Cannot be evaluated” | ||
| AND | ||
| Staining pattern is partly consistent with valid literature or consistent with limited literature | ||
| (iv) Paired antibodies show partly similar expression patterns | ||
| Uncertain | ONE OF THE FOLLOWING | 4411 |
| (i) Only multitargeting antibodies are available | ||
| (ii) At least one antibody has an RNA similarity score of low or very low consistency or is defined as “Cannot be evaluated” | ||
| AND | ||
| Staining pattern is inconsistent with valid literature, or there is no valid literature available | ||
| (iii) Staining pattern is inconsistent with valid literature, or there is no valid literature available | ||
| (iv) Paired antibodies show dissimilar expression patterns |
Definition of the criteria used to determine the reliability score for protein data based on the antibody performance in IHC.
Table 2. RNA Similarity Scorea.
| RNA similarity score | RNA category | definition |
|---|---|---|
| High consistency | Tissue enriched, Group enriched, or Tissue enhanced | Maximum one elevated tissue may be negative or show weak staining intensity; the remaining elevated tissues must show moderate or strong staining intensity |
| AND | ||
| Maximum 10% of nonelevated tissues may have higher staining intensity than the highest observed intensity of the elevated tissues | ||
| AND | ||
| Maximum 25% of nonelevated tissues may have the same intensity as the highest observed intensity of the elevated tissues | ||
| Low tissue specificity | Maximum 10% of the analyzed tissues with NX ≥ 1 are negative in IHC | |
| AND | ||
| Maximum 10% of the analyzed tissues with NX < 1 are positive in IHC | ||
| Medium consistency | Tissue enriched, Group enriched, or Tissue enhanced | Minimum one elevated tissue must show moderate or strong staining intensity |
| AND | ||
| Maximum 20% of nonelevated tissues may have higher staining intensity than the highest observed intensity of the elevated tissues | ||
| AND | ||
| Maximum 50% of nonelevated tissues may have the same intensity as the highest observed intensity of the elevated tissues | ||
| Low tissue specificity | Maximum 25% of the analyzed tissues with NX ≥ 1 are negative in IHC | |
| AND | ||
| Maximum 25% of the analyzed tissues with NX < 1 are positive in IHC | ||
| Low consistency | Tissue enriched, Group enriched, or Tissue enhanced | Minimum one elevated tissue must show at least weak staining intensity |
| AND | ||
| Maximum 40% of nonelevated tissues may have higher staining intensity than the highest observed intensity of the elevated tissues | ||
| AND | ||
| Maximum 60% of nonelevated tissues may have the same intensity as the highest observed intensity of the elevated tissues | ||
| Low tissue specificity | Maximum 50% of the analyzed tissues with NX ≥ 1 are negative in IHC | |
| AND | ||
| Maximum 50% of the analyzed tissues with NX < 1 are positive in IHC | ||
| Very low consistency | Any | None of the above categories and not defined as “Cannot be evaluated” |
| Cannot be evaluated | Any | All tissues were negative for IHC |
| OR | ||
| All tissues had NX < 1 | ||
| OR | ||
| Literature suggests complex dynamics between mRNA and protein levels due to, e.g., secreted proteins or isoforms |
Definition of the criteria used to determine the RNA similarity score, comparing the pattern of expression between mRNA levels and the IHC across 37 tissue types.
Orthogonal Validation
Orthogonal validation relies on the comparison of protein levels determined by IHC, with levels determined by an antibody-independent method across a panel of samples. mRNA levels for protein-coding genes were used as a proxy for where to expect high versus low expression of the corresponding protein. Here 3427 proteins were orthogonally validated and showed similar patterns of expression when comparing protein levels based on IHC with mRNA expression levels across 37 different tissues and organs. Protein expression levels were manually annotated in 75 main organ-specific cell types in these 37 organs, but most samples also included a large proportion of general cell types present in most organs that have not been annotated in detail, including immune cells and mesenchymal cells, for example, endothelial cells, fibroblasts, and smooth muscle cells. Therefore, the comparison of the trends in expression patterns between protein and mRNA levels was determined manually, taking into consideration all cell types present in these organs, and divided into the following RNA similarity scores: “High consistency”, “Medium consistency”, “Low consistency”, “Very low consistency”, and “Cannot be evaluated”. The trend was evaluated based on the relative relationship between RNA and protein levels across all tissues where data from both sources were available. The exact definitions for the RNA similarity scores are presented in Table 2. Of the 15 308 analyzed proteins, 8601 proteins were validated with at least one antibody with the RNA similarity scores “High consistency” or “Medium consistency”. These proteins underwent a second evaluation to determine if they should qualify for orthogonal validation, where it must be possible to select representative images of different staining intensities reflected by at least four-fold differences in mRNA expression levels. Because many of the 8601 proteins were expressed in all tissues with low variation of expression levels, these antibodies did not meet the criteria for orthogonal validation. The majority of the antibodies generated within the HPA are denoted single-targeting antibodies, and because orthogonal validation can only be performed using single-targeting antibodies, 412 proteins with high consistency with RNA levels for which only multitargeting antibodies exist were excluded. To avoid highlighting unreliable staining patterns, orthogonal validation was not considered for proteins where there was an independent antibody showing a dissimilar pattern of expression (subcellular localization and/or cell-type specificity), while both antibodies were equally consistent between mRNA and protein expression levels, and there was no available literature to guide the decision on which antibody was correct. Similarly, an antibody was not considered for orthogonal validation if the protein had evidence on the protein level with predicted subcellular localization and tissue specificity available in UniProt, and these data were based on human tissues and were contradictory to the observed immunohistochemical staining pattern.
In total, 3427 proteins were orthogonally validated using at least one antibody, and when comparing these 3427 proteins with RNA categories, it is clear that orthogonal validation is mostly suitable for genes defined as tissue elevated (Figure 2A). As many as 84% of the proteins with orthogonal validation based on RNAseq data have been classified as tissue elevated, divided into three different subcategories: (i) tissue enriched (at least four times higher mRNA level in one tissue compared with other tissues), (ii) group enriched (at least four times higher mRNA level in a group of two to five tissues compared with other tissues), or (iii) tissue enhanced (at least four times higher mRNA level in one tissue compared with the average level in all other tissues).
Figure 2.

Orthogonal validation. (A) Distribution of different RNA specificity categories across antibody validation reliability scores. (B) Box plot showing the distribution of Kendall tau values from the correlation of mRNA levels and protein expression values for different RNA similarity scores. (C) Distribution of Kendall tau values from the correlation of mRNA levels and protein expression values for the different reliability scores. (D) Distribution of Kendall tau values from the correlation of mRNA levels and protein expression values for orthogonally validated antibodies and antibodies without enhanced validation. (E–H) IHC examples showing RNA levels compared with protein expression in four different tissue types. (E) CLDN4 protein levels were visualized with the highest membranous expression in tight junctions of the colon followed by moderate membranous expression in the thyroid gland and kidney. CLDN4 was not detected in the testis. (F) HNF4A protein levels were visualized with the highest nuclear expression in glandular cells of the duodenum followed by moderate nuclear expression in liver hepatocytes and the ducts of the kidney. Lymph node expression was not detected. (G) HTN3 protein levels were visualized with high cytoplasmic expression in the glandular cells of the salivary gland. No protein was detected in the pancreas, rectum, or duodenum. (H) GRAP2 protein levels were visualized with high cytoplasmic expression in leukocytes in the lymph nodes, appendix, urinary bladder, and esophagus.
To confirm that the manual assessment of RNA similarity follows a certain trend, we also performed a Kendall rank correlation analysis between mRNA expression levels across 37 tissues and the semiquantitative protein expression levels based on IHC. In tissues where more than one cell type was evaluated, the highest protein expression value was used for the correlation. Even if the automated correlation analysis has limitations because it only takes into consideration cell types that have been annotated, it is evident that the analysis matches the manually assigned RNA similarity scores (Figure 2B). Also, there seems to be a slight shift in the distribution toward a higher correlation between the mRNA and protein expression levels for proteins with antibodies with enhanced validation compared with the other reliability scores (Figure 2C) and a clear separation between orthogonally validated antibodies compared with antibodies without enhanced validation (Figure 2D). Of the 3427 proteins with orthogonal validation, 3252 could be analyzed with Kendall rank correlation comparing mRNA and protein levels. Only 489 proteins had a correlation of >0.5, whereas 1595 proteins had a correlation between 0.2 and 0.5 and 1168 proteins had a correlation <0.2. This shows that despite a certain consistency between the statistical correlation analyses and the IHC RNA similarity scores and reliability scores, which supports the manual assessment, the manual evaluation identified a high proportion of proteins showing a similar pattern of expression between mRNA and protein levels that was missed in the correlation analysis.
Figure 2E–H shows examples of proteins with orthogonal validation. The tight junction protein Claudin 4 (CLDN4) (Figure 2E) and the transcriptional regulator Hepatocyte nuclear factor 4 alpha (HNF4A) (Figure 2F) both showed a high Kendall rank correlation of >0.7 and p.adj < 0.05. Both proteins consistently showed high protein expression levels in organs with high levels of mRNA, whereas the expression was significantly lower in organs with no or low mRNA levels. Two examples of proteins that were orthogonally validated but have a low (<0.4 and p.adj <0.05) or nonsignificant Kendall rank correlation are the saliva protein Histatin 3 (HTN3) (Figure 2G) and the GRB2-related adaptor protein 2 (GRAP2) (Figure 2H), involved in leukocyte-specific protein-tyrosine kinase signaling. Here it is evident that cases with a poor Kendall rank correlation may also represent highly validated antibodies. Whereas the expected selective expression of HTN3 in salivary gland is represented by both data sets, the IHC staining also showed faint unspecific staining in the intestinal tract, which would be neglected in a knowledge-based interpretation of the staining pattern but contributes to a poor correlation with mRNA levels. Furthermore, low levels of mRNA above the cutoff are found in a few organs, including the pancreas, but most likely represent noise that is not translated to detectable protein levels. Despite a poor Kendall rank correlation, it is evident that mRNA and protein levels follow the same trend, and HTN3 constitutes an example of a protein that is reliably detected in the salivary gland. Another example is GRAP2, which is abundantly expressed in the immune cells of most organs. Whereas these cells are manually evaluated in lymphoid organs where they represent a majority, they are also present in lower amounts in other organs where they have not been annotated and therefore do not constitute a part of the protein expression data set. Immunohistochemical images clearly show expression in a smaller subset of immune cells in the stroma in most tissues, which is consistent with low mRNA levels. This protein is therefore suggested to be reliably detected in immune cells despite a poor Kendall rank correlation.
Independent Antibody Validation
Another method for enhanced validation is the use of independent antibodies, defined as a similar expression pattern observed by an independent antibody targeting a nonoverlapping region of the same protein. To determine if the antibodies are independent, it is necessary to know the antigen sequence toward which the antibody has been raised. Of the 15 308 proteins analyzed, >6500 proteins were targeted by more than one antibody, but only 4084 of these corresponded to antibodies known to target nonoverlapping regions of the protein. These 4084 proteins were manually evaluated to determine similarity by taking into consideration the overall protein expression patterns across all 44 tissues and the cell types within these tissues. Antibodies showing a similar pattern in terms of cell-type specificity, spatial distribution (e.g., cell-to-cell variability), and subcellular localization qualified for independent antibody validation.
To validate the results for the 871 antibody pairs defined as independently validated based on manual evaluation, the Kendall rank correlation analysis was used. It was evident that these antibody pairs showed a higher correlation between the protein expression levels compared with antibody pairs that did not qualify for independent antibody validation (Figure 3A). A high proportion of these 871 proteins were also orthogonally validated, but as many as 348 proteins (40%) had only independent antibody validation (Figure 2A). As expected, most of the corresponding genes for these 348 proteins were defined as having low tissue specificity based on RNA expression levels (265 genes), that is, they were not elevated in any tissue, and as many as 304 genes were detected above the cutoff in all 37 tissues analyzed at the mRNA level. The Kendall rank correlation also showed a lower correlation between the mRNA and protein levels for independent antibodies compared with antibodies that were orthogonally validated (Figure 4B). This suggests that independent antibody validation constitutes an attractive approach for ubiquitously expressed proteins because even if the protein may be expressed in all tissues, it can still be localized to specific structures or with a certain spatial pattern within these tissues. Independent antibody validation is also suitable for proteins where a poor correlation between mRNA and protein levels is expected, for example, secreted proteins, or for rare tissues or structures where no mRNA data is available.
Figure 3.

Independent antibody validation. (A) Kendall rank correlation showed a higher correlation between mRNA and protein levels for proteins that were validated with the orthogonal method compared with proteins for which independent antibodies were used. (B) Kendall rank correlation showed that the correlation between corresponding protein levels across all tissues for paired antibodies were significantly higher for proteins that met the criteria for independent antibody validation compared with antibody pairs that were not independently validated. (C) IHC images showing the nuclear protein expression of ADAR with two independent antibodies in the skin, cerebral cortex, and kidney. Selective nuclear expression in the seminiferous ducts in the testis was detected. (D) IHC images showing the granular cytoplasmic protein expression of CLPB with two independent antibodies in the smooth muscle of the prostate, pyramidal neurons in the cerebral cortex, ducts in the kidney, and glandular cells in the salivary glands. (E) IHC images showing the membranous and cytoplasmic expression of FCHO2 with two independent antibodies in the placenta, endometrium, liver, and lymph node.
Figure 4.

Protein evidence in relation to antibody validation and expression. The barplots show the distribution of (A) IHC reliability scores and (B) the RNA abundance category across the different levels of neXtProt protein evidence, respectively. (C) Box plot showing the maximum level of RNA expression (NX) for tissue elevated genes having different levels of protein evidence. (D) Bar plot showing the distribution of protein evidence across the genes belonging to the different IHC validation categories.
In Figure 3C–E, IHC stainings of proteins with independent antibody validation are shown. The nuclear protein Adenosine deaminase, RNA specific (ADAR) (Figure 3C), the ATPase ClpB homologue, mitochondrial AAA ATPase chaperonin (CLPB) (Figure 3D), and the endocytosis-related protein FCH domain only 2 (FCHO2) (Figure 3E) were all ubiquitously expressed across many different cell types, but the analysis of the IHC staining pattern on consecutive sections clearly showed similar spatial distributions between the antibody pairs, with selective expression in certain cell types.
Protein Evidence Levels of the Human Proteome
Since the first release of the HPA in 2005, the number of human protein-coding genes in Ensembl has decreased from more than 34 000 to fewer than 20 000. This reflects that the human proteome is far from fully explored and indicates that not all of the current protein-coding genes necessarily will be considered as protein-coding in the future.
The neXtProt database is a resource built on top of the UniProt database with a focus on the characterization and functional annotation of the human proteins.14 neXtProt provides information on the evidence of existence for each of their 20 350 human proteins based on current experimental data, including data from high-throughput efforts and data from orthologs in related species. The levels of evidence and the number of proteins in each category are “Experimental evidence at protein level” (n = 17 874), “Experimental evidence at transcript level” (n = 1596), “Protein inferred by homology” (n = 253), “Protein predicted” (n = 50), and “Protein uncertain” (n = 577). The “Protein uncertain” category (PE5) has been excluded from the HPP missing proteins since 2013.22
Here we used evidence levels from neXtProt for the comparison with the results from IHC and mRNA in the HPA. The overlap between the two data sets is, however, not complete, mostly due to different Ensembl versions being used by the HPA and neXtProt. Furthermore, because of the different gene models, isoforms of a single Ensembl gene can be mapped to different neXtProt entries, or a single neXtProt entry may correspond to several Ensembl genes. Of the current 19 670 protein-coding genes in the HPA data set based on Ensembl v92, 18 936 were mapped to at least one neXtProt entry, whereas 734 genes do not have a corresponding entry in neXtProt. The major reason for this was genes having transcript sequences with minor differences from the neXtProt entry and instead corresponding to sequences of the unreviewed part of UniProt. There are also genes where the neXtProt identifier referred to in Ensembl has been removed or changed in the latest neXtProt version due to the use of different database versions. Furthermore, there are about 1500 neXtProt IDs that are not present among the cross-references of the HPA gene set. These entries correspond to (i) immunity-related genes excluded from the HPA gene set such as immunoglobulins and variable chain T-cell receptor genes, (ii) genes that are not protein-coding in the present Ensembl version, and (iii) genes mapped to different protein identifiers in Ensembl and neXtProt.
The barplot in Figure 4A shows the number of genes and the neXtProt evidence level across the different IHC reliability scores based on all 19 670 genes. For genes with isoforms having different neXtProt entries, the highest evidence level has been selected, and genes not mapped to neXtProt are annotated as “No evidence”. Antibodies have been successfully produced toward almost 80% of human proteins with evidence at the protein level but toward only ∼50% of the proteins in the transcript and no evidence categories. A reliable orthogonal or independent validation of an antibody needs the target protein to be expressed in the tissues used for the validation, and a protein widely expressed across tissues is easier to detect and thus find evidence for. When investigating the relationship between the protein evidence and the RNA expression pattern (Figure 4B), it was not surprising that the fraction of genes detected in all analyzed tissues was >50% among the genes with protein evidence (n = 17 442) but <10% among genes with only transcript evidence (n = 1341).
Genes not detected at the mRNA level are present in all evidence categories. This may seem contradictory, especially for the protein and transcript evidence categories, but in many cases, it can be explained by the expression of the protein in a tissue not included in the standard TMA setup, such as the eye, pituitary gland, or lactating breast.23 Among the not detected genes that have protein evidence are keratin-associated proteins, which reside in hair follicles, and taste receptors, whereas olfactory receptors found in the olfactory bulb are examples of not detected genes with transcript level evidence. Out of the 97 genes with evidence inferred from homology, 93 are not detected on the mRNA level, and 74 of those belong to the olfactory or taste receptor family.
Genes that show a tissue restricted expression, especially those expressed in only a single tissue, may be less explored and have a lower level of evidence unless they are present in a commonly studied tissue. On the basis of RNAseq data, the HPA has defined a set of 2845 tissue enriched genes, which are at least four times more highly expressed in one tissue compared with the highest expression of all other tissues. These are distributed across all evidence categories, and the vast majority (>80%) have evidence at the protein level, with more than half of them being enriched in the testis, brain, liver and lymphoid tissue, often with lower expression in other organs. The majority (89%) of the tissue enriched genes with transcript and homology evidence, on the contrary, are expressed in a single or a few tissues only, mainly the testis, brain, and skin. Some of these are uncharacterized open reading frame proteins, and many belong to the families of olfactory receptors, keratin-associated proteins, and defensins with expression only at low levels in the main human tissues, which may be the reason for their lacking evidence at the protein level. Interestingly, the mRNA expression levels for tissue enriched genes are significantly lower in the transcript evidence category compared with those with evidence at protein level, which is shown in Figure 4C.
Immunohistochemistry for the Exploration of “Missing Proteins”
“Missing proteins” are defined as proteins that lack experimental evidence at the protein level but have experimental evidence at the transcript level, that correspond to proteins inferred from homology based on orthologs in closely related species, or that are predicted but without evidence at the protein, transcript, or homology levels. On the basis of a 1–5 tier ranking system of protein existence (PE), “missing proteins” are also referred to as PE2–4. The overall goal of the HPP is to continue the quest of defining all of the gene products encoded by the human genome and increasing the number of proteins with experimental evidence at the protein level.22,24 There are 1899 proteins defined as “missing proteins” in neXtProt, out of which 1438 are represented in the current HPA gene set. These constitute interesting targets for further exploration by methods other than mass spectrometry, such as IHC. Because experimental data at the protein level are taken into consideration in the validation of antibodies for IHC, it is expected that the proteins with a lack of evidence at the protein level are targeted by a higher proportion of antibodies with uncertain reliability or, more often, are not yet analyzed by IHC. However, there are groups of interesting proteins that are highly validated by IHC but lack protein evidence (Figure 4D).
Of the 1438 “missing proteins” represented in the HPA gene set, 703 are targeted by at least one antibody, out of which 56 proteins have enhanced validation (Supplementary Table S2). All 56 of these proteins are elevated in particular tissues, and only 3 proteins were found above the detection limit based on mRNA levels in all 37 analyzed tissues. The elevated tissue with the highest number of “missing proteins” targeted by IHC was the testis (n = 20), followed by the brain (n = 15), epididymis (n = 6), and kidney (n = 6). Fifty-four of these proteins were validated with the orthogonal strategy, one with independent antibodies, and one with both methods. Representative IHC images of the 55 “missing proteins” targeted by antibodies validated by the orthogonal approach are displayed in Figure 5. The images clearly show not only that many of these targets were cell-type-specific but also that a large proportion were expressed in a smaller subset of cells or specific structures. The remaining 647 “missing proteins” that were targeted by antibodies did not meet the criteria for enhanced validation, and as many as 512 of the “missing proteins” with available antibodies were validated as uncertain. A majority (n = 506) of the “missing proteins” targeted by antibodies without enhanced validation are suggested to be elevated in particular tissues based on mRNA levels (Figure 6). Again, the testis (n = 186) and brain (n = 137) stand out, but many of these proteins were also elevated in other tissues. One of the reasons for the surprisingly low reliability of the antibody data for these proteins compared with that for other tissue elevated proteins in the HPA is that for almost one-third of these proteins (201 proteins), only multitargeting antibodies could be generated, which share identity of >80% with proteins from at least one more gene. Furthermore, 65 of these proteins are elevated in blood cells, which means that the expression of these proteins is difficult to evaluate in tissues; therefore, IHC may not be the optimal method for validation.
Figure 5.

Immunohistochemical staining patterns of “missing proteins” targeted by antibodies validated by the orthogonal strategy. The spatial localizations of the stainings are as follows: Cerebellum: DNAH100S, nuclei in granule cells; EGR4, astrocyte membranes. Cerebral cortex: HES5, neuronal nuclei; KLHL32, astrocyte membranes; SMIM17, neuropil; STRC, neuropil. Hippocampus: GRIK4, neuronal processes; NKAIN3, glial nuclei. Retina: ANKRD33, photoreceptor cytoplasm; SLC1A7, cytoplasm in nerve fibers. Adrenal gland: FGF11, cytoplasm in zona reticularis. Pituitary gland: anterior pituitary membranes. Skin: LCE6A, cytoplasm in cornified layer; SPRR4, cytoplasm in keratinocytes. Heart muscle: RD3L, intercalated disc membranes. Skeletal muscle: KLHL33 and RASL10B, cytoplasm in subset of myocytes. Pancreas: RBPJL, cytoplasm in islets of Langerhans. Thymus: FRMD1, cytoplasm in subset of medullary cells. Kidney: AQP6, cytoplasm in renal tubules; TMEM213, cytoplasm in distal tubules and collecting ducts; C21orf62 and SLC6A18, membranes in renal tubules; FXYD4, membranes in collecting ducts. Colon: TPSG1, cytoplasm in glandular cells. Duodenum: SLC22A18AS, cytoplasm in glandular cells. Small intestine: R3HDML, plasma in goblet cells. Stomach: SHISAL2B, cytoplasm in enteroendocrine cells. Epididymis: CLPSL1, cytoplasm in glandular cells; DEFB136 and RNASE12, cytoplasm in secretory granules; LCN9, cytoplasm and nuclei in glandular cells; ZMAT1, cytoplasm in connective tissue. Testis: ADAM20, SH2D7, SPATA12 and CHRNB3, cytoplasm in sperm flagella; ANKRD62, nuclei in spermatogonia; C1orf167, cytoplasm and membrane in Leydig cells; C3orf22, cytoplasm in preleptotene and spermatogonia; C9orf50, cytoplasm and membranes in spermatids and pachytene spermatocytes; C12orf56, cytoplasm in spermatids and nucleoli in Sertoli cells; C22orf42, cytoplasm in Leydig cells and spermatogonia; CC2D2B, cytoplasm in pachytene spermatocytes and spermatids; H1FOO, nuclei in spermatids; LRRC27, cytoplasm and membrane in seminiferous ducts; MGAT4D, cytoplasm in Leydig cells; PKDREJ, cytoplasm and nuclei in spermatogonia and preleptotene spermatocytes; SMIM21, cytoplasm and nuclei in Leydig cells; SPDYE4, cytoplasm in sertoli cells and spermatids; USP29, nuclei in Sertoli cells; VCX2, nuclei in germ cells; ZFAND4, cytoplasm in spermatids.
Figure 6.

Tissue specificity for 1438 proteins defined as “missing proteins”. The bar plot shows the number of genes that based on mRNA levels were elevated in a certain tissue as compared with other tissues, and the proportion of these proteins that have been targeted with antibodies corresponding to different reliability scores.
More than half of the “missing proteins” have not been targeted by any antibody (n = 735). Many of these (278 proteins, 38%) are olfactory receptors, but a large proportion also represents proteins elevated in organs that are easy to access, and only 112 proteins are below the detection limit in all 37 analyzed tissues. (Figure 6). These tissue restricted proteins not previously targeted by antibodies, together with “missing proteins” for which the used antibodies have not yet been analyzed with enhanced validation, constitute interesting targets for further tissue-specific studies aiming at determining the existence of these proteins in a particular tissue.
Immunohistochemistry for the Exploration of Proteins with an Unknown Function
Another important group of proteins that should be further evaluated are PE1 proteins that lack functional annotation. These proteins, referred to as uPE1,15,16 correspond to 1136 Ensembl genes in the HPA gene set (Supplementary Table S1) and to 1254 entries in the neXtProt 2020-01 release. Further knowledge on the expression of these proteins across different human tissues and organs constitutes an attractive starting point for further functional studies, as a protein’s function is closely linked to its expression. Here we provide the cell-type-specific localization for 899 of these proteins, out of which 171 were analyzed with “Enhanced” validation. As many as 154 of these 171 proteins showed elevated expression in particular organs at both the mRNA and protein levels, with the majority found in the testis (67 proteins), brain (21 proteins), and fallopian tube (17 proteins), highlighting particular tissues and cell types of certain interest for the functional characterization of these proteins.
Discussion
Spatial proteomics based on IHC constitutes the standard approach for the cell-type-specific localization of proteins in tissues.25 Today, IHC is a widely used method in both basic and clinical research and constitutes the standard strategy in routine diagnostic pathology for detecting cell-type-specific markers that define certain disease phenotypes or biological states.26 Whereas clinically used markers for IHC undergo strict validation and are constantly compared between different laboratories to ensure specificity and reproducibility, there is no widely acknowledged strategy for exactly how research antibodies for IHC should be validated. The field has been fueled by the exponential increase in commercial antibody production, from 10 000 to >3.8 million antibodies over the last 15 years.27 As an example, the antibody portal Antibodypedia (http://www.antibodypedia.com)28 lists almost 10 000 different antibodies directed toward the widely studied epidermal growth factor receptor (EGFR), but emerging scientific interest in certain proteins has quickly lead to an increase in available antibodies. Angiotensin I converting enzyme 2 (ACE2), which has been suggested as the main receptor for the SARS-CoV-2 virus causing the COVID-19 pandemic, has, within a few months, led to the availability of >900 antibodies from >40 providers. It is evident, however, that a lack of proper antibody validation may lead to completely different results, which was recently shown in the case of ACE2.29,30 There is a widely acknowledged demand to require higher standards by antibody providers,4 but even a specific antibody may produce false-positive results due to unspecific off-target binding if the protocol is not properly optimized, ultimately leaving the responsibility on the individual researcher to ensure that the antibodies have been properly validated. The IWGAV has suggested five main pillars for antibody validation that should be used in an application-specific manner, but there is still an urgent need for more exact criteria on how to validate antibodies for IHC.
IHC constitutes the main method for generating the tissue-based map of the human proteome, and 78% of the human protein-coding genome has been targeted by antibodies in the Tissue Atlas as part of the HPA effort with the overall aim to map the entire human proteome with a single-cell resolution.18 The HPA has implemented the approaches for antibody validation as suggested by the IWGAV. These enhanced validation strategies have been confidently applied to >10 000 antibodies targeting almost 7000 human proteins in at least one of the antibody applications (Western blot, IHC, or immunofluorescence). Despite the implementation of application-specific criteria for enhanced validation, it should, however, be noted that antibody-based proteomics is a challenging method due to the risk of cross-reactivity. To reduce this risk, all internally generated HPA antibodies have been generated toward sequences with the lowest possible identity to proteins of other genes, and the antibodies need to pass several additional quality controls before use. Such criteria include the sequencing of plasmid inserts ensuring cloning of the correct PrEST sequence, the analysis of the size of the resulting recombinant protein by mass spectrometry, followed by affinity purification and the analysis of the binding selectivity on a PrEST array.31 An antibody is approved for further use only if no cross-reactivity is observed among the other randomly selected protein fragments. These procedures still do not guarantee that cross-reactivity to other proteins will not occur, but they constitute important quality controls. Together with a thorough manual evaluation of the expected staining pattern at the tissue, cellular, and subcellular levels, strategies for enhanced validation, and the assignment of reliability scores, the HPA database is divided into comprehensive sets of information, highlighting which data have been most confidently validated. It should also be noted that multitargeting antibodies with known cross-reactivity toward other protein family members of highly homologous sequences are not considered for enhanced validation.
In the present study, we propose a comprehensive approach for the enhanced validation of antibodies by IHC, where 5981 antibodies covering 3775 human proteins were validated based on the orthogonal strategy or independent antibodies. The orthogonal validation was performed by the manual evaluation of IHC staining intensity across a large set of different tissue samples with quantitative mRNA expression levels in corresponding tissues. Correlation between mRNA and protein levels has been previously debated. Some studies comparing mRNA levels with mass spectrometry suggest a relatively low correlation,32−34 for example, r < 0.5, whereas other studies have shown that at the steady state, the levels of a specific transcript and the corresponding protein tend to be high across different tissues if a gene-specific RNA-to-protein conversion factor is introduced.11 It still remains to be confirmed which of the genes suggested to have a poor correlation between mRNA and protein levels depend on biology and which results can be explained by technological limitations. Furthermore, no large-scale studies have been performed comparing protein expression levels using both mass spectrometry and IHC, and it is thus not known which proteins may show a higher correlation with mRNA using IHC instead of mass spectrometry. Here 3378 proteins showed a similar pattern of expression when comparing mRNA levels with IHC, suggesting that for these proteins, the tissue specificity using IHC can be orthogonally validated by mRNA levels. For the remaining proteins where we do not see a similar pattern of expression, further experiments or methods are needed to determine if some of these antibodies should qualify for orthogonal validation. In this study, the manually graded consistency between RNA and protein expression patterns in many cases were in line with the Kendall rank statistical correlation, but it was evident that the Kendall rank correlation failed to identify many proteins manually evaluated as having similar patterns of protein and mRNA expression. This is expected because IHC levels are only semiquantitative based on staining intensity, and the tissue samples consist of a complex mixture of different cell types, of which only some have been annotated. Furthermore, minor differences between the data sets that are less important for the overall interpretation lead to a poor correlation despite a high consistency between mRNA and protein levels. A common example is the very weak unspecific staining in a few organs in addition to the staining that is considered true protein expression. Such background staining, which in some cases has the wrong subcellular localization, for example, the faint cytoplasmic staining observed for nuclear proteins, would be neglected by the human observer. Another example is the low levels of RNA in other organs for proteins where a certain tissue shows a very high expression in just one tissue, which is consistent with the IHC staining. For such cases where the IHC method is not sensitive enough to pick up very low levels or it is not clear if the low mRNA levels are translated into detectable protein levels, the analysis would lead to a poor Kendall rank correlation despite a high consistency between the data sets. Because of these difficulties, a manual correlation analysis taking into consideration all of the above factors leads to the fairest interpretation of consistency between mRNA levels and protein levels based on IHC. It should, however, also be noted that the manual interpretation of IHC staining patterns is highly subjective and requires long-term training and strict guidelines to ensure the evaluation of the correct cell types and the identification of artifacts. The rapidly evolving field of digital pathology with image analysis based on machine-learning algorithms will likely lead to higher fidelity spatial data and more quantitative measurements of protein signals.
The orthogonal strategy based on comparison with mRNA levels is an attractive approach for the validation of proteins with differential expression. The method may, however, be inconclusive for mRNAs or proteins present at low levels or in smaller subsets of cells, especially when exploring proteins for which the exact spatial localization is not known. In these situations, alternative methods, for example, RNAscope,35 to study the in situ mRNA expression could aid to validate the results on the protein level.
Orthogonal strategies are less suitable for ubiquitously expressed proteins, and such proteins may instead be validated using independent antibodies. Two independent antibodies of high quality may, however, still generate slightly different staining patterns in some analyzed samples due to inadequate protocol optimization. In addition to the 871 antibody pairs that qualified for independent antibody validation here, the HPA has published antibodies targeting nonoverlapping regions for almost 3200 additional proteins that currently do not qualify for independent antibody validation. It is an ongoing effort to continue to optimize these antibody pairs to identify more targets for which independent antibody validation can be used. Another technology related to independent antibodies that complements IHC for the in situ detection and cell-type-specific localization of proteins is the use of the proximity ligation assay (PLA), which requires the binding of two antibodies to generate a signal. This thus serves as an interesting approach for improving the specificity of detection.36
Over the past decade, the number of proteins that can be experimentally validated by mass spectrometry based on stringent criteria has increased significantly, and almost 85% of all human proteins have now been confidently identified by mass spectrometry. Mass spectrometry provides the standard for detecting and quantifying a targeted set of proteins in a sample but has a bias toward highly expressed proteins. It is evident from the present investigation that the success rate of confident protein detection is also lower based on IHC for proteins referred to as “missing proteins”. Still, IHC has the advantage of the sensitive detection of proteins present in smaller subsets of cells. In the analysis of such proteins, mRNA constitutes an important starting point in the identification of suitable samples.23 In the present investigation, we found that a high proportion of “missing proteins” were elevated in certain tissue types that are relatively easy to access, such as the testis, which has been previously described.37−41 Here as many as 395 “missing proteins” were elevated in the testis and not yet mapped by antibodies analyzed with enhanced validation, constituting important targets for further characterization. It should, however, be noted that the mRNA expression levels of testis elevated genes corresponding to “missing proteins” are significantly lower than those for testis elevated genes with protein evidence, and because several of these proteins belong to, for example, the olfactory receptor family, some of these proteins might actually be enriched in another not yet analyzed tissue. At the same time, almost all of the 56 “missing proteins” that were identified by antibodies with enhanced validation in the present investigation were expressed in smaller subsets of cells or localized to specific subcellular structures, which could explain the low mRNA abundance. Nevertheless, tissue elevated genes constitute important lists for the further analysis of “missing proteins”. It should also be pointed out that relatively many “missing proteins” were elevated in the blood according to mRNA levels or are expected to be secreted. Such proteins are less suitable for detection by IHC but may be traceable using other approaches. One option is to combine the sensitivity of antibody-based detection with the specificity of mass spectrometry read-out.42 Such analyses using immunoprecipitation-based approaches have been useful in antibody studies focusing on particular groups of proteins found in tissues43 as well as the plasma.44 The feasibility of the integration of mass spectrometry with IHC for “missing proteins” is indicated by 53 of the 56 “missing proteins” validated by antibodies in this study having at least one theoretical proteotypic peptide, and for five of the genes with elevated expression in male tissues, there has been at least one observation of a peptide with a single genomic location in the PeptideAtlas Testis build.
The main strategy for determining protein evidence is mass spectrometry, and currently, only a few of the 17 874 proteins that in neXtProt have experimental evidence at the protein level have received their PE1 status based on antibody data. The HPA is an integral part of the Antibody Resource Pillar and an HPP partner initiative,12,13 and further efforts aiming at integrating antibody data with results based on mass spectrometry are clearly warranted. One example of a study that could further elucidate the correlation between protein levels based on mass spectrometry and IHC would be to analyze the same tissue samples using both methods. This would ultimately lead to an increased understanding of which types of proteins are confidently detected by one method and not the other and also show how protein epitopes may be affected by formalin fixation as part of the IHC sample preparation.
Another emerging method that will likely lead to important implications for proteomics is single-cell RNA-seq (scRNA-seq).45 This constitutes an excellent approach for studying mRNAs that are expected to be expressed in smaller subsets of cells that may fall below the detection limit when mixed with other cell types in complex tissue samples. scRNA-seq is especially attractive for further exploration as an orthogonal approach for the validation of antibodies by IHC because the method allows for direct comparisons of cell-type-specific expression patterns, which is not possible when comparing with data obtained from a mixture of different cell types. In addition to the identification of “missing proteins”, single-cell methods or the spatial localization of proteins within a tissue also have the advantage of providing a functional context, as a protein’s function is tightly linked to its location. This is especially interesting for proteins that have an unknown function, for example, proteins that are referred to as uPE1.15,16 As an example, proteins shown to be expressed in sperm flagella constitute important targets for functional studies analyzing motility. Single-cell proteomic technologies are also being developed for mass spectrometry, and further advances in this field will likely lead to increased integrations between various data sets on both the mRNA and the protein level.
Here we present a comprehensive strategy for the enhanced validation of antibodies for IHC, which has important implications for large-scale efforts to map the human proteome. The streamlined workflow holds promise for integration with mass spectrometry and transcriptomics data sets for the spatiotemporal expression of human proteins in health and disease, and further discussions on the criteria for how antibody-based protein data can be used to determine evidence of protein existence are clearly warranted.
Acknowledgments
The project was funded by the Knut and Alice Wallenberg Foundation. Pathologists and staff at the Department of Clinical Pathology, Uppsala University Hospital are acknowledged for providing the tissues used for RNA-seq and immunohistochemistry. We also thank all staff of the Human Protein Atlas for their work.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jproteome.0c00486.
Supplementary Table S1. List of all 19 670 genes mapped to Ensembl in the HPA, with information on gene name, UniProt ID, protein evidence level, functional annotation status (uPE1), IHC reliability score, RNA specificity category, RNA distribution category, Kendall tau, and adjusted p value. Supplementary Table S2. List of the 56 missing proteins with enhanced validation with information on gene name, UniProt ID, protein evidence level, IHC reliability score, RNA specificity category, and elevated tissues (XLSX)
The authors declare no competing financial interest.
Supplementary Material
References
- Arthur G. Albert Coons: harnessing the power of the antibody. Lancet Respir. Med. 2016, 4 (3), 181–2. 10.1016/S2213-2600(16)00020-5. [DOI] [PubMed] [Google Scholar]
- Bradbury A.; Pluckthun A. Reproducibility: Standardize antibodies used in research. Nature 2015, 518 (7537), 27–9. 10.1038/518027a. [DOI] [PubMed] [Google Scholar]
- Bordeaux J.; Welsh A.; Agarwal S.; Killiam E.; Baquero M.; Hanna J.; Anagnostou V.; Rimm D. Antibody validation. BioTechniques 2010, 48 (3), 197–209. 10.2144/000113382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker M. Reproducibility crisis: Blame it on the antibodies. Nature 2015, 521 (7552), 274–6. 10.1038/521274a. [DOI] [PubMed] [Google Scholar]
- O’Hurley G.; Sjostedt E.; Rahman A.; Li B.; Kampf C.; Ponten F.; Gallagher W. M.; Lindskog C. Garbage in, garbage out: a critical evaluation of strategies used for validation of immunohistochemical biomarkers. Mol. Oncol. 2014, 8 (4), 783–98. 10.1016/j.molonc.2014.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sfanos K. S.; Yegnasubramanian S.; Nelson W. G.; Lotan T. L.; Kulac I.; Hicks J. L.; Zheng Q.; Bieberich C. J.; Haffner M. C.; De Marzo A. M. If this is true, what does it imply? How end-user antibody validation facilitates insights into biology and disease. Asian J. Urol 2019, 6 (1), 10–25. 10.1016/j.ajur.2018.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khoury J. D.; Wang W. L.; Prieto V. G.; Medeiros L. J.; Kalhor N.; Hameed M.; Broaddus R.; Hamilton S. R. Validation of Immunohistochemical Assays for Integral Biomarkers in the NCI-MATCH EAY131 Clinical Trial. Clin. Cancer Res. 2018, 24 (3), 521–531. 10.1158/1078-0432.CCR-17-1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhlen M.; Bandrowski A.; Carr S.; Edwards A.; Ellenberg J.; Lundberg E.; Rimm D. L.; Rodriguez H.; Hiltke T.; Snyder M.; Yamamoto T. A proposal for validation of antibodies. Nat. Methods 2016, 13 (10), 823–7. 10.1038/nmeth.3995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoek J. M.; Hepkema W. M.; Halffman W. The effect of journal guidelines on the reporting of antibody validation. PeerJ 2020, 8, e9300. 10.7717/peerj.9300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhlen M. Response to: Should we ignore western blots when selecting antibodies for other applications?. Nat. Methods 2017, 14 (3), 215–216. 10.1038/nmeth.4194. [DOI] [PubMed] [Google Scholar]
- Edfors F.; Hober A.; Linderback K.; Maddalo G.; Azimi A.; Sivertsson A.; Tegel H.; Hober S.; Szigyarto C. A.; Fagerberg L.; von Feilitzen K.; Oksvold P.; Lindskog C.; Forsstrom B.; Uhlen M. Enhanced validation of antibodies for research applications. Nat. Commun. 2018, 9 (1), 4130. 10.1038/s41467-018-06642-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omenn G. S.; Lane L.; Overall C. M.; Corrales F. J.; Schwenk J. M.; Paik Y. K.; Van Eyk J. E.; Liu S.; Pennington S.; Snyder M. P.; Baker M. S.; Deutsch E. W. Progress on Identifying and Characterizing the Human Proteome: 2019 Metrics from the HUPO Human Proteome Project. J. Proteome Res. 2019, 18 (12), 4098–4107. 10.1021/acs.jproteome.9b00434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legrain P.; Aebersold R.; Archakov A.; Bairoch A.; Bala K.; Beretta L.; Bergeron J.; Borchers C. H.; Corthals G. L.; Costello C. E.; Deutsch E. W.; Domon B.; Hancock W.; He F.; Hochstrasser D.; Marko-Varga G.; Salekdeh G. H.; Sechi S.; Snyder M.; Srivastava S.; Uhlen M.; Wu C. H.; Yamamoto T.; Paik Y. K.; Omenn G. S. The human proteome project: current state and future direction. Mol. Cell. Proteomics 2011, 10 (7), M111.009993. 10.1074/mcp.M111.009993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zahn-Zabal M.; Michel P.-A.; Gateau A.; Nikitin F.; Schaeffer M.; Audot E.; Gaudet P.; Duek P. D.; Teixeira D.; Rech de Laval V.; et al. The neXtProt knowledgebase in 2020: data, tools and usability improvements. Nucleic Acids Res. 2019, 48 (D1), D328–D334. 10.1093/nar/gkz995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paik Y. K.; Omenn G. S.; Hancock W. S.; Lane L.; Overall C. M. Advances in the Chromosome-Centric Human Proteome Project: looking to the future. Expert Rev. Proteomics 2017, 14 (12), 1059–1071. 10.1080/14789450.2017.1394189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paik Y. K.; Overall C. M.; Corrales F.; Deutsch E. W.; Lane L.; Omenn G. S. Toward Completion of the Human Proteome Parts List: Progress Uncovering Proteins That Are Missing or Have Unknown Function and Developing Analytical Methods. J. Proteome Res. 2018, 17 (12), 4023–4030. 10.1021/acs.jproteome.8b00885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fagerberg L.; Hallstrom B. M.; Oksvold P.; Kampf C.; Djureinovic D.; Odeberg J.; Habuka M.; Tahmasebpoor S.; Danielsson A.; Edlund K.; Asplund A.; Sjostedt E.; Lundberg E.; Szigyarto C. A.; Skogs M.; Takanen J. O.; Berling H.; Tegel H.; Mulder J.; Nilsson P.; Schwenk J. M.; Lindskog C.; Danielsson F.; Mardinoglu A.; Sivertsson A.; von Feilitzen K.; Forsberg M.; Zwahlen M.; Olsson I.; Navani S.; Huss M.; Nielsen J.; Ponten F.; Uhlen M. Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics. Mol. Cell. Proteomics 2014, 13 (2), 397–406. 10.1074/mcp.M113.035600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhlen M.; Fagerberg L.; Hallstrom B. M.; Lindskog C.; Oksvold P.; Mardinoglu A.; Sivertsson A.; Kampf C.; Sjostedt E.; Asplund A.; Olsson I.; Edlund K.; Lundberg E.; Navani S.; Szigyarto C. A.; Odeberg J.; Djureinovic D.; Takanen J. O.; Hober S.; Alm T.; Edqvist P. H.; Berling H.; Tegel H.; Mulder J.; Rockberg J.; Nilsson P.; Schwenk J. M.; Hamsten M.; von Feilitzen K.; Forsberg M.; Persson L.; Johansson F.; Zwahlen M.; von Heijne G.; Nielsen J.; Ponten F. Proteomics. Tissue-based map of the human proteome. Science 2015, 347 (6220), 1260419. 10.1126/science.1260419. [DOI] [PubMed] [Google Scholar]
- Uhlen M.; Karlsson M. J.; Zhong W.; Tebani A.; Pou C.; Mikes J.; Lakshmikanth T.; Forsström B.; Edfors F.; Odeberg J.; et al. A genome-wide transcriptomic analysis of protein-coding genes in human blood cells. Science 2019, 366 (6472), eaax9198. 10.1126/science.aax9198. [DOI] [PubMed] [Google Scholar]
- Kampf C.; Olsson I.; Ryberg U.; Sjöstedt E.; Pontén F. Production of tissue microarrays, immunohistochemistry staining and digitalization within the human protein atlas. J. Visualized Exp. 2012, (63), e3620. 10.3791/3620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team. R: A Language and Environment for Statistical Computing; Foundation for Statistical Computing: Vienna, Austria, 2017. [Google Scholar]
- Lane L.; Bairoch A.; Beavis R. C.; Deutsch E. W.; Gaudet P.; Lundberg E.; Omenn G. S. Metrics for the Human Proteome Project 2013–2014 and strategies for finding missing proteins. J. Proteome Res. 2014, 13 (1), 15–20. 10.1021/pr401144x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sjostedt E.; Sivertsson A.; Hikmet Noraddin F.; Katona B.; Nasstrom A.; Vuu J.; Kesti D.; Oksvold P.; Edqvist P. H.; Olsson I.; Uhlen M.; Lindskog C. Integration of Transcriptomics and Antibody-Based Proteomics for Exploration of Proteins Expressed in Specialized Tissues. J. Proteome Res. 2018, 17 (12), 4127–4137. 10.1021/acs.jproteome.8b00406. [DOI] [PubMed] [Google Scholar]
- Baker M. S.; Ahn S. B.; Mohamedali A.; Islam M. T.; Cantor D.; Verhaert P. D.; Fanayan S.; Sharma S.; Nice E. C.; Connor M.; Ranganathan S. Accelerating the search for the missing proteins in the human proteome. Nat. Commun. 2017, 8, 14271. 10.1038/ncomms14271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coons A. H.; Creech H. J.; Jones R. N. Immunological properties of an antibody containing a fluorescent group. Exp. Biol. Med. 1941, 47, 200–202. 10.3181/00379727-47-13084P. [DOI] [Google Scholar]
- Leong A. S. Diagnostic immunohistochemistry--problems and solutions. Pathology 1992, 24 (1), 1–4. 10.3109/00313029209063611. [DOI] [PubMed] [Google Scholar]
- Goodman S. L. The antibody horror show: an introductory guide for the perplexed. New Biotechnol. 2018, 45, 9–13. 10.1016/j.nbt.2018.01.006. [DOI] [PubMed] [Google Scholar]
- Bjorling E.; Uhlen M. Antibodypedia, a portal for sharing antibody and antigen validation data. Mol. Cell. Proteomics 2008, 7 (10), 2028–37. 10.1074/mcp.M800264-MCP200. [DOI] [PubMed] [Google Scholar]
- Hamming I.; Timens W.; Bulthuis M. L.; Lely A. T.; Navis G.; van Goor H. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J. Pathol. 2004, 203 (2), 631–7. 10.1002/path.1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hikmet F.; Méar L.; Edvinsson Å.; Micke P.; Uhlén M.; Lindskog C. The protein expression profile of ACE2 in human tissues. Mol. Syst. Biol. 2020, 16 (7), e9610. 10.15252/msb.20209610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sjoberg R.; Mattsson C.; Andersson E.; Hellstrom C.; Uhlen M.; Schwenk J. M.; Ayoglu B.; Nilsson P. Exploration of high-density protein microarrays for antibody validation and autoimmunity profiling. New Biotechnol. 2016, 33 (5), 582–92. 10.1016/j.nbt.2015.09.002. [DOI] [PubMed] [Google Scholar]
- Jiang L.; Wang M.; Lin S.; Jian R.; Li X.; Chan J.; Dong G.; Fang H.; Robinson A. E.; Aguet F.; et al. A quantitative proteome map of the human body. Cell 2020, 183 (1), 269–283.e19. 10.1016/j.cell.2020.08.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payne S. H. The utility of protein and mRNA correlation. Trends Biochem. Sci. 2015, 40 (1), 1–3. 10.1016/j.tibs.2014.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel C.; Marcotte E. M. Insights into the regulation of protein abundance from proteomic and transcriptomic analyses. Nat. Rev. Genet. 2012, 13 (4), 227–232. 10.1038/nrg3185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F.; Flanagan J.; Su N.; Wang L. C.; Bui S.; Nielson A.; Wu X.; Vo H. T.; Ma X. J.; Luo Y. RNAscope: a novel in situ RNA analysis platform for formalin-fixed, paraffin-embedded tissues. J. Mol. Diagn. 2012, 14 (1), 22–9. 10.1016/j.jmoldx.2011.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindskog C.; Backman M.; Zieba A.; Asplund A.; Uhlen M.; Landegren U.; Ponten F. Proximity Ligation Assay as a Tool for Antibody Validation in Human Tissues. J. Histochem. Cytochem. 2020, 68 (7), 515–529. 10.1369/0022155420936384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carapito C.; Duek P.; Macron C.; Seffals M.; Rondel K.; Delalande F.; Lindskog C.; Freour T.; Vandenbrouck Y.; Lane L.; Pineau C. Validating Missing Proteins in Human Sperm Cells by Targeted Mass-Spectrometry- and Antibody-based Methods. J. Proteome Res. 2017, 16 (12), 4340–4351. 10.1021/acs.jproteome.7b00374. [DOI] [PubMed] [Google Scholar]
- Jumeau F.; Com E.; Lane L.; Duek P.; Lagarrigue M.; Lavigne R.; Guillot L.; Rondel K.; Gateau A.; Melaine N.; Guevel B.; Sergeant N.; Mitchell V.; Pineau C. Human Spermatozoa as a Model for Detecting Missing Proteins in the Context of the Chromosome-Centric Human Proteome Project. J. Proteome Res. 2015, 14 (9), 3606–20. 10.1021/acs.jproteome.5b00170. [DOI] [PubMed] [Google Scholar]
- Pineau C.; Hikmet F.; Zhang C.; Oksvold P.; Chen S.; Fagerberg L.; Uhlen M.; Lindskog C. Cell Type-Specific Expression of Testis Elevated Genes Based on Transcriptomics and Antibody-Based Proteomics. J. Proteome Res. 2019, 18 (12), 4215–4230. 10.1021/acs.jproteome.9b00351. [DOI] [PubMed] [Google Scholar]
- Vandenbrouck Y.; Lane L.; Carapito C.; Duek P.; Rondel K.; Bruley C.; Macron C.; Gonzalez de Peredo A.; Coute Y.; Chaoui K.; Com E.; Gateau A.; Hesse A. M.; Marcellin M.; Mear L.; Mouton-Barbosa E.; Robin T.; Burlet-Schiltz O.; Cianferani S.; Ferro M.; Freour T.; Lindskog C.; Garin J.; Pineau C. Looking for Missing Proteins in the Proteome of Human Spermatozoa: An Update. J. Proteome Res. 2016, 15 (11), 3998–4019. 10.1021/acs.jproteome.6b00400. [DOI] [PubMed] [Google Scholar]
- Zhang Y.; Li Q.; Wu F.; Zhou R.; Qi Y.; Su N.; Chen L.; Xu S.; Jiang T.; Zhang C.; Cheng G.; Chen X.; Kong D.; Wang Y.; Zhang T.; Zi J.; Wei W.; Gao Y.; Zhen B.; Xiong Z.; Wu S.; Yang P.; Wang Q.; Wen B.; He F.; Xu P.; Liu S. Tissue-Based Proteogenomics Reveals that Human Testis Endows Plentiful Missing Proteins. J. Proteome Res. 2015, 14 (9), 3583–94. 10.1021/acs.jproteome.5b00435. [DOI] [PubMed] [Google Scholar]
- Marcon E.; Jain H.; Bhattacharya A.; Guo H.; Phanse S.; Pu S.; Byram G.; Collins B. C.; Dowdell E.; Fenner M.; Guo X.; Hutchinson A.; Kennedy J. J.; Krastins B.; Larsen B.; Lin Z. Y.; Lopez M. F.; Loppnau P.; Miersch S.; Nguyen T.; Olsen J. B.; Paduch M.; Ravichandran M.; Seitova A.; Vadali G.; Vogelsang M. S.; Whiteaker J. R.; Zhong G.; Zhong N.; Zhao L.; Aebersold R.; Arrowsmith C. H.; Emili A.; Frappier L.; Gingras A. C.; Gstaiger M.; Paulovich A. G.; Koide S.; Kossiakoff A. A.; Sidhu S. S.; Wodak S. J.; Graslund S.; Greenblatt J. F.; Edwards A. M. Assessment of a method to characterize antibody selectivity and specificity for use in immunoprecipitation. Nat. Methods 2015, 12 (8), 725–31. 10.1038/nmeth.3472. [DOI] [PubMed] [Google Scholar]
- Venkataraman A.; Yang K.; Irizarry J.; Mackiewicz M.; Mita P.; Kuang Z.; Xue L.; Ghosh D.; Liu S.; Ramos P.; Hu S.; Bayron Kain D.; Keegan S.; Saul R.; Colantonio S.; Zhang H.; Behn F. P.; Song G.; Albino E.; Asencio L.; Ramos L.; Lugo L.; Morell G.; Rivera J.; Ruiz K.; Almodovar R.; Nazario L.; Murphy K.; Vargas I.; Rivera-Pacheco Z. A.; Rosa C.; Vargas M.; McDade J.; Clark B. S.; Yoo S.; Khambadkone S. G.; de Melo J.; Stevanovic M.; Jiang L.; Li Y.; Yap W. Y.; Jones B.; Tandon A.; Campbell E.; Montelione G. T.; Anderson S.; Myers R. M.; Boeke J. D.; Fenyo D.; Whiteley G.; Bader J. S.; Pino I.; Eichinger D. J.; Zhu H.; Blackshaw S. A toolbox of immunoprecipitation-grade monoclonal antibodies to human transcription factors. Nat. Methods 2018, 15 (5), 330–338. 10.1038/nmeth.4632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fredolini C.; Bystrom S.; Sanchez-Rivera L.; Ioannou M.; Tamburro D.; Ponten F.; Branca R. M.; Nilsson P.; Lehtio J.; Schwenk J. M. Systematic assessment of antibody selectivity in plasma based on a resource of enrichment profiles. Sci. Rep. 2019, 9 (1), 8324. 10.1038/s41598-019-43552-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regev A.; Teichmann S. A.; Lander E. S.; Amit I.; Benoist C.; Birney E.; Bodenmiller B.; Campbell P.; Carninci P.; Clatworthy M.; et al. The Human Cell Atlas. eLife 2017, 6, e27041. 10.7554/eLife.27041. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
High-resolution images corresponding to immunohistochemically stained TMA cores of 44 different tissue types corresponding to all antibodies analyzed in the present investigation are available in the latest version 19.3 of the HPA (https://v19.proteinatlas.org). The normalized consensus transcript expression levels based on transcriptomics data from the HPA, GTEx, and FANTOM5 as well as the annotated protein expression levels based on IHC can be accessed under the download page (https://v19.proteinatlas.org/about/download).