

Abstract
Proteogenomic identification of translated small open reading frames in human has revealed thousands of microproteins, or polypeptides of fewer than 100 amino acids, that were previously invisible to geneticists. Hundreds of microproteins have been shown to be essential for cell growth and proliferation, and many regulate macromolecular complexes. One such regulatory microprotein is NBDY, a 68-amino acid component of the human cytoplasmic RNA decapping complex. Heterologously expressed NBDY was previously reported to regulate cytoplasmic ribonucleoprotein (RNP) granules known as P-bodies and reporter gene stability, but the global effect of endogenous NBDY on the cellular transcriptome remained undefined. In this work, we demonstrate that endogenous NBDY directly interacts with the human RNA decapping complex through EDC4 and DCP1A and localizes to P-bodies. Global profiling of RNA stability changes in NBDY knock out (KO) cells reveals dysregulated stability of over 1400 transcripts. DCP2 substrate transcript half-lives are both increased and decreased in NBDY KO cells, which correlates with 5′ UTR length. NBDY deletion additionally alters the stability of non-DCP2 target transcripts, possibly as a result of downregulated expression of nonsense-mediated decay (NMD) factors in NBDY KO cells. We present a comprehensive model of the regulation of RNA stability by NBDY.
Keywords: NBDY, microprotein, mRNA decapping complex, mRNA decay, processing body, protein-protein interaction, TimeLapse-seq
Graphical Abstract
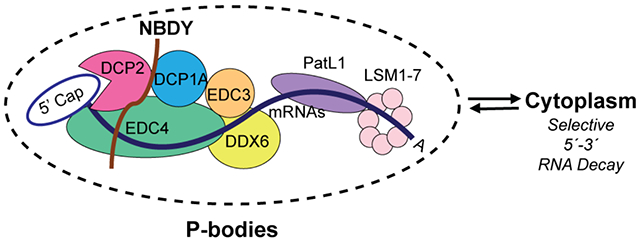
INTRODUCTION
The discovery of thousands of small open reading frames (smORFs, <100 codons) that previously escaped automated genome annotation has revolutionized our understanding of the molecular information encoded in the human genome1. The products of smORF translation have been referred to as small proteins, microproteins, micropeptides, and alt-proteins, and a recent review2 recommended standardized use of the term small proteins; herein we utilize “microprotein” for consistency with a prior report3. Hundreds of smORFs have been shown to be essential for cell proliferation4, and the functions of several dozen smORF-encoded microproteins that bind to and regulate proteins or macromolecular complexes have been elucidated across different species, including Toddler in zebrafish5, sarcolambin in Drosophila6, and BRAWNIN in human7. However, the vast majority of microproteins remain, in most cases, poorly characterized at the molecular and cellular level8. For example, we previously showed that ectopically expressed NBDY microprotein can bind to the human cytoplasmic RNA decapping complex to regulate decay of a target gene as well as the formation of phase-separated RNA-protein granules known as P-bodies3, but the effect of the endogenous protein on the human transcriptome has not yet been defined, the molecular basis of its regulation of P-bodies is unclear, and its direct interaction with decapping complex proteins has not been biochemically validated.
The 5′-to-3′ RNA decay pathway controls the stability of regulatory transcripts and plays a critical role in RNA quality control9. In metazoans, this process starts with removal of the 5′ 7-methylguanosine (m7G) cap of deadenylated transcripts by the decapping enzyme DCP2 in complex with its coactivators DCP1A and EDC4, followed by final exonucleolytic degradation by XRN110. Decapping proteins, in complex with deadenylated transcripts, can reversibly enter P-bodies, which, like other RNP granules, have properties of phase-separated liquid droplets11, 12. Live-cell single-molecule imaging demonstrated that 5′-to-3′ RNA decay occurs on decapping complexes freely diffusing in the cytosol, and not in macroscopically observable P-bodies13, 14, suggesting that P-bodies function in RNA storage rather than decay.
In this work, we show that NBDY is a regulator of the specificity of the human cytoplasmic RNA decapping complex via its interaction with the WD40 domain of the decapping accessory protein EDC4. Genetic ablation of NBDY globally dysregulates RNA stability and decapping rate of DCP2 substrates, as well as non-DCP2 substrates as a secondary result of gene expression changes. This dysregulation leads to increased P-body numbers and inhibited cell proliferation in NBDY knock out (KO) cells. The interaction of NBDY with EDC4 is required for correct half-life specification for DCP2 substrates, and NBDY ablation alters DCP2 substrate stability at the decapping step. Interestingly, DCP2 substrates can be either stabilized or destabilized by NBDY KO, and this effect correlates with 5′ UTR length. Loss of the NBDY-EDC4 interaction leads to increases in P-bodies and fails to rescue wild type RNA decay rates for DCP2 substrate transcripts. However, stabilization of non-DCP2 target transcripts in NBDY KO cells does not require the NBDY-EDC4 interaction, and likely results from decreased synthesis of transcripts encoding components of the nonsense-mediated decay (NMD) pathway. These results together demonstrate that NBDY is a regulator of global RNA stability that is required for correct specification of DCP2 target decay rates depending on properties of their 5′ UTRs, and furthermore that NBDY deletion leads to compensatory gene expression changes that stabilize non-DCP2 targets.
MATERIALS AND METHODS
Data analysis
Two-tailed and equal-variance Student’s t-test, Mann-Whitney U test, Kruskal-Wallis test and analysis of variance (ANOVA) with Dunnett’s test were performed using Excel, GraphPad Prism 7 or R for statistical significance with significance defined as a p-value<0.05. Equal variance between samples was established using an F-test. All data represent at least three biological replicates unless otherwise stated. Data are presented as the mean ± standard deviation. P-body counting was performed using ImageJ as previously described15.
Cell Culture
HEK 293T cells were purchased from ATCC, and early-passage stocks were maintained to ensure cell line provenance and sterility. HEK 293T was maintained in Dulbecco’s Modified Eagle Medium (DMEM, Corning) supplemented with 10% (vol/vol) fetal bovine serum (FBS, Sigma Aldrich) and 100 U/mL penicillin-streptomycin (Sigma Aldrich). Cells were verified to be mycoplasma-free using the ATCC Universal mycoplasma detection kit.
Antibodies
Primary antibodies used for Western blotting and/or immunofluorescence were as follows: mouse monoclonal anti-FLAG (Sigma, F3165); rabbit polyclonal anti-EDC4 (Sigma, SAB4200114); rabbit polyclonal anti-DCP2 (Novus Biologicals, NBP1-41070); mouse monoclonal anti-beta actin (Invitrogen, BA3R); rabbit polyclonal anti-DCP1A (Sigma, D5444); rabbit polyclonal anti-XRN1 (Bethyl, A300-443A; Sigma, PLA0105). Secondary antibodies for Western blotting were goat anti-rabbit peroxidase conjugate (Rockland, 611-103-122; Merck, AP132P) and goat anti-mouse peroxidase conjugate (Rockland, 610-1319). The secondary antibody for immunofluorescence was goat anti-rabbit Alexa Fluor 568 (Life Technologies, A-11011).
Specific synthetic peptides used to generate antibodies against NBDY comprised amino acids 8-28 of NBDY. The rabbit polyclonal antibody was generated by GenScript (Piscataway, NJ).
Generation of CRISPR knock out (KO) cells
NBDY KO HEK 293T cells were generated using guide RNAs (gRNAs) designed with the guide design tool from the Zhang lab (crispr.mit.edu) to target the NBDY genomic region (gRNAs: 5'-GATAATCCCAACGCGGAGCG-3' and 5'-CACAAGGTTGGTCTCCCATA-3'). Double-stranded DNA oligonucleotides corresponding to the gRNAs were inserted into pSpCas9(BB)-2A-GFP vector (Addgene, as a gift from F. Zhang, MIT, Cambridge, MA). In each case, an equal mixture of the two gRNA plasmids were transfected into HEK 293T cells using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s instructions, and GFP-positive cells were sorted with flow cytometry. Loss of NBDY expression was confirmed by Western blot, genomic DNA PCR and sequencing (primer sequences listed in Table S1). In the NBDY KO cell line used in this study, the two alleles were disrupted by a 236-nt homozygous deletion.
XRN1 KO HEK 293T cells were previously reported16 and were generated using previously validated gRNAs17 to target the XRN1 genomic region (gRNAs; 5'-taaaacgcctcccacgctgc-3' and 5'-ttaagagaagaagttcgatt-3'). Loss of XRN1 expression was confirmed by Western blot, genomic DNA PCR and sequencing. In the XRN1 KO cell line, the two alleles were disrupted with a 7-nt homozygous deletion. DCP2 knock out (KO) HEK 293T cells were previously reported16 and were generated using previously described guide RNAs (gRNAs)18. Loss of DCP2 expression was confirmed by Western blot, genomic DNA PCR and sequencing. In the DCP2 KO cell line used in this study, the two alleles were disrupted by a 36-nt homozygous deletion at the catalytic site spanning E147/148. XRN1/NBDY double knock out (DKO) cells were generated using previously reported gRNAs16 to target the XRN1 genomic region in the NBDY KO cells, with confirmation by Western blot, genomic DNA PCR and sequencing.
Lentivirus production and infection
Lentivirus was produced as previously described19. Briefly, HEK 293T cells were co-transfected with expression construct in pLJM1, along with pMD2.G and psPAX2, and growth media were replaced after 5 h. 48 h post-transfection, media containing viruses was harvested, filtered through a 0.45-μm filter, aliquoted and flash frozen.
Stable expression cell lines were generated by transducing wild type HEK 293T or HEK 293T NBDY KO cells with ~20 μl of lentiviral particle suspension at 75% confluency in 6-well plates. 48 h after 2 ng/ml puromycin selection, cells were harvested and stable transgene expression validated by western blotting.
Colony forming assay
HEK 293T, HEK 293T NBDY KO and HEK 293T NBDY rescue cells were plated at 2x104 cells/plate in a 6-well plate (triplicate) with fresh culture medium, which was replaced every 3 days. Surviving colonies were stained with 0.4% crystal violet (Sigma) in 50% methanol, and visible colonies were counted using Image-Pro 3D Suite software version 5.1.1.38 for Windows (Media Cybernetics, Inc., Bethesda, MD, USA).
TimeLapse-seq
Wild type (WT) HEK 293T and NBDY KO cells were treated in duplicate with 500 μM s4U for 2 h, and a single replicate control per cell line was not treated with s4U. Total RNA was isolated from cell pellets using TRIzol reagent, followed by phenol-chloroform extraction and isopropanol precipitation supplemented with 50 μM DTT. After washing twice with 75% ethanol, the RNA pellet was dried and resuspended in nuclease-free water and treated with Turbo DNase to remove genomic DNA. RNA was isolated using RNAclean beads (Beckman), treated with TimeLapse chemistry (2,2,2-trifluoroethylamine and sodium periodate) and purified as previously described20. Purified recoded RNA was then sequenced using the mammalian pico-input SMARTer stranded Total RNA-seq kit v2. Paired-end 100nt or 150nt RNA sequencing was performed using an Illumina Hiseq-4000 or Novaseq.
TimeLapse-seq analysis
TimeLapse-seq read trimming and alignment to mature RNA isoforms were performed as described previously20. Mutation calling was performed as described previously with the following modifications: for bases with overlapping coverage in a read pair, only the mutation with higher base quality is used for analysis. Reads were assigned as new or old based on a per-read 3.6% T-to-C mutation rate. The per-read mutation cutoff was determined through maximization of sensitivity and specificity for varying mutation rates, determined using the following equations:
For this analysis, “new” reads are intronic reads in highly expressed transcripts with s4U and TimeLapse chemistry treatment, and “old” reads are in highly expressed transcripts without s4U treatment.
Old and new read counts per gene were normalized per sample based on total RNA counts using EdgeR. Log2-fold change analysis was performed using DEseq2 on total, new, and old RNA counts, with scale factors from total RNA input into new and old RNA analyses. Change in synthesis and degradation rates were determined using the following equations:
where fnew is the fraction of new RNA and [RNA] is the normalized total RNA read count per gene.
Predominantly synthesis- or decay-driven changes were then determined using an experimentally validated cutoff:
Cell-based RNA decay assay by qRT-PCR
DCP2 KO, NBDY KO, NBDY rescue or WT HEK 293T cells were cultured in 12-well plates to 70% confluency. Cells were harvested on ice at the indicated time points after transcriptional arrest by actinomycin D (5 μg/mL). Total RNA was isolated with TRIzol (Invitrogen) following the manufacturer’s instructions, followed by DNase I (NEB) treatment. RNA levels at indicated time points relative to time = 0 hour were quantified from reverse-transcribed cDNA by real-time PCR using iTaq Universal SYBR Green Supermix (Bio-Rad) based on a comparative Ct method as described previously21. Half-lives were calculated from a linear regression model of ln (relative RNA remaining) against time using the following equation: t1/2 = ln(2)/slope22. Primer sequences are listed in Table S2.
Quantitative splinted ligation RT-PCR (qSL-RT-PCR)
The detection of decapped RNA by qSL-RT-PCR was performed in WT, XRN1 KO, NBDY/XRN1 or DCP2/XRN1 double knock out (DKO) HEK 293T cells as previously described23. In brief, HEK 293T WT, XRN1 KO, and NBDY/XRN1 double knock out (DKO) HEK 293T cells were cultured in 6-well plates to 70% confluency. Actinomycin D (5 μg/mL) was added, and cells were harvested on ice at the indicated time points. Total RNA was isolated with TRIzol, and 10 μg total RNA was mixed with 20 pmol of each splint oligonucleotide and 30 pmol of anchor RNA (5'-GCUGAUGGCGAUGAAUGAACACUGCGUUUGCUGGCUUUGAUG-3'). The oligos were annealed by sequential incubation for 5 min from 70 °C to 60 °C to 42 °C and to 25 °C, followed by the ligation step with 2 μl T4 DNA ligase (NEB) at 16 °C overnight. 20 U RNasin Plus (Promega) was included to prevent RNA degradation. 2 μl DNase I (NEB) was then added to digest genomic DNA for 1 h at 37 °C. The RNA was precipitated with 0.3 M sodium acetate (pH 5.2) and 2.5 volume of ethanol at −20 °C overnight, washed with 70% ethanol and resuspended in 30 μl of DEPC-treated water. cDNA was synthesized from 1 μg of the ligated RNA with GoScript kit (Promega) using gene specific reverse primers (Rev-2, see Table S2). Total RNA and splinted ligation product were then quantified by real-time PCR using gene specific Fwd-2/Rev-2 primers and Anchor Fwd/Rev-2 primers, respectively (Table S2). A ΔΔCt method was used to determine the relative amount of decapped RNA as previously described24, and the amounts of total RNA remaining at each time point relative to time 0 h were calculated from 2−ΔΔCt using ΔCt (t = 0) as the reference value.
In vitro decapping assays
HEK 293T WT and NBDY KO cells stably expressing FLAG-DCP1A25 were cultured in DMEM (Invitrogen) containing 10% fetal bovine serum (Invitrogen) in 150-mm-diameter dishes, and decapping complex was immunopurified by anti-FLAG M2 agarose (Sigma) with 4 h incubation at 4 °C. Beads were washed eight times with 1 ml of washing buffer (50 mM Tris-HCl pH 7.5, 150 mM NaCl, 0.05% Triton X-100), and FLAG-tagged protein was eluted by gently shaking the beads in 50 μl of washing buffer containing 1 mg/ml of FLAG peptide and 0.1 mg/ml of bovine serum albumin at 4 °C for 2 h. Eluates were stored at −80 °C prior to the decapping assay. A 5' m7G cap-labeled RNA substrate was produced by incubating a 24-nucleotide in vitro transcribed RNA (5'-GAAGAAGCGGGCAUGCGGCCAGCCA-3')26 with GTP, S-adenosyl-l-methionine (Sigma), and bacterially expressed vaccinia virus capping enzyme (NEB) as described by Lykke-Andersen et al[25]. Decapping reactions were carried out at 37 °C for 2 h by mixing 10 μl of capped RNA and 0.2 to 10 μl of immunopurified decapping complex in 50 mM Tris-HCl pH 8.0, 30 mM (NH4)2SO4, 1 mM MgCl2. In one experiment, the decapping reaction was subsequently incubated with 1 mM ATP at 30 °C for 30 min. All reactions were terminated by addition of EDTA to 10 mM, and resolved on RNA sequencing gels (20% TBE/urea).
Recombinant expression and purification of human NBDY
His-tagged human NBDY in pET28a was transformed into an E. coli. BL21(DE3) strain. Overnight cultures were diluted 1:100 in Luria-Bertani (LB) media supplemented with 50 μg/mL of kanamycin and grown at 37 °C. At OD600 0.6-0.8, expression was induced by addition of 0.1 mM of isopropyl β-d-1-thiogalactopyranoside (IPTG) followed by shaking at 18 °C overnight. After cell harvest and resuspension in 20 mM Tris-HCl pH 7.4, 150 mM NaCl, 0.2 mM DTT, cells were lysed by sonication (6x pulses of 15 s each at half maximal power on ice). The solution was clarified by centrifugation at 10,000 g for 30 min at 4 °C and loaded onto a column containing 200 μl of His-tagged resin (Amersham Biosciences) pre-equilibrated with 20 mM imidazole, 140 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.8 mM KH2PO4 pH 7.4. Following incubation for 20 min at 4 °C, the resin was washed 5 times with the column buffer, and proteins were eluted with 200 mM imidazole, 140 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.8 mM KH2PO4 pH 7.4.
Recombinant expression and purification of human EDC4 WD40 domain
The WD40 domain of EDC4 (residues 72-528) was cloned into pFastBac (Thermo Fisher) with an N-terminal GST tag (or N-terminal MBP tag) following the manufacturer’s instructions and expressed in Sf9 insect cells (Thermo Fisher) by Bac-to-Bac Baculovirus Expression System (Thermo Fisher). Briefly, pFastBac contained EDC4 WD40 domain was transformed into DH10Bac E.coli (Thermo Fisher) for transposition into the bacmid, then used blue/white agar plates (contained 50 μg/mL kanamycin, 7 μg/mL gentamicin, 10 μg/mL tetracycline, 100 μg/mL Bluo-gal, and 40 μg/mL IPTG) selection to identify colonies containing the recombinant bacmid. Sf9 cells (1.5 × 106 cells per milliliter) were transfected with 1 μg pFastBac EDC4 WD40 Bacmid and 10 μl Cellfectin II reagent (Thermo Fisher) to produce P1 recombinant baculovirus. After 72h transfection, P1 recombinant baculovirus were harvested and infected 10 mL Sf9 cell (1.5 × 106 cells per milliliter) to obtain the following P2 and P3 recombinant baculovirus. Sf9 cells (0.8 L; 4.5 × 106 cells per milliliter) were infected with 8 mL of P3 recombinant baculovirus and cultured in a 2.8-L flask at 27°C with shaking in SF-900™ II SFM insect serum medium (Thermo Fisher) containing 100 U/ml penicillin-streptomycin (Thermo Fisher). Infected cells were harvested after 48 h or when viability dropped to 80%–85%. Harvested cells were washed with ice-cold PBS buffer, flash-frozen in liquid nitrogen, and stored at −80°C prior to purification. The recombinant protein was purified on glutathione resin (GE Healthcare) or amylose resin (NEB) followed by Superdex 200 gel filtration column (GE Healthcare) in 20 mM Tris-HCl (pH 7.5), 150 mM NaCl, and 1 mM DTT (dithiothreitol). Purified EDC4 WD40 domain was concentrated to 15–20 mg/mL and stored at −80°C before use.
DCP1A and EDC4 immunopurification
FLAG-tagged DCP1A or EDC4 in pcDNA3 was transfected into HEK 293T cells in 10cm dishes using Lipofectamine2000 (Thermo Fisher) per the manufacturer’s instructions. 24 h post-transfection, cells were harvested and lysed on ice for 20 min using 400 μl TBS with 1% Triton X-100 (TBS-T) supplemented with Roche Complete protease inhibitor cocktail tablets and PhosSTOP (Sigma). 10 % cell lysate samples were saved for analysis of input. A 50 μl aliquot of anti-FLAG agarose beads (clone M2, Sigma) was pre-equilibrated with 1 ml TBS-T and suspended in cell lysate. Bead suspensions were rotated at 4 °C for 2 h, washed 3 times with TBS-T and 3 times with TBS. Elution was performed with 50 μl of 3× FLAG peptide (Sigma) at a final concentration of 1 mg/ml in TBS at 4 °C for 1 h. Beads were removed by centrifugation, and excess FLAG peptide was removed using a Bio-Spin 6 column (Bio-Rad) previously equilibrated in TBS. Eluates were diluted to the same protein concentration with lysis buffer and analyzed by SDS-PAGE and Western blotting.
P-body imaging using confocal microscopy
HEK 293T cells were grown on fibronectin-coated glass coverslips in a 48-well plate to 70% confluency. Cells were fixed with 10% neutral buffered formalin (Fisher Scientific), permeabilized with methanol at −20 °C, and blocked with blocking buffer (3% BSA in DPBS) for 1 h at room temperature. For examination of NBDY colocalization with P-bodies, cells were stained with rabbit anti-NBDY at a 1:100 dilution in blocking buffer overnight at 4 °C, followed by 3x PBS washes. Goat anti-rabbit Alexa Fluor 568 was applied at a 1:1000 dilution in blocking buffer for 1 to 4 h at room temperature in the dark, with 5 subsequent PBS washes. Anti-DCP1A Alexa Fluor 647 was then added at 1:1000 dilution and incubated for 1 h, followed by 3x PBS washes. Cells were post-fixed with 10% buffered formalin, stained with DAPI, and imaged by laser scanning confocal microscopy (Leica TCS SP8) with PL (field planarity) APO (apochromatic) 63x/1.40 oil, CS2 and PL APO 100x/1.44 oil, CORR (correction collar) CS (confocal scanning).
For quantifying P-bodies in different cell lines, cells were stained with rabbit anti-DCP1A at a 1:1000 dilution in blocking buffer overnight at 4 °C, followed by 3x PBS washes and incubation with goat anti-rabbit Alexa Fluor 568. The cells were post-fixed, stained with DAPI and imaged by laser scanning confocal microscopy.
RESULTS
Endogenous NBDY interacts with the RNA decapping complex via the WD40 domain of EDC4
Evidence to date that NBDY interacts with the RNA decapping complex has relied on over-expression and indirect assays. We therefore examined the concentration, localization and interactions of endogenous NBDY in human cells using a specific antibody, and examined P-body numbers and proliferation in a NBDY knock out (KO) cell line generated via CRISPR/Cas9 editing. Quantitative western blotting with an antibody to endogenous NBDY yielded a cellular concentration of 72000 ± 6000 molecules per cell, or approximately 72 nM (Figure 1a). NBDY is present at approximately 1:3 stoichiometry with its previously reported binding partner EDC4, which is present at 200000 ± 8000 copies per cell (Figure 1a)27, 28. Endogenous NBDY co-purifies with components of the RNA decapping complex (Figure 1b) and colocalizes with P-bodies (Figure 2a and Figure S1a), as previously reported for epitope-tagged NBDY3. Consistent with previously reported data using siRNA3, endogenous P-body numbers increase by 3-fold in NBDY KO cells, which can be rescued by stably reintroducing the NBDY coding sequence on the KO background, confirming that the increase in P-bodies is a result of specific NBDY deletion and not a result of off-target editing (Figure 2a-b and Figure S1b). The NBDY KO cell line exhibits decreased viability relative to wild type (WT) HEK 293T that is restored in the NBDY rescue cells (Figure 2c), suggesting a role for NBDY in cell proliferation. Endogenous NBDY therefore associates with the RNA decapping complex and P-bodies, and is required for maintenance of wild type P-body numbers in cells.
Figure 1∣. Endogenous NBDY quantitation and interaction with the human cytoplasmic RNA decapping complex.
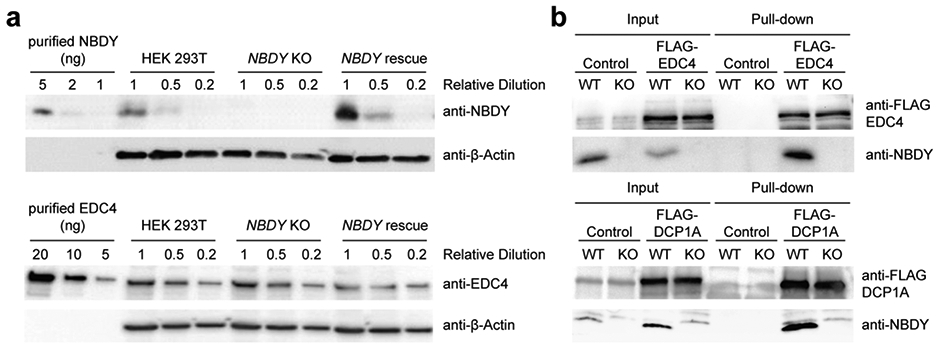
(a) Quantification of endogenous protein levels (NBDY and EDC4) in HEK 293T cells by Western blotting. (b) Endogenous NBDY co-immunopurified with FLAG-EDC4 (top) and FLAG-DCP1A (bottom) stably expressed in HEK 293T cells. WT, wild type; KO, NBDY knock out.
Figure 2∣. Endogenous NBDY localizes to and regulates P-bodies and affects cell proliferation.
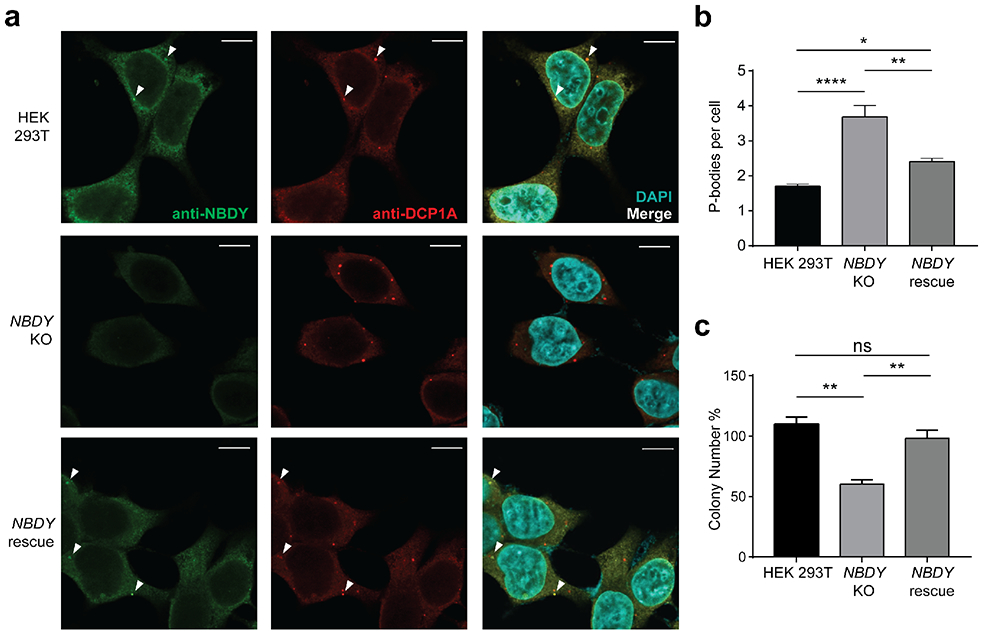
(a) Detection of endogenous NBDY by immunofluorescence. Fixed cells were stained with antibodies detecting NBDY and a P-body marker, DCP1A. (b) P-body numbers in HEK 293T, NBDY KO and NBDY rescue cell lines. Six fields of view for each cell line (>180 cells) were used to quantify average P-bodies (anti-DCP1A, P-body marker) per cell. Data represent mean ± s.e.m, and significance was evaluated with one-way ANOVA. *P < 0.05; **P < 0.01; ****P < 0.0001, Dunnett’s test. Scale bars, 10 μm. For additional field of view, see Figure S1a. (c) Cell proliferation assay for HEK 293T, NBDY KO and NBDY rescue cell lines. Significance was analyzed by one-way ANOVA. **P < 0.01, Dunnett’s test.
EDC4 is essential for assembly of the human RNA decapping complex as well as P-bodies29 and was previously identified as a NBDY binding partner via photo-cross-linking3, but the direct nature, specificity and functional significance of this interaction remained to be established. EDC4 truncation analysis confirms that the N-terminal WD40 domain (1-538) is sufficient for reciprocal co-immunoprecipitation with NBDY when both epitope-tagged proteins are over-expressed in HEK 293T cells (Figure 3a, b). Consistent with previous results from alanine scanning mutagenesis in cells3, interaction of the purified EDC4 WD40 domain with purified NBDY requires NBDY W34 (Figure 3c). Surface plasmon resonance for binding of purified EDC4 to immobilized NBDY (Figure 4a) and isothermal titration calorimetry of EDC4 against NBDY fit to a one-site binding model (Figure 4b) support a direct interaction between purified NBDY and EDC4 WD40 domain with essentially 1:1 stoichiometry and consistently derive a NBDY-EDC4 WD40 dissociation constant of ~1 μM. Furthermore, titration of EDC4 against the non-interacting W34A mutant of NBDY led to undetectable binding (Figure 4b, right), confirming the specificity of the NBDY-EDC4 interaction and the requirement of NBDY W34 for binding. Independently, the DCP1A EVH1 domain interacts with the C-terminal polyproline repeats of NBDY (Figure S2), as expected based on the binding specificity of EVH1 domains30, though deletion of the polyproline motif does not affect RNA stability and therefore may not be necessary for NBDY function (vide infra). These results demonstrate that NBDY interacts with EDC4 and DCP1A via two independent linear motifs.
Figure 3∣. NBDY directly contacts EDC4 via the WD40 domain in cells.
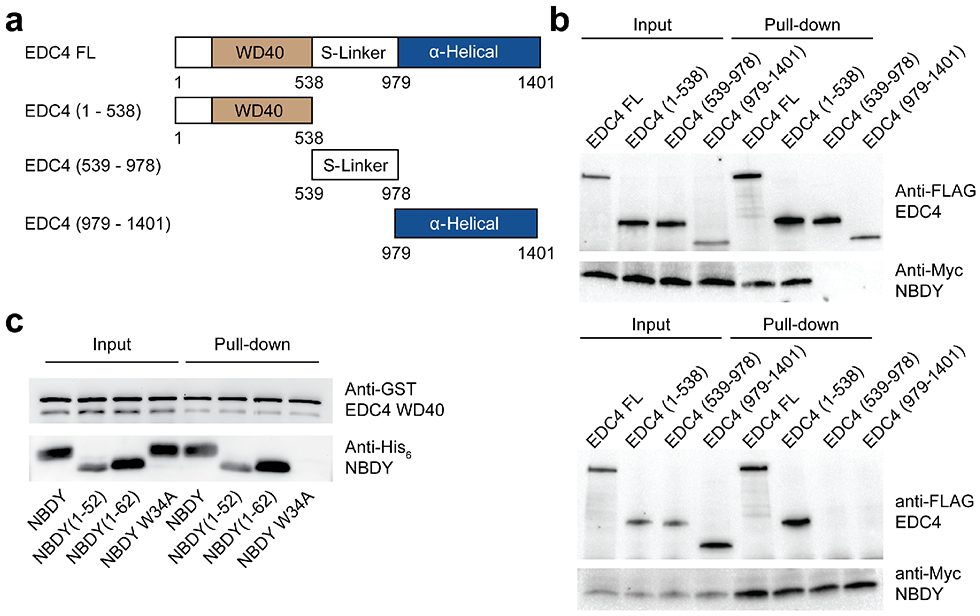
(a,b) Domain mapping of the interaction between NBDY and EDC4 (full length or indicated fragments, a) in HEK 293T cells, via reciprocal co-immunoprecipitation of myc epitope-tagged NBDY vs FLAG epitope-tagged EDC4 constructs from cell lysates and Western blotting. Top: Immunoprecipitation: FLAG-EDC4, Immunoblotting: Myc-NBDY; Bottom: Immunoprecipitation: Myc-NBDY, Immunoblotting: FLAG-EDC4. (c) Pulldown of recombinant GST-EDC4-WD40 (residues 72-538) with purified His6-tagged NBDY constructs.
Figure 4∣. Low-micromolar association of NBDY with EDC4 in vitro requires residue W34 of NBDY.
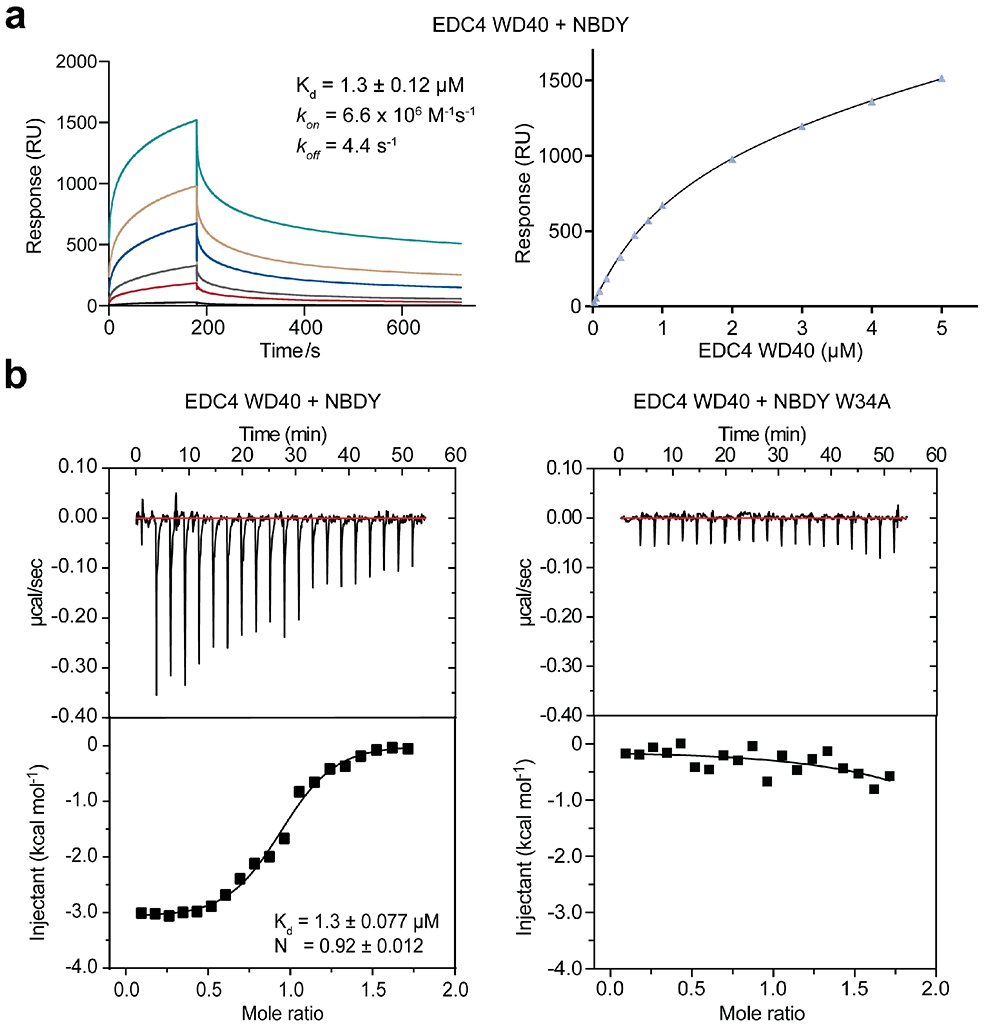
(a) Surface plasmon resonance response for purified NBDY binding to the purified EDC4 WD40 domain. (b) Affinity and specificity measurement of EDC4 WD40 domain binding to NBDY (left) or the non-interacting W34A NBDY mutant (right) by isothermal titration calorimetry. From low to high, curves represent EDC4 concentrations increasing from 0.025 μM to 5 μM.
NBDY regulates decay of DCP2 target and non-target RNAs via different mechanisms
Since changes in global RNA turnover rates affect P-body formation31 and cell growth32, we hypothesized that the observed effects of NBDY levels on P-body numbers and cell proliferation were reporting on NBDY-dependent changes in global RNA dynamics. To characterize the role of NBDY in the regulation of RNA stability transcriptome-wide, we globally profiled changes in RNA stability in the NBDY KO cell line using RNA metabolic labeling with T-to-C chemical recoding (TimeLapse-seq) (Figure 5a, b)20. NBDY KO dysregulates >1400 RNA half-lives, with 1200 cellular RNAs post-transcriptionally stabilized and 226 destabilized relative to WT; in comparison, knock out of the RNA decapping enzyme DCP2 dysregulates the stability of approximately 2000 transcripts16, demonstrating that NBDY is an important regulator of global RNA stability. The observation that 6-fold more transcripts are stabilized than destabilized in the NBDY KO is consistent with an increase in P-body numbers, because P-body formation depends on RNA concentration. RNA synthesis was also perturbed, with 776 and 1912 transcripts transcriptionally up- or down-regulated in NBDY KO (Figure S3a, b). A similar effect was previously observed in cells lacking the RNA decapping enzyme DCP2, which we propose is due to gene expression changes that compensate for loss of RNA decay factor activity16. RNAs related to regulation of cell growth were significantly enriched among transcripts that exhibit decreased synthesis in NBDY KO cells, consistent with the observed growth inhibitory effect of NBDY depletion (Figure 2c and Figure S3).
Figure 5 ∣. NBDY determines the stability of thousands of human transcripts.
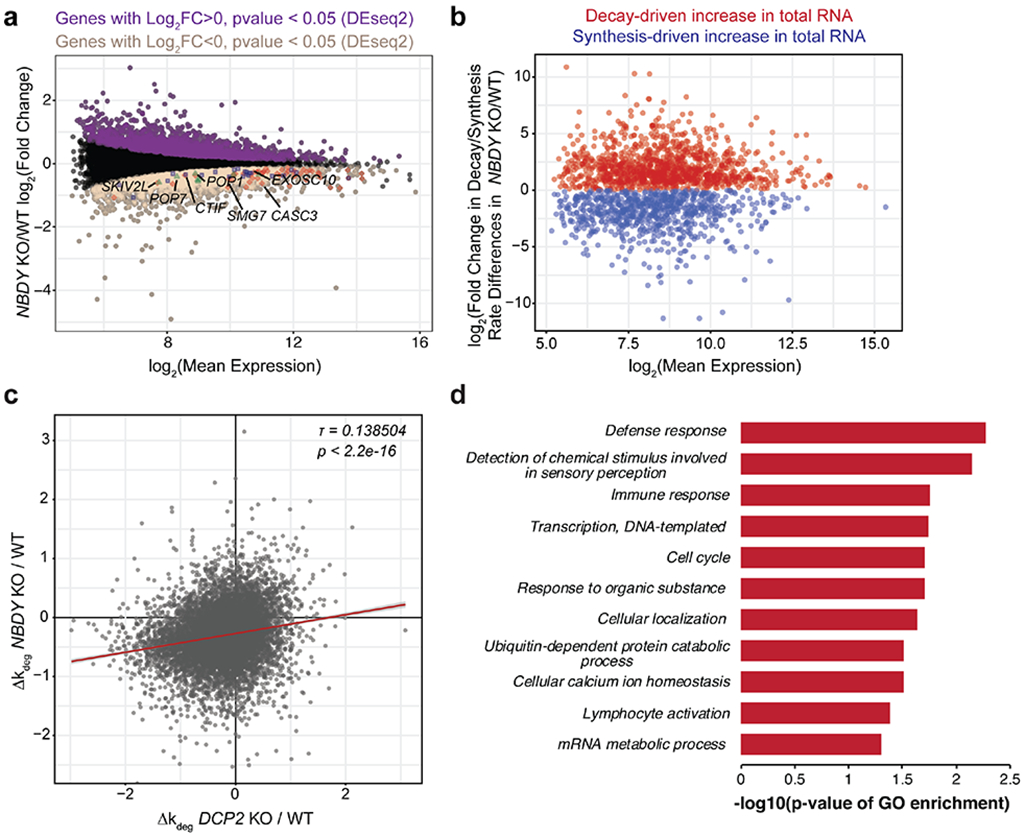
(a,b) Profiling changes in RNA dynamics in NBDY KO versus wild type (WT) HEK 293T cells via metabolic labeling and TimeLapse-seq. The M (log ratio) A (mean average) plot indicates significantly upregulated (purple) and downregulated genes (sienna) derived from gene-level analysis of total RNAs (a). Decay- vs synthesis-driven dynamics were modeled in NBDY KO versus WT HEK 293T cells based on observed T-to-C mutation rates (b). In (a), synthesis downregulated RNA decay machineries were highlighted; Red, nonsense-mediated decay; blue, miRNA-mediated decay; green, endonucleolytic decay. (c) NBDY and DCP2 globally regulate the stability of overlapping sets of RNAs, as shown by moderate correlation between the changes in estimated RNA degradation rate in each KO relative to WT cells (Kendall tau rank correlation coefficient τ=0.138504, p < 2.2e-16). (d) Significant biological process GO Slim terms of post-transcriptionally stabilized genes in NBDY KO versus WT HEK 293T cells. Fisher’s exact test was performed using PANTHER overrepresentation test with FDR<0.05.
A weak correlation was observed between RNA stability changes in cells lacking NBDY vs cells lacking DCP2 (Figure 5c); among NBDY KO-stabilized RNAs, 37% are previously reported DCP2 substrates16. Conversely, 24.5% of previously reported DCP2 substrates are stabilized in the NBDY KO. A subset of NBDY KO-stabilized RNAs correlate with biological processes previously described to be regulated by DCP2, including defense and immune response (Figure 5d)33, consistent with co-regulation of these transcripts by NBDY and DCP2 KO. However, not all DCP2 substrates are stabilized in the NBDY KO; 0.5% of DCP2 substrates (9 total) are destabilized in the NBDY KO, and the remainder of previously identified DCP2 substrates are unchanged in stability in the NBDY KO. NBDY KO can therefore stabilize, destabilize or leave unchanged the rate of decay of DCP2 substrates. More remarkably, the remaining 73% of NBDY KO-stabilized RNAs are not DCP2 targets, and are uniquely stabilized by NBDY KO. We therefore sought (1) to confirm that NBDY KO can dysregulate the decay rate of DCP2 targets in either direction, and that this effect requires interaction with EDC4; (2) to confirm that NBDY KO can stabilize non-DCP2 targets via an EDC4-independent mechanism, and (3) to identify the determinants or mechanism of these effects.
qRT-PCR measurement of RNA stability after transcriptional arrest validated that NBDY KO can either stabilize or destabilize DCP2 decapping substrates and that NBDY coding sequence complementation rescues WT RNA decay rates of both of these classes (Figure 6a and Figure S4a). For example, EPC2 and GJC1 (Figure 6a) are both DCP2 substrates, because they are stabilized in DCP2 KO cells, but EPC2 is stabilized in NBDY KO, whereas GJC1 is destabilized in NBDY KO. We examine the molecular basis for the differential effect of NBDY KO on specific DCP2 substrates in the next section (vide infra). NBDY KO also destabilizes some transcripts that decrease in stability in DCP2 KO cells, likely as a result of secondary, compensatory effects that are similar in both KO cell lines; for example, ATM and MRE11 are destabilized in both NBDY and DCP2 KO cells (Figure 6a and Figure S4a). Finally, we confirmed that NBDY KO increases the stability of some transcripts that are not DCP2 substrates. For example, the stability of ZNF84, a potential nonsense-mediated decay (NMD) target34, is not altered by DCP2 KO, but is increased in NBDY KO (Figure 6a). Overall, qRT-PCR confirmed the trends observed via TimeLapse-seq, and showed that NBDY complementation rescues wild type decay rates for all transcript classes.
Figure 6∣. NBDY regulates stability of DCP2 target transcripts at the decapping step.

(a) RNA stability measurement of selected genes belonging to the following classes: stabilized in both DCP2 KO and NBDY KO (EPC2), stabilized by DCP2 KO but destabilized in NBDY KO (GJC1), and destabilized in both DCP2 KO and NBDY KO (ATM), stabilized in NBDY KO and unaffected in DCP2 KO (ZNF84). Number of biological replicates: n=3. Error bars represent mean ± s.d. Significance was analyzed by ANOVA linear regression. **P < 0.01; ***P < 0.001; ****P < 0.0001, Dunnett’s test. (b) Splinted ligation RT-PCR (qSL-RT-PCR) in WT, XRN1 KO, XRN1/DCP2 or XRN1/NBDY double knock out (DKO) HEK 293T cell lines was performed to assay decapping of a transcript stabilized by both DCP2 and NBDY KO, EPC2 (top). Total RNA was quantified by qRT-PCR at each timepoint (bottom). Significance was analyzed by ANOVA linear regression. Error bars represent mean ± s.d. n=4 biological replicates. ****P < 0.0001, Dunnett’s test.
We hypothesized that both stabilization and destabilization of DCP2 substrates in the NBDY KO occur as a result of transcript-specific changes in the rate of decapping by DCP2 (either faster or slower) in the absence of NBDY. However, no direct measurement of the effect of NBDY on RNA decapping in cells or in vitro has yet been made. We applied a splinted ligation assay that measures decapped RNA isolated from cells lacking the downstream exonuclease XRN1 to EPC2, a transcript stabilized in both NBDY and DCP2 KO cells (Figure 6b and Figure S4b). A fraction of decapped EPC2 is detectable in XRN1 KO cells, because the decapped species that is normally rapidly degraded by XRN1 in wild type cells can build up. In cells lacking either DCP2 or NBDY in addition to XRN1, however, this decapped species is undetectable, providing evidence that both of these proteins are required for cellular EPC2 decapping and suggesting that NBDY KO alters the decay rate of at least one DCP2 target by affecting its access to or decapping by DCP2.
We subsequently examined whether NBDY affects the assembly or catalytic activity of the decapping complex toward a model RNA in an in vitro decapping assay. Briefly, active decapping complex was immunopurified from WT vs NBDY KO cells, and its activity toward a short, capped model RNA substrate was examined. However, no difference in the association of core protein components or in vitro decapping activity of decapping complexes with or without NBDY was observed (Figure S5)26. We concluded that NBDY does not affect the in vitro enzymatic activity of the decapping complex, and that the molecular basis for NBDY-dependent stabilization vs destabilization of DCP2 substrates, and regulation of non-DCP2 substrates in cells, must be more complex.
Given the observation that NBDY directly binds to EDC4, we hypothesized that NBDY must exert its effects on DCP2 targets – either stabilization or destabilization – through its interaction with EDC4. We further hypothesized that regulation of non-DCP2 targets by NBDY would proceed via an independent mechanism that does not require interaction with EDC4. Expression of the EDC4 interaction-deficient W34A mutant of NBDY on the NBDY KO background leads to an increased number of P-bodies relative to wild type NBDY (Figure 7a-b), similar to the NBDY KO and consistent with global perturbation of RNA stability. In NBDY KO cells in which W34A mutant NBDY has been stably reintroduced, EPC2 decays significantly more slowly than in NBDY rescue cells; GJC1 and ATM decay significantly more rapidly in the W34A complementation line than in NBDY rescue. These trends mirror the differences in stabilities for these transcripts in NBDY KO cells as compared to wild type HEK 293T, though these samples were not included in the experiment shown in Figure 7. We conclude that the NBDY-EDC4 interaction is required for specification of the stabilities of these transcripts, and propose that NBDY regulates the stabilities of DCP2 targets via its direct interaction with the decapping complex.
Figure 7∣. The NBDY-EDC4 interaction is required for RNA half-life specification of transcripts co-regulated by NBDY and DCP2.

(a,b) NBDY localization and P-body numbers were assayed in NBDY and NBDY W34A rescue cell lines. Quantitative data (right) represents mean values ± s.e.m, and significance was evaluated with one-way ANOVA. **P < 0.01, Dunnett’s test). Scale bars, 20 μm. (c) RNA stability measurement for selected genes in NBDY rescue versus NBDY W34A (non-EDC4-interacting mutant) complementation cell line. Selected genes fall into the following classes: stabilized in both DCP2 KO and NBDY KO (EPC2), stabilized by DCP2 KO but destabilized in NBDY KO (GJC1), destabilized in both DCP2 KO and NBDY KO (ATM), and exclusively stabilized in NBDY KO (ZNF84). Number of biological replicates: n=3 for EPC2 and n=4 for all other genes. Error bars represent mean ± s.d. **P < 0.01, linear regression t-test.
Interestingly, a transcript that is exclusively destabilized in NBDY KO cells that is not a DCP2 substrate, ZNF84, exhibits indistinguishable decay rates in cells expressing wild type or W34A NBDY, demonstrating that NBDY regulates its stability independent of its association with the DCP2 and EDC4-containing RNA decapping complex. We therefore conclude that non-DCP2 targets are stabilized by a distinct mechanism in NBDY KO cells (vide infra).
In contrast to EDC4, the interaction with DCP1A is not required for NBDY activity toward DCP2 substrates, because expression of the NBDY (1-52) truncation (that does not bind DCP1A) on the NBDY KO background supported a decay rate of the previously identified DCP2 target RRP41 that was indistinguishable from expression of the wild type NBDY sequence on the KO background (Figure S6)35. NBDY therefore exerts its effect on the rate of decay of DCP2 substrates through its interaction with EDC4.
Molecular determinants of RNA stability changes in NBDY KO
Having shown that NBDY KO can either stabilize or destabilize DCP2 substrates, and that NBDY KO also stabilizes a large number of non-DCP2 substrates, we wished to identify the molecular determinants that define these transcript populations. We previously showed that P-body enrichment is a strong correlate of DCP2 selectivity16, so we tested whether NBDY targets are determined by P-body localization. We found that non-DCP2 target RNAs exclusively stabilized in the NBDY KO are not enriched in P-bodies, while all DCP2 substrates, regardless of stability variation in the NBDY KO, showed P-body enrichment (Figure S7a). We conclude that (1) P-body localization does not differentiate whether NBDY KO stabilizes or destabilizes DCP2 targets, and (2) based on their lack of P-body enrichment, the non-DCP2 targets regulated by NBDY are a separate population and likely dysregulated in the NBDY KO through a distinct mechanism.
We hypothesized that these results supported different mechanisms of regulation of DCP2 substrate vs non-substrate transcripts by NBDY, and that stabilization of non-DCP2 targets is an indirect effect of NBDY KO. Specifically, we hypothesized that non-DCP2 target RNAs were stabilized in the NBDY KO cells as a result of compensatory changes in RNA synthesis (transcription and processing). Examination of genes downregulated by synthesis in NBDY KO revealed downregulation of transcripts encoding key regulators of other RNA decay machineries (Figure S3a), including nonsense-mediated decay (NMD) (Figure S3a) and miRNA-mediated decay (Figure S3a). Specifically, SKIV2L, CTIF, and POP1/7 are exclusively down-regulated by NBDY ablation, and not by DCP2 deletion (Figure S3c). Exclusively NBDY KO-stabilized RNAs are significantly enriched in NMD targets (17%), compared to all other genes and to transcripts stabilized in both DCP2 and NBDY KO or DCP2 KO alone (Figure S7b). Taken together, these results suggest that non-DCP2 target transcripts are stabilized in NBDY KO cells indirectly as a result of downregulated synthesis of RNAs encoding components of other RNA decay pathways including NMD.
We then turned our attention back to the mechanism of stabilization vs. destabilization of DCP2 substrates in NBDY KO. We hypothesized that specific properties of DCP2 substrate transcripts might determine the directionality of their stability change in NBDY KO cells. Because only a small number of DCP2 substrates are destabilized in NBDY KO cells (9 total, 0.5%), we considered them together with DCP2 targets that are unchanged in half-lives in NBDY KO, and compared the physical properties of this combined class to the properties of DCP2 substrates stabilized by NBDY KO. We found that stabilization by both NBDY and DCP2 KO is associated with longer 5′ UTRs, while length of the coding region, length of 3′ UTRs, and GC content had no significant effect (Figure S7c-f). A destabilizing 5′ cis element has been described for the RRP41 transcript, which contains a cap proximal stem loop that confers direct binding and efficient decapping by DCP2, providing precedent for the importance of 5′ UTR properties to DCP2 recognition25. We conclude that NBDY KO differentially regulates the stability of DCP2 targets based on their 5′ UTR properties.
DISCUSSION
The NBDY microprotein maintains the correct half-lives of >1400 cellular transcripts, nearly the same number of total targets of the RNA decapping enzyme DCP2, establishing NBDY as an important regulator of cellular RNA dynamics. Importantly, we demonstrated that the interaction between NBDY and EDC4 is required for NBDY-regulated stability of DCP2 targets. The observed bidirectional dysregulation of DCP2 substrates in NBDY KO cells is consistent with a role for NBDY in determining specificity of the cytoplasmic RNA decapping complex, and our results suggest that this discrimination depends on 5′ UTR length. NBDY preferentially regulates decapping of DCP2 targets that contain shorter 5′ UTRs – transcripts with longer 5′ UTR are degraded faster in the NBDY KO cells. It is possible that NBDY controls the accessibility of transcripts to DCP2 decapping based on their 5′ UTR length or structure. The molecular mechanism by which NBDY achieves this discrimination will be of future interest.
A significant number of transcripts stabilized in the NBDY KO are not previously identified DCP2 targets. We showed that this regulation occurs via compensatory changes secondary to NBDY deletion. The ability of the non-interacting NBDY W34A mutant to rescue wild type ZNF84 half-life further supports our proposal that NBDY regulates the stability of non-DCP2 substrates independent of its direct interaction with the decapping complex. Observation of downregulated expression of NMD factors that is specific to the NBDY KO, and correlation of NMD targets with NBDY KO-stabilized, non-DCP2 substrates, provides a mechanistic explanation of this secondary effect of NBDY KO. Overall, NBDY-mediated regulation of global RNA stability is complex. First, NBDY KO can either stabilize or destabilize DCP2 substrates, an effect that depends on its direct interaction with the decapping complex and is determined by 5′ UTR length. Second, NBDY KO stabilizes non-DCP2 target mRNAs via compensatory downregulation of alternative RNA decay pathways. We summarize this model in Figure 8.
Figure 8∣. NBDY in sculpting specificity of the RNA decapping complex: a model.
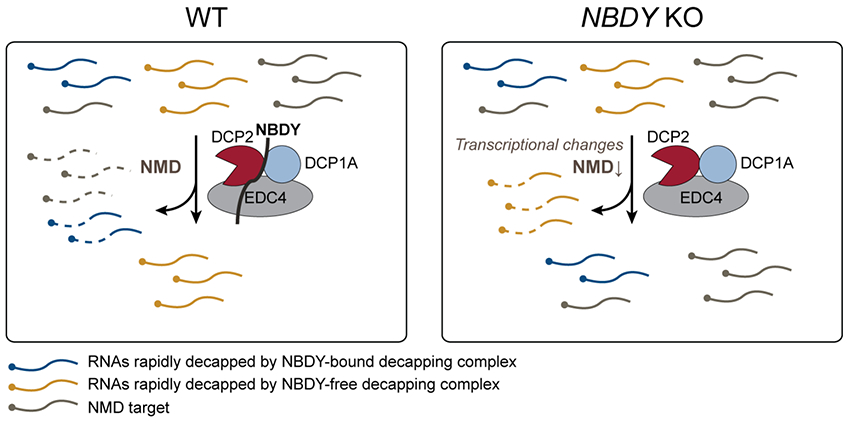
NMD targets (grey) can be degraded by endonucleolytic or exonucleolytic RNA decay from either direction. Mature cytoplasmic RNAs are targeted to rapid decay by NBDY-bound (blue) or by NBDY-free (yellow) decapping complex in wild type cells. The former is stabilized and the latter destabilized in NBDY KO. Meanwhile, NMD targets and substrates of alternative decay pathways are stabilized by secondary transcriptional changes during NBDY knockout.
NBDY expression level is anticorrelated with P-body numbers under basal conditions. This is consistent with a prior report3 in which NBDY over-expression largely eliminated P-bodies, and NBDY silencing increased P-bodies 3-fold. It is likely that experimental perturbation of NBDY expression level alters P-bodies due to changes in RNA stability, consistent with the TimeLapse-seq results reported in this study. Approximately 6-fold more RNAs are stabilized than destabilized in NBDY KO cells, whether via direct or indirect mechanisms, and these stabilized transcripts have the potential to nucleate additional P-bodies via liquid-liquid phase separation. Similar observations have been made in the case of DCP2 inactivation, which leads to an increase in P-body numbers or sizes as a result of increased abundance of deadenylated, but not decapped, RNAs in complex with decay factors31, 36.
Taken together, our data demonstrate that endogenous NBDY serves as a regulator of global RNA stability and P-body formation. These results suggest that the thousands of yet-uncharacterized protein products of smORFs in human cells may harbor additional important regulators of essential cellular processes.
Supplementary Material
ACKNOWLEDGMENT
We thank Dahyana A. Escayola, Nadia G. D’Lima, Lauren Winkler and Christopher Rudeen for early work on NBDY function. We thank all current members of the Slavoff lab for helpful discussions.
Funding Sources
This work was supported by the Searle Scholars Program, an Odyssey Award from the Richard and Susan Smith Family Foundation, NIH R01GM122984, and Yale University West Campus start-up funds (to S.A.S.), as well as NIH DP2 HD083992-01 (to M.D.S). A. K. (5T32GM067543-12) and J. A. S. (T32GM007223) were in part supported by NIH Predoctoral Training Grants.
ABBREVIATIONS
- NMD
nonsense-mediated decay
- RNP
ribonucleoprotein
- smORF
small open reading frames
Footnotes
REFERENCES
- [1].Andrews SJ, and Rothnagel JA (2014) Emerging evidence for functional peptides encoded by short open reading frames, Nat Rev Genet 15, 193–204. [DOI] [PubMed] [Google Scholar]
- [2].Orr MW, Mao Y, Storz G, and Qian SB (2020) Alternative ORFs and small ORFs: shedding light on the dark proteome, Nucleic Acids Res 48, 1029–1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].D'Lima NG, Ma J, Winkler L, Chu Q, Loh KH, Corpuz EO, Budnik BA, Lykke-Andersen J, Saghatelian A, and Slavoff SA (2017) A human microprotein that interacts with the mRNA decapping complex, Nat Chem Biol 13, 174–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Chen J, Brunner A, Cogan J, Nunez J, Fields A, Adamson B, Itzhak D, Li J, Mann M, Leonetti M, and Weissman J (2020) Pervasive functional translation of noncanonical human open reading frames, Science 367, 1140–1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Pauli A, Norris M, Valen E, Chew G, Gagnon J, Zimmerman S, Mitchell A, Ma J, Dubrulle J, Reyon D, Tsai S, Joung J, Saghatelian A, and Schier A (2014) Toddler: An Embryonic Signal That Promotes Cell Movement via Apelin Receptors, Science 343, 1248636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Magny E, Pueyo J, Pearl F, Cespedes M, Niven J, Bishop S, and Couso J (2013) Conserved Regulation of Cardiac Calcium Uptake by Peptides Encoded in Small Open Reading Frames, Science 341, 1116–1120. [DOI] [PubMed] [Google Scholar]
- [7].Zhang S, Reljić B, Liang C, Kerouanton B, Francisco JC, Peh JH, Mary C, Jagannathan NS, Olexiouk V, Tang C, Fidelito G, Nama S, Cheng RK, Wee CL, Wang LC, Duek Roggli P, Sampath P, Lane L, Petretto E, Sobota RM, Jesuthasan S, Tucker-Kellogg L, Reversade B, Menschaert G, Sun L, Stroud DA, and Ho L (2020) Mitochondrial peptide BRAWNIN is essential for vertebrate respiratory complex III assembly, Nat Commun 11, 1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Makarewich CA, and Olson EN (2017) Mining for Micropeptides, Trends Cell Biol 27, 685–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Mugridge JS, Coller J, and Gross JD (2018) Structural and molecular mechanisms for the control of eukaryotic 5'-3' mRNA decay, Nat Struct Mol Biol 25, 1077–1085. [DOI] [PubMed] [Google Scholar]
- [10].Garneau NL, Wilusz J, and Wilusz CJ (2007) The highways and byways of mRNA decay, Nat Rev Mol Cell Biol 8, 113–126. [DOI] [PubMed] [Google Scholar]
- [11].Decker CJ, and Parker R (2012) P-bodies and stress granules: possible roles in the control of translation and mRNA degradation, Cold Spring Harb Perspect Biol 4, a012286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Luo Y, Na Z, and Slavoff S (2018) P-Bodies: Composition, Properties, and Functions, Biochemistry 57, 2424–2431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Horvathova I, Voigt F, Kotrys AV, Zhan Y, Artus-Revel CG, Eglinger J, Stadler MB, Giorgetti L, and Chao JA (2017) The Dynamics of mRNA Turnover Revealed by Single-Molecule Imaging in Single Cells, Mol Cell 68, 615–625.e619. [DOI] [PubMed] [Google Scholar]
- [14].Tutucci E, Vera M, Biswas J, Garcia J, Parker R, and Singer RH (2018) An improved MS2 system for accurate reporting of the mRNA life cycle, Nat Methods 15, 81–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Nissan T, and Parker R (2008) Analyzing P-bodies in Saccharomyces cerevisiae, Methods Enzymol 448, 507–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Luo Y, Schofield JA, Simon MD, and Slavoff SA (2020) Global Profiling of Cellular Substrates of Human Dcp2, Biochemistry. DOI: 10.1021/acs.biochem.0c00069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Sanjana NE, Shalem O, and Zhang F (2014) Improved vectors and genome-wide libraries for CRISPR screening, Nat Methods 11, 783–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Mauer J, Luo X, Blanjoie A, Jiao X, Grozhik A, Patil D, Linder B, Pickering B, Vasseur J, Chen Q, Gross S, Elemento O, Debart F, Kiledjian M, and Jaffrey S (2017) Reversible methylation of m(6)A(m) in the 5 ' cap controls mRNA stability, Nature 541, 371–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Tiscornia G, Singer O, and Verma IM (2006) Production and purification of lentiviral vectors, Nat Protoc 1, 241–245. [DOI] [PubMed] [Google Scholar]
- [20].Schofield JA, Duffy EE, Kiefer L, Sullivan MC, and Simon MD (2018) TimeLapse-seq: adding a temporal dimension to RNA sequencing through nucleoside recoding, Nat Methods 15, 221–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Blewett N, Coller J, and Goldstrohm A (2011) A quantitative assay for measuring mRNA decapping by splinted ligation reverse transcription polymerase chain reaction: qSL-RT-PCR, RNA 17, 535–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Chen CY, Ezzeddine N, and Shyu AB (2008) Messenger RNA half-life measurements in mammalian cells, Methods Enzymol 448, 335–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, and Zhang F (2013) Genome engineering using the CRISPR-Cas9 system, Nat Protoc 8, 2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Schmittgen TD, and Livak KJ (2008) Analyzing real-time PCR data by the comparative C(T) method, Nat Protoc 3, 1101–1108. [DOI] [PubMed] [Google Scholar]
- [25].Lykke-Andersen J, Shu MD, and Steitz JA (2000) Human Upf proteins target an mRNA for nonsense-mediated decay when bound downstream of a termination codon, Cell 103, 1121–1131. [DOI] [PubMed] [Google Scholar]
- [26].Warminski M, Sikorski PJ, Warminska Z, Lukaszewicz M, Kropiwnicka A, Zuberek J, Darzynkiewicz E, Kowalska J, and Jemielity J (2017) Amino-Functionalized 5' Cap Analogs as Tools for Site-Specific Sequence-Independent Labeling of mRNA, Bioconjug Chem 28, 1978–1992. [DOI] [PubMed] [Google Scholar]
- [27].Ponomarenko EA, Poverennaya EV, Ilgisonis EV, Pyatnitskiy MA, Kopylov AT, Zgoda VG, Lisitsa AV, and Archakov AI (2016) The Size of the Human Proteome: The Width and Depth, Int J Anal Chem 2016, 7436849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Wisniewski JR, Hein MY, Cox J, and Mann M (2014) A "proteomic ruler" for protein copy number and concentration estimation without spike-in standards, Mol Cell Proteomics 13, 3497–3506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Chang CT, Bercovich N, Loh B, Jonas S, and Izaurralde E (2014) The activation of the decapping enzyme DCP2 by DCP1 occurs on the EDC4 scaffold and involves a conserved loop in DCP1, Nucleic Acids Res 42, 5217–5233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Lai T, Cho H, Liu Z, Bowler MW, Piao S, Parker R, Kim YK, and Song H (2012) Structural basis of the PNRC2-mediated link between mRNA surveillance and decapping, Structure 20, 2025–2037. [DOI] [PubMed] [Google Scholar]
- [31].Sheth U, and Parker R (2003) Decapping and decay of messenger RNA occur in cytoplasmic processing bodies, Science 300, 805–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Mazzoni C, and Falcone C (2011) mRNA stability and control of cell proliferation, Biochem Soc Trans 39, 1461–1465. [DOI] [PubMed] [Google Scholar]
- [33].Li Y, Dai J, Song M, Fitzgerald-Bocarsly P, and Kiledjian M (2012) Dcp2 decapping protein modulates mRNA stability of the critical interferon regulatory factor (IRF) IRF-7, Mol Cell Biol 32, 1164–1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Colombo M, Karousis E, Bourquin J, Bruggmann R, and Muhlemann O (2017) Transcriptome-wide identification of NMD-targeted human mRNAs reveals extensive redundancy between SMG6-and SMG7-mediated degradation pathways, RNA 23, 189–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Li Y, Song MG, and Kiledjian M (2008) Transcript-specific decapping and regulated stability by the human Dcp2 decapping protein, Mol Cell Biol 28, 939–948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Aizer A, Kalo A, Kafri P, Shraga A, Ben-Yishay R, Jacob A, Kinor N, and Shav-Tal Y (2014) Quantifying mRNA targeting to P-bodies in living human cells reveals their dual role in mRNA decay and storage, J Cell Sci 127, 4443–4456. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.