


Abstract
The accessibility of target gene, a factor critical for gene regulation, is controlled by epigenetic fine-tuning of chromatin organization. While there are multiple experimental techniques to study change of chromatin architecture with its epigenetic state, measurements from them are not always complementary. A qualitative discrepancy is noted between recent super-resolution imaging studies, particularly on Polycomb-group protein repressed domains in Drosophila cell. One of the studies shows that Polycomb-repressed domains are more compact than inactive domains and are segregated from neighboring active domains, whereas Hi-C and chromatin accessibility assay as well as the other super-resolution imaging studies paint a different picture. To examine this issue in detail, we analyzed Hi-C libraries of Drosophila chromosomes as well as distance constraints from one of the imaging studies, and modeled different epigenetic domains by employing a polymer-based approach. According to our chromosome models, both Polycomb-repressed and inactive domains are featured with a similar degree of intra-domain packaging and significant intermixing with adjacent active domains. The epigenetic domains explicitly visualized by our polymer model call for extra attention to the discrepancy of the super-resolution imaging with other measurements, although its precise physicochemical origin still remains to be elucidated.
INTRODUCTION
Post-translational histone modifications and chromatin modifying proteins regulate gene expressions in eukaryote (1–4). Genome-wide enrichment patterns, acquired from chromatin immunoprecipitation (ChIP)-array, associate chromatin loci into different ‘states’, each with distinct histone modification pattern (5–8). Analysis of chromatin state provides insights into regulatory elements, circuits and their cell-type specificities (6,8), offering glimpses into the complex chromatin landscape. Recent advances in high-resolution Hi-C (9–12) and super-resolution fluorescence in situ hybridization (FISH) techniques (13–15) have greatly enriched our knowledge of 3D chromosome organizations, and raised the question of how chromosomes fold in different epigenetic states.
The super-resolution FISH imaging by Boettiger et al. (13) is a seminal study that has offered direct visualization of 46 distinct genomic domains in Drosophila Kc167 cells (Figure 1A, Supplementary Figure S2A). On the basis of the enrichment profiles of H3K4me2, unmodified H3, and H3K27me3, the domains were classified into (i) transcriptionally active (A-type); (ii) inactive (I-type) and (iii) Polycomb group (PcG) protein repressed (R-type) domains (See Supplementary Information for other classification of chromatin states). A special spotlight was cast on Polycomb-repressed domains that demonstrated remarkably high degrees of intra-domain chromatin compaction (see R-07 domain visualized in Figure 1A). The analysis of Polycomb-repressed domains indicated an unusual scaling of radius of gyration (Rg) with domain length (N), Rg ∼ Nc with c = 0.22, which was smaller than the exponent for the compact packing (c = 1/3) (13).
Figure 1.

Super-resolution imaging data from the three different studies. (A) Three epigenetic domains visualized by Boettiger et al. From the left to the right shown are the image of A-16 (active), I-06 (inactive), and R-07 (repressed) domains. (B) The radius of gyration for each of three domain types shown in (A) as a function of domain length. The data in blue fitted to dashed line is for the Ph knocked-down repressed domains. The figures in (A) and (B) were both taken from Ref. (13). (C) Density of three types of epigenetic domains. Blue, black and red correspond to the R-, I- and A-domains, respectively. The figure was taken from Figure 1H in Szabo et al. (14). (D) Contact probability as a function of genomic distance. The figure was taken from Supplementary Figure S2C in Cattoni et al. (27).
The gene silencing associated with Polycomb-repressed domains during development is regulated by two main complexes of PcG proteins, Polycomb repressive complexes 1 and 2 (PRC1 and PRC2) (16–23). Subunits of PRC1 induce condensation of nucleosomal arrays in vitro (24,25). Deletion of Polyhomeotic 1 (Phc1, a component of PRC1) in mouse decompacts small PRC1-bound domains (<140 kb) and increases the expression level of PcG-silenced genes (16). In Drosophila cells, PcG proteins aggregate into nano-clusters mediated by the Ph subunit of PRC1 (26). These results suggest that PcG proteins tightly pack gene loci and silence the gene expression, which is also consistent with the finding that decompaction of Polycomb-repressed domain results from the knockdown of Ph (Drosophila homolog of Phc1) (13) (from the blue solid line to the dashed line in Figure 1B).
However, recent chromatin accessibility measurements (ATAC-seq, DNase-seq and FAIRE-seq) (28) carried out by King et al. (29) on the PcG protein mediated gene silencing have pointed to a different conclusion. It was shown that the reduced chromatin accessibility in vivo could still be maintained after knocking out PcG proteins. In addition, little difference in accessibility was identified in distal elements of both PcG-bound and PcG-free promoters (Supplementary Figure S2 in (29)). Instead of PcG complexes, chromatin remodeling factors or the low levels of histone acetylation was proposed as the factor contributing to the maintenance of limited accessibility.
Imaging data associated with Polycomb-repressed domains are also available from two other super-resolution imaging studies on Drosophila chromosomes, one by Szabo et al. (Figure 1C) (14) and the other by Cattoni et al. (Figure 1D) (27). Whereas the focus of their study was on examining the effect of epigenetic modification on the topologically associated domains and higher-order chromosome organization, there is clear indication in their data that Polycomb-repressed domains are similar with or even slightly more open than inactive domains (see Figure 1C, D).
In a nutshell, the unusually compact structures of PcG-occupied domain observed in the super-resolution study by Boettiger et al. (Figure 1A, B) (13) do not fully conform to those implied from other super-resolution imaging studies (Figure 1C, D) (14,27) as well as to the chromatin accessibility measurements by King et al. (29). To gain better understanding to different epigenetic states, 3D modeling and visualization of each state would be of great help. To this end, we applied a recently developed, polymer-based chromosome modeling approach, termed the heterogeneous loop model (HLM) (30) (https://github.com/leiliu2015/HLM, see Supplementary Information and Supplementary Figure S1), on Hi-C data of Drosophila Kc167 cells (9) and generated 3D structures of active, inactive, and Polycomb-repressed domains.
In this paper, we first analyze Hi-C data of different epigenetic domains, and next build the corresponding 3D chromosome models to study their structural properties. According to our 3D models, Polycomb-repressed domains are not so compact as indicated by Boettiger et al.’s super-resolution imaging (13). Instead, they are similar with inactive domains in terms of intra-domain compaction and intermixing with spatially juxtaposed active domains, which is better aligned with the chromatin accessibility assays (5,6,29) and with other super-resolution imaging studies on epigenetic domains (14,27).
MATERIALS AND METHODS
Construction of chromatin conformational ensemble
To generate a conformational ensemble of a genomic region of interest, we used HLM, the key parameters of which were determined based on the contact probabilities of the corresponding region of Hi-C. Details about the original HLM can be found in Ref. (30). Each monomer of our chromatin polymer model in this study represents 5 kb genome. The genomic position of simulated chromatin, the total number of monomers (N), and the Pearson correlation which assesses the similarity between the contact probabilities from Hi-C and HLM are summarized in Supplementary Table S1. Detailed information on the genomic regions of epigenetic (sub)domains studied by super-resolution microscopy (13,14), which are modeled in this work, is given in Supplementary Tables S2 and S3.
Hi-C, epigenetic and DNA accessibility data
For the 3D chromatin structure modeling of Drosophila Kc167 cells, we chose sub-kb resolution Hi-C data by Eagen et al. (GSE89112) (9). The contact frequency matrix, normalized using Knight-Ruiz (KR) method (31), was rescaled such that the contact probability P(s) = 1 as a function of genomic distance s along the chromatin chain was satisfied at s = 1, and was used as the input of HLM. We also analyzed the Hi-C data of Kc167 cells from other two experiments (GSE63515 (32), GSE38468 (17)), as well as S2R+ cells (embryonic cell lines; late embryonic stage; GSE99104 (14)).
The ChIP-chip (chromatin immunoprecipitation followed by microarray hybridization) data of histone modification H3K4me2, H3K4me3, unmodified H3, and the DamID (DNA adenine methyltransferase identification) binding profile of the PcG protein Polycomb (Pc, a subunit of PRC1) are available for Kc167 cells (GSE22069) (5). The log2(fold enrichment) at different loci, within a genomic window of 5 kb, is plotted in the tracks in Figure 2, Supplementary Figures S2 and S6, aligned with heatmaps of contact probabilities. For comparison, presented in Supplementary Figure S6 are the enrichment profiles of H3K4me3 (ChIP-chip; modENCODE914 (33)) and Pc (ChIP-seq; GSM604723 (34)) in the ∼3 Mb region of S2-DRSC cells.
Figure 2.

Three different types of epigenetic domains inferred from Hi-C. Heatmap of contact probabilities from Hi-C (upper diagonal region) and from HLM (lower diagonal region) for the (A) active domain A-23, (B) inactive domain I-14 and (C) Polycomb-repressed domain R-11, each of which is the largest of the domain type. Enrichment profiles of H3K4me2 (red), unmodified H3 (black), and Pc (blue) are shown on the left in each map. (D) Contact probability P(s) as a function of genomic distance s for the three largest domains. (E) (top) P(s) for all 46 domains (13). (bottom) 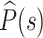 is the contact probability averaged over the domain of the same epigenetic type. (F) γ versus chromatin accessibility. γ is the value of exponent determined from the scaling relationship P(s) ∼ s−γ. The domain accessibility is calculated from DNase I hypersensitivity assay (6) (see Materials and Methods for details). The histograms of γ and domain accessibility are shown on the side and top of the panel, respectively. Significance of similarity in histograms between the values of γ and between domain accessibilities is shown on the top of histograms with the notation: ns (not significant, P > 0.05), * (P < 0.05), ** (P < 0.01), and ***** (P < 1 × 10−5).
is the contact probability averaged over the domain of the same epigenetic type. (F) γ versus chromatin accessibility. γ is the value of exponent determined from the scaling relationship P(s) ∼ s−γ. The domain accessibility is calculated from DNase I hypersensitivity assay (6) (see Materials and Methods for details). The histograms of γ and domain accessibility are shown on the side and top of the panel, respectively. Significance of similarity in histograms between the values of γ and between domain accessibilities is shown on the top of histograms with the notation: ns (not significant, P > 0.05), * (P < 0.05), ** (P < 0.01), and ***** (P < 1 × 10−5).
The chromatin accessibility is based on the DNase I hypersensitivity assay of Kc167 cells (6) (https://compbio.hms.harvard.edu/kharchenko-et-al-nature-2011), which is given as the log2 ratio of the read density at individual loci and the mean density. In addition, the mean value of the loci accessibility within the domain is used to represent the domain accessibility (Figure 2 and Supplementary Figure S5). All relevant experimental data were assembled with respect to the reference genome assembly dm3 of Drosophila melanogaster.
Characterization of domain structures
The 3D conformational ensemble of epigenetic domains generated using HLM was characterized by means of the following structural properties (see Supplementary Information for the precise mathematical expression for each measure):
The mean radius of gyration (rg) was calculated as a function of genomic distance s for the subchains in a genomic domain of interest, and it was fitted to rg(s) ∼ sν, where the scaling exponent ν characterizes the chromatin chain organization inside the domain. The chromatin density inside the domain can be approximated as
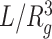 , where L is the total number of monomers in the domain whose radius of gyration is Rg.
, where L is the total number of monomers in the domain whose radius of gyration is Rg.The asphericity (Asp.) was calculated to characterize the overall shape of chromatin domain in reference to a perfect sphere. Asp. = 0 is for the sphere, and Asp. > 0 quantifies the extent of deviation from a spherical shape.
The density and surface roughness of a domain were evaluated by means of the Voronoi tessellation (35), which offers a well-defined volume V and surface area S of the domain. The surface roughness was quantified by calculating the surface area S relative to that of a perfect sphere (S0). By definition, S/S0 ≥ 1 should be satisfied, and a rougher domain surface would give rise to a larger value of S/S0. Given the volume from the Voronoi tessellation, the domain density is calculated as L/V.
The overlap fraction of X domain with Y domain is defined as the number of monomers in X domain that are within a distance 2a from any monomer in the domain Y relative to the total number of monomers comprising X domain (13), where a is the van der Waals radius or the diameter of each monomer. The overlap factor between X and Y domains is determined as the ratio of the number of inter-domain monomer pairs to the number of all inter-domain monomer pairs.
To visualize a structural ensemble of modeled chromatin domain (30), the geometric centers of three or four (sub)domains, if any, were first selected from the whole domain, and next the distribution of the interdomain distances were computed based on the ensemble of structures. Several chromatin conformations were then randomly selected from the most populated cluster determined based on the interdomain distances, and were aligned and rendered.
RESULTS
Epigenetic state-dependent intra-domain organization of chromatin inferred from Hi-C
For each of 46 domains examined in Ref (13), we first analyzed the corresponding regions of Hi-C data (9) (Figure 2A–C) and calculated the intra-domain contact probability, P(s), as a function of the genomic distance s. P(s) shows power-law decay, P(s) ∼ s−γ, over the range of s ≃ (0.01−0.4) Mb. The largest active (A-23), inactive (I-14) and Polycomb-repressed domains (R-11) among 46 domains are characterized with different exponent γ (Figure 2D). In terms of γ, A-domains are clearly discerned from I- and R-domains (Figure 2E. See also Supplementary Figure S2B). In general, a larger value of γ is indicative of less compact and more sparsely organized structure (36–38), and hence more accessible to the protein factors or nucleases. Thus, A-23 (γ = 1.27 ± 0.06) is more accessible than I-14 (γ = 0.63 ± 0.05) and R-11 (γ = 0.78 ± 0.05). The mean accessibility and γ value evaluated over all 46 domains are positively correlated (Spearman corr. = 0.81, Pearson corr. = 0.83) (Figure 2F), These two measures not only distinguish A-domains from I- and R-domains, but also indicate that I-domains are comparable to R-domains (Figure 2F). Analysis of two other Hi-C data (17,32), where chromatin of the same cell line was digested with different type of restriction enzymes, DpnII (17) and HindIII (32), offers the same conclusion (Supplementary Figures S3 and S4).
The above results based on Hi-C data are at odds with the Boettiger et al.’s super-resolution imaging (13) (Figure 1A) which indicates that Polycomb-repressed chromatin is featured with the densest intra-domain packing among the three epigenetic types.
Comparison of HLM-generated epigenetic domains
To elaborate more on the differences between three epigenetic states other than γ, we modeled 3D structures of A-23, I-14 and R-11 domains by employing the HLM approach (30) and visualized them (Figure 3A).
Figure 3.
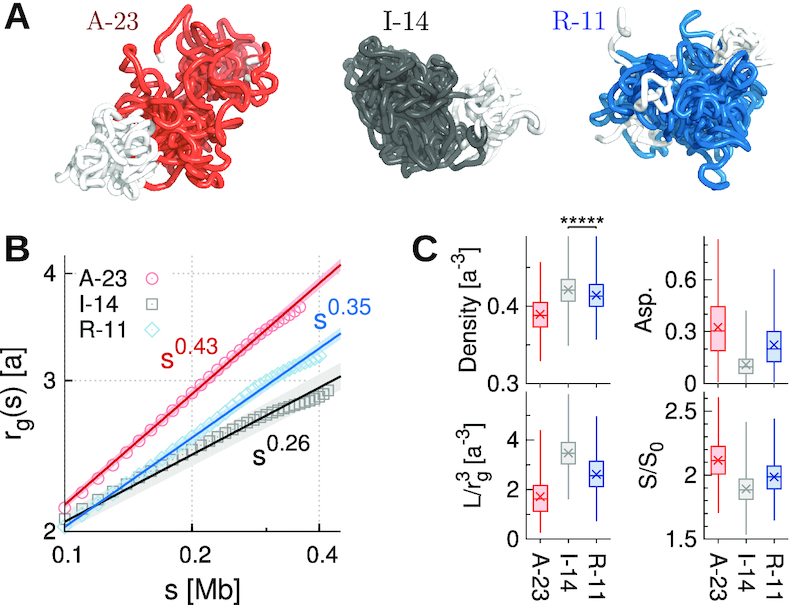
Structural properties of the three types of largest epigenetic domains modeled by HLM. (A) Ensembles of 3D structures of A-23, I-14 and R-11 domains. (B) The mean gyration radius rg(s) as a function of genomic distance s in log-log scale. (C) Density, 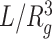 , asphericity, and surface roughness S/S0 of different domains. The density of monomers in I-14 is significantly greater than that in R-11 (Mann–Whitney U test, P < 1 × 10−5).
, asphericity, and surface roughness S/S0 of different domains. The density of monomers in I-14 is significantly greater than that in R-11 (Mann–Whitney U test, P < 1 × 10−5).
As shown in Figure 3A, the A-23 domain is aspherical and loosely packed. I-14 is more spherical and compact than R-11 domain. The average radius of gyration rg(s) increases with the subchain length (s) as rg(s) ∼ sν (Figure 3B). A-23 is characterized with the largest exponent ν (=0.43), followed by R-11 (ν = 0.35) and I-14 (ν = 0.26). A smaller ν is expected for a more densely packed polymer chain (13). In terms of the monomer number density evaluated based on Voronoi tessellation (35), as well as its crude estimate 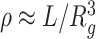 where Rg is the radius of gyration of the whole domain of size L, we obtain ρI-14 ≳ ρR-11 > ρA-23 (Figure 3C). In addition, I-14 has the most spherical shape with the smoothest domain surface. All these features for the three different epigenetic domains are aligned with those implied by γ (Figure 2D) and the chromatin accessibility (as labeled in Figure 2F). Based on the 3D HLM structures of all domains, we find that chromatin accessibility is negatively correlated with the domain density (Spearman corr. = −0.78 with P < 1 × 10−5. See Supplementary Figure S5B).
where Rg is the radius of gyration of the whole domain of size L, we obtain ρI-14 ≳ ρR-11 > ρA-23 (Figure 3C). In addition, I-14 has the most spherical shape with the smoothest domain surface. All these features for the three different epigenetic domains are aligned with those implied by γ (Figure 2D) and the chromatin accessibility (as labeled in Figure 2F). Based on the 3D HLM structures of all domains, we find that chromatin accessibility is negatively correlated with the domain density (Spearman corr. = −0.78 with P < 1 × 10−5. See Supplementary Figure S5B).
The two independently calculated exponents γ and ν enabled us to extract the effective dimension (deff) in which the chromatin chains are organized. The contact probability between two points separated by the genomic separation s along the chromatin chain (P(s)) would be inversely proportional to the effective volume Veff(s) explored by the chain segment between the two, namely, 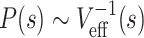 . In addition, the effective volume is calculated as a power of space dimension of the segment as
. In addition, the effective volume is calculated as a power of space dimension of the segment as 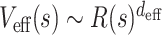 where R(s) scales as R(s) ∼ sν (37). From this theoretical consideration,
where R(s) scales as R(s) ∼ sν (37). From this theoretical consideration, 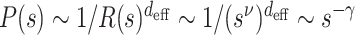 , we obtain the relation γ = deffν. From the analysis in Figure 4, I- and R-domains are characterized with deff ≈ 2, significantly smaller than deff ≈ 2.9 for A-domains. This suggests that unlike A-domain whose effective dimension is close to 3, I- and R-domains are characterized with the dimension close to 2. The values of deff ≈ 2 for I- and R-domains indicates that chromatin chains of I- and R-domains are effectively confined and organized in two dimensions. This is consistent with the knowledge that BLACK and BLUE chromatins, which are the two classes of the five-colored chromatin states (see Supplementary Information) corresponding to inactive and repressed chromatins, display extensive colocalization with Lamin, the protein expressed in nuclear envelope (Figure 3A in (5)).
, we obtain the relation γ = deffν. From the analysis in Figure 4, I- and R-domains are characterized with deff ≈ 2, significantly smaller than deff ≈ 2.9 for A-domains. This suggests that unlike A-domain whose effective dimension is close to 3, I- and R-domains are characterized with the dimension close to 2. The values of deff ≈ 2 for I- and R-domains indicates that chromatin chains of I- and R-domains are effectively confined and organized in two dimensions. This is consistent with the knowledge that BLACK and BLUE chromatins, which are the two classes of the five-colored chromatin states (see Supplementary Information) corresponding to inactive and repressed chromatins, display extensive colocalization with Lamin, the protein expressed in nuclear envelope (Figure 3A in (5)).
Figure 4.
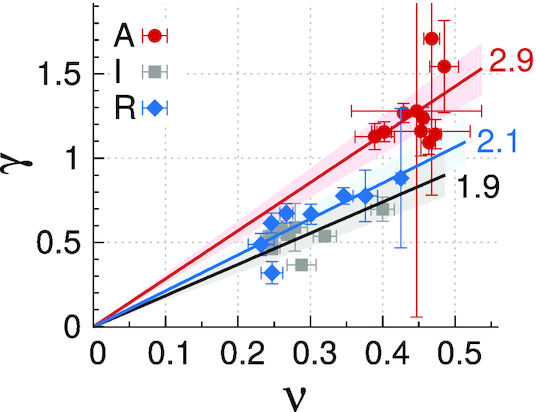
γ versus ν for three different types of epigenetic domains. The data are fitted to a relation γ = deffν to determine the effective dimension deff for each domain type. (deff = 2.85 ± 0.13 (A-domains), 1.85 ± 0.11 (I-domains), 2.13 ± 0.11 (R-domains)).
Epigenetic domains have also been visualized by Szabo et al. using super-resolution microscopy in a 3 Mb genomic region of Drosophila S2R+ cells (14). Their data shows that chromatin density of inactive domains is greater than that of Polycomb-repressed domains (see Figure 1B). Because the chromatin states and Hi-C data of the 3-Mb region of Kc167 and S2R+ cells, both derived from late embryos (39), are remarkably similar (see Supplementary Figure S6A), we modeled the associated genomic region of S2R+ cells by taking advantage of the Hi-C library of Kc167 cells that retains higher resolution. The density of chromatin chain (ρ) in three domains shows ρI ≳ ρR > ρA (Supplementary Figure S6F), which is consistent with the observation by Szabo et al. (14) (Figure 1 B).
Finally, it is of particular note that there is apparent discrepancy between rg(s)’s of R-11 measured from FISH (filled diamonds) and from HLM-generated ensemble (empty diamonds in Figure 5A). R-11 domain is clearly more compact in the Boettiger’s super-resolution FISH imaging than that based on our polymer model. The exponent ν = 0.22 is unusual and difficult to rationalize given that ν = 1/3 would be the best situation in which growing subchain can fill the space as compact as possible. The exponent ν = 0.22 means that the packing density of a domain made of subchain increases with the subchain size s as ρ(s) ∼ s/rg(s)3 ∼ s1 − 3ν with 1 − 3ν > 0. The ‘sticky’ polymer model proposed by Boettiger et al. (13) could generate polymer conformation with multiple loops which might represent the situation created by PRC1 complexes, partly explaining the exponent ν = 0.22; however, the domain length giving rise to ν = 0.22 was limited to a narrow range (Figure 4c in (13) and explanation therein). Furthermore, it is unlikely that the structures generated using the sticky polymer model reproduce the corresponding heatmap of contact probability from Hi-C data. For the exponent c defined in the scaling relationship between the gyration radii of whole domains and their lengths, Rg(L) ∼ Lc, the greatest difference is also found in R-domains, i.e. c = 0.22 for FISH (Figure 1B) and c = 0.38 for Hi-C (Figure 5B).
Figure 5.
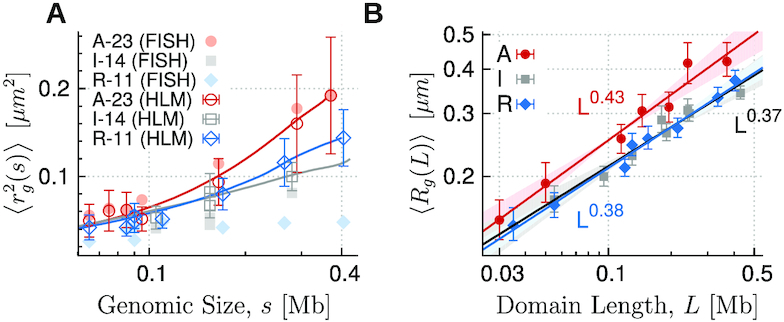
(A) The squared radius of gyration 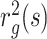 of subchains of size (length) s calculated from HLM is compared with those measured with FISH. (B) The gyration radius as a function of domain length in log-log scale for each type of epigenetic domain. Data points are obtained for individual domains by modeling HLMs of 7 A-, 8 I- and 8 R-domains, the size of which is greater than 25 kb (L > 25 kb). By fitting the data to the scaling relation Rg ∼ Lc, we obtain the exponent c = 0.43 ± 0.03, 0.37 ± 0.03, and 0.38 ± 0.02 for the A-, I- and R-type domains, respectively.
of subchains of size (length) s calculated from HLM is compared with those measured with FISH. (B) The gyration radius as a function of domain length in log-log scale for each type of epigenetic domain. Data points are obtained for individual domains by modeling HLMs of 7 A-, 8 I- and 8 R-domains, the size of which is greater than 25 kb (L > 25 kb). By fitting the data to the scaling relation Rg ∼ Lc, we obtain the exponent c = 0.43 ± 0.03, 0.37 ± 0.03, and 0.38 ± 0.02 for the A-, I- and R-type domains, respectively.
Intermixing between two different epigenetic domains inferred from Hi-C
Next, to study the intermixing between two different epigenetic domains spatially juxtaposed, we used sub-kb resolution Hi-C data (9) and modeled 3D structures for the genomic regions that encompass several epigenetic domains (see Supplementary Figures S6–S10 and Table S1). The overlap fraction of I- and R-domains with respect to their adjacent A-domains, the definition of which was taken from Ref. (13) (see Materials and Methods for the details), are summarized in Figure 6A. Both I- and R-domains show significant amount of intermixing with neighboring A-domains (A-17, A-03 and A-04), displaying comparable levels of interdomain overlap with A-domain, i.e., ϕRA ≈ ϕIA (Figure 6A). The HLM-generated chromatin conformations (Supplementary Figures S6D and S7D) show this more explicitly. It is again at odds with Boettiger et al.’s super-resolution imaging results (Figure 3 in (13)), which pointed out that Polycomb-repressed domains have little intermixing with A-domains.
Figure 6.
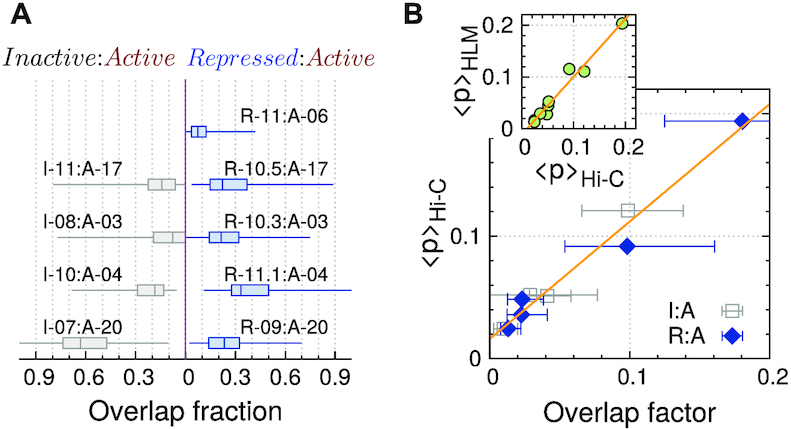
Comparison of spatial overlap of inactive and repressed domains with adjacent active domains. (A) Overlap fraction. (B) The inter-domain mean contact probability measured from Hi-C, 〈p〉Hi-C, versus overlap factor (corr. =0.99). (Inset) 〈p〉Hi-C is plotted against the same quantity measured from chromosome models 〈p〉HLM (corr. = 0.98).
The correlation between Hi-C and HLM-derived contact maps is sufficiently good. But, to further ensure that our 3D model correctly captures the features of Hi-C inter-domain contacts, we calculated the average inter-domain contact probabilities between two different domain types (I and A, R and A) based on Hi-C (〈p〉Hi-C) and 3D models (〈p〉HLM). They are highly correlated (the inset of Figure 6B. See also Supplementary Figures S6A, S7A and S8A). In addition, there is a strong correlation between 〈p〉Hi-C and the inter-domain overlap factor calculated using 3D structures (40) (see Figure 6B and its definition in Materials and Methods).
DISCUSSION
As long as Hi-C and super-resolution imaging data are complementary to each other, analysis based on integration of two distinct experiments would offer promising results (41–46); however, contradictory data would give rise to discordant outcome. Here, we underlie the incompatibility between the Hi-C and Boettiger et al.’s FISH measurement (13) by combining the data from two measurements and using them as input data for HLM.
We extended HLM, such that the information of FISH measurement can be incorporated into the model as well, along with Hi-C data to generate 3D polymer models (Figure 7). Specifically, FISH measurement by Boettiger et al. offers information of gyration radii of subchains, which can be converted as the distance restraints between pairs of genomic loci in the framework of HLM and enables us to incorporate them into HLM straightforwardly. Information from two distinct measurements, Hi-C and FISH, can be linearly combined via a weighting factor f as follows.
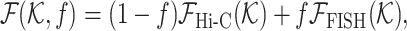 |
(1) |
where 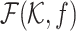 is a f-dependent objective function that mixes the information from Hi-C and FISH, with which to determine the stiffness matrix (
is a f-dependent objective function that mixes the information from Hi-C and FISH, with which to determine the stiffness matrix (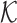 -matrix) required to build a polymer model of chromatin using HLM (see Supplementary Information for technical details).
-matrix) required to build a polymer model of chromatin using HLM (see Supplementary Information for technical details).
Figure 7.
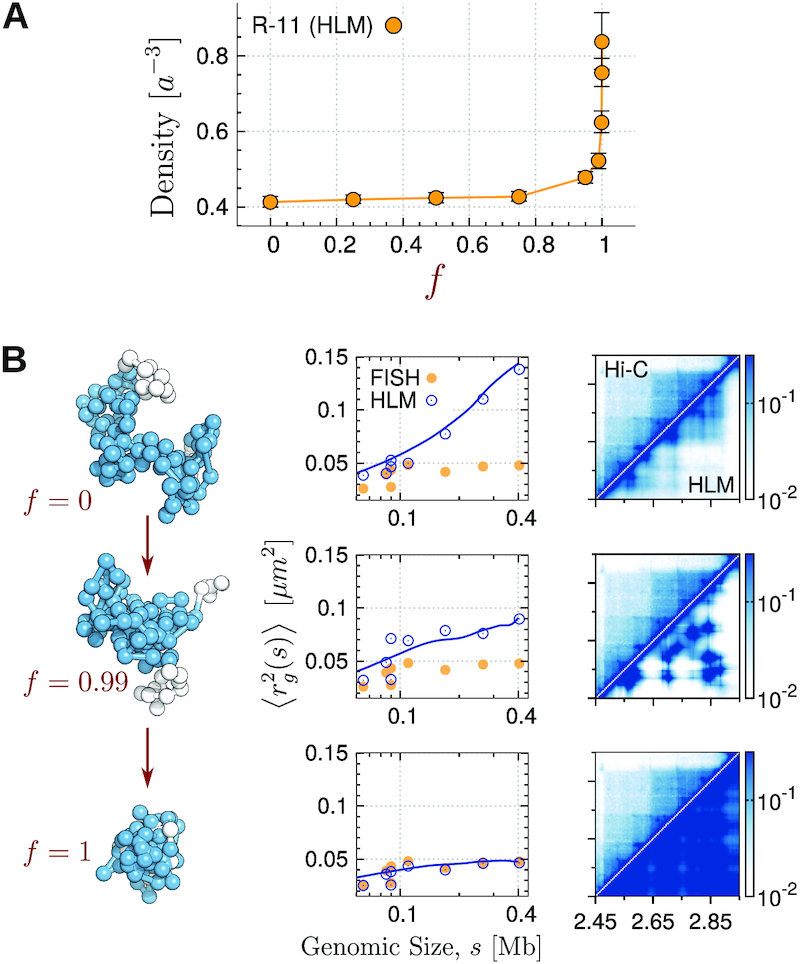
Comparison between the intra-domain chromatin folding inferred from Hi-C (9) and that measured with FISH (13). (A) Density of R-11 domain as a function of a parameter f, which was used to incorporate the restraints from two different experiments into HLM. The parameter f can be varied between 0 and 1 with f = 0 corresponding to Hi-C and f = 1 to FISH. (B) (Left panels) A randomly selected conformation of R-11 at each f value. (Middle panels) 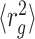 of subchains of R-11 between specific two loci measured by FISH and HLM are shown with orange filled circles and blue open circles, respectively; the blue lines are
of subchains of R-11 between specific two loci measured by FISH and HLM are shown with orange filled circles and blue open circles, respectively; the blue lines are 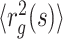 of subchains with varying s calculated by using HLM-generated structures of R-11 domain. (Right panels) Heatmap of contact probabilities from Hi-C and HLM (Hi-C/HLM is in the upper/lower diagonal of the panels) at f = 0, 0.99 and 1, from the top to bottom.
of subchains with varying s calculated by using HLM-generated structures of R-11 domain. (Right panels) Heatmap of contact probabilities from Hi-C and HLM (Hi-C/HLM is in the upper/lower diagonal of the panels) at f = 0, 0.99 and 1, from the top to bottom.
We find that the resulting polymer model of R-11 domain, which incorporates the restraints exclusively from Boettiger et al.’s FISH measurement (13) (f = 1), is twice denser than the one modeled solely based on Hi-C (f = 0) (Figure 7B). As f is varied from 0 to 1, the chromatin chain exhibits striking condensation (Figure 7B, left panels). At f = 1, the HLM-generated chromatin conformations are characterized with severe overlap between the monomers (the structure depicted at the bottom on the left panel of Figure 7B). A high degree of intra-domain packaging within the repressed domain bear close resemblance to the imaging data by Boettiger et al. (see the image of R10 in Figure 2b in (13)). Although the gyration radii of subchains in the 3D model better match to their values measured with FISH with increasing f (Figure 7 B, middle panels), the intra-domain contact probabilities deviate further from Hi-C data (Figure 7B, right panels).
In stark contrast to Boettiger et al.’s report on R-domain (13), an unusually expanded Polycomb-regulated gene (HoxD) cluster was also found in FISH measurement (47). Knockout of Ring1B (a subunit of PRC1) only slightly weakened 5C contacts, whereas the distance between two probes enclosing the region increased significantly in the FISH measurement. It was surmised that this bias was due to a protein network made of remaining PRC2 cross-linked by fixation in 5C which kept DNA loci in proximity (47).
It is noteworthy that there is fundamental difference between Hi-C and FISH data (40,48), one is averaged over a population of millions of cells masking the extensive cell-to-cell variability and the other is based on much smaller number of cells. The FISH-Hi-C paradox resulting from this difference has recently been well addressed using a polymer model by Shi and Thirumalai (49). In principle, the extensive cell-to-cell variability could have contributed to the apparent discrepancy between Hi-C and FISH data on Polycomb-repressed domains (see Supplementary Information for conformational variations in domain structures and Supplementary Figure S11); however, it is still puzzling that the discordant conclusions on Polycomb-repressed domains are drawn from even the same type of super-resolution FISH measurements (Figure 1). Our Hi-C derived structures of Polycomb-repressed domains conform to the super-resolution FISH measurements by Szabo et al. (14) and Cattoni et al. (27), but not to that by Boettiger et al. (13).
Critical difference between the super-resolution FISH imagings of Boettiger et al. (13) and Szabo et al. (14) could have been originated from the homologous pairing in the tetraploid nucleus of Drosophila cells. Particularly, homologous R-domains could have been bundled together more tightly than the other two domain types in the Boettiger et al’s super-resolution FISH imaging. Szabo et al. (14) confined their analysis to single-domain nanocompartments in a haploid context, carefully eliminating the chance of homologous pairing in their sample image, and reported the 3D density of oligopaint signals; yet, such careful separation of haploid from tetraploid was not carried out in the Boettiger et al’s super-resolution FISH imaging. Recently, Lesage et al. (50) have noted the issue of homologous pairing and proposed a deconvolution procedure to remove the effect of homologous pairing from Boettiger et al.’s data. As an alternative to the sticky polymer model with strong self-attraction (13), they further considered a mildly self-attracting homopolymer model which forms bundles via homologous pairings, and predicted a crossover for L > 100 kb in the scaling law of single R-domains in a haploid context, so that only large R-domains form compact globules, whereas A-, I- and small R-type domains are open coils. The HLM based on Hi-C, in contrast, predicts that the largest R-domain (R-11) is more open than the largest I-domain (I-14) (Figure 3), and does not find any crossover in the scaling of gyration radius for any type of domains (Figure 5 B).
Meanwhile, Lesage et al. quantified epigenetic domain type-dependent Kuhn segment lengths in units of nm and base pairs (bp), enabling to map the monomer unit (a), corresponding to 5 kb genomic size, of the HLM model in this study to a physical length in a domain type-dependent manner, such that a = knm(5 kb/kbp)c nm, where the values for Kuhn segment, knm and kbp, for each domain are estimated in the Table 3 of Ref. (50). This allows us to reassess the gyration radius for each epigenomic domain by using the relation Rg(L) = knm(L/kbp)c instead of Rg(L) = a(L/5kb)c used in Figure 5B. However, even when we used the Kuhn segment lengths extracted from Boettiger et al’s data, our conclusion as to the compactness of R-domain in comparison with I-domains remains the same. As shown in Supplementary Figure S12, the degrees of intra-domain compaction in R- and I-type domains are still comparable.
Of particular note is great variation in the biochemical and structural properties among R-domains. In light of chromatin accessibility it appears that there are at least two classes of R-type domains (Figure 2F). All three R-domains with DNase fold enrichments larger than 0.5 are those with domain lengths shorter than 50 kb; unlike large R-domains, small R-domains are as open as the active ones (50). It has also been pointed out that intra-domain distribution of PRC1-bound loci can affect chromatin intra-domain folding, giving rise to two different modes of R-domain (16). The first mode has large loops bridged by PRC1 (e.g. Blue1 domain in Supplementary Figure S6D), yielding relatively small γ, ν and density. The second mode is generated by densely spaced PRC1, and R-11 domain in Figure 3 corresponds to this second mode. Together with the heterogeneity of nuclear localization of repressive domains (27), all of these observations call for a refined classification of Polycomb-repressed domains.
Lastly, fixation of cell culture and in situ hybridization are in general carried out under chemically harsh condition, going through multiple cycles of heating/cooling, washing with chemical buffers, and variable incubation time (9,13,14,17,27,32). It has been known that even if the same chemical agents and buffer condition are used, morphology of fixed cell is sensitive to the incubation time as well as to the concentration range of formaldehyde and other chemical agents being added, engendering formation of bubble-like structure at a whole cell scale and changes in both nucleoplasm and cytoplasm (51,52).
Based on the gyration radii of labeled telomeres as well as STORM image of fixed Kc167 and HEK293 (human embryonic kidney) cells in comparison with live cells, Boettiger et al. assumed that their fixation condition introduced no detectable change (Supplementary Figure S2d in (13)). However, neither the Hi-C (9,17,32) nor other imaging studies (14,27) had followed this particular fixation protocol; in addition, temperature and incubation time to hybridize FISH probes to the denatured DNA were not identical among the super-resolution imaging studies. Physicochemical effect of the hybridization protocol as well as the fixation buffer on PcG-bound chromatin in Drosophila cells remains to be investigated.
Supplementary Material
ACKNOWLEDGEMENTS
We thank the Center for Advanced Computation in KIAS for providing computing resources.
Contributor Information
Lei Liu, Department of Physics, Zhejiang Sci-Tech University, Hangzhou 310018, People’s Republic of China.
Changbong Hyeon, Korea Institute for Advanced Study, Seoul 02455, Republic of Korea.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
KIAS Individual Grant [CG035003 to C.H.], at Korea Institute for Advanced Study. Funding for open access charge: Intramural grant.
Conflict of interest statement. None declared.
REFERENCES
- 1. Kouzarides T. Chromatin modifications and their function. Cell. 2007; 128:693–705. [DOI] [PubMed] [Google Scholar]
- 2. Bannister A.J., Kouzarides T.. Regulation of chromatin by histone modifications. Cell Res. 2011; 21:381–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Greer E.L., Shi Y.. Histone methylation: a dynamic mark in health, disease and inheritance. Nature Rev. Genet. 2012; 13:343–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Allis C.D., Jenuwein T.. The molecular hallmarks of epigenetic control. Nat. Rev. Genet. 2016; 17:487–500. [DOI] [PubMed] [Google Scholar]
- 5. Filion G.J., van Bemmel J.G., Braunschweig U., Talhout W., Kind J., Ward L.D., Brugman W., de Castro I.J., Kerkhoven R.M., Bussemaker H.J. et al.. Systematic protein location mapping reveals five principal chromatin types in Drosophila cells. Cell. 2010; 143:212–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kharchenko P.V., Alekseyenko A.A., Schwartz Y.B., Minoda A., Riddle N.C., Ernst J., Sabo P.J., Larschan E., Gorchakov A.A., Gu T. et al.. Comprehensive analysis of the chromatin landscape in Drosophila melanogaster. Nature. 2011; 471:480–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hon G., Wang W., Ren B.. Discovery and annotation of functional chromatin signatures in the human genome. PLoS Comput. Biol. 2009; 5:e1000566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ernst J., Kheradpour P., Mikkelsen T.S., Shoresh N., Ward L.D., Epstein C.B., Zhang X., Wang L., Issner R., Coyne M. et al.. Mapping and analysis of chromatin state dynamics in nine human cell types. Nature. 2011; 473:43–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Eagen K.P., Aiden E.L., Kornberg R.D.. Polycomb-mediated chromatin loops revealed by a subkilobase-resolution chromatin interaction map. Proc. Natl. Acad. Sci. U.S.A. 2017; 114:8764–8769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Rao S. S.P., Huntley M.H., Durand N.C., Stamenova E.K., Bochkov I.D., Robinson J.T., Sanborn A.L., Machol I., Omer A.D., Lander E.S. et al.. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell. 2014; 159:1665–1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sexton T., Yaffe E., Kenigsberg E., Bantignies F., Leblanc B., Hoichman M., Parrinello H., Tanay A., Cavalli G.. Three-dimensional folding and functional organization principles of the Drosophila genome. Cell. 2012; 148:458–472. [DOI] [PubMed] [Google Scholar]
- 12. Wang Q., Sun Q., Czajkowsky D.M., Shao Z.. Sub-kb Hi-C in D. melanogaster reveals conserved characteristics of TADs between insect and mammalian cells. Nat. Commun. 2018; 9:188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Boettiger A.N., Bintu B., Moffitt J.R., Wang S., Beliveau B.J., Fudenberg G., Imakaev M., Mirny L.A., Wu C.-T., Zhuang X.. Super-resolution imaging reveals distinct chromatin folding for different epigenetic states. Nature. 2016; 529:418–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Szabo Q., Jost D., Chang J.-M., Cattoni D.I., Papadopoulos G.L., Bonev B., Sexton T., Gurgo J., Jacquier C., Nollmann M. et al.. TADs are 3D structural units of higher-order chromosome organization in Drosophila. Sci. Adv. 2018; 4:eaar8082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Beliveau B.J., Kishi J.Y., Nir G., Sasaki H.M., Saka S.K., Nguyen S.C., Wu C.-T., Yin P.. OligoMiner provides a rapid, flexible environment for the design of genome-scale oligonucleotide in situ hybridization probes. Proc. Natl. Acad. Sci. U.S.A. 2018; 115:E2183–E2192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kundu S., Ji F., Sunwoo H., Jain G., Lee J.T., Sadreyev R.I., Dekker J., Kingston R.E.. Polycomb repressive complex 1 generates discrete compacted domains that change during differentiation. Molecular Cell. 2017; 65:432–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Li L., Lyu X., Hou C., Takenaka N., Nguyen H., Ong C.-T., Cubeñas-Potts C., Hu M., Lei E., Bosco G. et al.. Widespread rearrangement of 3D chromatin organization underlies polycomb-mediated stress-induced silencing. Mol. Cell. 2015; 58:216–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Schuettengruber B., Bourbon H.-M., Croce L.D., Cavalli G.. Genome regulation by polycomb and trithorax: 70 years and counting. Cell. 2017; 171:34–57. [DOI] [PubMed] [Google Scholar]
- 19. Schwartz Y.B., Kahn T.G., Nix D.A., Li X.-Y., Bourgon R., Biggin M., Pirrotta V.. Genome-wide analysis of Polycomb targets in Drosophila melanogaster. Nature Genet. 2006; 38:700–705. [DOI] [PubMed] [Google Scholar]
- 20. Entrevan M., Schuettengruber B., Cavalli G.. Regulation of genome architecture and function by polycomb proteins. Trends Cell Biol. 2016; 26:511–525. [DOI] [PubMed] [Google Scholar]
- 21. Ogiyama Y., Schuettengruber B., Papadopoulos G.L., Chang J.-M., Cavalli G.. Polycomb-dependent chromatin looping contributes to gene silencing during Drosophila development. Mol. Cell. 2018; 71:73–88. [DOI] [PubMed] [Google Scholar]
- 22. Klocko A.D., Ormsby T., Galazka J.M., Leggett N.A., Uesaka M., Honda S., Freitag M., Selker E.U.. Normal chromosome conformation depends on subtelomeric facultative heterochromatin in Neurospora crassa. Proc. Natl. Acad. Sci. U.S.A. 2016; 113:15048–15053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Xiong J., Zhang Z., Zhu B.. Polycomb ‘polypacks’ the chromatin. Proc. Natl. Acad. Sci. U.S.A. 2016; 113:14878–14880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Francis N.J., Kingston R.E., Woodcock C.L.. Chromatin compaction by a polycomb group protein complex. Science. 2004; 306:1574–1577. [DOI] [PubMed] [Google Scholar]
- 25. Grau D.J., Chapman B.A., Garlick J.D., Borowsky M., Francis N.J., Kingston R.E.. Compaction of chromatin by diverse Polycomb group proteins requires localized regions of high charge. Genes Dev. 2011; 25:2210–2221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wani A.H., Boettiger A.N., Schorderet P., Ergun A., Münger C., Sadreyev R.I., Zhuang X., Kingston R.E., Francis N.J.. Chromatin topology is coupled to Polycomb group protein subnuclear organization. Nat. Commun. 2016; 7:10291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Cattoni D.I., Gizzi A. M.C., Georgieva M., Di Stefano M., Valeri A., Chamousset D., Houbron C., Déjardin S., Fiche J.-B., González I. et al.. Single-cell absolute contact probability detection reveals chromosomes are organized by multiple low-frequency yet specific interactions. Nat. Commun. 2017; 8:1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tsompana M., Buck M.J.. Chromatin accessibility: a window into the genome. Epigenet. Chromatin. 2014; 7:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. King H.W., Fursova N.A., Blackledge N.P., Klose R.J.. Polycomb repressive complex 1 shapes the nucleosome landscape but not accessibility at target genes. Genome Res. 2018; 28:1494–1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Liu L., Kim M.H., Hyeon C.. Heterogeneous loop model to infer 3D chromosome structures from Hi-C. Biophys. J. 2019; 117:613–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Knight P.A., Ruiz D.. A fast algorithm for matrix balancing. IMA J. Numer. Anal. 2013; 33:1029. [Google Scholar]
- 32. Hou C., Li L., Qin Z., Corces V.. Gene density, transcription, and insulators contribute to the partition of the Drosophila genome into physical domains. Mol. Cell. 2012; 48:471–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. The modENCODE Consortium Identification of functional elements and regulatory circuits by Drosophila modENCODE. Science. 2010; 330:1787–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Enderle D., Beisel C., Stadler M.B., Gerstung M., Athri P., Paro R.. Polycomb preferentially targets stalled promoters of coding and noncoding transcripts. Genome Res. 2011; 21:216–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Rycroft C.H. VORO++: a three-dimensional Voronoi cell library in C++. Chaos. 2009; 19:041111. [DOI] [PubMed] [Google Scholar]
- 36. Halverson J.D., Smrek J., Kremer K., Grosberg A.Y.. From a melt of rings to chromosome territories: the role of topological constraints in genome folding. Rep. Prog. Phys. 2014; 77:022601. [DOI] [PubMed] [Google Scholar]
- 37. Liu L., Hyeon C.. Contact statistics highlight distinct organizing principles of proteins and RNA. Biophys. J. 2016; 110:2320–2327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Peng C., Fu L.-Y., Dong P.-F., Deng Z.-L., Li J.-X., Wang X.-T., Zhang H.-Y.. The sequencing bias relaxed characteristics of Hi-C derived data and implications for chromatin 3D modeling. Nucleic Acids Res. 2013; 41:e183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Cherbas L., Willingham A., Zhang D., Yang L., Zou Y., Eads B.D., Carlson J.W., Landolin J.M., Kapranov P., Dumais J. et al.. The transcriptional diversity of 25 Drosophila cell lines. Genome Res. 2011; 21:301–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Giorgetti L., Heard E.. Closing the loop: 3C versus DNA FISH. Genome Biol. 2016; 17:215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Li Q., Tjong H., Li X., Gong K., Zhou X.J., Chiolo I., Alber F.. The three-dimensional genome organization of Drosophila melanogaster through data integration. Genome Biol. 2017; 18:145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Dixon J.R., Xu J., Dileep V., Zhan Y., Song F., Le V.T., Yardimci G.G., Chakraborty A., Bann D.V., Wang Y. et al.. Integrative detection and analysis of structural variation in cancer genomes. Nat. Genet. 2018; 50:1388–1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Nir G., Farabella I., Estrada C.P., Ebeling C.G., Beliveau B.J., Sasaki H.M., Lee S.H., Nguyen S.C., McCole R.B., Chattoraj S. et al.. Walking along chromosomes with super-resolution imaging, contact maps, and integrative modeling. PLoS genetics. 2018; 14:e1007872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Abbas A., He X., Zhou B., Zhu G., Ma Z., Gao J.-T., Zhang M.Q., Zeng J.. Integrating Hi-C and FISH data for modeling 3D organizations of chromosomes. Nat. Commun. 2019; 10:2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Bintu B., Mateo L.J., Su J.-H., Sinnott-Armstrong N.A., Parker M., Kinrot S., Yamaya K., Boettiger A.N., Zhuang X.. Super-resolution chromatin tracing reveals domains and cooperative interactions in single cells. Science. 2018; 362:eaau1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Cardozo Gizzi A.M., Cattoni D.I., Fiche J.-B., Espinola S., Gurgo J., Messina O., Houbron C., Ogiyama Y., Papadopoulos G.-L., Cavalli G. et al.. Microscopy-based chromosome conformation capture enables simultaneous visualization of genome organization and transcription in intact organisms. Mol. Cell. 2019; 74:212–222. [DOI] [PubMed] [Google Scholar]
- 47. Williamson I., Berlivet S., Eskeland R., Boyle S., Illingworth R.S., Paquette D., Dostie J., Bickmore W.A.. Spatial genome organization: contrasting views from chromosome conformation capture and fluorescence in situ hybridization. Genes Dev. 2014; 28:2778–2791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Fudenberg G., Imakaev M.. FISH-ing for captured contacts: towards reconciling FISH and 3C. Nature Methods. 2017; 14:673–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Shi G., Thirumalai D.. Conformational heterogeneity in human interphase chromosome organization reconciles the FISH and Hi-C paradox. Nat. Commun. 2019; 10:3894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Lesage A., Dahirel V., Victor J.-M., Barbi M.. Polymer coil–globule phase transition is a universal folding principle of Drosophila epigenetic domains. Epigenet. Chromatin. 2019; 12:28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Crawford C., Barer R.. The action of formaldehyde on living cells as studied by phase-contrast microscopy. J. Cell Sci. 1951; 3:403–452. [Google Scholar]
- 52. Hoffman E.A., Frey B.L., Smith L.M., Auble D.T.. Formaldehyde crosslinking: a tool for the study of chromatin complexes. J. Biol. Chem. 2015; 290:26404–26411. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.