

ABSTRACT KCNV2
-associated retinopathy or “cone dystrophy with supernormal rod responses” is an autosomal recessive cone-rod dystrophy with pathognomonic ERG findings. This gene encodes Kv8.2, a voltage-gated potassium channel subunit that acts as a modulator by shifting the activation range of the K+ channels in photoreceptor inner segments. Currently, no treatment is available for the condition. However, there is a lack of prospective long-term data in large molecularly confirmed cohorts, which is a prerequisite for accurate patient counselling/prognostication, to identify an optimal window for intervention and outcome measures, and ultimately to design future therapy trials. Herein we provide a detailed review of the clinical features, retinal imaging, electrophysiology and psychophysical studies, molecular genetics, and briefly discuss future prospects for therapy trials.
KEYWORDS: Gene therapy, potassium channels, molecular genetics, KCNV2, ERG, cone dystrophy, cone-rod dystrophy, retinal dystrophy, supernormal rod responses
Introduction
KCNV2-associated retinopathy (OMIM #610356) is an unusual, autosomal recessive cone-rod dystrophy with pathognomonic electroretinogram (ERG) findings (1–4). It was first described by Gouras et al in 1983 as a cone dystrophy with nyctalopia and supernormal rod responses (5). In the USA, it has an estimated frequency of 1/865,000 inhabitants and an incidence of 5 new cases per year (6). Wu et al. linked “cone dystrophy with supernormal rod responses” (CDSRR) to a 1.5 Mb region on chromosome 9p24, and subsequently identified disease-causing sequence variants in the KCNV2 gene (7,8). KCNV2 encodes a voltage-gated potassium channel, which sets vertebrate photoreceptor resting potential and voltage response (9).
This article aims to provide a detailed overview of the current clinical literature regarding KCNV2 retinopathy, review our current understanding of the molecular genetics, and discuss potential novel treatments.
Clinical presentation
Patients often present in the first or second decades of life with central scotoma, poor visual acuity, variable photophobia, and red-green axis dyschromatopsia with relative tritan sparing (3,4,10,11). Younger children may display an abnormal head posture, head shaking, and/or nystagmus, which can improve over time (12). Nyctalopia may also be reported at presentation, and patients often have mild to moderate myopia (13). A significant proportion of patients report both notable night blindness and photophobia, a combination of symptoms that is unusual in the early stages of a cone-rod dystrophy.
Retinal imaging
Fundus examination often reveals a relatively normal retinal periphery and a range of macular abnormalities, which vary from discrete accentuation of the foveal reflex to more pronounced macular retinal pigment epithelial (RPE) atrophy (10,13). Fundus autofluorescence (FAF) imaging reveals a wide range of findings including ring-like or bull’s-eye changes, increased foveal AF, and reduced central signal in keeping with atrophy, have all been reported (Figure 1). A parafoveal ring of increased AF is a common finding in younger patients, which may initially involve a broader area or multiple foci forming a concentric pattern in the second decade of life, and ultimately evolves into concentric areas of decreased signal indicative of RPE/photoreceptor dysfunction/loss (3,4,13).
Figure 1.
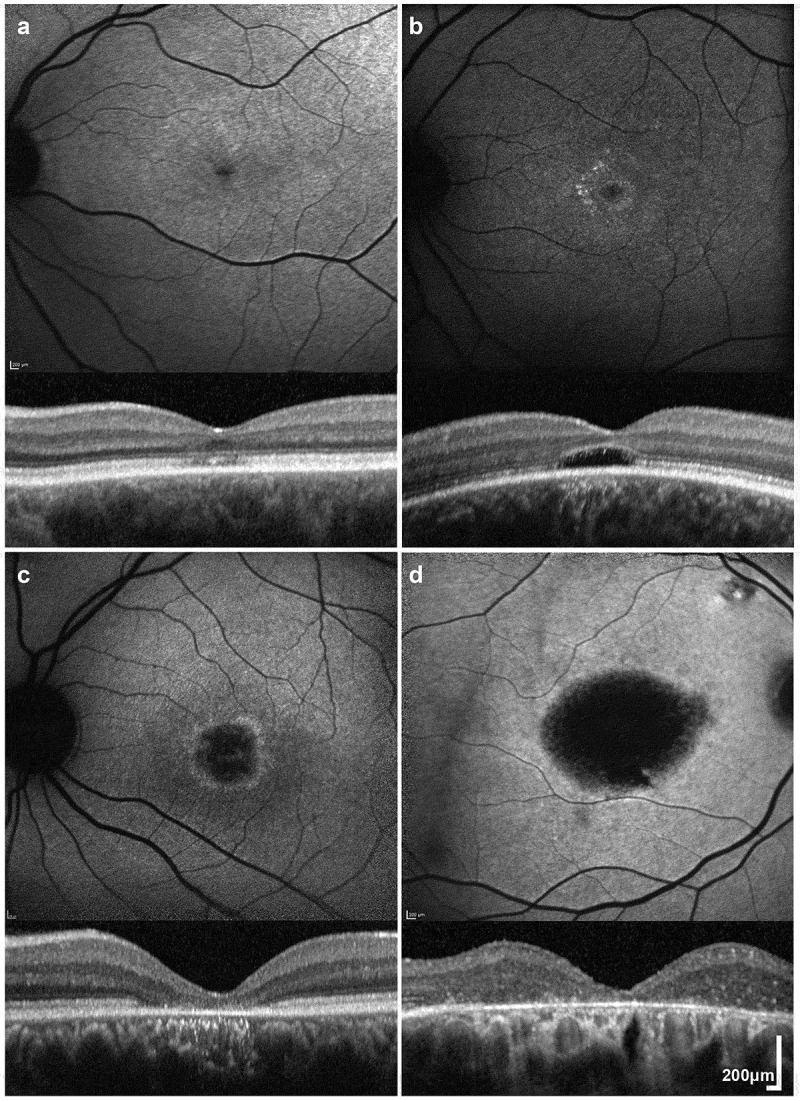
Retinal Imaging in KCNV2-Retinopathy. (a-d) Fundus autofluorescence (FAF) imaging with corresponding horizontal trans-foveal optical coherence tomography (OCT) scans of four patients with disease-causing KCNV2 variants (a, b, c and d; 49, 25, 28 and 71 years of age respectively). A wide range of FAF patterns is observed: increased foveal signal (a), bull’s-eye maculopathy (b), perifoveal ring of increased signal with central atrophy (c and d). Corresponding OCT images show: small discontinuities and attenuation of the foveal ellipsoid zone (EZ) (a), a hyporeflective zone (b), and more extensive loss of the EZ and retinal pigment epithelial atrophy (c-d).
Optical coherence tomography (OCT) identifies variable outer retinal integrity, from mild discontinuous reflectivity to more extensive loss of the ellipsoid zone (EZ), including a central hyporeflective zone (HRZ) in some patients (Figure 1) (4,14). Although foveal EZ changes are evident even in the earliest stages of the disease, there appears to be a relatively wide temporal window before significant atrophy is evident (14).
Adaptive optics scanning light ophthalmoscopy (AOSLO) is a non-invasive imaging modality that enables the visualization of photoreceptors at a microscopic level by correcting for ocular aberrations (15). AOSLO in KCNV2 retinopathy reveals cone photoreceptor mosaic disruption with patches of absent and non-waveguiding cones and overall reduced cone density, but significant residual photoreceptors that could be therapeutically targeted (4).
Electrophysiology, pupillometry and psychophysics
KCNV2-retinopathy has a pathognomonic ERG signature (Figure 2) (13,16). International Society for Clinical Electrophysiology of Vision (ISCEV) – standard (17) light-adapted (LA) ERGs are reduced and delayed, and photopic On-Off ERGs (18) typically show abnormalities of both cone-mediated On- and Off- systems (13). Under dark-adapted (DA) conditions, the rod-mediated dim flash (DA0.01) ERG is severely delayed and typically of subnormal amplitude; whereas to a strong flash the (DA10.0) ERG a-wave has a characteristic flattened trough of normal or near-normal amplitude with a late negative component, and the b-wave is of relatively high amplitude (and may be supernormal). Although not needed for diagnosis, a stimulus-response series reveals no detectable response to a very dim white flash (e.g. 0.002 cd.sm−2; detectable in healthy subjects), but there is a disproportionate increase in the ERG b-wave with increasing intermediate flash strengths. Pattern ERG P50 (19) is invariably undetectable, irrespective of age or fundus appearance, in keeping with severe macular dysfunction (13). In the largest cross-sectional study to date (n = 24), the ERG findings did not correlate with age, which suggests that the progressive structural macular degeneration can occur in the presence of relatively stable peripheral retinal function (13).
Figure 2.
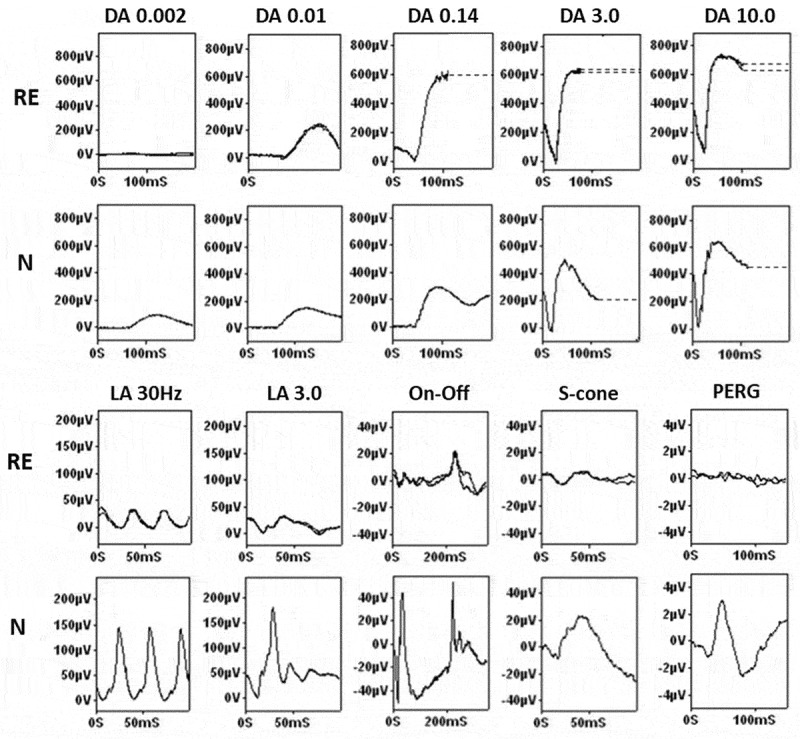
Electroretinography in KCNV2-Retinopathy. Full-field ERGs and PERG recorded from a patient with KCNV2-retinopathy (right eye; RE), compared with representative control recordings from an unaffected subject (N). Dark-adapted (DA) responses are shown for flash strengths of between 0.002 and 10.0 cd.s.m−2 (DA 0.002 – DA 10.0). In the case of KCNV2-retinopathy the DA 0.002 ERG is undetectable; DA 0.01 ERG is delayed and subnormal; the DA 10.0 ERG a-wave trough has a relatively broad shape of mildly subnormal amplitude with a late negative component; the DA 10 ERG b-wave is of relatively high amplitude. Light-adapted (LA) ERGs are shown for a flash strength 3.0 cd.s.m−2 (LA 30 Hz and 2 Hz); responses are reduced and delayed. Photopic On-Off ERGs show delay and reduction affecting both On and Off responses and S-cone ERGs are subnormal. The PERG P50 component is undetectable. Recordings were symmetrical and are shown for the right eye only. Abnormal traces are superimposed to demonstrate reproducibility with exception of DA 0.14 (single trace recorded). Broken lines replace blink artefacts that occur after the b-waves.
In 2019, Collison et al. performed pupillometry in two unrelated patients with molecularly confirmed KCNV2-retinopathy. They detected pupillary responses to moderate to high-luminance stimuli, including responses to high-luminance short-wavelength stimuli that were within normal limits. The normal sustained pupillary responses suggest an outer retinal locus and are consistent with ERG evidence of relatively preserved inner retinal function (20).
A detailed psychophysical investigation of 5 patients with KCNV2-retinopathy, concluded that the defect in the voltage-gated potassium channel produces a nonlinear distortion of the photoreceptor response after otherwise normal phototransduction (21). The authors thereby suggested that the previous name of the disorder (cone dystrophy with ‘supernormal’ rod ERG) to be potentially misleading, given their identification of comparable loss of both cone and rod photoreceptor function; also consistent with the mildly reduced DA10.0 ERG a-waves seen in most cases (13).
The combination of clinical, imaging, and ERG findings that characterize the phenotype of KCNV2-retinopathy are highly suggestive of the disease. Nevertheless, it remains possible that the condition is underdiagnosed due to a lack of clinical awareness of this particular phenotype, limited access to specialist ERG testing or failure to recognize the pathognomonic ERG features, which are not always associated with a DA strong flash ERG b-wave of abnormally high amplitude, and also a lack of genetic testing (13,16,22,23).
Molecular genetics
KCNV2 is a 2-exon gene, encoding a 545 amino acid protein, that was first cloned in 2002 (8). It is predominantly expressed in the heart and retina (24). When first described, the protein product was named Kv11.1, rather than Kv8.2, as it is known now; with the nomenclature change being that Kv11.1 was reassigned to a pore-forming subunit of a rapidly activating-delayed rectifier K+ channel, a product of the KCNH2 gene (OMIM #152427). Kv8.2 is a regulatory subunit, which is known to be an electrically “silent” K+ channel subunit when expressed as a homotetramer. Initially, Ottschytsch et al. suggested that it combines with other proteins in heterotetrameric complexes. Indeed, Kv2.1 was found to generate current and promote trafficking of Kv6.3, Kv10.1 and Kv8.2, which supported his hypothesis (8). Through obligatory heteromerization with Kv2.1, Kv8.2 affects cellular excitability potential and alters the K+ current.
Four years later (2006), Wu et al. linked CDSRR to a 1.5 Mb region on chromosome 9p24 in a large multiply consanguineous family from UAE, and identified a homozygous nonsense variant in KCNV2. In situ hybridization using a KCNV2 antisense riboprobe demonstrated its expression in the inner segments of human rod and cone photoreceptors (PR) (7). The importance of this gene in the visual cycle was further supported by Czirják et al., when it was suggested that the Kv2.1/Kv8.2 complex contributed to photoreception, which further explains why variants in KCNV2 lead to a visual disorder (24). More recently, it has been proposed that the presence of Kv8.2 in the heteromeric complex regulates the function of the Kv2.1/Kv8.2 complex by shifting the activation range of the K+ channels in photoreceptor inner segments. Otherwise, as in the case of a dysfunctional KCNV2 gene, the absence/reduced function of the subunit Kv8.2 in the potassium channels, would shift and depolarize the resting potential of the cells, which might account for the pathognomonic ERG findings in KCNV2-retinopathy (9).
In preparations of micro-dissected retinal neurons, the transcript levels of Kv8.2 and Kv2.1 were found to display daily rhythms, with elevated values during the night. It has been proposed that the transcriptional regulation of Kv8.2 and Kv2.1 is a mechanism by which the ‘retinal clock’ drives visual function according to different environmental lighting conditions (25).
Using chromatin immunoprecipitation and bioinformatic prediction analysis, two cone-rod homeobox (CRX) binding sites and one NRL binding site have been identified in the KCNV2 promoter. Interestingly, shRNA-mediated knockdown of CRX binding sites in mouse models, resulted in reduced KCNV2 promoter activity and low endogenous KCNV2 mRNA expression in the retina, suggesting that retina-specific expression of KCNV2 is controlled by the transcription factor CRX (26). These findings may be helpful in designing future gene therapy for KCNV2-retinopathy.
Protein structure
Voltage-dependent K+ channels are composed of alpha-subunits, which determine the structure of the channel, and beta-subunits which modulate its properties (27). KCNV2 encodes Kv8.2, which is an alpha-subunit (8). Each channel subunit consists of: (i) an N-terminus with a highly conserved tetramerization domain known as N-terminal A and B box (NAB or T1) that facilitates interaction between compatible alpha-subunits; (ii) 6 transmembrane domains (S1-S6) with a positively-charged S4 that forms the voltage sensor domain (VSD); (iii) extracellular and intracellular loop segments; and (iv) an ultra-conserved potassium selective motif (Gly-Tyr-Gly) in the pore forming loop between S5-S6 (P loop), which forms the selective filter (28–33). A graphical representation of the protein and its domains is presented in Figure 3. Variants located in the intracellular amino-terminal region (T1) of Kv8.2 are very likely to be pathogenic as this domain lends stability to the channel structure. Variants in this region have produced elevated levels of non-functional monomers in a yeast model that were then degraded (34). However, using a different yeast model, it has been shown that sequence variants in T1 do not result in misfolding or fast degradation of the protein, but robustly prevent and disrupt interaction between the T1 domains of Kv8.2 and Kv2.1 (35).
Figure 3.
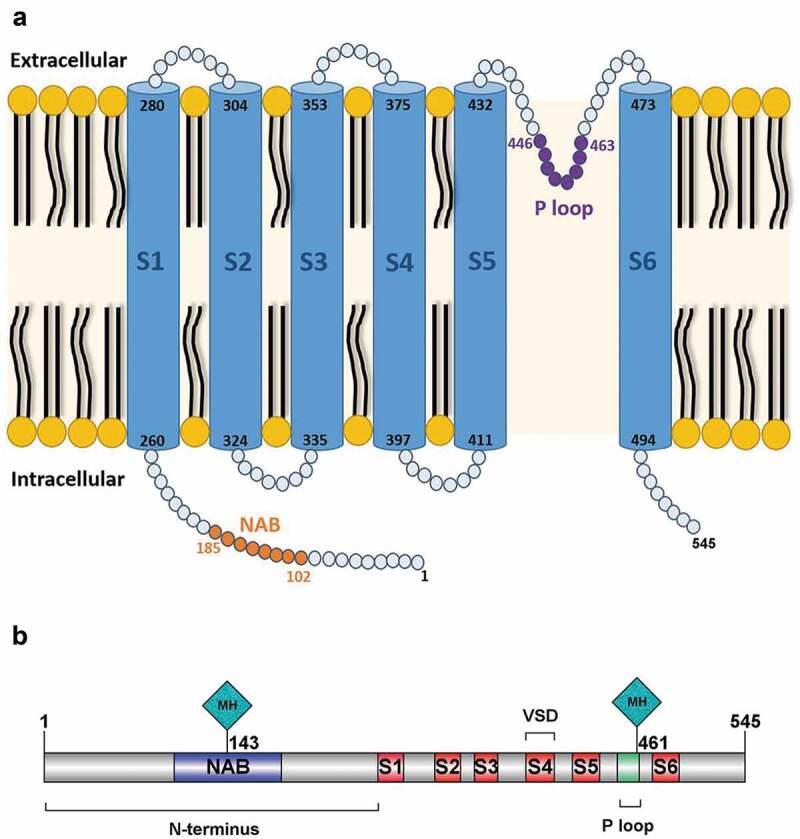
KCNV2 Protein Structure. (a) Graphical representation of the alpha-subunit of the potassium channel (Kv8.2) encoded by KCNV2. The subunit consists of: (i) a highly conserved tetramerization domain; N-terminal A and B box (NAB) that facilitates interaction between compatible alpha-subunits; (ii) 6 transmembrane domains (S1-S6); (iii) extracellular and intracellular loop segments; and (iv) an ultra-conserved potassium selective motif in the pore-forming loop between S5-S6 (P loop). (b) The two most frequently reported variants in patients with KCNV2-retinopathy are c.1381 G > A (p.Gly461Arg) and c.427 G > T (p.Glu143X), and may thereby represent mutational hotspots (MH); both locations are represented with a lozenge-shaped site in the gene annotation.
Reported sequence variants
More than 100 patients and 95 different variants have been reported across 22 studies (4,7,10–14,20-23,36–46). Supplementary table summarizes the previously described variants, including conservation, in silico prediction and frequency assessment. Of these, 46 are missense variants (two of which are located in the last codon and generate an extension of 61 amino acids), 21 nonsense variants, 14 intragenic deletions (13 causing a frameshift), 3 out-of-frame insertions (with subsequent frameshift), 4 duplications (2 causing frameshift), 6 gross deletions of an entire exon or the whole gene, and 1 complex rearrangement (c.19_1356 + 9571 delinsCATTTG; p.Arg7HisfsX57). Approximately two thirds of these variants are located in the amino-terminal region (N-terminus and NAB domains).
The most frequently reported variant is c.1381 G > A (p.Gly461Arg), located in the third residue of the ultra-conserved GYG-tripeptide motif (47). It has been reported as a disease-causing variant in 35 patients, either in the homozygous or compound heterozygous state (4,10,14,23,36–40). Moreover, it accounts for approximately 83% of all disease-causing variants reported in the P loop domain. Based on its frequency, p.Gly461Arg may represent a mutational hotspot (Figure 3). Another possible mutational hotspot is located in the amino-terminal A and B box (NAB), c.427 G > T (p.Glu143X). This nonsense variant causes premature protein termination and is predicted to cause loss of function. It has been reported in 31 patients (7,12,14) and accounts for approximately 41% of all disease-causing variants in the highly conserved NAB domain.
Directions for therapy
There is currently no approved treatment for KCNV2-retinopathy, apart from symptomatic supportive measures including tinted spectacles/contact lenses and access to low visual aids/assistive technologies.
Gene supplementation therapy offers the possibility to improve the outcomes of several forms of monogenic inherited retinal disorders (IRD). It aims to deliver a “normal” copy of a defective gene that is no longer able to produce viable protein. Data from long-term follow-up studies of the pivotal gene therapy trials for RPE65-related retinal dystrophy (RPE65-RD) (OMIM #204100) are promising (48–50) and have resulted in the first FDA/EMA approved gene therapy for an ocular condition. One of the main factors that prompted interest in using gene therapy for RPE65 was that despite the profound visual loss in animal models and humans, there was a wide window of structural photoreceptor preservation for therapeutic intervention (51).
KCNV2-retinopathy may also be a suitable target for gene therapy. Firstly, KCNV2 is a small gene that can readily be packaged within the viral vector of choice, AAV. Secondly, there is a mouse model that recapitulates human disease very closely (including the ERG phenotype) and so can be targeted for therapeutic intervention (52). Thirdly, there are favourable functional and structural phenotypic features. In 1984, Alexander and Fishman reported three cases, of which two had the ‘typical’ supernormal rod ERGs but without nyctalopia, suggestive of good rod function despite abnormal scotopic ERG (53). Further functional studies have suggested that inner-retinal function and the phototransduction cascade are relatively normal in KCNV2-retinopathy (16,20). Structurally, although morphological changes at the fovea are evident on OCT in early stages of the disease, there appears to be a broad window of opportunity for therapeutic intervention before advanced structural changes and marked photoreceptor cell loss have occurred (13). This is supported by findings in the KCNV2 knock-out mouse, where approximately 80% of cones are still intact by six months of age as compared to wild type, which if similar to humans, may allow for relatively late photoreceptor-directed treatment (52). However, further clinical and pre-clinical research, including prospective natural history studies, are needed to establish the optimal window for intervention, appropriate structural and functional (both retinal and visual) end-points to monitor both safety and efficacy, and identify participants most likely to benefit.
There are three main routes being explored to deliver a gene therapy product to the retina: via (i) intravitreal, (ii) subretinal or (iii) suprachoroidal injection. Although intravitreal injections are less invasive than subretinal injections and may be readily delivered by non-specialist surgeons, most currently available AAVs are unable to efficiently and reproducibly reach the outer retina – mainly due to the inner limiting membrane (ILM) acting as a physical barrier, thereby limiting transduction to the inner retinal layers (54). Modified AAVs – particularly serotype 2 – are proposed to be more effective in penetrating the ILM and allow broader transduction (55–59). However, in the case of KCNV2-retinopathy, which primarily affects photoreceptors, subretinal delivery of a gene therapy product is currently likely to be the most effective approach.
Pharmacological approaches with potassium channel modulators may provide a promising option for the treatment of several conditions, including cardiac arrhythmias, epilepsy, depression, autoimmune diseases and many others (60–63). Chemical agents that affect potassium channel functions may either activate or block current flow or alter channel gating (61). In theory, certain patients with KCNV2-retinopathy (depending on the effect of specific sequence variants on protein/channel structure/function) may also benefit from potassium channel modulators. How these might be safely delivered long term would also need to be addressed.
Conclusions and future directions
Evidence from animal models and clinical studies identify KCNV2-retinopathy as a severe early onset retinal dystrophy with slowly progressive maculopathy, that might be amenable to future treatments. Phenotypic studies suggest that there is indeed relative structural preservation of retinal architecture and intact phototransduction (16,20,21,52). Multiple gene therapy trials for IRDs are ongoing (48,64–67), with the first approved gene therapy for RPE65-RD now available (NCT00999609). Further pre-clinical work in animal models and iPSC-derived models is needed to explore safety, efficacy and dosing of potential gene or drug therapy to facilitate translation to human clinical trials.
In the UK, KCNV2 retinopathy accounts for 0.7% of the pedigrees with IRDs (68). Prospective data in large molecularly confirmed cohorts are the cornerstone for understanding the natural history of the disease. This is a prerequisite for the best-informed design of future therapy trials, as well as for patient counselling and advice on prognosis. Detailed phenotyping of patients with KCNV2-retinopathy will facilitate the identification of an optimal window for intervention, provide specific parameters to quantify treatment effects and define clinical endpoints, and help identify suitable patients for therapeutic intervention.
Supplementary Material
Funding Statement
This research was supported by grants from the National Institute for Health Research Biomedical Research Centre at Moorfields Eye Hospital NHS Foundation Trust and UCL Institute of Ophthalmology, Onassis Foundation, Leventis Foundation, The Wellcome Trust (099173/Z/12/Z), Moorfields Eye Hospital Special Trustees, Moorfields Eye Charity, Retina UK, and the Foundation Fighting Blindness (USA).
Declaration of interest
The authors alone are responsible for the content and writing of this article not an official position of the institution. MM and MG consult for MeiraGTx Ltd.
Contributors
TACG, MG reviewed the literature, drafted the manuscript and provided critical revision. AGR, MM conceived, supervised, and revised the manuscript.
Supplementary material
Supplemental data for this article can be accessed in the journal website.
References
- 1.Michaelides M, Hardcastle AJ, Hunt DM, Moore AT.. Progressive cone and cone-rod dystrophies: phenotypes and underlying molecular genetic basis. Surv Ophthalmol. 2006. May-Jun;51(3):232–58. doi: 10.1016/j.survophthal.2006.02.007. [DOI] [PubMed] [Google Scholar]
- 2.Gill JS, Georgiou M, Kalitzeos A, Moore AT, Michaelides M. Progressive cone and cone-rod dystrophies: clinical features, molecular genetics and prospects for therapy. Br J Ophthalmol. 2019. January 24;103(5):711–20. pii: bjophthalmol-2018-313278. doi: 10.1136/bjophthalmol-2018-313278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Michaelides M, Holder GE, Webster AR, Hunt DM, Bird AC, Fitzke FW, Mollon JD, Moore AT. A detailed phenotypic study of “cone dystrophy with supernormal rod ERG”. Br J Ophthalmol. 2005. March;89(3):332–39. doi: 10.1136/bjo.2004.050567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vincent A, Wright T, Garcia-Sanchez Y, Kisilak M, Campbell M, Westall C, Héon E. Phenotypic characteristics including in vivo cone photoreceptor mosaic in KCNV2-related “cone dystrophy with supernormal rod electroretinogram”. Invest Ophthalmol Vis Sci. 2013. January 30;54(1):898–908. doi: 10.1167/iovs.12-10971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gouras P, Eggers HM, MacKay CJ. Cone dystrophy, nyctalopia, and supernormal rod responses. A new retinal degeneration. Arch Ophthalmol. 1983. May;101(5):718–24. doi: 10.1001/archopht.1983.01040010718003. [DOI] [PubMed] [Google Scholar]
- 6.Stone EM, Andorf JL, Whitmore SS, DeLuca AP, Giacalone JC, Streb LM, Braun TA, Mullins RF, Scheetz TE, Sheffield VC, et al. Clinically focused molecular investigation of 1000 consecutive families with inherited retinal disease. Ophthalmology. 2017. September;124(9):1314–31. doi: 10.1016/j.ophtha.2017.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wu H, Cowing JA, Michaelides M, Wilkie SE, Jeffery G, Jenkins SA, Mester V, Bird AC, Robson AG, Holder GE, et al. Mutations in the gene KCNV2 encoding a voltage-gated potassium channel subunit cause “cone dystrophy with supernormal rod electroretinogram” in humans. Am J Hum Genet. 2006. September;79(3):574–79. doi: 10.1086/507568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ottschytsch N, Raes A, Van Hoorick D, Snyders DJ. Obligatory heterotetramerization of three previously uncharacterized Kv channel alpha-subunits identified in the human genome. Proc Natl Acad Sci U S A. 2002. June 11;99(12):7986–91. doi: 10.1073/pnas.122617999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gayet-Primo J, Yaeger DB, Khanjian RA, Puthussery T. Heteromeric Kv2/Kv8.2 channels mediate delayed rectifier potassium currents in primate photoreceptors. J Neurosci. 2018. April 4;38(14):3414–27. doi: 10.1523/JNEUROSCI.2440-17.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wissinger B, Dangel S, Ja¨gle H, Hansen L, Baumann B, Rudolph G, Wolf C, Bonin M, Koeppen K, Ladewig T, et al. Cone dystrophy with supernormal rod response is strictly associated with mutations in KCNV2. Invest Ophthalmol Vis Sci. 2008. February;49(2):751–57. doi: 10.1167/iovs.07-0471. [DOI] [PubMed] [Google Scholar]
- 11.Zobor D, Kohl S, Wissinger B, Zrenner E, Jägle H.Rod and cone function in patients with KCNV2 retinopathy. PLoS One. 2012;7(10):e46762.doi: 10.1371/journal.pone.0046762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Khan AO, Alrashed M, Alkuraya FS. ‘Cone dystrophy with supranormal rod response’ in children. Br J Ophthalmol. 2012. March;96(3):422–26. doi: 10.1136/bjophthalmol-2011-300271. [DOI] [PubMed] [Google Scholar]
- 13.Robson AG, Webster AR, Michaelides M, Downes SM, Cowing JA, Hunt DM, Moore AT, Holder GE. “Cone dystrophy with supernormal rod electroretinogram”: a comprehensive genotype/phenotype study including fundus autofluorescence and extensive electrophysiology. Retina. 2010. January;30(1):51–62. doi: 10.1097/IAE.0b013e3181bfe24e. [DOI] [PubMed] [Google Scholar]
- 14.Sergouniotis PI, Holder GE, Robson AG, Michaelides M, Webster AR, Moore AT. High-resolution optical coherence tomography imaging in KCNV2 retinopathy. Br J Ophthalmol. 2012. February;96(2):213–17. doi: 10.1136/bjo.2011.203638. [DOI] [PubMed] [Google Scholar]
- 15.Georgiou M, Kalitzeos A, Patterson EJ, Dubra A, Carroll J, Michaelides M. Adaptive optics imaging of inherited retinal diseases. Br J Ophthalmol. 2018. August;102(8):1028–35. doi: 10.1136/bjophthalmol-2017-311328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vincent A, Robson AG, Holder GE. Pathognomonic (diagnostic) ERGs. A review and update. Retina. 2013. January;33(1):5–12. doi: 10.1097/IAE.0b013e31827e2306. [DOI] [PubMed] [Google Scholar]
- 17.McCulloch DL, Marmor MF, Brigell MG, Hamilton R, Holder GE, Tzekov R, Bach M. ISCEV Standard for full-field clinical electroretinography (2015 update). Doc Ophthalmol. 2015. February;130(1):1–12. doi: 10.1007/s10633-014-9473-7. [DOI] [PubMed] [Google Scholar]
- 18.Sustar M, Holder GE, Kremers J, Barnes CS, Lei B, Khan NW, Robson AG. ISCEV extended protocol for the photopic On-Off ERG. Doc Ophthalmol. 2018. June;136(3):199–206. doi: 10.1007/s10633-018-9645-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bach M, Brigell MG, Hawlina M, Holder GE, Johnson MA, McCulloch DL, Meigen T, Viswanathan S. ISCEV standard for clinical pattern electroretinography (PERG): 2012 update. Doc Ophthalmol. 2013. February;126(1):1–7. doi: 10.1007/s10633-012-9353-y. [DOI] [PubMed] [Google Scholar]
- 20.Collison FT, Park JC, Fishman GA, Stone EM, McAnany JJ. Two-color pupillometry in KCNV2 retinopathy. Doc Ophthalmol. 2019. August;139(1):11–20. doi: 10.1007/s10633-019-09691-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stockman A, Henning GB, Michaelides M, Moore AT, Webster AR, Cammack J, Ripamonti C. Cone dystrophy with “supernormal” rod ERG: psychophysical testing shows comparable rod and cone temporal sensitivity losses with no gain in rod function. Invest Ophthalmol Vis Sci. 2014. February 10;55(2):832–40. doi: 10.1167/iovs.13-12919. [DOI] [PubMed] [Google Scholar]
- 22.Zelinger L, Wissinger B, Eli D, Kohl S, Sharon D, Banin E. Cone dystrophy with supernormal rod response: novel KCNV2 mutations in an underdiagnosed phenotype. Ophthalmology. 2013. November;120(11):2338–43. doi: 10.1016/j.ophtha.2013.03.031. [DOI] [PubMed] [Google Scholar]
- 23.Wissinger B, Schaich S, Baumann B, Bonin M, Jägle H, Friedburg C, Varsányi B, Hoyng CB, Dollfus H, Heckenlively JR, et al. Large deletions of the KCNV2 gene are common in patients with cone dystrophy with supernormal rod response. Hum Mutat. 2011. December;32(12):1398–406. doi: 10.1002/humu.21580. [DOI] [PubMed] [Google Scholar]
- 24.Czirják G, Tóth ZE, Enyedi P. Characterization of the heteromeric potassium channel formed by kv2.1 and the retinal subunit kv8.2 in Xenopus oocytes. J Neurophysiol. 2007. September;98(3):1213–22. doi: 10.1152/jn.00493.2007. [DOI] [PubMed] [Google Scholar]
- 25.Hölter P, Kunst S, Wolloscheck T, Kelleher DK, Sticht C, Wolfrum U, Spessert R. The retinal clock drives the expression of Kcnv2, a channel essential for visual function and cone survival. Invest Ophthalmol Vis Sci. 2012. October 5;53(11):6947–54. doi: 10.1167/iovs.12-10234. [DOI] [PubMed] [Google Scholar]
- 26.Aslanidis A, Karlstetter M, Walczak Y, Jägle H, Langmann T. RETINA-specific expression of Kcnv2 is controlled by cone-rod homeobox (Crx) and neural retina leucine zipper (Nrl). Adv Exp Med Biol. 2014;801:31–41. [DOI] [PubMed] [Google Scholar]
- 27.Tian C, Zhu R, Zhu L, Qiu T, Cao Z, Kang T. Potassium channels: structures, diseases, and modulators. Chem Biol Drug Des. 2014. January;83(1):1–26. doi: 10.1111/cbdd.12237. [DOI] [PubMed] [Google Scholar]
- 28.Kuang Q, Purhonen P, Hebert H. Structure of potassium channels. Cell Mol Life Sci. 2015. October;72(19):3677–93. doi: 10.1007/s00018-015-1948-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pongs O, Schwarz JR. Ancillary subunits associated with voltage-dependent K+ channels. Physiol Rev. 2010. April;90(2):755–96. doi: 10.1152/physrev.00020.2009. [DOI] [PubMed] [Google Scholar]
- 30.Shen NV, Pfaffinger PJ. Molecular recognition and assembly sequences involved in the subfamily-specific assembly of voltage-gated K+ channel subunit proteins. Neuron. 1995. March;14(3):625–33. doi: 10.1016/0896-6273(95)90319-4. [DOI] [PubMed] [Google Scholar]
- 31.Kreusch A, Pfaffinger PJ, Stevens CF, Choe S. Crystal structure of the tetramerization domain of the Shaker potassium channel. Nature. 1998. April 30;392(6679):945–48. doi: 10.1038/31978. [DOI] [PubMed] [Google Scholar]
- 32.Lecar H, Larsson HP, Grabe M. Electrostatic model of S4 motion in voltage-gated ion channels. Biophys J. 2003. November;85(5):2854–64. doi: 10.1016/S0006-3495(03)74708-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Catterall WA. Ion channel voltage sensors: structure, function, and pathophysiology. Neuron. 2010. September 23;67(6):915–28. doi: 10.1016/j.neuron.2010.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Strang C, Cushman SJ, DeRubeis D, Peterson D, Pfaffinger PJ. A central role for the T1 domain in voltage-gated potassium channel formation and function. J Biol Chem. 2001. July 27;276(30):28493–502. doi: 10.1074/jbc.M010540200. [DOI] [PubMed] [Google Scholar]
- 35.Smith KE, Wilkie SE, Tebbs-Warner JT, Jarvis BJ, Gallasch L, Stocker M, Hunt DM. Functional analysis of missense mutations in Kv8.2 causing cone dystrophy with supernormal rod electroretinogram. J Biol Chem. 2012. December 21;287(52):43972–83. doi: 10.1074/jbc.M112.388033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Thiagalingam S, McGee TL, Weleber RG, Sandberg MA, Trzupek KM, Berson EL, Dryja TP. Novel mutations in the KCNV2 gene in patients with cone dystrophy and a supernormal rod electroretinogram. Ophthalmic Genet. 2007. September;28(3):135–42. doi: 10.1080/13816810701503681. [DOI] [PubMed] [Google Scholar]
- 37.Ben Salah S, Kamei S, Sénéćhal A, Lopez S, Bazalgette C, Bazalgette C, Eliaou CM, Zanlonghi X, Hamel CP. Novel KCNV2 mutations in cone dystrophy with supernormal rod electroretinogram. Am J Ophthalmol. 2008. June;145(6):1099–106. doi: 10.1016/j.ajo.2008.02.004. [DOI] [PubMed] [Google Scholar]
- 38.Thiadens AA, Phan TM, Zekveld-Vroon RC, Leroy BP, van den Born LI, Hoyng CB, Klaver CC, Roosing S, Pott JW, van Schooneveld MJ, et al. Clinical course, genetic etiology, and visual outcome in cone and cone-rod dystrophy. Ophthalmology. 2012. April;119(4):819–26. doi: 10.1016/j.ophtha.2011.10.011 [DOI] [PubMed] [Google Scholar]
- 39.Fujinami K, Tsunoda K, Nakamura N, Kato Y, Noda T, Shinoda K, Tomita K, Hatase T, Usui T, Akahori M, et al. Molecular characteristics of four Japanese cases with KCNV2 retinopathy: report of novel disease-causing variants. Mol Vis. 2013. July;20(19):1580–90. [PMC free article] [PubMed] [Google Scholar]
- 40.Friedburg C, Wissinger B, Schambeck M, Bonin M, Kohl S, Lorenz B. Long-term follow-up of the human phenotype in three siblings with cone dystrophy associated with a homozygous p.G461R mutation of KCNV2. Invest Ophthalmol Vis Sci. 2011. November 7;52(12):8621–29. doi: 10.1167/iovs.11-8187. [DOI] [PubMed] [Google Scholar]
- 41.Oishi M, Oishi A, Gotoh N, Ogino K, Higasa K, Iida K, Makiyama Y, Morooka S, Matsuda F, Yoshimura N. Next-generation sequencing-based comprehensive molecular analysis of 43 Japanese patients with cone and cone-rod dystrophies. Mol Vis. 2016. February;20(22):150–60. [PMC free article] [PubMed] [Google Scholar]
- 42.Huang L, Xiao X, Li S, Jia X, Wang P, Sun W, Xu Y, Xin W, Guo X, Zhang Q. Molecular genetics of cone-rod dystrophy in Chinese patients: new data from 61 probands and mutation overview of 163 probands. Exp Eye Res. 2016. May;146:252–58. doi: 10.1016/j.exer.2016.03.015. [DOI] [PubMed] [Google Scholar]
- 43.Kutsuma T, Katagiri S, Hayashi T, Yoshitake K, Iejima D, Gekka T, Kohzaki K, Mizobuchi K, Baba Y, Terauchi R, et al. Novel biallelic loss-of-function KCNV2 variants in cone dystrophy with supernormal rod responses. Doc Ophthalmol. 2019. June;138(3):229–39. doi: 10.1007/s10633-019-09679-6. [DOI] [PubMed] [Google Scholar]
- 44.Lenis TL, Dhrami-Gavazi E, Lee W, Mukkamala SK, Tabacaru MR, Yannuzzi L, Gouras P, Tsang SH. Novel compound heterozygous mutations resulting in cone dystrophy with supernormal rod response. JAMA Ophthalmol. 2013. November;131(11):1482–85. doi: 10.1001/jamaophthalmol.2013.4681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Grigg JR, Holder GE, Billson FA, Korsakova M, Jamieson RV. The importance of electrophysiology in revealing a complete homozygous deletion of KCNV2. J Aapos. 2013. December;17(6):641–43. doi: 10.1016/j.jaapos.2013.08.006. [DOI] [PubMed] [Google Scholar]
- 46.Xu D, Su D, Nusinowitz S, Sarraf D. Central ellipsoid loss associated with cone dystrophy and KCNV2 mutation. Retin Cases Brief Rep. 2018. Fall;12(Suppl 1):S59–S62. doi: 10.1097/ICB.0000000000000661 [DOI] [PubMed] [Google Scholar]
- 47.Heginbotham L, Lu Z, Abramson T, MacKinnon R. Mutations in the K+ channel signature sequence. Biophys J. 1994. April;66(4):1061–67. doi: 10.1016/S0006-3495(94)80887-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bainbridge JW, Mehat MS, Sundaram V, Robbie SJ, Barker SE, Ripamonti C, Georgiadis A, Mowat FM, Beattie SG, Gardner PJ, et al. Long-term effect of gene therapy on Leber’s congenital amaurosis. N Engl J Med. 2015. May 14;372(20):1887–97. doi: 10.1056/NEJMoa1414221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Testa F, Maguire AM, Rossi S, Pierce EA, Melillo P, Marshall K, Banfi S, Surace EM, Sun J, Acerra C, et al. Three-year follow-up after unilateral subretinal delivery of adeno-associated virus in patients with Leber congenital Amaurosis type 2. Ophthalmology. 2013. June;120(6):1283–91. doi: 10.1016/j.ophtha.2012.11.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Weleber RG, Pennesi ME, Wilson DJ, Kaushal S, Erker LR, Jensen L, McBride MT, Flotte TR, Humphries M, Calcedo R, et al. Results at 2 years after gene therapy for RPE65-deficient leber congenital amaurosis and severe early-childhood-onset retinal dystrophy. Ophthalmology. 2016. July;123(7):1606–20. doi: 10.1016/j.ophtha.2016.03.003. [DOI] [PubMed] [Google Scholar]
- 51.Chung DC, Traboulsi EI. Leber congenital amaurosis: clinical correlations with genotypes, gene therapy trials update, and future directions. J Aapos. 2009. December;13(6):587–92. doi: 10.1016/j.jaapos.2009.10.004. [DOI] [PubMed] [Google Scholar]
- 52.Hart NS, Mountford JK, Voigt V, Fuller-Carter P, Barth M, Nerbonne JM, Hunt DM, Carvalho LS. The role of the voltage-gated potassium channel proteins Kv8.2 and Kv2.1 in vision and retinal disease: insights from the study of mouse gene knock-out mutations. eNeuro. 2019. February 25;6(1):E.NEURO.0032-19.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Alexander KR, Fishman GA. Supernormal scotopic ERG in cone dystrophy. Br J Ophthalmol. 1984. February;68(2):69–78. doi: 10.1136/bjo.68.2.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dalkara D, Kolstad KD, Caporale N, Visel M, Klimczak RR, Schaffer DV, Flannery JG. Inner limiting membrane barriers to AAV-mediated retinal transduction from the vitreous. Mol Ther. 2009. December;17(12):2096–102. doi: 10.1038/mt.2009.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Reid CA, Ertel KJ, Lipinski DM. Improvement of photoreceptor targeting via intravitreal delivery in mouse and human retina using combinatory rAAV2 capsid mutant vectors. Invest Ophthalmol Vis Sci. 2017. December 1;58(14):6429–39. doi: 10.1167/iovs.17-22281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yin L, Greenberg K, Hunter JJ, Dalkara D, Kolstad KD, Masella BD, Wolfe R, Visel M, Stone D, Libby RT, et al. Intravitreal injection of AAV2 transduces macaque inner retina. Invest Ophthalmol Vis Sci. 2011. April 25;52(5):2775–83. doi: 10.1167/iovs.10-6250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jüttner J, Szabo A, Gross-Scherf B, Morikawa RK, Rompani SB, Hantz P, Szikra T, Esposti F, Cowan CS, Bharioke A, et al. Targeting neuronal and glial cell types with synthetic promoter AAVs in mice, non-human primates and humans. Nat Neurosci. 2019. August;22(8):1345–56. doi: 10.1038/s41593-019-0431-2. [DOI] [PubMed] [Google Scholar]
- 58.Ali RR, Reichel MB, De Alwis M, Kanuga N, Kinnon C, Levinsky RJ, Hunt DM, Bhattacharya SS, Thrasher AJ. Adeno-associated virus gene transfer to mouse retina. Hum Gene Ther. 1998. January 1;9(1):81–86. doi: 10.1089/hum.1998.9.1-81. [DOI] [PubMed] [Google Scholar]
- 59.Hellström M, Ruitenberg MJ, Pollett MA. Cellular tropism and transduction properties of seven adeno-associated viral vector serotypes in adult retina after intravitreal injection. Gene Ther. 2009. April;16(4):521–32. doi: 10.1038/gt.2008.178. [DOI] [PubMed] [Google Scholar]
- 60.Robertson DW, Steinberg MI. Potassium channel modulators: scientific applications and therapeutic promise. J Mec Chem. 1990. June;33(6):1529–41. doi: 10.1021/jm00168a001. [DOI] [PubMed] [Google Scholar]
- 61.Judge SI, Smith PJ, Stewart PE, Bever Jr. CT Jr.. Potassium channel blockers and openers as CNS neurologic therapeutic agents. Recent Pat CNS Drug Discov. 2007. November;2(3):200–28. doi: 10.2174/157488907782411765. [DOI] [PubMed] [Google Scholar]
- 62.Wulff H, Zhorov BS. K+ channel modulators for the treatment of neurological disorders and autoimmune diseases. Chem Rev. 2008. May;108(5):1744–73. doi: 10.1021/cr078234p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lei M, Wu L, Terrar DA, Huang CL. Modernized classification of cardiac antiarrhythmic drugs. Circulation. 2018. October 23;138(17):1879–96. doi: 10.1161/CIRCULATIONAHA.118.035455. [DOI] [PubMed] [Google Scholar]
- 64.Hirji N, Aboshiha J, Georgiou M, Bainbridge J, Michaelides M. Achromatopsia: clinical features, molecular genetics, animal models and therapeutic options. Ophthalmic Genet. 2018. April;39(2):149–57. doi: 10.1080/13816810.2017.1418389. [DOI] [PubMed] [Google Scholar]
- 65.Maguire AM, Russell S, Wellman JA, Chung DC, Yu ZF, Tillman A, Wittes J, Pappas J, Elci O, Marshall KA, et al. Efficacy, safety, and durability of voretigene neparvovec-rzyl in RPE65 mutation-associated inherited retinal dystrophy: results of phase 1 and 3 trials. Ophthalmology. 2019. September;126(9):1273–85. doi: 10.1016/j.ophtha.2019.06.017. [DOI] [PubMed] [Google Scholar]
- 66.MacLaren RE, Groppe M, Barnard AR, Cottriall CL, Tolmachova T, Seymour L, Clark KR, During MJ, Cremers FP, Black GC, et al. Retinal gene therapy in patients with choroideremia: initial findings from a phase 1/2 clinical trial. Lancet. 2014. March 29;383(9923):1129–37. doi: 10.1016/S0140-6736(13)62117-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Rahman N, Georgiou M, Khan KN, Michaelides M. Macular dystrophies: clinical and imaging features, molecular genetics and therapeutic options. Br J Ophthalmol. 2020. Apr;104(4):451–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Pontikos N, Arno G, Jurkute N, Schiff E, Ba-Abbad R, Malka S, Gimenez A, Georgiou M, Wright G, Armengol M, et al. Genetic basis of inherited retinal disease in a molecularly characterised cohort of over 3000 families from the United Kingdom. Ophthalmology. 2020. doi: 10.1016/j.ophtha.2020.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.