

SUMMARY
Current models suggest that chromosome domains segregate into either an active (A) or inactive (B) compartment. B-compartment chromatin is physically separated from the A compartment and compacted by the nuclear lamina. To examine these models in the developmental context of C. elegans embryogenesis, we undertook chromosome tracing to map the trajectories of entire autosomes. Early embryonic chromosomes organized into an unconventional barbell-like configuration, with two densely folded B compartments separated by a central A compartment. Upon gastrulation, this conformation matured into conventional A/B compartments. We used unsupervised clustering to uncover subpopulations with differing folding properties and variable positioning of compartment boundaries. These conformations relied on tethering to the lamina to stretch the chromosome; detachment from the lamina compacted, and allowed intermingling between, A/B compartments. These findings reveal the diverse conformations of early embryonic chromosomes and uncover a previously unappreciated role for the lamina in systemic chromosome stretching.
Graphical Abstract
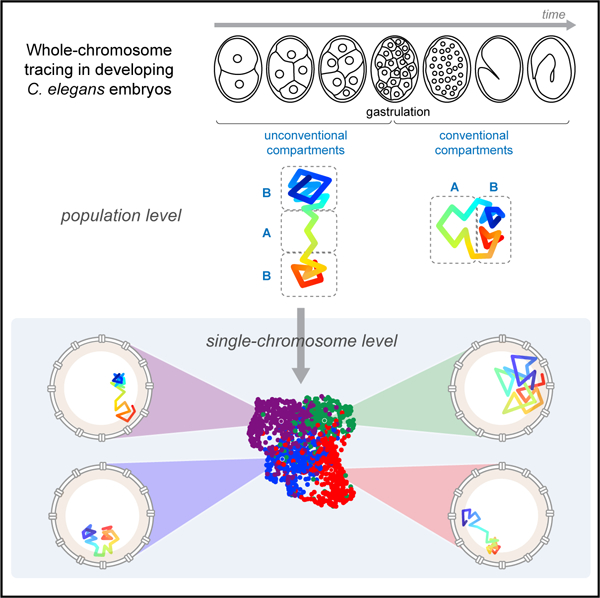
In Brief
Sawh et al. use chromosome tracing to map whole-chromosome architecture during C. elegans embryogenesis. They show conventional A/B compartments arise at gastrulation. Prior to this developmental milestone, chromosomes adopt an extended barbell-like configuration, which requires stretching by the nuclear lamina. Unsupervised clustering further uncovers the prevalent conformations in single chromosomes.
INTRODUCTION
Since the experiments of Emil Heitz, it has been recognized that the nucleus is heterogeneous in its organization (Heitz, 1928), but only recently have tools become available to precisely determine the three-dimensional (3D) arrangement of chromosomes. The advent of chromosome conformation capture and advanced DNA fluorescence in situ hybridization (FISH) technologies led to the discovery that chromosomes occupy territories within nuclei, where they fold into large compartments and smaller topologically associated domains (TADs) (Beliveau et al., 2015; Cremer and Cremer, 2001; Dekker et al., 2002; Dixon et al., 2012; Hou et al., 2012; Lieberman-Aiden et al., 2009; Nora et al., 2012; Sexton et al., 2012). A-type compartments are gene dense and enriched in open, active chromatin (euchromatin). B compartments reflect compact chromatin (heterochromatin) marked by repetitive sequences, repressive histone modifications, and lamina association (Bonev and Cavalli, 2016; van Steensel and Belmont, 2017). In addition, B compartments are identified by physical association between noncontiguous regions of heterochromatin along the chromosome (Ahringer and Gasser, 2018; Lieberman-Aiden et al., 2009).
The mechanisms that govern the formation of compartment boundaries are beginning to emerge. Compartments reflect transcriptional status, but transcription is neither necessary nor sufficient for their establishment (Hug and Vaquerizas, 2018). Cohesin and CCCTC-binding factor (CTCF) promote looping to form TADs but suppress some compartments (Nora et al., 2017; Nuebler et al., 2018; Schwarzer et al., 2017). B compartments also depend on the interaction of DNA with the nuclear lamina to promote and radially sequester heterochromatin in lamina-associated domains (LADs) (Falk et al., 2019; Luperchio et al., 2014; van Steensel and Belmont, 2017). Recent genome-wide chromosome conformation capture (Hi-C) studies of cultured cells have suggested that the lamina interacts with B-compartment chromatin to promote its compaction and inhibit interactions with A compartments (Ulianov et al., 2019; Zheng et al., 2018), but it is unclear whether the lamina performs these roles in animals and at all stages of life.
One complication of studying chromosome architecture is the variability of conformations that exists both in populations of cultured cells and in embryos. TAD and compartment structures vary from cell to cell, but consensus boundaries are often defined by population averages (Bintu et al., 2018; Finn et al., 2019; Flyamer et al., 2017; Nagano et al., 2013, 2017; Ramani et al., 2017; Stevens et al., 2017; Szabo et al., 2018; Tan et al., 2018; Wang et al., 2016). Recent Hi-C studies have shown that TAD and compartment structures are particularly dynamic in embryos. In zebrafish, TADs and compartments are present prior to zygotic genome activation (ZGA), but they disappear during ZGA and recover only later in development (Kaaij et al., 2018). In Drosophila and mouse early embryonic development, TADs and compartments arise progressively following ZGA (Du et al., 2017; Flyamer et al., 2017; Hug et al., 2017; Ke et al., 2017; Stadler et al., 2017). In C. elegans, the architecture of early embryonic chromosomes is unknown. However, maturing embryos have well-delineated TADs on the X chromosome, set by the dosage compensation machinery (Anderson et al., 2019; Crane et al., 2015; Brejc et al., 2017). Weaker TADs and compartments along the autosomes have also been identified (Crane et al., 2015), but it is unknown how or when they are generated. These collective observations have begun to elucidate embryonic structures and raise the questions of which chromosome conformations predominate in vivo and how they arise during development. New approaches are needed to reveal the diversity and shared properties of chromosome conformation in developing cell populations.
Here, we adapt high-throughput multiplexed DNA FISH to trace entire individual chromosomes in intact C. elegans embryos. Chromosome tracing by multiplexed FISH has the advantage of precisely and directly determining chromosome trajectories in single cells while preserving cellular and tissue integrity (Bintu et al., 2018; Cardozo Gizzi et al., 2019; Mateo et al., 2019; Nir et al., 2018; Wang et al., 2016). We examine multiple autosomal chromosomes during early embryogenesis and find that, on average, they are organized in a barbell-like arms-apart configuration, where epigenetically similar domains are physically separated. This organization then matures into conventional A/B compartments at the onset of gastrulation. We find that the early embryo signature is caused by two factors, the presence of structural heterogeneity in the population and the influence of the nuclear lamina. To gain a deeper understanding of the heterogeneity, we introduce unsupervised clustering to uncover and define groups of chromosomes that share similar conformations within the population. These groups are distinguished by regions of the chromosomes with greater variability. The subpopulations vary in their compartment boundaries and fundamental folding patterns; none are identical to the population average. Finally, we uncover a surprising role for the nuclear lamina to separate similar domains and thereby override the self-attraction typical of epigenetically similar regions.
RESULTS
Visualization of Embryonic Chromosomes with Chromosome Tracing
To define the higher-order architecture of interphase chromosomes of early embryos, we adapted chromosome tracing by multiplexed FISH (Bintu et al., 2018; Wang et al., 2016) for C. elegans (see STAR Methods). We generated labeled primary FISH probes that recognized 100-kb-sized regions spanning 22 TADs on the largest autosome, chromosome V (chrV), which were previously defined by Hi-C of mixed/late-stage embryos (Figure 1A; Crane et al., 2015). When probed en masse, primary FISH probes revealed the whole chromosome territory (Figure 1B). Primary probes also carried TAD-specific readout sequences in their tails (Figure 1A), enabling us to identify individual TADs in sequential hybridizations on the same sample using unique secondary probes. We segmented the images in 3D using contiguous primary probe signal as the marker of chromosome territories and traced the conformation of chromosomes within each volume (Figures 1A–1E and S1; see STAR Methods). Direct comparison of mean distances measured between TADs in chromosome traces to inverse contact frequencies from previous Hi-C measurements (Crane et al., 2015) yielded an overall high correlation (Pearson correlation coefficient = 0.90), corroborating the tracing results (Figure 1F). To test the robustness of our measurements, we performed down-sampling of the chromosome traces, which produced similar measurements (Figure S2A).
Figure 1. Whole-Chromosome Tracing in C. elegans Embryos by Multiplexed FISH.

(A) Schematic of chrV annotated with TAD boundaries (orange ticks), Hi-C compartments (gray), boundaries of the chromosome arms and center defined by epigenetic state and LADs, FISH probe positions (colored dots), and experimental design.
(B) Example of raw data z-projections on an ~12-cell embryo. Scale bar, 5 µm. Foci from the same interphase nucleus are circled in blue as an example.
(C) All foci detected in (B) colored by TAD identity (left), and the primary probe reconstruction (right).
(D) Z-projection of 3D-segmentated chromosome territories by contiguous primary probe signal, where each volume is colored differently. A zoom-in of a region with detected segments and primary probe signal shown.
(E) Examples chromosome traces, colored blue to red as in (A).
(F) Pearson correlation of mean distances between TADs measured by FISH and inverse contact frequencies between the same genomic regions measured by Hi-C.
See also Figure S1.
Conventional Compartments Arise at the Onset of Gastrulation
To define early embryonic chromosome architecture, we plotted the mean distances between each TAD pair in a matrix (Figures 2A and S2A). We normalized the raw distances with their genomic distances while considering the polymer nature of the chromosome (where adjacent sequences are expected to be closest together in space) by fitting the data to a power-law function, as done previously (Wang et al., 2016). The best-fit curve generated represents the expected spatial distances (Figure 2B), and the position of each data point relative to the curve represents the observed/expected distance (Figure 2C). TAD pairs in red were closer in space than expected (TAD1–TAD8 and TAD18–TAD22), while those in blue were further apart than expected (TAD9–TAD17). These data reveal areas of highly folded DNA.
Figure 2. Conventional Compartments Arise during Gastrulation.

(A) ChrV mean distance matrix in early embryos (2–40 cells), colored by distance (in µm).
(B) Mean distances for TAD pairs as a function of genomic distance; the best-fit line represents the expected spatial distance for each genomic distance.
(C) Normalized distance matrix, plotted as observed spatial distance as a function of genomic distance for each TAD pair. Red denotes pairs that are closer than expected and blue the opposite.
(D) Distance matrix of pre-gastrula embryos (top). Distance matrix of gastrulating embryos (middle). Contact frequency (CF) matrix of late-stage embryos (bottom). Data are from Crane et al. (2015).
(E) Eigenvector decomposition of the inter-TAD matrices in (D). Note the gradual appearance of the conventional B-A-B compartment configuration (white-black-white).
(F) Each TAD from the inter-TAD matrices in (D) plotted in 2D principal-component space. In the pre-gastrula (top), the right and left arms are similar by PC2 but distinct by PC1. In gastrula (middle), the arms are similar by PC1 and PC2, and in late embryos (bottom), the arms are similar by PC1 but different by PC2. TADs of the same compartment are labeled by shape (○,□, x); compartment calls are presented as white/gray/black bars. Unconventional compartment borders (pre-gastrula) were defined by spectral clustering, and conventional compartment borders (later stages) were defined by standard eigenvector value (see STAR Methods).
See also Figures S2 and S3.
We performed eigenvector decomposition on the normalized spatial distance matrix to define compartments (Lieberman-Aiden et al., 2009; Wang et al., 2016). This standard method assumes two compartments exist and uses the sign of the first principal component (PC1) to designate TADs to one compartment or the other. Previous studies with mature embryos suggested that all C. elegans autosomes are divided into three compartments: two ‘‘arms’’ that resemble B compartments, with repetitive DNA, extensive lamina attachments and histone modifications associated with gene silencing; and a central region that carries many unique genes and more active modifications, characteristic of an A compartment (Figure 1A) (Ahringer and Gasser, 2018; Crane et al., 2015; Ikegami et al., 2010; Liu et al., 2011).
The eigenvector analysis revealed that compartments arose gradually during development. In early embryos (2–24 cells), chrV left and right arms displayed opposite PC1 signs, suggesting they belonged to different compartments, and the center was not identified as its own compartment (Figure S2B; Figures 2D and 2E, top). During gastrulation (25+ cells), the PC1 profile changed markedly, and classified the arms as similar, distinct from most of the center region (Figures 2D and 2E, middle), and reminiscent of the epigenetic profile. Calculating the PC1 profile of chrV in late-stage embryos using Hi-C-derived contact frequencies between TADs (Crane et al., 2015) generated a pattern similar to post-gastrulation chromosomes, with B compartments in the arms and a central A compartment (Figures 2D and 2E, bottom). These results indicate that gastrulation is a key developmental milestone for the emergence of conventional compartments, which are absent from early embryos.
To test the universality of the early embryo organization, we traced the structure of chrI using 14 TAD-specific regions (Figure S3). The PC1 profile was similar to that of chrV, with opposing PC1 signs for the two arms (Figure S3E). We conclude that this organization may be a general feature of autosomes in early embryos and that these chromosomes lack conventional compartmentalization.
We asked what specific structural features distinguished early versus gastrula chromosomes. One possibility was that changes in long-range interactions between the arms were drivers of conventional compartment assignments. Another possibility was that changes in local folding between neighboring TADs explained the compartment differences. To test these ideas, we created toy datasets with altered interactions between the left and right arms but constant local folding. Adding close interactions between the arms to the early-embryo spatial distance matrix resulted in similar PC1 values in the arms (Figure S2C). Conversely, removing high-contact frequencies in the Hi-C matrix between the left and right arms led to complete loss of this pattern (Figure S2D). These results indicate that long-range interactions between the arms have a dominant effect on the PC1 profile, driving the standard compartment calls. Furthermore, these results indicate that during gastrulation, the critical structural changes that generate conventional compartments by PC1 are the gains in arm-arm proximity (Figures S2E and S2F).
To achieve a more nuanced understanding of chromosome architecture in early embryos, we increased the dimensionality of compartment analysis by plotting both the first and second principal components for each TAD. This analysis showed that the left and right arm TADs were similar by PC2 and were distinct from most of the central TADs, even in early embryogenesis (Figures 2F and S2B). In fact, PC2 accounted for almost as much variance in inter-TAD pairwise distance patterns as PC1 (32% PC1, 22% PC2). These results suggest that PC2 may be more reflective of the local profile of the chromosome, while PC1 reflects the extended distances between the arms.
To define groups of similar TADs unbiasedly and with increased sensitivity to the dimensionality of the data, we used graph-based methods to cluster TADs directly from the normalized distance matrix and plotted the results in 2D PC space (see STAR Methods). This analysis in early embryos identified three sections corresponding to the left arm, center, and right arm (Figures 2F and S2B). These collective findings indicate that chrV displays unconventional compartments, meaning two similarly folded B-compartment-like arms that are separated in space by an A-compartment-like central region. These unconventional compartments then mature into conventional ones, with associating B regions, at the onset of gastrulation (Figures S2E and S2F).
Early Embryonic Chromosomes Adopt a Barbell-like Organization
We examined early embryonic chromosomes to gain a better understanding of their unconventional configuration. We found that the closest TAD pairs were within the arms; TAD1–TAD8 and TAD18–TAD22 contained low normalized distances, while the central TADs (TAD9–TAD17) were more widely spaced (Figure 2C). These results suggested that the chromosome arms were highly folded over multiple contiguous TADs, while the center was more extended at the inter-TAD scale.
We confirmed these results with independent experiments at higher resolution using tracing libraries below the TAD scale (adjacent 100-kb regions), one to cover the center (TAD12–TAD14) and two others to span sections of the left and right arms (TAD2–TAD7 and TAD17–TAD19; Figure 3A). Higher-resolution tracing revealed that the center had large pairwise distances over most genomic ranges, and folds <1 µm were confined to nearest neighbors, while the arms contained tighter domain-like structures (Figure 3B). We compared traces between the left arm and the similarly sized center and found that center traces produced significantly larger pairwise distances and larger radii of gyration (Figures 3C and 3D).
Figure 3. Early Embryonic Chromosomes Adopt a Barbell-like Organization.

(A) Schematic of high-resolution tracing regions (consecutive 100-kb regions) of the left arm, center, and right arm. Regions 15–18 contain repetitive sequences and were not uniquely probe-able. TAD boundaries are represented by orange ticks.
(B) Mean distance matrices between regions in the left arm, center, and right arm. (C and D) Comparison of the center and left arm in overall mean pairwise distances (C) and radii of gyration.
(D) Data are presented by raincloud plot: individual points, the distribution curve, and boxplots representing the median, boxed by the 25th to 75th percentiles. Whiskers extend to the highest and lowest data points, not including outliers (gray circles). Significance calculated by one-way ANOVA.
(E) Power-law scaling of spatial distance to genomic distance for the left arm, right arm, and center. s (scaling exponent) and a (step size) are derived from the best fit curve (solid lines). The 95% confidence intervals of the fits are as follows: left a (0.937, 0.946), left s (0.138, 0.149), center a (1.121, 1.128), center s (0.101, 0.108), right a (1.00, 1.033), and right s (0.159, 0.185).
(F) Schematic of angle measurements between consecutive TADs (i.e., at the vertex of TAD (n — 1)-TAD(n)-TAD(n + 1)) in all individual traces.
(G) Histogram of the angles generated between all consecutive TADs. The data are fit to a kernel distribution (solid line), contrasted with the expected values for a random walk polymer (dashed line).
(H) The angles data in (G) split by section: the left arm, center, and right arm. The means of the arms are significantly smaller than the center by two-sample t test without assuming equal variances.
To define the folding pattern of each section better, we fit the raw data to a power law function of genomic distance versus spatial distance for each region pair and derived the scaling exponents (s) and folding step sizes (a) for each chromosome section from these measurements (Figure 3E; see STAR Methods) (Mirny, 2011). These variables provide quantitative measures of how the length of the polymer relates to the volume it occupies, where a describes the compaction and s describes the path/directionality of the fiber. The center of chrV had the largest step size (larger a value), indicating it was less compact than the left or right arms (i.e., the green line above the red/blue, Figure 3E). However, with increasing genomic distance, the growth of the spatial distance between regions in the center was slower than that of the arm regions, leading to a smaller scaling exponent (s). The smaller s value may be caused by pairwise distances in the center of the chromosome approaching a global confinement (e.g., the territory or nuclear edge) due to their intrinsically larger step sizes. The chromosome arms contained slightly smaller angles measured at the vertices between three consecutive TADs compared to the center (Figures 3F–3H). More acute angles between TADs are consistent with tighter folding.
These data reinforce the notion that the arms resembled one another by local folding in early embryos, before they were designated as conventional B compartments by eigenvector analysis. Thus, chromosome tracing at both the inter-TAD and intra-TAD scale demonstrated that the arms and center of early embryonic chromosomes possessed different local topologies and suggested that the chromosomes assumed a barbell-like configuration of an extended central region separating two more densely folded arms. The similar folding of chrV arms versus center is consistent with their similar epigenetic profiles (Ahringer and Gasser, 2018; Gaydos et al., 2014; Ikegami et al., 2010; Liu et al., 2011; Mutlu et al., 2018). However, the overall barbell organization in early embryos showed that not all epigenetically similar domains were consolidated in space (i.e., TADs in the arms). This result indicates that long-range homotypic interactions do not dominate the architecture of early C. elegans embryos.
Lamina Association Stretches Chromosomes and Overrides B-Compartment Self-Association
We asked what properties of early embryonic nuclei generated the barbell arrangement. Mitotic chromosomes, which are stiff and rod-like, might be expected to display this behavior (Gibcus et al., 2018); however, the interphase chromosome conformations found here did not resemble the compact and uniform helical winding of mitotic chromosomes (Figures 2 and 3). Alternatively, tethering to the nuclear lamina, via LADs, could dictate chromosome conformation (Figure 4A). To test this idea, we disrupted lamina attachment by mutation of cec-4. cec-4 encodes a chromodomain protein that links methylated H3K9 regions to the lamina (Gonzalez-Sandoval et al., 2015) (Figure 4A). Importantly, this mutation maintains both heterochromatin and lamina integrity, allowing us to disrupt chromosome tethering selectively (S. Gasser, personal communication).
Figure 4. CEC-4 Stretches Chromosomes and overrides B-Compartment Self-Association.

(A) ChIP-seq signal for LEM-2 (lamina association) across chrV, plotted as Z score of ChIP-input for two replicates (blue lines) (top). Z score difference (cec-4 — wild-type [WT]) of LEM-2 ChIP (average of two replicates) (bottom). Regions in red indicate loss of lamina association in the mutant. Data are from Gonzalez-Sandoval et al. (2015).
(B) Mean distance matrix between TADs in cec-4 mutants. (C) Mean distances between TADs in WT and cec-4. (D) Radii of gyration in WT and cec-4 traces. Data are presented by raincloud plot as in Figure 3. Significance was calculated by one-way ANOVA.
(E) TAD-specific changes in normalized mean distance from WT to cec-4 embryos. WT LADs are displayed by purple bars.
(F) Scaling of mean distance versus genomic distance for WT (blue) and cec-4 (red). The 95% confidence intervals are as follows: WT a (1.024, 1.055), WT s (0.179, 0.194), cec-4 a (0.767, 0.824), and cec-4 s (0.196, 0.228).
(G) Example cec-4 chromosome traces in xy projection (grid size = 500 nm). TADs of the left (blue) and right (red) arms were enclosed in 3D Delaunay tri-angulations to visualize their volumes. The distances between arm centroids were measured and plotted with kernel distribution fits (right), indicating chrV arms are closer in cec-4.
(H) Schematic representation of chrV conformations in WT and cec-4 mutants.
See also Figure S4.
Lamina detachment via loss of CEC-4 reduced pairwise distances overall in chrV (Figures 4B and 4C). Individual cec-4 chromosome traces also produced smaller radii of gyration, indicating that the chromosome occupied less space in the nucleus (Figure 4D). All TAD pairs were closer in the mutant, including TADs from the central A-like compartment, where no association with the lamina has been previously detected (Figure 4E). Similarly, chrI traces also displayed compaction in cec-4 mutants, although the phenotype was less severe (Figure S4). Differences in the severity of structural effects may be due to the specific genetic makeup of each chromosome or different dependencies on CEC-4. We conclude that stretching by CEC-4 is a common feature of both autosomes chrV and chrI.
We examined the scaling exponent and step size of cec-4 chromosomes. The whole-chrV step size (a) was markedly reduced in cec-4 (0.80) compared to wild-type (1.04), while the scaling exponent of spatial distance to genomic distance (s) was slightly larger (Figure 4F). These results suggest that cec-4 chromosomes rely on smaller folding steps to become more compact than wild type. Since lamina attachment occurs preferentially in the arms but compaction was systemic in cec-4 mutants (Figures 4A, 4E, and S4), stretching occurred beyond regions of direct physical contact. Moreover, loss of CEC-4 allowed the left and right B-compartment-like arms to come into close proximity (Figures 4B and 4G), indicating that lamina tethering overrides homotypic B-compartment interactions in early embryos. We conclude that tethering to the lamina stretches chromosomes over both long and short genomic distances, thereby contributing to the barbell organization of the early embryo (Figure 4H).
Single Chromosomes Fold into Multiple Higher-Order Conformations
Current approaches using Hi-C and DNA FISH determine either population-average or single-molecule configurations, but neither approach can identify and quantify prevalent conformations or detect regions of variance within a population. To address these issues, we analyzed individual traces by adapting Seurat, an unsupervised, graph-based clustering toolkit that was originally developed to identify populations of similar cells from single-cell RNA-sequencing (scRNA-seq) data (Butler et al., 2018; Macosko et al., 2015; Satija et al., 2015). We reasoned that each pairwise measurement from chromosome tracing was analogous to a gene expression value, while each chromosome trace was analogous to a cell. Like RNA expression, we expect some variables to be interdependent (e.g., neighboring regions) and others to be independent, which would be uncovered by unsupervised clustering. We performed single-chromosome clustering based on the pairwise distance patterns of 1,629 wild-type whole-chrV traces, which contained 231 variables corresponding to the unique measurements between the 22 TADs.
Analyzing the traces by pairwise distance patterns using 20 principal components (i.e., dimensions) followed by graph-based clustering yielded four subpopulations (clusters α–δ; see STAR Methods). Clusters α–δ were visualized in two dimensions by uniform manifold approximation and projection (UMAP) or t-Distributed Stochastic Neighbor Embedding (t-SNE) plots, where each dot represents a single chromosome trace (Figures 5A and S5A). Examination of the clusters by individual experiments eliminated the possibility that any cluster arose due to batch effects (Figure S5B).
Figure 5. Single Chromosomes Fold into Multiple Higher-Order Conformations.

(A) UMAP plot of chrV clusters, where each dot represents a single trace (left). UMAP of clusters α-γ-β, showing a possible transition (right). Colors represent clusters defined in 20D space.
(B) Dispersion of TAD pairs versus mean distance (see STAR Methods). TAD pairs with lowest variance have low dispersion, and pairs with high variance have high dispersion (top and bottom 5% in red).
(C) Distance matrices of each cluster in (A).
(D) Difference matrices between each cluster and the population average (red, compaction; blue, separation).
(E) Normalized distance matrices for each cluster as observed/expected values. Compartment calls are presented as in Figure 2. Only cluster γ displays conventional compartmentalization.
(F) Each TAD from the cluster distance matrices plotted in 2D principal-component space. TADs of the same compartment are labeled by shape (○,□, x). A simplified cartoon for each cluster is shown to the right, colored by TAD.
See also Figures S5 and S6.
We classified TAD pairs by the level of variance in the total dataset and identified pairs that were markers of unique variability within each cluster. This analysis revealed that lowest total variance among all traces occurred between select pairs of the left arm and between the left arm and center (Figure 5B). Thus, chrV contained distinct regions of relatively stable folding. Markers of unique variability (measurements that distinguished chromosomes in one cluster versus all other clusters) were concentrated in TAD pairs within and between the left and right arms (Figure S5D), indicating these relationships were the drivers of differential clustering.
To analyze the conformations of the subpopulations, we derived spatial distance matrices and difference matrices between each cluster and the population average (Figures 5C and 5D). We further generated normalized distance matrices and compartment assignments for each cluster (Figures 5E and 5F; see STAR Methods). Cluster α (29% of traces) was characterized by a tightly packaged TAD1–TAD5, a transitional TAD6– TAD12 (mixed high and low normalized distances), and a loosely packaged TAD13–TAD22 (Figures 5E and 5F). Cluster β (24% of traces) resembled a near-mirror image of cluster α and contained a loosely packaged TAD1–TAD10, a transitional TAD11–TAD13, and a tightly packaged TAD14–TAD22. A unifying feature of clusters α and β was the separation of the two arms, with corresponding PC1 profiles, similar to the population average structure (Figures 2A and S5E).
In contrast, cluster γ contained traces with evenly folded left and right arms in close proximity, suggesting a barbell chromosome folded in two (Figures 5C–5E). Only cluster γ produced a plaid pattern on the normalized distance matrix and a PC1 profile, where the arms were classified similarly and the center was distinct (Figures 5E and S5E). Cluster γ therefore displayed conventional compartmentalization with increased long-range homotypic interactions, similar to the population average of post-gastrulation chromosomes (Figure 2). These results explain why conventional compartments were not detected in the early-embryo population average (Figures 2 and S2B), since they represent only 23% of individual traces.
The conformation of cluster δ was the most dissimilar from the others, with large pairwise distances and long-distance loops. Cluster δ was characterized by large, positive changes in distances relative to the average for almost all TAD pairs (Figure 5D), large radii of gyration (Figure S5C), and a lack of close multi-domain structures. From compartment analysis, cluster δ was highly disorganized (alternating positive and negative PC1 values) and may have been too stretched to reveal correlations between many contiguous TADs (Figure S5E).
We confirmed the results of the sub-population analysis by examining arm-arm distances in individual traces from each cluster (Figures S5F–S5H). These results indicate that single-chromosome clustering yielded bona fide sub-populations, which differed in their inter-arm proximities. Together, these analyses revealed that chromosomes retained a barbell configuration in three of the four clusters (i.e., all but cluster δ). In each case, however, the barbell deviated from the population average either by the extent of folding in the arms (α and β) or by having a bent bar (γ).
We performed similar analysis for chrI. Five higher-order conformations were uncovered by clustering, demonstrating that structural diversity and variable compartment boundaries are broad features of early embryonic chromosomes (Figure S6). These results collectively demonstrate that there is no absolute boundary between compartments from cell to cell. The variable boundaries may help explain why previous studies failed to detect sharp boundary transitions between Hi-C compartments or LADs (Crane et al., 2015; Ikegami et al., 2010), as they were based on population averages, which would dilute the effect of each distinct conformation.
Fractal Globule Folding Patterns Reflect up to Half of Endogenous Conformations
The fractal globule model describes one possible chromosome architecture in which the DNA fiber is packed tightly into globules at all scales (meaning sequences close in genomic distance are preferentially close in spatial distance), the chain does not cross itself, and it has a scaling exponent of spatial distance to genomic distance of one-third (Mirny, 2011). The existence of this pattern in vivo has been supported by Hi-C evidence but questioned by recent multiplexed FISH data in human cells (Lieberman-Aiden et al., 2009; Mirny, 2011; Wang et al., 2016). We examined the folding schemes of the chrV clusters through power-law function fit to define the folding patterns of the chromosomes and determine if any resembled a fractal globule polymer (Figure 6A).
Figure 6. Single-Chromosome Analysis Reveals Subpopulations with Unique Folding Patterns.

(A) Power-law scaling of spatial distance to genomic distance for each cluster (blue, best fit curves of the raw data). s = scaling exponent of spatial distance, a = step size. Values in brackets are 95% confidence intervals of the fit. Dotted line represents the fractal globule model prediction with s = 1/3.
(B) Radial-normalized mean distance (RND) to nuclear boundary for each TAD in individual traces (see STAR Methods). Red dotted line indicates RND = 0.15 (lamina contact). Error bars represent ±SEM.
Clusters α and β had larger scaling exponents (s = 0.24–0.28) than the population average (s = 0.19, Figure 4G) or clusters γ and δ (s = 0.11–0.14). Conversely, the step size for clusters γ and δ were larger than those for clusters α and β (Figure 6A). These parameters suggested that chromosomes in clusters α and β folded similarly to fractal globules at the whole-chromosome 21-Mb scale (blue curve similar to dotted curve, Figure 6A). However, chromosomes in clusters γ and δ did not fold in a fractal manner but instead favored long-range genomic interactions with large step sizes. The clusters of chrI also displayed divergent spatial distance scaling, with chrI cluster 4 most closely resembling the fractal globule model predictions (Figure S6E). These results show that the different chromosome subpopulations have fundamentally different folding schemes, some of which conform to the fractal globule model.
Lamina Association Is Required for Open Morphology
Traditionally, the lamina is a site of DNA compaction, but our analysis of the average chromosome configuration suggested a role in chromosome stretching. To gain further insight, we investigated the effects of lamina tethering on the chrV clusters. First, we asked whether the variable morphology reflected differential tethering locations. Second, we determined the repercussions of removing lamina attachments on structure.
The location of tethering was variable, as the distance of any TAD to the nuclear boundary in individual traces varied considerably (Figure 6B). This variability may be a result of dynamic on-off events of chromosome regions, as seen in other studies (Kind et al., 2013, 2015; van Steensel and Belmont, 2017). Intriguingly, distended regions typically coincided with proximity to the lamina. For example, the extended right arm of cluster α chromosomes (TAD22), and the center of cluster γ chromosomes (TAD11) were closest to the lamina (Figure 6A). Cluster δ chromosomes were closer to the lamina than all others throughout their lengths, particularly at TAD6, TAD10, and TAD21. This result suggests that cluster δ chromosomes have more tethering sites than other configurations and are consequently stretched to a greater degree.
To test this model of lamina stretching, we clustered cec-4 chromosome traces to define what conformations exist upon loss of lamina association. At this single-chromosome level, comparison of chrV wild-type and cec-4 traces showed a marked separation by genotype in tSNE space, indicating that few specific conformations were shared (Figure 7A). chrV cec-4 traces segregated into three clusters (clusters 0–2; Figures 7A–7C). Difference matrices between clusters 0–2 and the wild-type total revealed that all cec-4 clusters showed widespread compaction at the inter-TAD level, with the greatest proximity gains in the mutant occurring between TADs of the left and right arms, particularly in cluster 2 (Figure 7D). Individual traces in cec-4 clusters also displayed smaller radii of gyration than all wild-type clusters (Figure 7B). ChrV cec-4 clusters 0–2 resembled more compact versions of wild-type clusters α–γ (Figures 7C and 5B), supporting our previous population average conclusion that the lamina stretched the chromosomes. ChrI in cec-4 mutants also showed an increase in short and long-range interactions in two out of three subpopulations (65% of traces; Figures S7A–S7C). Furthermore, a distinct subpopulation similar to wild-type chrV cluster δ (extended and long-range looping) was not resolved in cec-4 mutants. These results suggest that lamina tethering was required to form conformations similar to those of cluster δ, but not clusters α–γ. Moreover, they indicate that large, open chromosome morphology relies on lamina attachment during early embryogenesis.
Figure 7. Lamina Association Is Required for Open Morphology.

(A) t-SNE plot comparing chrV WT (blue) and cec-4 (red) traces (left). UMAP plot of chrV cec-4 clusters (right).
(B) Radii of gyration of WT chromosome clusters (as in Figure S5C for direct comparison) and cec-4 clusters, indicating subpopulations of cec-4 chromosomes are consistently more compact than WT. Data presented by raincloud plot, as in Figure 3.
(C) Distance matrices of cec-4 clusters 0–2.
(D) Difference matrices between each cec-4 cluster and the WT population average (red, compaction; blue, separation).
(E) Normalized distance matrices for each cluster as observed/expected values. Compartment calls are presented as in Figure 2. Only cluster 2 displays conventional compartmentalization.
(F) Each TAD from the cluster distance matrices plotted in 2D principal-component space. TADs of the same compartment are labeled by shape (○,□, x).
See also Figure S7.
Finally, we also observed effects of cec-4 on chromosome compartmentalization. ChrV cec-4 cluster 2 (27% of the population) displayed the strongest conventional compartmentalization of any wild-type or mutant cluster (Figures 7E and 7F). Thus, lamina attachment was not required to generate compartments per se. However, compartment boundaries in cec-4 were not identical to wild type (Figure 7E). Loss of cec-4 also resulted in increased disorganization on both autosomes, where neighboring TADs no longer correlated with each other (chrV cec-4 cluster 1 and chrI cec-4 clusters 1–2; Figures 7E and S7D). Collectively, these data indicate that CEC-4 exerts an extensive influence on structural diversity and the positioning and strength of compartment boundaries.
DISCUSSION
This study makes four contributions toward understanding chromosome architecture. First, we developed methods to trace entire chromosomes and define higher-order architecture in intact animals. Second, we introduced single-chromosome clustering to identify genomic regions with high and low variance. This approach enabled us to assess heterogeneity and to classify individual chromosomes into subpopulations, which will be broadly useful for analyzing conformational diversity from chromosome tracing or single-cell Hi-C. Third, we defined the unique features of early embryonic chromosomes. These include prevalent compartment-level diversity with variable boundaries and separated, unconventional B compartments to generate a barbell configuration. Fourth, we uncovered a role for the lamina to stretch chromosomes in early embryos. Stretching prevented interactions between A-B and B-B compartments, overriding B compartment self-association.
To assess chromosome structure in situ at high throughput, chromosome tracing is rapidly emerging as a powerful technique. Recent studies using broadly similar FISH methods have elegantly revealed and studied the origins of domain-level structures (Bintu et al., 2018; Boettiger et al., 2016; Cardozo Gizzi et al., 2019; Mateo et al., 2019; Nir et al., 2018) and whole-territory volumes (Fields et al., 2019). Chromosome tracing, first detailed in Wang et al. (2016), enables single-cell analysis and direct observation of chromosome conformations, thereby complementing Hi-C methods. In this work, we adapt whole-chromosome tracing to an animal model to study the emergence of compartment structure during development. Our study differs from others in both genomic scale and resolution and the application of single-chromosome clustering.
A Barbell-Shaped Chromosome in Early Embryos
Compartment-level structure is an important and understudied area of chromosome biology. We examined compartment formation as embryos transitioned from the earliest stages through gastrulation. In the early embryo, chrV resembled a barbell, with separated nascent B compartments and an extended A compartment. 53% of chrV traces and 80% of chrI traces reflected the arms-apart organization, indicating this was the most prevalent in early embryogenesis. This morphology likely depends on the contribution of the extended nature of the A compartment to physically separate the flanking B compartments and tethering to the lamina to keep the chromosome distended.
Chromosome arms consolidated at the onset of gastrulation. One key difference between the stages is the change in cell-cycle timing. The early embryonic cell cycle is extremely fast in C. elegans (<60 min) and progressively slows during development, beginning at gastrulation (Kipreos and van den Heuvel, 2019). It is possible that similar compartments need sufficient time during interphase to find each other and cluster in space. A second difference between early and gastrula embryos is the state of heterochromatin. Previous studies have implicated histone modifications for differential folding and compaction, particularly the Polycomb complex, which mediates facultative heterochromatin (Boettiger et al., 2016; Eskeland et al., 2010; Sexton et al., 2012). Polycomb and other silencing machinery may promote phase separation via homotypic chromatin attraction and/or the association of inactive regions to the lamina (Larson et al., 2017; Nuebler et al., 2018; van Steensel and Belmont, 2017; Strom et al., 2017). Polycomb modifications are inherited from the parental generation and present on the arm regions of C. elegans chromosomes at the earliest stages of embryogenesis (Celniker et al., 2009; Gaydos et al., 2014; Liu et al., 2011; Mutlu et al., 2018), which may explain the segregation of A from B compartments and B-B proximity in some clusters. However, early C. elegans embryos lack mature constitutive heterochromatin, which appears during gastrulation (Mutlu et al., 2018, 2019). Since heterochromatin is thought to have strong self-association properties (Falk et al., 2019), less constitutive heterochromatin may weaken B compartment self-association in early embryos.
Increasing the dimensionality of compartment analysis allowed for the discovery of unconventional compartments. It is possible that similar compartments in other animals were previously overlooked by standard eigenvector decomposition, which can be dominated by long-range interactions, and fail to reflect more local genome organization. The absence of conventional compartments has been observed at particular development stages, including in mouse oocytes (Flyamer et al., 2017), zebrafish embryos during ZGA (Kaaij et al., 2018), and Drosophila embryos prior to ZGA (Hug et al., 2017). We find in C. elegans embryos that conventional compartments arise post-ZGA and at the onset of gastrulation. In other organisms, the underlying cause of the loss of conventional compartmentalization has not been determined. Our data suggest that an absence of long-range interactions between locally similar regions may underlie the lack of detectable compartments, similar to the population average C. elegans chrV. Additionally, chromosomes could also contain a heterogeneous mixture of compartment-level structures, as observed in this study, which would be undetectable by population studies and standard single-dimension compartment assignment methods.
Chromosome Stretching by the Nuclear Lamina
Recent Hi-C studies have suggested that the lamina compacts chromatin in B compartments, with variable effects on the A compartment in cultured cells (Ulianov et al., 2019; Zheng et al., 2018). In contrast, our data show that the lamina is required to stretch the entire chromosome and affects all inter-TAD relationships. Regions that were the most distended in our wild-type clusters were closest to the lamina and vice versa. We envision three possible explanations for the different results between our work and other studies. First, the cell populations differed between the studies. Prior studies used cultured cells (Drosophila S2 cells and mouse embryonic stem cells [ESCs]) while ours used intact embryos, and it is unknown whether chromosomes behave similarly in these different biological contexts. Early embryonic chromosome organizations differ from those in more differentiated cells (Flyamer et al., 2017; Kaaij et al., 2018) (this work). In support of different organization during development, disruption of lamina attachment by cec-4 mutations led to decompaction of the X chromosomes in differentiated, larval C. elegans (Snyder et al., 2016).
Second, lamina dissociation by cec-4 mutation differs from complete lamin inactivation. Loss of cec-4 is relatively gentle, as it does not disrupt histone modifications associated with heterochromatin (Gonzalez-Sandoval et al., 2015) (S. Gasser, personal communication). Inactivation of lamin, on the other hand, disrupts heterochromatin and gene silencing, which may have secondary structural effects (Ulianov et al., 2019; Zheng et al., 2018).
Third, different methods were used to deduce compaction. FISH allows direct measurements of distance between loci, whereas Hi-C infers compaction based on the frequency of ligation between loci. Lower contact frequency implies less compact chromatin, but comparisons of data from Hi-C and FISH indicate that these methods do not always agree (Giorgetti and Heard, 2016; Hakim et al., 2011; Williamson et al., 2014). Changes in protein binding or biophysical properties may alter the frequency of ligation between two points independent of changes in compaction. In support of this idea, FISH analysis after lamina disruption in cultured mESCs failed to reveal whole-chromosome decondensation that had been predicted by Hi-C in the same samples (Zheng et al., 2018). Taken together, these findings imply that the extent and qualitative effects of lamina influence on chromosome organization is highly dependent on biological and methodological context.
Another important consideration is that different chromosomes may be affected differently by lamina association. We identified varied degrees of cec-4 dependence between chrV and chrI by tracing. chrV is the largest chromosome in the genome (~21 Mb), and its size possibly contributes to the large degree of change seen once it is released from the lamina. Consistent with this idea, chrI is ~15 Mb, and showed less severe changes in structure. The genetic makeup of chrV and chrI also differ. chrI is over-enriched, while chrV is under-enriched, for genes with lethal phenotypes along its length (Kamath et al., 2003). chrV bears the only gene that significantly changes expression in cec-4 embryos (Gonzalez-Sandoval et al., 2015) and whose global gene expression increases in cec-4 mutant larvae (Snyder et al., 2016). Collectively, these observations indicate that heterogeneity of chromosome size or sequence may contribute toward the lamina’s influence.
Implications for Diverse Higher-Order Conformations
Unsupervised clustering enabled the discovery of conformational details that were otherwise masked by population averages. Compartment structure was highly variable; most conformations shared certain elements with the population average, but none were identical. These conformations could not have been deduced from previous bulk Hi-C or chromatin immunoprecipitation sequencing (ChIP-seq) data, which rarely offer single-cell information. Together with other recent studies on the variability of TAD structure (Bintu et al., 2018; Flyamer et al., 2017; Nagano et al., 2017; Stevens et al., 2017; de Wit, 2019), our findings highlight the need to assess conformational heterogeneity in cell populations through single-cell analysis to make informed conclusions regarding the presence or absence of chromosome structures.
Clustering enabled us to parse multiple modes of polymer packaging. The fractal globule model resembled the packaging of roughly half of the higher-order chrV conformations and 14% of chrI. The other clusters and the population average had much smaller scaling exponents, similar to chromosomes in human IMR90 cells (Wang et al., 2016). This result provides validation for the fractal globule model for large genomic length scales in vivo but also shows that the model does not apply to all endogenous conformations in interphase nuclei. Simulations have shown that fractal globule polymers retain open topology, are unknotted, and are able to rapidly unfold under conditions such as gene activation (Mirny, 2011). We therefore speculate that chrV clusters α and β and chrI cluster 4 may also be more rapidly responsive than other conformations.
Chromosome traces belonging to different clusters sometimes overlapped in t-SNE/UMAP space, and it is tempting to speculate that these represent dynamic transitions between conformations. Possible transitions were most visible from chrV α-γ-β and chrI 2–3-4 (Figures 5A and S6B). This would imply that higher-order chromosome structures of interphase nuclei exist in a continuum rather than completely discrete states. Alternatively, the clusters could reflect variable structures after cell division, as previous studies have shown that lamina attachment varies between mother and daughter cells (Kind et al., 2013). As different sections of a chromosome attach to the lamina, they may also engender different morphologies. The picture that emerges is of a dynamic system where lamina attachment and higher-order conformations intimately intertwine.
STAR★METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for reagents may be directed to and will be fulfilled by the Lead Contact, Susan E. Mango (susan.mango@unibas.ch). There are restrictions to the availability of probe library stock due to the lack of an external centralized repository for its distribution and our need to maintain the stock. We are glad to share libraries with reasonable compensation by requestor for its processing and shipping.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
C. elegans strains were cultured as in (Brenner, 1974), at 20°C. Bristol strain N2 was used as wild-type. The cec-4 mutant strain used was RB2301 (F32E10.2(ok3124))(Gonzalez-Sandoval et al., 2015). Data were gathered from embryos 2–40-cells in age.
METHODS DETAILS
General description of chromosome tracing method
We designed FISH probes to C. elegans chromosomes to map inter-TAD distances on entire autosome lengths, and to map finer-scale structure on sections of chrV. In both cases, we use a dual-probe scheme: labeled primary probes to coat the genome in specific regions (e.g., to define chromosome territories), and labeled secondary probes to detect specific sub-groups of those primary probes in sequential imaging rounds. Each round of imaging is separated by a photo-bleaching step to quench the signal of the previous round. In each tracing experiment, the primary probes share the same fluorophore label, so their total signal marks the total volume of the chromosome (or chromosome section). Within this volume, individual TAD/regions are resolved from one another using unique secondary probes, which are hybridized and imaged in sequential rounds on the same sample.
Probe design and synthesis
Whole-chromosome tracing probes
Primary hybridization probes for whole chromosome DNA FISH targeted one 100kb region per TAD, as described in detail below. Primary probe sequences consisted of 100nt-long oligonucleotides containing 4 sections: a 20nt 5′ PCR-priming sequence, a 30nt genome homologous sequence, a 30nt readout sequence (to bind to its corresponding secondary probe), and a 20nt 3′ PCR-priming sequence. This design is based on the whole-chromosome tracing in human cells from (Wang et al., 2016).
We manually set 100kb-long target genomic regions internal to 22 of 24 TADs in chromosome V that were originally identified in (Crane et al., 2015), omitting the two smallest TADs. For chromosome I, we used 100kb-long target regions corresponding to 14 of the 15 TADs, omitting the smallest TAD. The genomic coordinates for the target regions can be found in Data S1. Oligo pools were generated to target the 100kb regions at a density of 10probes/kb. For each of the 100kb regions (each TAD), we extracted the DNA sequence from the C. elegans genome assembly (ce10) and split each 100kb region into 1kb fragments (2200 fragments total) for probe design. The 30nt genome homologous section for each probe was generated by OligoArray2.1 (Rouillard et al., 2003) (which uses NCBI-BLAST 2.2.26 and OligoArrayAux secondary structure predictor) on the Harvard FAS Odyssey high performance computing cluster. The parameters used were: melting temperature 60–100°C, no cross-hybridization or predicted secondary structure with a melting temperature greater than 70°C, GC content 30%–90%, no stretches of 7 or more identical nucleotides.
Next, the 30nt sequences were further checked for specificity by NCBI BLAST+ 2.7.1, and unique sequences were then fused to the priming and readout sequences, with each of the 100kb-long target regions using a unique readout sequence. The library of 100nt-long probes was synthesized by CustomArray, Inc (Bothell, WA). The sequences for these probes are provided in Data S2 and S3. The sequences for PCR-priming and readout sequences are provided in Data S1.
Sub-TAD scale tracing probes
Primary probes targeting consecutive 100kb genomic regions spanning sections of the chrV left arm, center (A compartment), and right arm were designed using a method similar to the whole-chromosome tracing probes, unless otherwise noted. The genomic locations for these probes are provided in Data S1. The sequences for these probes are provided in Data S4, S5, and S6. We designed probes for 39 regions in the left arm (4.2Mb total), 40 regions in the center (4Mb total), and 13 regions in the right arm (1.3Mb total) to increase specificity (longer homology region), resolution (consecutive 100kb regions without gaps), and signal intensity (dual readout regions) compared to the whole-chromosome tracing. In the left arm, regions 15–18 were omitted from tracing because they contained repetitive sequences, and were not uniquely probe-able. These probes consisted of 142nt-long sequences containing 5 sections: a 20nt 5′ PCR-priming sequence, a 30nt readout sequence, a 42nt genome homologous sequence, a 30nt readout sequence (identical to the first readout sequence), and a 20nt 3′ PCR-priming sequence. The OligoArray2.1 parameters used were: melting temperature 78–100°C, no cross-hybridization or predicted secondary structure with a melting temperature greater than 76°C, GC content 30%–90%, no stretches of 7 or more identical nucleotides.
Probe amplification
The oligo libraries were amplified and labeled with fluorophores in limited cycle PCR (Phusion Hot Start Master Mix, Life Technologies), high yield in vitro transcription (HiScribe T7 Quick High Yield RNA Synthesis Kit, NEB), and cDNA synthesis reactions (Maxima H Minus Reverse Transcriptase, Fisher Scientific), as previously described without modification (Wang et al., 2016). Probes were purified using: the DNA Clean & Concentrator kit with Spin IC columns after PCR, and Spin V columns using Oligo Binding Buffer after cDNA synthesis (all reagents from Zymo). Primary probes for whole-chrV, chrI tracing, and chrV left arm sub-TAD tracing were 5′ labeled with ATTO 565 (IDT). Primary probes for the center and right arm sub-TADs were 5′ labeled with Alexa Fluor 647 (IDT).
Embryo sample preparation and primary probe in situ hybridization
C. elegans embryos were hand dissected from young gravid adults grown at 20°C and adhered to poly-L-lysine-coated 40mm round coverslips. Embryos were fixed in 1% paraformaldehyde/ 0.05% Triton X-100(Sigma Aldrich)/ 1XPBS (Life Technologies) for 5 minutes at room temperature, permeabilized by freeze-cracking the eggshell (Kiefer et al., 2007) and submerged in ice-cold methanol for 5 minutes. At room temperature, coverslips were washed once in 1X PBS for 5 minutes, and three times in 1X PBS/0.5%Triton X-100 for 15 minutes each. Samples were then treated with 0.05mg/ml RNase A (ThermoFisher) in 2X SSC (Life Technologies) at 37°C for 30 minutes. Embryos were blocked in hybridization buffer (10% dextran sulfate (Fisher Scientific)/ 0.1% Tween-20 (Sigma Aldrich)/ 2X SSC/ 50% formamide (OmniPur)) for one hour. Primary probes were diluted to ~1µM in hybridization buffer, applied to the embryos on the round coverslip, and the coverslip was inverted onto a glass slide. To allow the probes to intercalate genomic DNA, the slides were placed on an 80°C heat block in a humid chamber for 10 minutes. The samples were then allowed to hybridize for at least 16 hours in a humid chamber. For the whole-chromosome tracing probes, steps from blocking onward were performed at 37°C, while for the sub-chromosome fine tracing probes (longer homology) these steps were performed at 47°C. After hybridization, the coverslips were dislodged from the glass slide and washed in 2XSSC/50% formamide for 30 minutes, followed by two washes each in 2XSSC and 0.5XSSC, each for 30 minutes. Washes were performed using pre-warmed buffers. Samples were either used immediately for imaging, or stored at 4°C. Prior to imaging, 0.1µm TetraSpeck beads (ThermoFisher) in 2XSSC were adhered to the coverslip to correct for sample drift during imaging rounds, and nuclei were stained with SYTOX Green nucleic acids stain or DAPI (ThermoFisher) according to the manufacturer’s instructions.
Multiplexed FISH protocol
Experiments were performed at Harvard University or the University of Basel using an automated protocol with an epifluorescence microscope coupled to a custom microfluidics system at room temperature, as previously described (Wang et al., 2016) unless otherwise stated. Embryos adhered to the coverslip were selected as fields of view (FOV) by strong primary probe signal. For each FOV, the first round of imaging consisted of capturing the signal of the primary probe (561nm or 647nm illumination), nuclear stain (488nm), and fiducial beads (488nm/561nm/647nm) in 200nm z-steps for a total range of 12–30µm. The beads on the surface of the coverslip were clearly distinguishable by z-position from all other fluorescent targets within the embryo nuclei. The primary probe signal was then photobleached to undetectable levels. Secondary hybridizations (using probes complementary to the readout sequences of the primary probes) were performed for 30 minutes in 2XSSC/25% ethylene carbonate (Sigma Aldrich) at 8nM. The samples were then washed for 3 minutes in 2XSSC/25% ethylene carbonate, then 2.5 minutes in 2XSSC. The next round of imaging consisted of capturing the secondary probe signals (561nm or 647nm) and fiducial beads (488nm) before photobleaching of the secondary probe signals. Different secondary probes used in the same round are distinguished using different fluorophores. The secondary hybridizations, imaging, and bleaching steps were then repeated sequentially for all the remaining genomic segments to be imaged, for all fields of view on the coverslip.
Data analysis
Foci fitting and assignment
All foci detected in DNA FISH were fit to 3D Gaussian functions and their centroid positions were calculated, as previously described without modification (Wang et al., 2016). Each focus was assigned a TAD or region identification number based on which hybridization round it appeared in and which fluorophore it bore. The 3D positions of each focus were corrected for sample drift between hybridization rounds by calculating the centroid 3D movements of the fiducial beads between hybridization rounds and subtracting these values. We used 3D Gaussian fitting of the 100nm fiducials in each field of view to obtain sub-pixel super resolution in drift correction. The average localization precision of our method was calculated to be 50nm (Wang et al., 2016).
Volumetric image segmentation and chromosome tracing
Fiducial bead fluorescent signal was disregarded by excluding the initial z slices from the downstream analysis. For each embryo, nuclei were identified using the general nuclear stain as a marker, and territories were identified using the primary probe signal as a marker. These volumes were determined using marker-controlled watershed segmentation in MATLAB vR2016b (https://ch.mathworks.com/help/images/marker-controlled-watershed-segmentation.html), optimized for each embryo. We discarded clearly mitotic nuclei from further analysis by general DNA morphology. Embryonic cells are diploid and replicating, and we detected 2–4 foci per TAD/region per nucleus as expected. For any particular TAD/region, there may be more than one focus per contiguous primary probe signal if replicated (< 5% of the sample). Within each territory volume (defined by primary probe signal), we connected the detected foci to each other by closest distance in ascending order to create the chromosome trace (i.e., TAD1 connected to the closest TAD2 focus, TAD2 connected to the closest TAD3 focus). We eliminated any volumes that were too ambiguous to trace from further analysis (< < 5% of our samples). Each TAD/region is not always detected in each territory (see Figures S1 and S3 for quantitation). If no foci were present in a particular hybridization round inside the territory volume, that region was skipped and the trace was connected to the next TAD/region in the sequence. The median length of traces was six foci for wild-type whole-chr tracing probes, therefore we increased the number of experiments to reach enough sampling of all TADs/regions to build complete mean pairwise distance matrices. We note that we obtained the same distance matrix values for data using ≥10 foci per chromosome as for data using all measurements. For all pairwise measurements, we achieved a median SEM of 0.05 µm, and a maximum SEM of 0.25µm, well below the mean distance of any pairwise measurement. The number of each pairwise measurement was 240 for chrI and 194 for chrV on average, however pairs with chrV TAD11 were more difficult to detect compared to all other probed regions for unknown reasons. We calculated the number of chromosome copies present in the identified territories by counting the number of foci per TAD/region in each territory, and found that 80% of territories contained a single copy, 17% contained two copies (ie replicated or adjacent alleles), and less than 3% contained three or four (ie replicated and adjacent alleles). Therefore, the majority of the data presented originate from chromosomes that are well-separated in space (C. elegans chromosomes do not pair at interphase). Almost identical results were obtained considering only those volumes with a single copy (80%). In each chromosome trace, the distances between all the foci were measured, and then used to create the mean spatial distance matrices. The number of individual chromosomes profiled in each dataset and genotype are shown in the corresponding mean pairwise distance figures. Random down-sampling of the data was done using the MATLAB datasample function.
Normalization of distances by power-law function fit
As in (Wang et al., 2016), data was fit to
where x = genomic distance, y = mean spatial distance, a = step size, and s = scaling exponent. The power law fit represents the expected spatial distance of end-to-end measurements versus genomic distance, taking into account the polymer nature of the chromosome. The deviation of a particular measurement to the fit line is its normalized (observed/expected) distance.
We note that chrV TAD17 contains a high amount of repetitive sequences including the 5S rRNA gene cluster. Since repetitive sequences are difficult to map to the genome in general, it is possible that this region is underestimated in size in current genome annotations of wild-type C. elegans, so there is some uncertainty associated with its normalized distance. A sister wild-type strain was recently extensively re-sequenced using long reads and showed a larger genomic size in this region (Yoshimura et al., 2019).
The step size is the expected end-to-end distance of each ~1Mb of chromatin fiber, and is a general readout of the level of compaction of chromatin folding. The step size is conceptually similar to persistence length (Mirny, 2011), but for much larger-scale structure. The scaling exponent describes how the polymer is packed in space. A random walk polymer without spatial confinement has a scaling exponent of 1/2. The fractal globule polymer model, a crumpled and unknotted model, predicts a scaling exponent of 1/3 (Mirny, 2011). Scaling exponents less than 1/3 describe more intermixed polymer folding schemes.
Comparison with Hi-C
The Hi-C defined(Crane et al., 2015) chrV compartments are: TADs 1–7 (B compartment), TADs 8–17 (A compartment), and TADs 18–22 (B compartment). Contact frequency measurements (ICE-corrected) from late/mixed-stage wild-type (N2) C. elegans embryos were downloaded from NCBI GSE63717. We used the 50kb binned Hi-C genome-wide matrix as the data source, and extracted the contact frequencies between each of the 100kb regions covered by the DNA FISH probes. We then calculated the Pearson correlation between the mean pairwise distances and the inverse contact frequencies.
Lamina-associated domains
Lamina-associated domains are defined from ChIP-seq of the lamina protein LEM-2 (Gonzalez-Sandoval et al., 2015; Ikegami et al., 2010). chrV LADs are enriched in: TADs 1–7, TAD 10, and TADs 16–22. chrI LADs are enriched in: TADs1–3 and TADs10–14. LEM-2 ChIP-seq data were downloaded from NCBI GSE74133, z-scores were calculated (IP-input) for each genotype (wild-type and cec-4), and plotted against genomic location.
Compartment calling
TAD groups were identified from the spatial positions of individual TADs as first described in (Wang et al., 2016). First, the observed/expected values were calculated for each TAD pair in the mean pairwise distance matrix through power-law scaling. This creates the normalized distance matrix. Standard compartment assignment via eigenvector decomposition uses the first principal component (PC1) to find groups of similar TADs from the normalized distance matrix. PC1 can be used to assign TADs to a maximum of two different groups, where TADs with a positive PC1 share a group and TADs with a negative PC1 share a separate group. A clear plaid pattern on the normalized distance matrix is a known indicator of two compartments (conventional compartmentalization), and the sign of PC1 can be accurately used to call compartment identity.
Toy datasets to test the structural origins of standard compartment assignment results were generated by changing the pairwise distances or contact frequencies between TADs1–7 (left arm) and TADs18–22 (right arm). These values were set to 1.4mm in Figure S2C to increase arm-arm proximity in early embryos, or set to 4 inverse contact frequency units in Figure S2D to decrease arm-arm contact frequency in late stage embryos.
Chromosomes without a plaid pattern and containing more than two distinct groups by normalized spatial distance matrix (unconventional compartmentalization) did not use the sign of PC1 to call similar TADs. Instead, the spectralcluster function in MATLAB was used to unbiasedly define groups of similar TADs (unconventional compartments) using the normalized distance matrix as the input (i.e., each TAD’s pairwise distance pattern to all other TADs).
Polarization of chromosome arm regions
In individual chromosome traces, the left and right arm regions were enclosed in convex hulls (using the MATLAB function convhull), and the polarization indexes were calculated as in (Wang et al., 2016), using the following equation:
A polarization index equal to 1 occurs when the two arms of a chromosome trace have no shared volume. The centroid position of each arm was also calculated (mean in xyz of all the foci contained in the arm), and the distances between these centroids in each trace was measured.
Distance to nuclear boundary
The distance of each TAD in a trace to the nuclear boundary was calculated as the distance to the closest edge of the nuclear marker in mm (determined by watershed segmentation) using the boundary function in MATLAB. This distance was then normalized to the radius of the detected nuclear volume in mm (radial-normalized distance (RND)), similar to recent work (Ulianov et al., 2019). RND values of 0.15 have been previously shown to be in contact with the nuclear lamina (Ulianov et al., 2019).
Single chromosome clustering analysis
Spatial distance matrices for all individual chromosome traces were constructed, and the fields corresponding to unique pairwise measurements (upper triangle of the matrix) were vectorized. All the distance vectors were then concatenated into a single file where each trace was assigned an identification number that could be tracked back to the original trace file with foci xyz coordinates. To identify clusters of individual chromosome traces with similar architecture, we adapted the single-cell transcriptomic clustering analysis R toolkit Seurat version 2.3 (Butler et al., 2018). Each chromosome trace was treated as a single cell, with pairwise distance measurements used as features equivalent to gene expression levels. For whole chromosome V tracing of 22 TADs, this yielded 231 unique variables. Since not all traces encompassed all TADs due to the sample complexity of embryos, we replaced any missing values in traces with the non-zero averages for each pairwise comparison. ChrV TAD 11 pairs had higher technical variation than other pairs, so they were removed from influencing the clustering before standard Seurat normalization and scaling methods. The pairwise distance variables were then used to identity 20 principal components (PCs) which explain the majority of the variation between traces. Seurat provides statistical tests to inform you how many principal components capture the majority of variance in the data – this told us that many more than 30 PCs were significant. We used 20 principal components for the final analysis, but similar results were obtained using as low as 8 or more than 20 PCs. Traces are then plotted in a graph structure to identity K-nearest neighbors based on similarity in Euclidean PC space. Finally, clusters were defined using the Louvain algorithm using the FindClusters command. The Louvain algorithm, and Seurat, supports the use of a resolution parameter, which sets the ‘granularity’ of the clustering (the number of clusters in the dataset). Clustering using too low a resolution fails to identify the heterogeneity in the dataset. Clustering using too a high resolution can lead to ‘over-clustering’, where bona fide clusters are split into multiple highly similar clusters. Optimal resolution was determined through trial and error (as in scRNA-seq analyses), using low to high resolution, until all clusters represented visually distinct structures. We performed Seurat clustering using 4 resolutions (0.6, 0.8, 1.0, 1.2) and analyzed the chromosome structures for all clusters for all resolutions. Resolution 1.0 was used for all datasets, and consistently outputted clusters with the most distinct structures, with minimal over-clustering (which can be corrected by merging two highly similar sub-populations). For example, resolution 0.8 failed to separate chrV β and γ sub-populations, so it was too low. Resolution 1.2 on chrV data split both α and δ into two highly similar sub-clusters each, so it was too high. t-distributed stochastic neighbor embedding (t-SNE) and Uniform Manifold Approximation and Projection (UMAP) dimensionality reduction was used for visualization purposes in 2D and 3D only. t-SNE and UMAP are not used to define the clusters. Notably, the different chromosome clusters did not appear as the completely distinct clouds in UMAP or tSNE space, as is typical of distinct cell types in scRNA-seq data. We note that the chromosome data inputs are more self-similar (same pairwise distance measurements in each trace, just different levels), compared to scRNA-seq data of mixed cell types each having divergent gene expression patterns. The differences within single chromosome data are therefore more analogous to variable gene expression patterns within a cell type rather than between cell types, and are not expected to generate completely separate clouds upon dimensionality reduction. As a control to test the robustness of the cluster analysis, we also repeated the clustering using fewer traces of a minimum length (≥10 foci for wild-type and ≥6 foci for cec-4). This higher-threshold analysis yielded a very similar segregation of conformations compared to the total. The FindAllMarkers command in Seurat was used to identity pairwise comparisons that were significantly enriched within trace clusters (markers). Variability in pairwise distance measures was compared using the FindVariableGenes function in Seurat. This function calculates the dispersion in pairwise distances while controlling for the mean pairwise distance and produces a scaled dispersion metric for each TAD pair. The top (most variable) and bottom (least variable) pairs were highlighted.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical tests are described in the appropriate figure legends. Throughout the manuscript, ‘‘n’’ refers to the number of individual chromosomes analyzed.
DATA AND CODE AVAILABILITY
All data is available in the main text or the supplementary materials. Original/source imaging data for Figures 1 and S3 are available through Mendeley Data (https://doi.org/10.17632/5tzxfssddm.1). Code used in this study was modified from Zhuang lab protocols (Chen et al., 2015; Wang et al., 2016). The distribution of some derivative works is prohibited by the original license to the public, but these will be made available upon request for academic use as permitted.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, Peptides, and Recombinant Proteins | ||
| Dextran Sulfate | Fisher Scientific | bp1585100 |
| PureLink™ RNase A (20 mg/mL) | Thermo Fisher Scientific (Invitrogen) | 12091021 |
| OmniPur® Formamide, Deionized | Merck Millipore | 4650–500ML |
| Ethylene carbonate 98% | Sigma Aldrich | E26258 |
| Critical Commercial Assays | ||
| Phusion Hot Start Master Mix | Life Technologies | Thermo Scientific F565S |
| NEB HiScribe T7 Quick High Yield RNA Synthesis Kit | BioConcept | E2050S |
| Zymo-Spin V Columns | LucernaChem | C1012–25 |
| DNA Clean & Concentrator-5 w/ Zymo-Spin IC Columns (Capped) | LucernaChem | D4013 |
| Zymo Oligo Binding Buffer | LucernaChem | D4060–1-10 |
| Maxima H Minus Reverse Transcriptase (200 U/µL) | Fisher Scientific | EP0751 |
| Deposited Data | ||
| Raw imaging data from Figure 1 and Figure S3 | Mendeley Data | doi:10.17632/5tzxfssddm.1 |
| C. elegans reference genome (UCSC version ce10/ WormBase Release WS220) | WormBase | ftp://ftp.wormbase.org/pub/wormbase/releases/WS220/species/c_elegans/ |
| Hi-C dataset, wild-type late/mixed stage embryos | (Crane et al., 2015) | NCBI GSE63717 |
| LEM-2 ChIP datasets, wild-type and cec-4 embryos | (Gonzalez-Sandoval et al., 2015) | NCBI GSE74133 |
| Experimental Models: Organisms/Strains | ||
| C. elegans Bristol strain N2 (wild-type) | Caenorhabditis Genetics Center | WB strain: N2 |
| C. elegans cec-4 strain RB2301 (F32E10.2(ok3124)) | Caenorhabditis Genetics Center | WB strain: RB2301; WBVar00094202 |
| Oligonucleotides | ||
| Sequences for probe amplification, see Data S1 | This paper | S1 |
| Readout (secondary probe) sequences, see Data S1 | This paper | S1 |
| Primary probe sequences for chrV, see Data S2 | This paper | S2 |
| Primary probe sequences for chrV left arm, see Data S4 | This paper | S4 |
| Primary probe sequences for chrV center, see Data S5 | This paper | S5 |
| Primary probe sequences for chrV right arm, see Data S6 | This paper | S6 |
| Primary probe sequences for chrI, see Data S3 | This paper | S3 |
| Software and Algorithms | ||
| MATLAB R2016b, R2019b | MathWorks | https://mathworks.com |
| OligoArray 2.1 | (Rouillard et al., 2003) | http://berry.engin.umich.edu/oligoarray2_1/OligoArrayInst.exe |
| NCBI-BLAST 2.2.26 | NCBI | https://ftp.ncbi.nlm.nih.gov/blast/executables/legacy.NOTSUPPORTED/ |
| NCBI BLAST+ 2.7.1 | NCBI | https://ftp.ncbi.nlm.nih.gov/blast/executables/blast+/ |
| OligoArrayAux | The UNAFold Web Server | http://unafold.rna.albany.edu/?q=DINAMelt/OligoArrayAux |
| Seurat 2.3 | (Butler et al., 2018) | https://satijalab.org/seurat/ |
| Raincloud-shiny | (Allen et al., 2019) |
https://gabrifc.shinyapps.io/raincloudplots/ https://micahallen.org/2018/03/15/introducing-raincloud-plots/ |
Highlights.
Chromosomes from early embryos resemble a barbell
Lamina interactions stretch chromosomes and separate compartments
Conventional compartments arise during gastrulation via long-distance associations
Single-chromosome clustering uncovers prevalent conformations
ACKNOWLEDGMENTS
We are grateful to E. Levine and all Mango and Zhuang lab members for insightful discussions. We thank R. Losick and A. Schier for comments on the manuscript and the Biozentrum Imaging Core Facility (IMCF) for technical assistance. Strains were provided by the CGC (NIH grant P40 OD010440). M.E.R.S. is supported by a postdoctoral fellowship from the CIHR. Funding to S.E.M. from the NIH (grant R37GM056264), Harvard University, and the Biozentrum of the University of Basel. X.Z. received funding from the NIH (grant R35GM122487) and HHMI. S.W. is supported by NIH Director’s New Innovator Award 1DP2GM137414.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.molcel.2020.02.006.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Ahringer J, and Gasser SM (2018). Repressive chromatin in Caenorhabditis elegans: establishment, composition, and function. Genetics 208, 491–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen M, Poggiali D, Whitaker K, Marshall TR, Kievit RA, et al. (2019). Raincloud plots: a multi-platform tool for robust data visualization. Wellcome Open Res 10.12688/wellcomeopenres.15191.1. [DOI] [PMC free article] [PubMed]
- Anderson EC, Frankino PA, Higuchi-Sanabria R, Yang Q, Bian Q, Podshivalova K, Shin A, Kenyon C, Dillin A, and Meyer BJ (2019). X chromosome domain architecture regulates Caenorhabditis elegans lifespan but not dosage compensation. Dev. Cell 51, 192–207.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beliveau BJ, Boettiger AN, Avendaño MS, Jungmann R, McCole RB, Joyce EF, Kim-Kiselak C, Bantignies F, Fonseka CY, Erceg J, et al. (2015). Single-molecule super-resolution imaging of chromosomes and in situ haplotype visualization using Oligopaint FISH probes. Nat. Commun 6, 7147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bintu B, Mateo LJ, Su J-H, Sinnott-Armstrong NA, Parker M, Kinrot S, Yamaya K, Boettiger AN, and Zhuang X (2018). Super-resolution chromatin tracing reveals domains and cooperative interactions in single cells. Science 362, eaau1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boettiger AN, Bintu B, Moffitt JR, Wang S, Beliveau BJ, Fudenberg G, Imakaev M, Mirny LA, Wu CT, and Zhuang X (2016). Super-resolution imaging reveals distinct chromatin folding for different epigenetic states. Nature 529, 418–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonev B, and Cavalli G (2016). Organization and function of the 3D genome. Nat. Rev. Genet 17, 661–678. [DOI] [PubMed] [Google Scholar]
- Brejc K, Bian Q, Uzawa S, Wheeler BS, Anderson EC, King DS, Kranzusch PJ, Preston CG, and Meyer BJ (2017). Dynamic control of X chromosome conformation and repression by a histone H4K20 demethylase. Cell 171, 85–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner S (1974). The genetics of Caenorhabditis elegans. Genetics 77, 71–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler A, Hoffman P, Smibert P, Papalexi E, and Satija R (2018). Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol 36, 411–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardozo Gizzi AM, Cattoni DI, Fiche J-B, Espinola SM, Gurgo J, Messina O, Houbron C, Ogiyama Y, Papadopoulos GL, Cavalli G, et al. (2019). Microscopy-based chromosome conformation capture enables simultaneous visualization of genome organization and transcription in intact organisms. Mol. Cell 74, 212–222.e5. [DOI] [PubMed] [Google Scholar]
- Celniker SE, Dillon LAL, Gerstein MB, Gunsalus KC, Henikoff S, Karpen GH, Kellis M, Lai EC, Lieb JD, MacAlpine DM, et al. ; modENCODE Consortium (2009). Unlocking the secrets of the genome. Nature 459, 927–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen KH, Boettiger AN, Moffitt JR, Wang S, and Zhuang X (2015). Spatially resolved, highly multiplexed RNA profiling in single cells. Science 348, aaa6090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crane E, Bian Q, McCord RP, Lajoie BR, Wheeler BS, Ralston EJ, Uzawa S, Dekker J, and Meyer BJ (2015). Condensin-driven remodelling of X chromosome topology during dosage compensation. Nature 523, 240–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cremer T, and Cremer C (2001). Chromosome territories, nuclear architecture and gene regulation in mammalian cells. Nat. Rev. Genet 2, 292–301. [DOI] [PubMed] [Google Scholar]
- de Wit E (2019). TADs as the caller calls them. J. Mol. Biol Published online October 23, 2019. 10.1016/j.jmb.2019.09.026. [DOI] [PubMed]
- Dekker J, Rippe K, Dekker M, and Kleckner N (2002). Capturing chromosome conformation. Science 295, 1306–1311. [DOI] [PubMed] [Google Scholar]
- Dixon JR, Selvaraj S, Yue F, Kim A, Li Y, Shen Y, Hu M, Liu JS, and Ren B (2012). Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 485, 376–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du Z, Zheng H, Huang B, Ma R, Wu J, Zhang X, He J, Xiang Y, Wang Q, Li Y, et al. (2017). Allelic reprogramming of 3D chromatin architecture during early mammalian development. Nature 547, 232–235. [DOI] [PubMed] [Google Scholar]
- Eskeland R, Leeb M, Grimes GR, Kress C, Boyle S, Sproul D, Gilbert N, Fan Y, Skoultchi AI, Wutz A, and Bickmore WA (2010). Ring1B compacts chromatin structure and represses gene expression independent of histone ubiquitination. Mol. Cell 38, 452–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falk M, Feodorova Y, Naumova N, Imakaev M, Lajoie BR, Leonhardt H, Joffe B, Dekker J, Fudenberg G, Solovei I, and Mirny LA (2019). Heterochromatin drives compartmentalization of inverted and conventional nuclei. Nature 570, 395–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fields BD, Nguyen SC, Nir G, and Kennedy S (2019). A multiplexed DNA FISH strategy for assessing genome architecture in Caenorhabditis elegans. eLife 8, e42823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finn EH, Pegoraro G, Brandaño HB, Valton A-L, Oomen ME, Dekker J, Mirny L, and Misteli T (2019). Extensive heterogeneity and intrinsic variation in spatial genome organization. Cell 176, 1502–1515.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flyamer IM, Gassler J, Imakaev M, Brandaño HB, Ulianov SV, Abdennur N, Razin SV, Mirny LA, and Tachibana-Konwalski K (2017). Single-nucleus Hi-C reveals unique chromatin reorganization at oocyte-tozygote transition. Nature 544, 110–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaydos LJ, Wang W, and Strome S (2014). Gene repression. H3K27me and PRC2 transmit a memory of repression across generations and during development. Science 345, 1515–1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibcus JH, Samejima K, Goloborodko A, Samejima I, Naumova N, Nuebler J, Kanemaki MT, Xie L, Paulson JR, Earnshaw WC, et al. (2018). A pathway for mitotic chromosome formation. Science 359, eaao6135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giorgetti L, and Heard E (2016). Closing the loop: 3C versus DNA FISH. Genome Biol 17, 215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Sandoval A, Towbin BD, Kalck V, Cabianca DS, Gaidatzis D, Hauer MH, Geng L, Wang L, Yang T, Wang X, et al. (2015). Perinuclear anchoring of H3K9-methylated chromatin stabilizes induced cell fate in C. elegans embryos. Cell 163, 1333–1347. [DOI] [PubMed] [Google Scholar]
- Hakim O, Sung M-H, Voss TC, Splinter E, John S, Sabo PJ, Thurman RE, Stamatoyannopoulos JA, de Laat W, and Hager GL (2011). Diverse gene reprogramming events occur in the same spatial clusters of distal regulatory elements. Genome Res 21, 697–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heitz E (1928). Das heterochromatin der moose. Jahrb. Wiss. Bot 69, 762–818. [Google Scholar]
- Hou C, Li L, Qin ZS, and Corces VG (2012). Gene density, transcription, and insulators contribute to the partition of the Drosophila genome into physical domains. Mol. Cell 48, 471–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hug CB, and Vaquerizas JM (2018). The birth of the 3D genome during early embryonic development. Trends Genet 34, 903–914. [DOI] [PubMed] [Google Scholar]
- Hug CB, Grimaldi AG, Kruse K, and Vaquerizas JM (2017). Chromatin architecture emerges during zygotic genome activation independent of transcription. Cell 169, 216–228.e19. [DOI] [PubMed] [Google Scholar]
- Ikegami K, Egelhofer TA, Strome S, and Lieb JD (2010). Caenorhabditis elegans chromosome arms are anchored to the nuclear membrane via discontinuous association with LEM-2. Genome Biol 11, R120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaaij LJT, van der Weide RH, Ketting RF, and de Wit E (2018). Systemic loss and gain of chromatin architecture throughout zebrafish development. Cell Rep 24, 1–10.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamath RS, Fraser AG, Dong Y, Poulin G, Durbin R, Gotta M, Kanapin A, Le Bot N, Moreno S, Sohrmann M, et al. (2003). Systematic functional analysis of the Caenorhabditis elegans genome using RNAi. Nature 421, 231–237. [DOI] [PubMed] [Google Scholar]
- Ke Y, Xu Y, Chen X, Feng S, Liu Z, Sun Y, Yao X, Li F, Zhu W, Gao L, et al. (2017). 3D chromatin structures of mature gametes and structural reprogramming during mammalian embryogenesis. Cell 170, 367–381.e20. [DOI] [PubMed] [Google Scholar]
- Kiefer JC, Smith PA, and Mango SE (2007). PHA-4/FoxA cooperates with TAM-1/TRIM to regulate cell fate restriction in the C. elegans foregut. Dev. Biol 303, 611–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kind J, Pagie L, Ortabozkoyun H, Boyle S, de Vries SS, Janssen H, Amendola M, Nolen LD, Bickmore WA, and van Steensel B (2013). Single-cell dynamics of genome-nuclear lamina interactions. Cell 153, 178–192. [DOI] [PubMed] [Google Scholar]
- Kind J, Pagie L, de Vries SS, Nahidiazar L, Dey SS, Bienko M, Zhan Y, Lajoie B, de Graaf CA, Amendola M, et al. (2015). Genome-wide maps of nuclear lamina interactions in single human cells. Cell 163, 134–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kipreos ET, and van den Heuvel S (2019). Developmental control of the cell cycle: Insights from Caenorhabditis elegans. Genetics 211, 797–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson AG, Elnatan D, Keenen MM, Trnka MJ, Johnston JB, Burlingame AL, Agard DA, Redding S, and Narlikar GJ (2017). Liquid droplet formation by HP1α suggests a role for phase separation in heterochromatin. Nature 547, 236–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieberman-Aiden E, van Berkum NL, Williams L, Imakaev M, Ragoczy T, Telling A, Amit I, Lajoie BR, Sabo PJ, Dorschner MO, et al. (2009). Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 326, 289–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu T, Rechtsteiner A, Egelhofer TA, Vielle A, Latorre I, Cheung M-S, Ercan S, Ikegami K, Jensen M, Kolasinska-Zwierz P, et al. (2011). Broad chromosomal domains of histone modification patterns in C. elegans. Genome Res 21, 227–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luperchio TR, Wong X, and Reddy KL (2014). Genome regulation at the peripheral zone: lamina associated domains in development and disease. Curr. Opin. Genet. Dev 25, 50–61. [DOI] [PubMed] [Google Scholar]
- Macosko EZ, Basu A, Satija R, Nemesh J, Shekhar K, Goldman M, Tirosh I, Bialas AR, Kamitaki N, Martersteck EM, et al. (2015). Highly parallel genome-wide expression profiling of individual cells using nanoliter droplets. Cell 161, 1202–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mateo LJ, Murphy SE, Hafner A, Cinquini IS, Walker CA, and Boettiger AN (2019). Visualizing DNA folding and RNA in embryos at single-cell resolution. Nature 568, 49–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirny LA (2011). The fractal globule as a model of chromatin architecture in the cell. Chromosome Res 19, 37–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mutlu B, Chen H-M, Moresco JJ, Orelo BD, Yang B, Gaspar JM, Keppler-Ross S, Yates JR, Hall DH, Maine EM, et al. (2018). Regulated nuclear accumulation of a histone methyltransferase times the onset of heterochromatin formation in C. elegans embryos. Sci. Adv 4, eaat6224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mutlu B, Chen H-M, Gutnik S, Hall DH, Keppler-Ross S, and Mango SE (2019). Distinct functions and temporal regulation of methylated histone H3 during early embryogenesis. Development 146, dev174516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagano T, Lubling Y, Stevens TJ, Schoenfelder S, Yaffe E, Dean W, Laue ED, Tanay A, and Fraser P (2013). Single-cell Hi-C reveals cell-to-cell variability in chromosome structure. Nature 502, 59–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagano T, Lubling Y, Vá rnai C, Dudley C, Leung W, Baran Y, Mendelson Cohen N, Wingett S, Fraser P, and Tanay A (2017). Cell-cycle dynamics of chromosomal organization at single-cell resolution. Nature 547, 61–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nir G, Farabella I, Pé rez Estrada C, Ebeling CG, Beliveau BJ, Sasaki HM, Lee SD, Nguyen SC, McCole RB, Chattoraj S, et al. (2018). Walking along chromosomes with super-resolution imaging, contact maps, and integrative modeling. PLoS Genet 14, e1007872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nora EP, Lajoie BR, Schulz EG, Giorgetti L, Okamoto I, Servant N, Piolot T, van Berkum NL, Meisig J, Sedat J, et al. (2012). Spatial partitioning of the regulatory landscape of the X-inactivation centre. Nature 485, 381–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nora EP, Goloborodko A, Valton A-L, Gibcus JH, Uebersohn A, Abdennur N, Dekker J, Mirny LA, and Bruneau BG (2017). Targeted degradation of CTCF decouples local insulation of chromosome domains from genomic compartmentalization. Cell 169, 930–944.e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nuebler J, Fudenberg G, Imakaev M, Abdennur N, and Mirny LA (2018). Chromatin organization by an interplay of loop extrusion and compartmental segregation. Proc. Natl. Acad. Sci. USA 115, E6697–E6706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramani V, Deng X, Qiu R, Gunderson KL, Steemers FJ, Disteche CM, Noble WS, Duan Z, and Shendure J (2017). Massively multiplex single-cell Hi-C. Nat. Methods 14, 263–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouillard J-M, Zuker M, and Gulari E (2003). OligoArray 2.0: design of oligonucleotide probes for DNA microarrays using a thermodynamic approach. Nucleic Acids Res 31, 3057–3062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satija R, Farrell JA, Gennert D, Schier AF, and Regev A (2015). Spatial reconstruction of single-cell gene expression data. Nat. Biotechnol 33, 495–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarzer W, Abdennur N, Goloborodko A, Pekowska A, Fudenberg G, Loe-Mie Y, Fonseca NA, Huber W, Haering CH, Mirny L, and Spitz F (2017). Two independent modes of chromatin organization revealed by cohesin removal. Nature 551, 51–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sexton T, Yaffe E, Kenigsberg E, Bantignies F, Leblanc B, Hoichman M, Parrinello H, Tanay A, and Cavalli G (2012). Three-dimensional folding and functional organization principles of the Drosophila genome. Cell 148, 458–472. [DOI] [PubMed] [Google Scholar]
- Snyder MJ, Lau AC, Brouhard EA, Davis MB, Jiang J, Sifuentes MH, and Csankovszki G (2016). Anchoring of heterochromatin to the nuclear lamina reinforces dosage compensation-mediated gene repression. PLoS Genet 12, e1006341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stadler MR, Haines JE, and Eisen MB (2017). Convergence of topological domain boundaries, insulators, and polytene interbands revealed by high-resolution mapping of chromatin contacts in the early Drosophila melanogaster embryo. eLife 6, e29550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens TJ, Lando D, Basu S, Atkinson LP, Cao Y, Lee SF, Leeb M, Wohlfahrt KJ, Boucher W, O’Shaughnessy-Kirwan A, et al. (2017). 3D structures of individual mammalian genomes studied by single-cell Hi-C. Nature 544, 59–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strom AR, Emelyanov AV, Mir M, Fyodorov DV, Darzacq X, and Karpen GH (2017). Phase separation drives heterochromatin domain formation. Nature 547, 241–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabo Q, Jost D, Chang J-M, Cattoni DI, Papadopoulos GL, Bonev B, Sexton T, Gurgo J, Jacquier C, Nollmann M, et al. (2018). TADs are 3D structural units of higher-order chromosome organization in Drosophila. Sci. Adv 4, eaar8082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan L, Xing D, Chang C-H, Li H, and Xie XS (2018). Three-dimensional genome structures of single diploid human cells. Science 361, 924–928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulianov SV, Doronin SA, Khrameeva EE, Kos PI, Luzhin AV, Starikov SS, Galitsyna AA, Nenasheva VV, Ilyin AA, Flyamer IM, et al. (2019). Nuclear lamina integrity is required for proper spatial organization of chromatin in Drosophila. Nat. Commun 10, 1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Steensel B, and Belmont AS (2017). Lamina-associated domains: links with chromosome architecture, heterochromatin, and gene repression. Cell 169, 780–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Su J-H, Beliveau BJ, Bintu B, Moffitt JR, Wu CT, and Zhuang X (2016). Spatial organization of chromatin domains and compartments in single chromosomes. Science 353, 598–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson I, Berlivet S, Eskeland R, Boyle S, Illingworth RS, Paquette D, Dostie J, and Bickmore WA (2014). Spatial genome organization: contrasting views from chromosome conformation capture and fluorescence in situ hybridization. Genes Dev 28, 2778–2791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshimura J, Ichikawa K, Shoura MJ, Artiles KL, Gabdank I, Wahba L, Smith CL, Edgley ML, Rougvie AE, Fire AZ, et al. (2019). Recompleting the Caenorhabditis elegans genome. Genome Res 29, 1009–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng X, Hu J, Yue S, Kristiani L, Kim M, Sauria M, Taylor J, Kim Y, and Zheng Y (2018). Lamins organize the global three-dimensional genome from the nuclear periphery. Mol. Cell 71, 802–815.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data is available in the main text or the supplementary materials. Original/source imaging data for Figures 1 and S3 are available through Mendeley Data (https://doi.org/10.17632/5tzxfssddm.1). Code used in this study was modified from Zhuang lab protocols (Chen et al., 2015; Wang et al., 2016). The distribution of some derivative works is prohibited by the original license to the public, but these will be made available upon request for academic use as permitted.