

Abstract
P/Q-type (Cav2.1) calcium channels support a host of Ca2+-driven neuronal functions in the mammalian brain. Alternative splicing of the main α1A(α12.1) subunit of these channels may thereby represent a rich strategy for tuning the functional profile of diverse neurobiological processes. Here, we applied a recently developed “transcript-scanning” method for systematic determination of splice variant transcripts of the human α12.1 gene. This screen identified seven loci of variation, which together have never been fully defined in humans. Genomic sequence analysis clarified the splicing mechanisms underlying the observed variation. Electrophysiological characterization and a novel analytical paradigm, termed strength–current analysis, revealed that one focus of variation, involving combinatorial inclusion and exclusion of exons 43 and 44, exerted a primary effect on current amplitude and a corollary effect on Ca2+-dependent channel inactivation. These findings significantly expand the anticipated scope of functional diversity produced by splice variation of P/Q-type channels.
Keywords: alternative splicing, P/Q-type calcium channel, α1A subunit, α12.1 subunit, transcript scanning, human brain, Ca2+-dependent inactivation, calmodulin
P/Q-type calcium channels, potentially the most abundant voltage-gated calcium channels in the mammalian brain (Mori et al., 1991), constitute prominent pathways of Ca2+ entry at both presynaptic and somatodendritic loci throughout the CNS (Catterall, 1998). As such, these channels not only represent a predominant trigger for neurotransmitter release in the CNS but also support a host of essential neuronal responses (Wheeler and Tsien, 1999). Moreover, spontaneous mutations within these channels underlie a growing list of inherited forms of ataxia, epilepsy, and migraine (Ophoff et al., 1996;Zhuchenko et al., 1997). Fitting with the diverse roles of these channels, their function can be fine-tuned by numerous mechanisms rather than being limited to a single profile. On the moment-to-moment time scale, gating can be modulated by channel phosphorylation (Zamponi et al., 1997) and G-protein interactions (Colecraft et al., 2001). More enduring functional adjustments ensue from variations in the hetero-oligomeric composition of channels, comprising a principal pore-forming α1A (Cav2.1) subunit, complexed with auxiliary β, α2δ, and possibly γ subunits (Dunlap et al., 1995). P/Q-type channels may assemble from several different β or α2δ gene products, yielding channels with distinctive functional properties (Patil et al., 1998). Another potential strategy for generating longer-term functional diversity is alternative splicing of transcripts for the principal α12.1 subunit. Because the human α12.1 gene comprises >47 exons (Ophoff et al., 1996), alternate splicing at just a few exon boundaries could yield an enormous number of variants, especially when considering the combinatorial possibilities.
Previous studies of α12.1 have established instances of splice variation and related functional effects. For example, in human brain, splicing results in optional translation of amino acids encoded by exon 47, which includes a poly-glutamine tract whose abnormal expansion triggers an inherited form of cerebellar ataxia (SCA6; Zhuchenko et al., 1997). However, a limitation of studies to date arises from the method used to discover splice variants of α12.1, which involves explicit sequencing of various partial-length cDNA clones of the α12.1 subunit. Because isolation of each clone from a conventional cDNA library is labor-intensive, extensive screening for splice variation becomes impractical. Accordingly, it is possible that only a fraction of splice variants has been revealed by traditional methods.
Here, we applied a recently developed, transcript-scanning approach (Mittman et al., 1999a,b) to the systematic identification of splice variation of the human α12.1 gene. This PCR-based approach rapidly amasses cDNA replicas of all transcript variants in a specified gene region and thus promises practical detection of most, if not all splice variants of the transcripts of this gene. This screen identified seven loci of variation, some of which are novel and the complete set of which has never been fully defined in humans. Electrophysiological experiments and a novel paradigm of analysis revealed that combinatorial inclusion and exclusion of exons 43 and 44 exerted a primary effect on current amplitude and a corollary effect on Ca2+-dependent channel regulation.
Parts of this paper have been published previously in abstract form (Soong et al., 2000).
MATERIALS AND METHODS
Transcript scanning by PCR and sequence analysis. The overall transcript-scanning approach has been described previously, as applied to T-type Ca2+ channels (Mittman et al., 1999a,b). Here, to scan cDNA replicas of human α1A (α12.1) transcripts, we designed PCR oligonucleotide primers to span 1–4 exons of this gene (Figs. 1A,B), using Oligo 6.1 software (Molecular Biology Insights, Cascade, CO). The sequence of oligonucleotides used for all scanning reactions (Fig. 1C) is detailed in Table S-1 (available as on-line supplementary information at www.jneurosci.org). PCR amplification from whole-brain (7400-1; Clontech, Palo Alto, CA), cerebellar (7401-1), thalamus (7188-1), substantia nigra (7193-1), hippocampus (7169-1), cerebral cortex (7110-1), amygdala (7190-1), and hypothalamus (7429-1) cDNA libraries produced products that were electrophoresed on an agarose gel, purified, and cloned into the pCR2.1-TOPO vector by high-efficiency topoisomerase-based ligation (TOPO TA cloning kit; Invitrogen, Carlsbad, CA). The ligation products were efficiently transformed into TOP bacterial cells (TOPO TA cloning kit). Splice variation sometimes produced gel bands with different mobility, and these were purified separately before TA subcloning. Eight to 28 individual bacterial colonies from each TA subcloning reaction were screened by PCR with exon scanning primers (supplemental on-line information, Table S-1, at www.jneurosci.org), followed by automated sequencing and display using Prism DNA Sequencing software (Applied Biosystems, Foster City, CA). Sequences were compared against the National Center for Biotechnology Information (NCBI) public databases by using the BLAST program. DNA sequence alignments to detect splicing of exons or alternate use of junctional sites were performed using the Lasergene Software (DNAStar, Madison, WI). In some instances, an initial analysis based on size differentiation during gel electrophoresis was performed.
Fig. 1.

Schematic representation of P/Q-type calcium channel α1A (α12.1) subunit and PCR scanning reactions used to detect splice variation therein.A, Diagram of α12.1 channel backbone structure, showing four homologous domains (I–IV), each with six transmembrane-spanning regions (1–6). The C-terminal tail contains structures postulated to be important for Ca2+regulation of the channel. EF, EF-hand;IQ, IQ-like CaM interaction domain; CBD, CaM interaction domain. B, Locations of exon transcripts corresponding to the backbone diagram in A.C, Locations of scanning reactions to detect splice variation (for details, see on-line Table S-1, atwww.jneurosci.org). D, Size differentiation of splice variants (±16/17), revealed by scanning reaction 5.
Intronic sequence analysis. Human genomic DNA (Clontech) was used to PCR amplify introns 9, 31, and 46 to determine or confirm the intron and exon organization across some sites for alternative splicing. The primers used for these introns are as follows: for intron 9, int9 forward (F), 5′-TCG CCG AGG ATG AAA CTG AC-3′; and int9 reverse (R), 5′-CAG CCT CTT CGG GGT TGA GC-3′); for intron 31, int31F, 5′-CGA TAT CCT CGT GAC TGA-GT-3′; and int31R, 5′-AGC AGA CAG ACA TAA GGC AG-3′); and for intron 46, int46F, 5′-GGG CCG CTA CAC CGA TGT GG-3′; and int46R, 5′-GTT GAG GGG GCT GGG CTT CC-3′). For other introns, data mining of the NCBI/European Molecular Biology Library (EMBL) database proved sufficient to specify key sites controlling splice variation. In particular, partial intronic sequences at most exonal boundaries were deposited (Ophoff et al., 1996) under accession numbers Z80114–Z80155. Partial intronic sequence surrounding exon 37b is available under accession number AF144098. Finally, large stretches of the genomic sequence spanning the human α12.1 gene are available under accession numbers AC098781 (exon 4), AC022436 (exons 5–10), AC026805 (exons 11–22), AC005305 (exons 22–31), and AC011446(exons 31*-47); all contained within the consensus reference sequence NT-031915.
Generation and characterization of full-length α1A(α12.1) clones by PCR. A full-length, single-gene α12.1 cDNA library was isolated by heminested PCR amplification of whole-brain and cerebellar cDNA libraries (Clontech; Regan et al., 2000). Two sets of primers were used: (1) F1, 5′-CCG GCA GCC TCA GCA TCA GC-3′; and R1, 5′-GGA TCA CAG GGG AAT AGG AC-3′; and (2) F2, 5′-GCG TAA CCC GGA GCC CTT TG-3′; and R2, 5′-CGG ATC ACA GGG GAA TAG GAC-3′. The first round of PCR, using oligonucleotides F1 and R1, followed a touch-down protocol, which was initiated by denaturation at 94°C for 2 min. This was followed by one cycle of PCR comprising denaturation at 94.5°C for 25 sec, annealing at 65°C for 30 sec, and extension at 68°C for 8 min. In the next five cycles, the annealing temperature was initially 63°C and progressively decreased by 1°C on each cycle. The terminal 28 cycles maintained the annealing temperature at 58°C. The buffer contained 1× pCRx and 2 mm Mg2+ (Elongase; Invitrogen, Gaithersburg, MD). The second round of PCR (performed with flanking oligonucleotides) was performed similarly, except that the initial annealing temperature was 67°C, and the touch-down protocol was programmed to decrease annealing temperature by 2°C over the next five cycles until reaching 55°C. The terminal 15 cycles of PCR maintained the annealing temperature at 55°C. The ∼7 kb PCR product was subcloned into the pCRXL vector (Invitrogen), transformed into TOP bacterial cells, and picked into 96-well microtiter plates for PCR screening and analyses. Individual clones were verified to be full-length by performing restriction enzyme digestions withBamHI and XbaI, and observing a resultant band of ∼7 kb.
The particular type of splice variant encoded by a given full-length α1A (α12.1) clone was determined by performing custom PCRs spanning the exonal region of interest on individual clones (Fig. 1C). Size or direct sequence analysis of scanning reaction products or both uniquely identified the type of splice variant. Compilation of like results for many such clones yielded the aggregate summary in Table 1.
Table 1.
Distribution of splice variants in a single-gene (α1A) library derived from human cerebellum
| Splice locus | % Variants at specified splice locus | Nclones analyzed | |||
|---|---|---|---|---|---|
| 10 (+VG) | Δ10A/Δ10B (+G or −) | ||||
| 10/Δ10A/Δ10B | 17% | 83% | 450 | ||
| 16+/17+ | 16−/17− | ||||
| ±16/17 | 100% | ∼0% | 340 | ||
| 17 (+VEA) | Δ17A (−VEA) | ||||
| 17/Δ17A | 0.4% | 99.6% | 540 | ||
| +31* (+NP) | −31* (−NP) | ||||
| ±31* | 95% | 5% | 1485 | ||
| 37a (EFa) | 37b (EFb) | ||||
| 37a/37b | 40.5% | 59.5% | 111 | ||
| 43+/44+ | 43+/44− | 43−/44+ | 43−/44− | ||
| ±43/±44 | 92.6% | 6.9% | 2% | 2% | 756 |
| 47 | Δ47 | ||||
| 47/Δ47 | 65% | 35% | 20 | ||
Construction of α1A (α12.1) cDNA clones with variable splicing at exons 43 and 44. The parental human α1A (α12.1) clone (Sutton et al., 1999) was a gift from Dr. Terry Snutch (University of British Columbia), and its splice variant content at the seven loci (Fig. 2) is the following: Δ10A (+G); 16+/17+; 17 (–VEA), 31* – (–NP); 37a (EFa); 43+/44−; 47ΔA. To generate α1A(α12.1) clones containing the other exon 43/44 splice combinations, a human brain cerebellar cDNA library (7401-1; Clontech) was amplified using a forward primer (5′-GAA AGC GGC CTC AAG GAG AG-3′) and a reverse primer containing an XbaI site (5′-tgc g tc tag aTC GCC CGG GCT TAG CAC CAA-3′). The PCR products were cloned into pCR2.1-TOPO vectors (TOPO TA cloning kit), and TOP bacterial transformants were placed into 96-well plates for PCR screening to determine the presence or absence of exons 43 and 44. Fifteen clones were then selected for DNA sequencing, and appropriate clones were substituted by BstEII and XbaI restriction enzyme sites into the original full-length α1A (α12.1) clone to generate the various constructs subjected to electrophysiological study. In the context of electrophysiological results, these four constructs are referred to as 43+/44+, 43−/44+, 43+/44−, and 43−/44−.
Fig. 2.

Seven loci of α12.1 splice variation detected by transcript scanning. The postulated schematic diagram (top) shows a more detailed secondary structure of α12.1, along with loci of splice variation (1–7), labeled according to transcript variant names. CI, Ca2+ inactivation region, containing structures believed important for CDI such as the EF-hand and IQ domain. Detailed changes in amino acid composition resulting from splice variation at each of seven loci are shown below. Atlocus 3, deletion of exons 16 and 17 would remove half the P-loop and the entire IIS6 segment, ostensibly producing a nonfunctional channel.
Transient expression of Cav2.1 splice variants in HEK293 cells. cDNA for one of four human α1A subunits (43+/44+, 43−/44+, 43+/44−, and 43−/44−) was transiently cotransfected with auxiliary β2a and α2bδ subunits in HEK293 cells, according to previously described methods (Peterson et al., 1999). β2a minimized voltage inactivation (Patil et al., 1998), thereby enhancing resolution of Ca2+-dependent regulation. Two to 3 d later, whole-cell recordings were obtained at room temperature.
Whole-cell electrophysiology and analysis. The bath solution contained (in mm): 140 TEA-MeSO3, 10 HEPES, pH 7.3, and 5 CaCl2 or BaCl2, 300 mOsm, adjusted with glucose. The internal pipette solution contained (in mm): 135 Cs-MeSO3, 5 CsCl2, 0.5 EGTA, 1 MgCl2, 4 MgATP, and 10 HEPES, pH 7.3, 290 mOsm, adjusted with glucose. Reported voltages are uncorrected for a –11 mV junction potential, and true voltage may be obtained by subtracting 11 mV from reported values. Whole-cell currents, obtained under voltage clamp with an Axopatch 200A amplifier (Axon Instruments), were filtered at 2 kHz and sampled at 10 kHz. Series resistance was typically 1–2 MΩ after >70% compensation. Leaks and capacitive transients were subtracted by a P/8 protocol. Test pulse depolarizations were delivered every 60 sec (facilitation protocol) or 100 sec (inactivation protocol).
Ca2+-dependent facilitation was determined from the normalized charge difference, ΔQ, obtained by integrating the difference between normalized traces ± prepulse (see Fig. 4, middle). Test pulse current traces were normalized to unity at the end of 50 msec depolarizing pulses to 0 or 5 mV. The fraction of channels facilitated by prepulse (Ffacilitated) is directly proportional to ΔQ divided by the time constant (τ) of facilitation, yielding relative facilitation (RF = ΔQ/τ). This follows by assuming that all channels are initially in the normal mode at test pulse onset, and subsequent shifts to the facilitated mode occur monoexponentially with time constant τ. Then, RF =Ffacilitated × [Po,facilitated −Po,normal]/Po,facilitated, where Po,facilitated andPo,normal are steady-state open probabilities in facilitated and normal modes, respectively. τ was explicitly determined from Ca2+ traces in each cell before calculation of RF. Ba2+RF was calculated by using τ values determined from Ca2+ traces in the same cell. All average data are presented as mean ± SEM after analysis by custom-written software in MATLAB 6.1 (MathWorks, Natick, MA). Smooth-curve fits to data are by eye.
Fig. 4.

Splicing at exons 43 and 44 does not affect CDF properties. A–D, Top, Prepulse voltage protocol used to reveal facilitation, with fixed test pulse depolarization to 0–5 mV and 30 msec prepulse depolarization.Middle, Exemplar Ba2+ and Ca2+ current traces corresponding to specific voltage pulses diagrammed at top. Thegray trace corresponds to the trial without a prepulse. The arrow in A marks slow activation phase characteristic of CDF. Bottom, Population averages (from n cells) of strength of CDF (RF) plotted as a function of prepulse potential. Ca2+ data are plotted as filled symbols; Ba2+ data are plotted asopen symbols. g is a metric for the strength of pure Ca2+-dependent facilitation, and mean values and SEM are shown.
RESULTS
The entire α1A(α12.1) cDNA, encoding the principal pore-forming subunit of P/Q-type calcium channels (Fig.1A), was systematically screened for alternative splicing of its constituent exons (Fig.1B). We exploited a transcript-scanning strategy (Mittman et al., 1999a,b), the crux of which was to perform multiple PCRs that amplified overlapping segments spanning the entire α12.1 cDNA (Fig. 1C), as represented in human brain cDNA libraries. Each “scanning reaction” serves to rapidly pool and amplify numerous copies of all splice variations within a specified region of the α12.1 subunit, thus greatly improving the practicality and sensitivity of discovering splice variants compared with traditional cloning and identification of partial-length channel cDNAs from a brain library. Furthermore, because of the overlapping and all-encompassing coverage of the scanning reactions, most of which spanned at least two exon boundaries, analysis of the resulting set of PCR products should in principle reveal most, if not all, splice variations. To identify splice variations represented within a given scanning reaction, the PCR product was cloned in bacteria, and individual bacterial colonies were subject to PCR with exon scanning primers (supplemental on-line information, Table S-1, at www.jneurosci.org). The identity of the splice variant composition contained in individual clones was then determined by heterogeneity of product size, specificity of primers used, and direct sequencing. Figure 1D illustrates the size analysis for PCR 5, spanning exons 16 and 17. The larger species (650 bp) results from amplification of transcripts containing both exons 16 and 17 (+16/17), and the smaller species (454 bp) results from variant transcripts lacking both exons 16 and 17 (–16/17).
The transcript-scanning strategy revealed a total of seven loci for alternative splicing of the α12.1 gene, as summarized in Figure 2: (1) at the beginning of exon 10 in the I–II interdomain loop (10/Δ10A/Δ10B), there can be insertion of a valine and glycine, insertion of a glycine alone, or no insertion of either amino acid; (2) in the IIS6 region, exons 16 and 17 can be present or absent (±16/17); (3) near the end of exon 17 in the II–III interdomain loop (17/Δ17A), there can be optional omission of the tripeptide VEA; (4) near the beginning of exon 32 in the IVS3–IVS4 loop (±31*), there can be insertion of the dipeptide NP; (5) at the proximal end of the C-terminal tail, there is mutually exclusive use of exon 37a or 37b(37a/37b), resulting in channels with one of two versions of an EF-hand motif (Kretsinger, 1976), which often represents a canonical Ca2+ binding site; (6) downstream of the CBD domain, exons 43 and 44 can be present or absent in all four combinations (43±/44±); and (7) at the distal end of the C terminus, there can be insertion of a pentanucleotide GGCAG 5′ of the beginning of exon 47, thus allowing in-frame translation of exon 47 to produce a long version of the C terminus (47). Otherwise, omission of the GGCAG in variant transcripts causes a frameshift, leading to stop codon termination of channel subunits near the beginning of exon 47 (Δ47). Although isolated subsets of these splice variants have been reported for rat, mouse, and human (see Discussion), specific variations at loci 2 and 6 are novel, and the full set has not been delineated in human.
To infer the mechanisms underlying the observed splice variation, we determined pertinent intronic sequences from the genomic DNA sequences corresponding to α1A(α12.1). With regard to 10/Δ10A/Δ10B variation in the I–II interdomain loop (Fig.3A), we obtained a 2.8 kb amplicon from PCR amplification of human genomic DNA across intron 9. Sequencing of the exon–intron boundaries suggests a possible mechanism for the three possible outcomes of splicing, involving alternative nonconsensus splice acceptors. The 10 (+VG) and Δ10A (+G) scenarios appear to exploit an unconventional GT/TG acceptor–donor site pair, whereas the no insertion (–) Δ10B case uses a more common GT/AG pair. Although much less common, the use of a TG acceptor has been found to represent ∼0.04% of 22,489 mammalian expressed sequence tag-verified splice pairs (Burset et al., 2000). Our sequencing result differs from that provided by Ophoff et al. (1996) (accession numberZ80123) regarding the junction between intron 9 and exon 10, where their database information indicates the sequence ccattgtagGAG instead of the ccattgttgGAG that we have determined. However, our sequence is identical to deposited genomic sequence found in GenBank AC022436. Concerning the dual cassette, ±16/17 splicing (Fig. 2, locus 2), the published sequence for introns 15 and 17 (accession numbers Z80127–Z80129 and AC026805) points to customary alternative use of a rather canonical GT/AG donor–acceptor site pair (Fig.3B). With respect to 17/Δ17A (Fig. 2, locus 3), the published intronic sequence (accession numbers Z80128–Z80129 andAC026805) suggests that there is alternative use of junctional donor sites after exon 17 (Fig. 3C). Turning to ±31* splicing (Fig. 2, locus 4), EMBL partial sequence for the margins of intron 31 (accession numbers Z80142–Z80143) failed to reveal alternative use of acceptor–donor sites near exon–intron boundaries as a plausible basis for this splice variation (Fig.3D). However, data mining the interior region of intron 31 (accession number AC011446) revealed a potential six-nucleotide exon (exon 31*) that encodes NP, and the putative exon 31* is flanked by canonical GT/AG acceptor–donor sites (Fig. 3D). Furthermore, a pyrimidine-rich tract resides just upstream of the acceptor AG site, further supporting this region as a legitimate exon (Sharp and Burdge, 1997; Zhang, 1998). Thus, cassette inclusion of a putative exon 31* appeared to be the mechanism of optional NP insertion. Nonetheless, database annotations failed to acknowledge this region as a potential exon, and there was no independent confirmation of the putative exon 31* sequence. Accordingly, we used exonic primers from exons 31 and 32 to amplify an ∼10 kb fragment encompassing intron 31 in its entirety. Nested PCR of this large, intronic DNA fragment enabled the cloning of a 0.9 kb fragment, the sequencing of which confirmed the essential features of the presumed exon 31* sequence. Hence, we conclude that ±31* splice variation involves optional exclusion and inclusion of a novel exon 31* encoding NP. To understand mutually exclusive cassette splicing of exons 37a and 37b (accession numbers Z80146–Z80148, AF144098, and AC011446) (Fig. 2,locus 5), as well as combinatorial splicing at exons 43 and 44 (accession numbers Z80150–Z80153 and AC011446) (Fig. 2, locus 6), data mining of exon–intron border sequences revealed alternative use of canonical GT/AG acceptor–donor site pairs (Fig.3E,F). Finally, with regard to locus 7 (Fig. 2), we used PCR to explicitly confirm published intron–exon boundaries among exon 46, intron 46, and exon 47 (accession numbers Z80154–Z80155 andAC011446). The sequence (Fig. 3G) suggests that alternate use of a canonical acceptor site just 5′ of exon 47 can generate the insertion of a pentanucleotide of GGCAG, which would result in a frameshift whereby exon 47 would be read in-frame after exon 46. Otherwise, GT/AG splicing precisely at the exon boundaries would yield an in-frame stop codon immediately after exon 46, yielding an α1A (α12.1) subunit with a shorter C-terminal tail. In summary, these results indicate that alternative splicing produces a rich ensemble of channel customizations, exploiting both traditional and rather unexpected mechanisms that include a six-nucleotide exon and noncanonical splice acceptor–donor site pairs.
Fig. 3.

Postulated mechanisms underlying splice variation of α12.1. A–G, Top row, Nucleotide sequence of relevant exon–intron boundaries.Bottom row, resultant transcript and encoded amino acids of each variant. Mechanisms for splicing were (A,G, alternate acceptors (A, G), cassette (B, D–F), and alternate donor (C).
Beyond establishing the spectrum of possible splice variations, it was important to gauge how frequently the various splice configurations occur. Moreover, because our exon scanning was performed on a cDNA library whose constituent clones need not span complete channel transcripts, it was also worth establishing whether the observed variants were present in full-length α1A(α12.1) cDNAs. To address these issues, we reexamined key transcript-scanning reactions, this time performed on a different library as a template, a full-length, single-gene α1A (α12.1) library, obtained by long PCR amplification (Regan et al., 2000) of the original human cerebellar library. Results of the analysis are summarized in Table 1, inspection of which reveals clear preferences for certain splice variants. For example, the –16/17 variant is undetectable or extremely rare, whereas Δ17A (–VEA) and +31* (+NP) variants heavily predominate. Other loci demonstrate more even representation of splice options, such as the distribution between 37a (EFa) and 37b (EFb) variants. Although two of the variants, 43−/44+ and 43−/44−(Table 1), are just detectable, it is quite possible that such “rare” splice variants could be more prevalent within specific types of neurons of the cerebellum. Furthermore, it would be quite interesting if these variants were more prevalent in other portions of the brain, fitting with a general theme in which regional distribution profiles of splice variants are customized by development or external and internal cues. In fact, this latter possibility was strongly supported by transcript scans across exons 43 and 44 in human brain region-specific cDNA libraries (Table 2). Here, the fraction of 43−/44+transcripts reached 10% in amygdala, whereas the fraction of 43−/44−transcripts exceeded 20% in amygdala, thalamus, and substantia nigra. Overall, the analysis establishes that, except for the 16−/17−variant, the ensemble of observed splice variants (Fig. 2) contains legitimate customizations of full-length channel transcripts, and that variations in the distribution within the ensemble of possibilities represents a potentially rich dimension for functional regulation.
Table 2.
Distribution of exon 43/44 splice variants in human brain region-specific cDNA libraries
| Brain region | % Variants at specified splice locus | Nclones analyzed | |||
|---|---|---|---|---|---|
| 43+/44+ | 43+/44− | 43−/44+ | 43−/44− | ||
| Amygdala | 33% | 31% | 10% | 26% | 222 |
| Cerebral cortex | 75% | 16% | 5% | 4% | 162 |
| Hippocampus | 44% | 40% | 8% | 8% | 167 |
| Hypothalamus | 37% | 48% | 3% | 12% | 182 |
| Thalamus | 42% | 32% | 4% | 22% | 189 |
| Substantia nigra | 32% | 43% | 3% | 22% | 201 |
Splice variation of α1A (α12.1) channels could have important functional correlates, as already shown by previous studies of partial sets of variants (Bourinet et al., 1999;Hans et al., 1999; Krovetz et al., 2000; Tsunemi et al., 2002). The more exhaustive suite of variants delineated here by systematic exon scanning raises expectations that splice-related functional diversity may well be considerably larger than currently appreciated. As a first step in defining the potentially broader spectrum of functional sequelae, we focused on novel splice variations in the α1A C-terminal tail, because this region contains critical structural determinants for Ca2+ feedback regulation of corresponding channels. In particular, Ca2+/CaM binding to an IQ-like binding motif in exon 40 (DeMaria et al., 2001), or possibly to a CBD binding motif in exon 42 (Lee et al., 1999, 2000), initiates both channel facilitation and inactivation by Ca2+ (DeMaria et al., 2001). The proximity of these structures to exons 43 and 44 (Figs. 1, 2), which are frequent sites of alternative splicing, suggested that combinatorial splice variation at this locus might tune Ca2+regulation, especially given the significant representation of each combinatorial possibility (Table 1).
Figure 4 compares the Ca2+ facilitation properties of α1A (α12.1) channels with all the possible variations at exons 43 and 44, with background splice variant structure delineated in Materials and Methods. Figure4A shows the behavior for the 43+/44+construct, using our previously reported methods of characterization (DeMaria et al., 2001), Facilitation was readily resolved in exemplar test pulse currents (+0 mV) with Ca2+ as the charge carrier (middle traces). Without a prepulse (to +20 mV), the test pulse waveform showed an initial rapid component of activation, followed by a slower phase of Ca2+ current increase (gray trace, arrow). This overall waveform morphology provides a direct indication of facilitation, because the biphasic kinetics arises from fast activation in a normal mode of gating, followed by more gradual Ca2+-driven conversion to a facilitated gating mode with greater open probability (Takahashi et al., 2001). For test pulse waveforms preceded by a prepulse (middle, black trace), channels were initially “prefacilitated” by Ca2+ entry during the prepulse. Hence, these currents activated rapidly to the fully facilitated level, as would be expected for fast activation of channels that reside predominantly within the facilitated gating mode at the test pulse onset. With Ba2+ as the charge carrier, exemplar test pulse currents (top traces) activated rapidly, with no appreciable slow phase, regardless of the presence or absence of a prepulse, consistent with “trapping” of channels in the normal mode of gating. To quantify facilitation, we integrated the difference between normalized test pulse currents in the absence and presence of a prepulse (see Materials and Methods), and this integral was used to determine the RF induced by a voltage prepulse. With Ca2+, relative facilitation demonstrated a bell-shaped dependence on prepulse voltage, providing a hallmark of a Ca2+-driven process. By contrast, the corresponding relationship with Ba2+ was far smaller, reflecting weak background G-protein modulation (DeMaria et al., 2001). The difference between Ba2+and Ca2+ relationships for a +20 mV prepulse, g, then provides a convenient quantifier of pure Ca2+-dependent facilitation. With respect to the effects of splice variation at exons 43 and 44, Figure4B–D revealed no detectable change in the profile of Ca2+ facilitation.
By contrast, splice variation appeared to have substantial effects on Ca2+-dependent inactivation (CDI) of channels, as well as on the overall amplitude of the current (Fig.5). Figure 5A shows the method of characterization for the 43+/44+construct. When viewed on a longer time base than used to resolve facilitation, specimen test pulse currents evoked by step depolarization to +10 mV could be seen to exhibit marked CDI, because currents decayed clearly faster with Ca2+versus Ba2+ as the charge carrier (top). The Ba2+ trace has been scaled downward approximately two times to enhance visual comparison of kinetics here and throughout. We quantified inactivation by determining the fraction of the peak current remaining after depolarizing for 800 msec (r800), plotted as a function of test pulse voltage (middle). Population means for these relationships confirmed a clearly deeper decay ofr800 with Ca2+ versus Ba2+, and the unmistakable U shape of the relationship with Ca2+ provided a classic hallmark for a Ca2+-driven process. The comparatively modest decline of the Ba2+relationship is believed to reflect a separate voltage-dependent inactivation process (Jones, 1999), so the distance between Ca2+ and Ba2+relationships at +10 mV, f, furnished a convenient index of pure CDI. Overall current size was assessed by averages of peak Ca2+ current amplitude plotted as a function of the test pulse voltage. Inspection of the corresponding analysis for the other splice variants (Fig. 5B–D) revealed two effects. First, CDI was clearly more pronounced in the 43−/44−construct, with f values of ∼0.4. Other constructs showed less CDI, with f values hovering between 0.2 and 0.3. Second, the 43−/44−construct produced twofold larger currents than the 43+/44−construct (Fig. 5, compare A, D, bottom), whereas the other two constructs manifested intermediate current amplitudes (Fig.5B,C, bottom). No clear differences in the voltage dependence of activation or inactivation were observed among the variants (Fig. 5 legend). One plausible mechanism for the effects on current size is that exons 43 and 44 may regulate the number of expressed channels, possibly by altered turnover rates secondary to complexation of this region of the channel with adaptor molecules (Maximov et al., 1999). Overall, splice variant effects on CDI and levels of current hold enormous potential biological impact for synaptic plasticity (von Gersdorff and Borst, 2002) and neurodegenerative disease (Pietrobon, 2002).
Fig. 5.

Splicing at exons 43 and 44 affects Ca2+ current amplitude and CDI.A–D, Top, One second depolarizing voltage pulse used to reveal CDI, along with exemplar Ba2+ and Ca2+ currenttraces. The gray trace was obtained with Ca2+ as the charge carrier; the black trace was obtained with Ba2+. Scale bar corresponds to the Ca2+ trace; Ba2+ traces were scaled downward to facilitate comparison of decay kinetics. Middle, Population averages (from n cells) for inactivation properties, as gauged by r800, the fraction of peak current remaining after an 800 msec depolarization plotted as a function of test pulse potential. Ca2+ data are plotted as filled symbols; Ba2+ data are plotted as open symbols. f is a metric for the strength of pure Ca2+-dependent inactivation, and mean values and SEM are shown. Bottom, Peak current versus test pulse potential with Ca2+as the charge carrier. Data were averaged from same cells as ther800 plot above. A versusD, Ca2+ current amplitude (bottom) and CDI are clearly greater in the 43−/44− versus 43+/44+ variant, with other splice forms showing intermediate behavior (B,C). No significant difference in the voltage dependence of activation or inactivation was observed. Half-activation voltages, as determined from current–voltage relationships (bottom) with Ca2+ as the charge carrier, were (in mV): −6.0 ± 1.6 (43+/44+; n = 8), 1.8 ± 0.7 (43−/44+;n = 4), −1.7 ± 1.3 (43+/44−; n= 5), and −5.6 ± 1.3 (43−/44−; n= 11). Half-inactivation voltages, as determined from 1 sec prepulse inactivation protocols with Ca2+ as the charge carrier, were (in mV): −2.4 ± 2.3 (43+/44+; n = 8), 6.5 ± 1.6 (43−/44+;n = 4), 0.1 ± 1.3 (43+/44−; n= 5), and −2.1 ± 1.1 (43−/44−; n= 11).
Before concluding that splice variation at exons 43 and 44 entails two independent functional outcomes, we questioned whether the effects might be coupled through a single mechanism. We had postulated previously that α1A(Cav2.1) CDI is driven by a “global” Ca2+ concentration signal that integrates Ca2+ influx through multiple channels (DeMaria et al., 2001). By contrast, we also postulated that Ca2+-dependent facilitation (CDF) responds primarily to a “local” Ca2+ signal that predominantly reflects the activity of individual channels. If these postulates hold true, then simply augmenting the number of expressed channels could increase the global Ca2+ signal and thereby enhance CDI, even without any change in the intrinsic properties of individual channels. Thus, the only direct effect of splice variation may be to increase the number of expressed channels, and the enhanced CDI may result as a corollary outcome of elevated channel number. Alternatively, splice variation could change the inherent propensity for each channel to undergo CDI, given the same global Ca2+signal. Finally, a combination of the above two scenarios would also accord with the functional results presented thus far.
To distinguish among these, we pursued two sets of experiments that consolidated and refined our global Ca2+–CDI hypothesis in a manner permitting discrimination among the three possible scenarios. In the first, we examined the effects of a high intracellular concentration (10 mm) of the rapid Ca2+chelator BAPTA on CDF and CDI. Under these conditions, processes driven by global Ca2+ signals should be strongly attenuated, whereas those responding with extreme selectivity to local Ca2+ influx might be mostly spared (Deisseroth et al., 1996). Figure 6,A and B, displays striking results in this regard for the 43+/44+construct. Exemplar traces (Fig. 6A) suggest complete sparing of CDF in the face of virtual elimination of CDI. Population data (Fig. 6B) entirely confirmed these trends, clearly demonstrating that CDF responds very selectively to local Ca2+ influx through individual channels, and that CDI requires a more global Ca2+signal. This experiment did not, however, exclude the possibility that, although the locus of Ca2+ sensing for CDI lies somewhat more distant from the channel pore than does the sensor for CDF (accounting for BAPTA sensitivity), a single channel could still manage to inactivate itself. Moreover, the first experiments did not gauge how sensitively different splice variants might undergo CDI in response to a given global Ca2+ signal, and splice variation could have produced CDI effects via changes in such sensitivity.
Fig. 6.
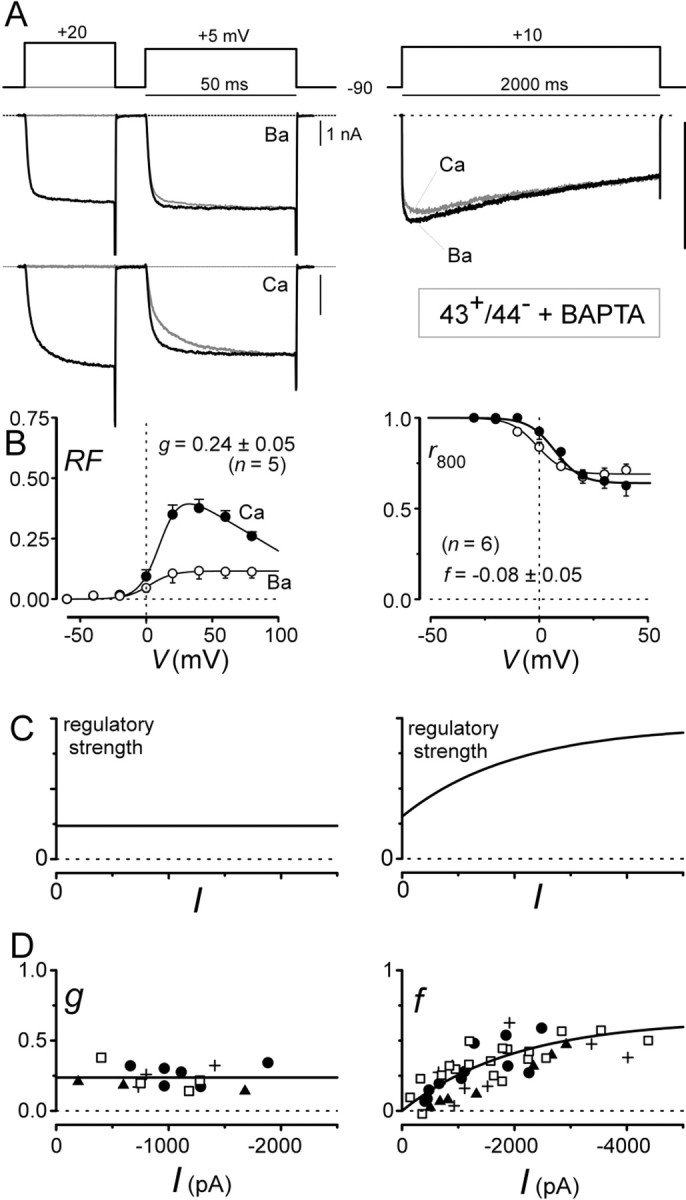
CDF responds selectively to local Ca2+ influx through individual channels, whereas CDI responds selectively to global Ca2+ influx through many channels. A, Exemplar tracesillustrating complete sparing of CDF and total elimination of CDI by intracellular 10 mm BAPTA. Format is identical to that of Figures 4 and 5. B, Population data, corresponding to exemplar traces in A, confirm selective elimination of CDI by BAPTA. Format is identical to that of Figures 4and 5. C, Theoretical predictions of strength–current analysis for Ca2+ regulatory processes with different selectivities for local versus global Ca2+influx. The y-axis plots the strength of Ca2+ regulation; the x-axis plots peak Ca2+ current; curves are schematic. Left, Scenario for a Ca2+regulatory process selective for local Ca2+ influx through individual channels. Right, Case for a Ca2+ regulatory process triggered by both local Ca2+ influx through individual channels (positivey-axis intercept) and global Ca2+influx through many channels (increasing regulatory strength with growing I). D, Strength–current analysis for CDF and CDI properties of various splice variants. Eachsymbol corresponds to one cell. Differentsymbols correspond to splice variants as follows:filled circle, 43+/44+; filled triangle, 43−/44+;plus sign, 43+/44−; and open square, 43−/44−.Left, CDF follows prediction of a process completely selective for local Ca2+ influx through individual channels, fitting with insensitivity to BAPTA in A andB. Right, CDI follows prediction of a process entirely selective for global Ca2+ influx through many channels (with 0-valued y-intercept), fitting with elimination of CDI by BAPTA in A andB. To resolve the y-intercept of therightCDI Iplot, some 43−/44− (open square) data here were obtained from cells only 12–18 hr after transfection rather than the usual 48–72 hr used for the bulk of the data, including those in Figure 5.
To address the remaining ambiguities, a novel experimental paradigm was devised. We reasoned that the strength of channel regulation would respond differently to progressive increases in the global Ca2+ signal, depending on the selectivity and sensitivity of the modulatory process for global versus local sources of Ca2+ influx (Fig.6C). As a practical measure of global Ca2+ levels, we chose to measure peak Ca2+ current amplitudes (I). If a process were completely selective for Ca2+ influx through individual channels, without regard for influx through adjacent or neighboring channels, then regulatory strength should be independent of overall Ca2+ current amplitude (Fig. 6C, left). Alternatively, if a regulatory process were sensitive to both local and global Ca2+ influxes, then regulatory strength should increase with increasing Ca2+ current amplitude (Fig. 6C, right), and the y-axis intercept of the relationship should indicate the regulatory strength resulting from Ca2+ influx through an individual channel. Finally, if a process were selectively responsive to global Ca2+ influx, whereas the influx via an isolated channel alone were unable to inactivate itself, then the regulatory strength relationship would be as shown in Figure6C (right), except that the y-axis intercept value would be 0.
Figure 6D shows the results for such a “strength–current” experiment, in which the strength of CDF (or CDI) and peak Ca2+ current amplitudes for individual cells contribute separate data points. Plots of the metricg (CDF strength) versus I (Fig. 6D, left) reveal a completely flat relationship, indicating entirely selective reliance on local Ca2+, a result fitting with deductions from the BAPTA experiments above (Fig.6B, left). The ability of a single flat relationship to fit data from all splice variants, plotted as different symbols, suggests that the CDF sensitivity to local Ca2+ influx is the same among splice variants, as might be expected from the similarity of aggregate averages for CDF analysis (Fig. 4A–D, bottom). By contrast, plots of the descriptor f (CDI strength) versusI strikingly demonstrate a positive correlation (Fig.6D, right), with a 0-valued y-axis intercept. This indicates that CDI is selectively reliant on global Ca2+ influx, and that an isolated channel would be incapable of inactivating itself. Because data from all splice variants, plotted as different symbols, appear to define a single relationship, CDI sensitivity to global Ca2+ influx is probably identical among splice variants, at least within the resolution of this experimental paradigm.
In summary, in-depth biophysical analysis (Fig. 6) indicates that the direct effect of splice variation at exons 43 and 44 is to regulate overall current amplitude, possibly by variations in channel number. Variations in CDI, especially prominent with the 43−/44−construct (Fig. 6D), result as corollary outcomes of fluctuations in current amplitude playing through an intrinsic sensitivity of CDI for global Ca2+levels.
DISCUSSION
Transcript scanning compared with previous screens for splice variation
Transcript scanning (Mittman et al., 1999a,b) was applied to screen systematically for splice variants of the human α1A (α12.1) gene. Locus 1 (Fig. 2) is novel for human, although there are rat analogs (Bourinet et al., 1999). Valine insertion slowed voltage inactivation but enhanced G-protein inhibition and PKC upregulation (Bourinet et al., 1999).
Locus 2 (±16/17) is new, although there are human analogs in α1B (α12.2, N-type) and α1E (α12.3, R-type) (Mittman and Agnew, 2000). Absence of the –16/17 variant in full-length α12.1 transcripts (Table 1) suggests that this variant may only be present in P/Q-type hemichannels (Scott et al., 1998). Mutant α12.1 hemichannels may cause episodic ataxia 2 (Ophoff et al., 1996), and there are hemichannel analogs for α1C(α11.2, L-type; Wielowieyski et al., 2001) and α12.2 (Mittman and Agnew, 2000), the latter of which may dominant negatively suppress full-length subunits (Raghib et al., 2001).
Optional VEA insertion (locus 3), within the “synprint” region for channel–SNARE–complex interaction (Rettig et al., 1996; Zhong et al., 1999), has not been explicitly linked to splice variation, but suggestions of such have been raised (Hans et al., 1999).
Locus 4 splicing has been found in rat brain (Bourinet et al., 1999) and human spinal cord (Krovetz et al., 2000), although the hypothesized mechanism used obscured splice acceptor–donor pairs (Bourinet et al., 1999). Identification of candidate exon 31* provides a simple explanation involving conventional GT/AG splice pairs. NP insertion decreases ω-Aga IVA sensitivity (Bourinet et al., 1999; Hans et al., 1999) and shifts the voltage dependence of activation (Bourinet et al., 1999) and inactivation (Hans et al., 1999; Toru et al., 2000). In α12.2, an analogous variant, encoding ET insertion in the corresponding IVS3-S4 loop, also affects activation and inactivation, and a similar cassette–exon mechanism was advanced (Lin et al., 1999).
Mutually exclusive splicing at locus 5 was detected in α12.1 clones from rat brain (Bourinet et al., 1999), human brain (Zhuchenko et al., 1997), and human spinal cord (Krovetz et al., 2000). Such splicing yields two versions of an EF-hand-like structure, typically supporting Ca2+ binding (Kretsinger, 1976). The analogous EF-hand motif in α11.2 is essential for its CDI (DeLeon et al., 1995; Zuhlke and Reuter, 1998;Peterson et al., 2000), but the motif probably transduces CDI rather than binds Ca2+ (Peterson et al., 2000). Instead, the trigger for Ca2+ regulation of either α11.2 or α12.1 is Ca2+/CaM interaction with a distinct IQ-like region (Peterson et al., 1999; Qin et al., 1999; Zuhlke et al., 1999; DeMaria et al., 2001). Hence, there is reason to wonder whether EF-hand splicing could alter Ca2+ regulation of P/Q-type channels (Bourinet et al., 1999; Chaudhuri et al., 2001).
Splicing of both exons 43 and 44 (locus 6) is novel; previous reports describe optional inclusion of exon 44 alone (Zhuchenko et al., 1997;Krovetz et al., 2000). Omitting exon 44 may diminish voltage-dependent inactivation (Krovetz et al., 2000), although we did not detect this effect (Fig. 5). The differing results may arise from our use of the auxiliary β2a subunit to diminish voltage-dependent inactivation and enhance quantification of Ca2+-dependent regulation (Bourinet et al., 1999; DeMaria et al., 2001). A previous study (Krovetz et al., 2000) used β1a, β1b, β3, and β4 subunits; all would enhance voltage-dependent inactivation (Patil et al., 1998).
Locus 7 (47/Δ47) is well documented in human brain (Zhuchenko et al., 1997; Hans et al., 1999), human spinal cord (Krovetz et al., 2000), and mouse (Toru et al., 2000). In the Δ47 variant (Mori et al., 1991;Ophoff et al., 1996), a stop codon terminates translation before exon 47. Pentanucleotide insertion in the 47 form causes a frameshift permitting translation of exon 47. Expansion of a poly-glutamine tract encoded in exon 47 triggers SCA6 (Zhuchenko et al., 1997), and we speculate that developmental enhancement of the 47 variant could help explain the delayed adult onset of this disease.
Finally, still other variants probably exist. Transcript scanning poorly resolves alternate versions of bracketing exons (1 and 47), because little is known about the respective upstream or downstream sequences against which scanning reaction oligonucleotides can be designed. Thus, the upstream oligonucleotide for scanning reaction 1 (Fig. 1C) was designed against the 5′ untranslated region of the known version of exon 1. Likewise, the downstream oligonucleotide for reaction 15 (Fig. 1C) was designed against the 3′ untranslated region of the known exon 47. These reactions would detect use of alternate splice acceptor–donor sites with the known exons (e.g., Δ47/47) but not of potential alternate versions of exons 1 and 47 (Tsunemi et al., 2002). Also, rare cell-specific RNA editing restricted to certain cells (Tsunemi et al., 2002) could elude our screen of cell population libraries.
Strength–current analysis
According to strength–current analysis, CDF fits with a process completely selective for Ca2+ influx through individual channels (Fig. 6C,D, left), and CDI fits with a process completely selective for global Ca2+ (Fig. 6C,D, right). Moreover, splice variation of exons 43 and 44 did not cause deviation from a baseline relationship, arguing against changes in intrinsic Ca2+ sensitivity.
To refine this interpretation and broaden understanding, we explore two alternative scenarios. Consider first a hypothetical outcome in which splice variation increases the intrinsic sensitivity of channel CDI to global Ca2+ (Fig.7A). Ca2+ current (I) produces the same global Ca2+ signal, but channels inactivate more strongly in response to the same global Ca2+, causing an upward-shifted relation. Also, the y-axis intercept would probably shift upward, because increased CDI sensitivity for global Ca2+ would likely pertain to Ca2+ influx through individual channels.
Fig. 7.
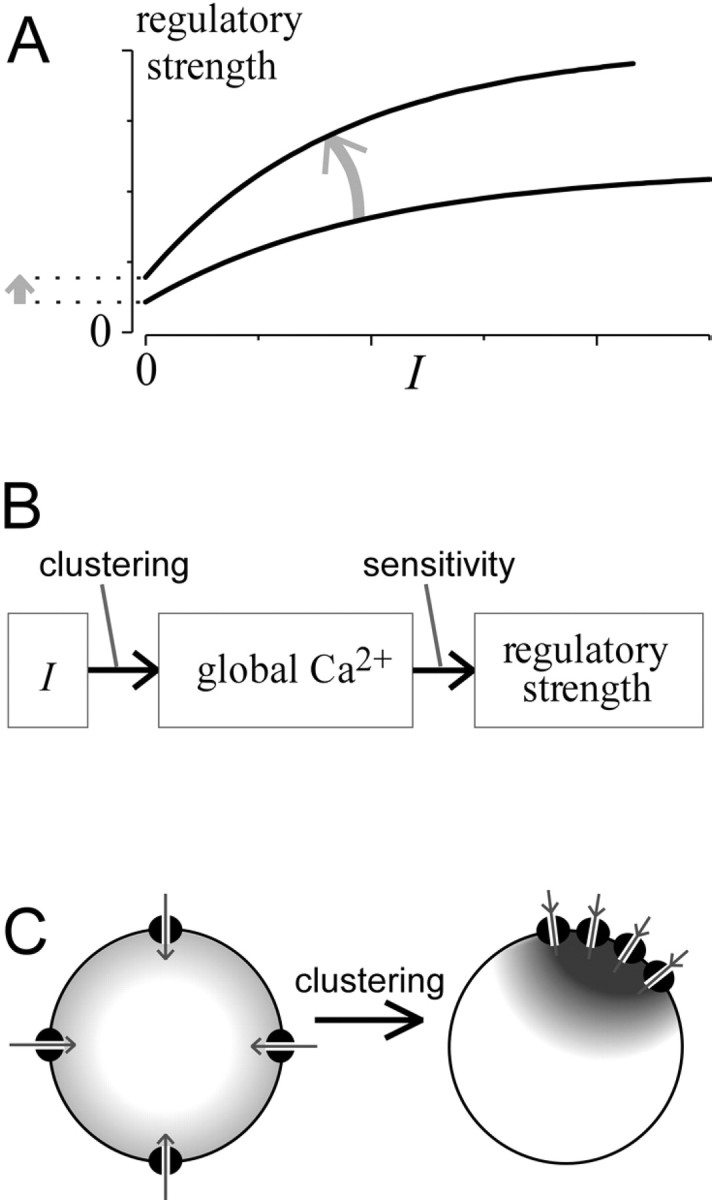
In-depth consideration of strength–current analysis. A, Theoretical curvesillustrating the anticipated shift (arrow) that would result from increased intrinsic Ca2+ sensitivity of a regulatory process for global Ca2+. The curvature of relationships reflects saturation of Ca2+responsiveness, the metric quantifying regulatory strength, or both.B, Underlying transformations connecting peak Ca2+ current amplitude (I) and regulatory strength. Increased intrinsic Ca2+ sensitivity would affect the second transformation, whereas cell-geometric factors such as channel clustering would affect the first. Changes in either transformation could impact the overall strength–current relationship.C, Diagram illustrating how increased channel clustering could enhance the transformation between I and the global Ca2+ signal relevant to channel regulation.
Considering the subtransformations linking CDI and I (Fig.7B) reveals another interpretation of the upward shift (Fig.7A). Channel Ca2+ influx must first be transformed into a relevant global Ca2+ signal, and the latter signal drives Ca2+ regulation (e.g., CDI). The first transformation reflects cell channel geometry, such as channel clustering (Fig. 7C); the second pertains to intrinsic Ca2+ sensitivity of channels for global Ca2+. Hence, enhanced channel clustering (Fig. 7C) could amplify the transformation from Ito global Ca2+, thereby causing the net effect in Figure 7A. In addition, if channels are only expressed as tightly packed clusters but not as individual channels, the y-intercept would reflect CDI resulting from Ca2+ influx through an individual channel cluster rather than through one channel. Also, increasing cluster size would possibly elevate the y-intercept.
Fortunately, our CDI data (Fig. 6D, right) indicate no shift in the relationship with splice variation. This outcome has only one likely interpretation: that splice variation changed neither Ca2+ sensitivity of channels for CDI nor channel clustering. The differences in CDI among splice variants solely reflect variations in the amplitude of the current.
Mechanism of current enhancement by exon 43/44 splicing
The remaining uncertainty concerns the basis for alterations of current amplitude with exon 43/44 splicing. Because I =N Poi, where N is channel number per cell, Po is the open probability, and i is the unitary current, changes inN, Po, and icould underlie the variation in I with exonal splicing. Which mechanism(s) underlies the data?
Fortunately, the invariance of CDF with splice variation (Fig.6D, left) favors a single mechanism, as follows. Given the complete insensitivity of CDF to buffering by 10 mm BAPTA (Fig. 6B, left) and the flat relationship between the strength of CDF and I(Fig. 6D, right), CDF must be selectively triggered by a local Ca2+ domain driven by Ca2+ influx through an individual channel (Deisseroth et al., 1996; Peterson et al., 2000). In this “local domain regimen,” the relevant domain Ca2+ concentration would be virtually synchronized in time with single-channel openings and directly proportional to i (Sherman et al., 1990), such that [Ca2+]domain ∝i when the channel is open or 0 when closed. The strength of CDF (g) should be a function of the “effective rate constant” governing the transition from nonfacilitated to facilitated channels at +10 mV (Peterson et al., 2000). In the local domain regimen, this rate constant would be a function of [Ca2+]domain andPo, wherePo is the peak open probability of unfacilitated channels at +10 mV. Specifically, the strength of CDF (g) should be closely proportional toPo × [Ca2+]domain2, where the square term reflects triggering of CDF by two Ca2+ ions binding to the C-terminal lobe of CaM (Peterson et al., 1999; DeMaria et al., 2001). Considering that [Ca2+]domain ∝i, we have that g ∝Poi2. The invariance ofg with splice variation (Fig. 6D, left) thus implies the invariance of both Poand i (absent unlikely cancellation of changes inPo andi2). By exclusion, variation in channel number (N) alone is the most likely mechanism underlying splice variant effects on I. One explanation for changes in N could be that the channel turnover rate is affected by channel complexation at or near exons 43 and 44 (Maximov et al., 1999) with as-yet-unknown adaptor molecules. Customization of channel interaction with adaptor molecules would be a most intriguing dimension for splice variant modulation.
Footnotes
The work was supported by the National Medical Research Council, Singapore (T.W.S.), a National Institutes of Health National Research Service Award fellowship (C.D.D.), National Institute of Mental Health Grant RO1-MH65531 (D.T.Y.), and a Johns Hopkins Singapore travel grant (T.W.S. and D.T.Y.). We thank T. P. Snutch for the gift of the human α12.1 cDNA clone.
Correspondence should be addressed to David T. Yue, Calcium Signals Laboratory, Departments of Biomedical Engineering and Neuroscience, Johns Hopkins University School of Medicine, Ross Building, Room 713, 720 Rutland Avenue, Baltimore, MD 21205. E-mail: dyue@bme.jhu.edu.
REFERENCES
- 1.Bourinet E, Soong TW, Sutton K, Slaymaker S, Mathews E, Monteil A, Zamponi GW, Nargeot J, Snutch TP. Splicing of a1A subunit gene generates phenotypic variants of P- and Q-type calcium channels. Nat Neurosci. 1999;2:407–415. doi: 10.1038/8070. [DOI] [PubMed] [Google Scholar]
- 2.Burset M, Seledtsov IA, Solovyev VV. Analysis of canonical and non-canonical splice sites in mammalian genomes. Nucleic Acids Res. 2000;28:4364–4375. doi: 10.1093/nar/28.21.4364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Catterall WA. Structure and function of neuronal Ca2+ channels and their role in neurotransmitter release. Cell Calcium. 1998;24:307–323. doi: 10.1016/s0143-4160(98)90055-0. [DOI] [PubMed] [Google Scholar]
- 4.Chaudhuri D, DeMaria CD, Alvania RS, Yue DT, Soong TW. Splice variation of human P/Q-type (Cav2.1) Ca2+ channels as an on/off switch for Ca2+-dependent facilitation. Soc Neurosci Abstr. 2001;27:14.15. [Google Scholar]
- 5.Colecraft HM, Brody DL, Yue DT. G-protein inhibition of N- and P/Q-type calcium channels: distinctive elementary mechanisms and their functional impact. J Neurosci. 2001;21:1137–1147. doi: 10.1523/JNEUROSCI.21-04-01137.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Deisseroth K, Bito H, Tsien RW. Signaling from synapse to nucleus: postsynaptic CREB phosphorylation during multiple forms of hippocampal synaptic plasticity. Neuron. 1996;16:89–101. doi: 10.1016/s0896-6273(00)80026-4. [DOI] [PubMed] [Google Scholar]
- 7.DeLeon MS, Wang Y, Jones LP, Perez-Reyes E, Wei X, Soong TW, Snutch TP, Yue DT. Essential Ca2+-binding motif for Ca2+-sensitive inactivation of L-type Ca2+ channels. Science. 1995;270:1502–1506. doi: 10.1126/science.270.5241.1502. [DOI] [PubMed] [Google Scholar]
- 8.DeMaria CD, Soong TW, Alseikhan BA, Alvania RS, Yue DT. Calmodulin bifurcates the local Ca2+ signal that modulates P/Q-type Ca2+ channels. Nature. 2001;411:484–489. doi: 10.1038/35078091. [DOI] [PubMed] [Google Scholar]
- 9.Dunlap K, Luebke JI, Turner TJ. Exocytotic Ca2+ channels in mammalian central neurons. Trends Neurosci. 1995;18:89–98. [PubMed] [Google Scholar]
- 10.Hans M, Urrutia A, Deal C, Brust PF, Stauderman K, Ellis SB, Harpold MM, Johnson EC, Williams ME. Structural elements in domain IV that influence biophysical and pharmacological properties of human a1A-containing high-voltage-activated calcium channels. Biophys J. 1999;76:1384–1400. doi: 10.1016/S0006-3495(99)77300-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jones LP. N-type calcium channel inactivation probed by gating-current analysis. Biophys J. 1999;76:2530–2552. doi: 10.1016/S0006-3495(99)77407-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kretsinger RH. Calcium-binding proteins. Annu Rev Biochem. 1976;45:239–266. doi: 10.1146/annurev.bi.45.070176.001323. [DOI] [PubMed] [Google Scholar]
- 13.Krovetz HS, Helton TD, Crews AL, Horne WA. C-terminal alternative splicing changes the gating properties of a human spinal cord calcium channel a1A subunit. J Neurosci. 2000;20:7564–7570. doi: 10.1523/JNEUROSCI.20-20-07564.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee A, Wong ST, Gallagher D, Li B, Storm DR, Scheuer T, Catterall WA. Ca2+/calmodulin binds to and modulates P/Q-type calcium channels. Nature. 1999;13:107–108. doi: 10.1038/20194. [DOI] [PubMed] [Google Scholar]
- 15.Lee A, Scheuer T, Catterall WA. Ca2+/calmodulin-dependent facilitation and inactivation of P/Q-type Ca2+ channels. J Neurosci. 2000;20:6830–6838. doi: 10.1523/JNEUROSCI.20-18-06830.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lin Z, Lin Y, Schorge S, Pan JQ, Beierlein M, Lipscombe D. Alternative splicing of a short cassette exon in alpha1B generates functionally distinct N-type calcium channels in central and peripheral neurons. J Neurosci. 1999;19:5322–5331. doi: 10.1523/JNEUROSCI.19-13-05322.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Maximov A, Sudhof TC, Bezprozvanny I. Association of neuronal calcium channels with modular adaptor proteins. J Biol Chem. 1999;274:24453–24456. doi: 10.1074/jbc.274.35.24453. [DOI] [PubMed] [Google Scholar]
- 18.Mittman S, Agnew WS. Three new alternative spliced exons of the calcium channel a1B subunit gene CACNA1B. Soc Neurosci Abstr. 2000;26:100. [Google Scholar]
- 19.Mittman S, Guo J, Emerick MC, Agnew WS. Structure and alternative splicing of the gene encoding alpha1I, a human brain T calcium channel alpha1 subunit. Neurosci Lett. 1999a;269:121–124. doi: 10.1016/s0304-3940(99)00319-5. [DOI] [PubMed] [Google Scholar]
- 20.Mittman S, Guo J, Agnew WS. Structure and alternative splicing of the gene encoding alpha1G, a human brain T calcium channel alpha1 subunit. Neurosci Lett. 1999b;274:143–146. doi: 10.1016/s0304-3940(99)00716-8. [DOI] [PubMed] [Google Scholar]
- 21.Mori Y, Friedrich T, Kim MS, Mikami A, Nakai J, Ruth P, Bosse E, Hofmann F, Flockerzi V, Furuichi T, Mikoshiba K, Imoto K, Tanabe T, Numa S. Primary structure and functional expression from complementary DNA of a brain calcium channel. Nature. 1991;350:398–402. doi: 10.1038/350398a0. [DOI] [PubMed] [Google Scholar]
- 22.Ophoff RA, Terwindt GM, Vergouwe MN, Van Eijk R, Oefner PJ, Hoffman SMG, Lamerdin JE, Mohrenweiser HW, Bulman DE, Ferrari M, Han J, Lindhout D, Van Ommen GJB, Hofker MH, Ferrari MD, Frants RR. Familial hemiplegic migraine and episodic ataxia type-2 are caused by mutations in the Ca2+ channel gene CACNL1A4. Cell. 1996;87:543–552. doi: 10.1016/s0092-8674(00)81373-2. [DOI] [PubMed] [Google Scholar]
- 23.Patil PG, Brody DL, Yue DT. Preferential closed-state inactivation of neuronal calcium channels. Neuron. 1998;20:1027–1038. doi: 10.1016/s0896-6273(00)80483-3. [DOI] [PubMed] [Google Scholar]
- 24.Peterson BZ, DeMaria CD, Yue DT. Calmodulin is the Ca2+ sensor for Ca2+-dependent inactivation of L-type calcium channels. Neuron. 1999;22:549–558. doi: 10.1016/s0896-6273(00)80709-6. [DOI] [PubMed] [Google Scholar]
- 25.Peterson BZ, Lee JS, Mulle JG, Wang Y, deLeon MS, Yue DT. Critical determinants of Ca2+-dependent inactivation within an EF-hand motif of L-type Ca2+ channels. Biophys J. 2000;78:1906–1920. doi: 10.1016/S0006-3495(00)76739-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pietrobon D. Calcium channels and channelopathies of the central nervous system. Mol Neurobiol. 2002;25:31–50. doi: 10.1385/MN:25:1:031. [DOI] [PubMed] [Google Scholar]
- 27.Qin N, Olcese R, Bransby M, Lin T, Birnbaumer L. Ca2+-induced inhibition of the cardiac Ca2+ channel depends on calmodulin. Proc Natl Acad Sci USA. 1999;96:2435–2438. doi: 10.1073/pnas.96.5.2435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Raghib A, Bertaso F, Davies A, Page KM, Meir A, Bogdanov Y, Dolphin AC. Dominant-negative synthesis suppression of voltage-gated calcium channel Cav2.2 induced by truncated constructs. J Neurosci. 2001;21:8495–8504. doi: 10.1523/JNEUROSCI.21-21-08495.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Regan MR, Emerick MC, Agnew WS. Full-length single-gene cDNA libraries: applications in splice variant analysis. Anal Biochem. 2000;286:265–276. doi: 10.1006/abio.2000.4819. [DOI] [PubMed] [Google Scholar]
- 30.Rettig J, Sheng ZH, Kim DK, Hodson CD, Snutch TP, Catterall WA. Isoform-specific interaction of the alpha1A subunits of brain Ca2+ channels with the presynaptic proteins syntaxin and SNAP-25. Proc Natl Acad Sci USA. 1996;93:7363–7368. doi: 10.1073/pnas.93.14.7363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Scott VE, Felix R, Arikkath J, Campbell KP. Evidence for a 95 kDa short form of the alpha1A subunit associated with the omega-conotoxin MVIIC receptor of the P/Q-type Ca2+ channels. J Neurosci. 1998;18:641–647. doi: 10.1523/JNEUROSCI.18-02-00641.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sharp PA, Burdge CB. Classification of introns: U2-type or U12-type. Cell. 1997;91:875–879. doi: 10.1016/s0092-8674(00)80479-1. [DOI] [PubMed] [Google Scholar]
- 33.Sherman A, Keizer J, Rinzel J. Domain model for Ca2+-inactivation of Ca2+ channels at low channel density. Biophys J. 1990;58:985–995. doi: 10.1016/S0006-3495(90)82443-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Soong TW, DeMaria CD, Mittman S, Agnew WS, Yue DT. Systematic determination of splice variation in human P/Q-type Ca2+ channels: functional impact of C-terminal variants. Soc Neurosci Abstr. 2000;26:2093. [Google Scholar]
- 35.Sutton KG, McRory JE, Guthrie H, Murphy TH, Snutch TP. P/Q-type calcium channels mediate the activity-dependent feedback of syntaxin-1A. Nature. 1999;401:800–804. doi: 10.1038/44586. [DOI] [PubMed] [Google Scholar]
- 36.Takahashi SX, DeMaria CD, Colecraft HM, Yue DT. Kinetic mechanism underlying Ca2+-dependent facilitation of P/Q-type Ca2+ channels. Soc Neurosci Abstr. 2001;27:151.155. [Google Scholar]
- 37.Toru S, Murakoshi T, Ishikawa K, Saegusa H, Fujigasaki H, Uchihara T, Nagayama S, Osanai M, Mizusawa H, Tanabe T. Spinocerebellar ataxia type 6 mutations alters P-type calcium channel function. J Biol Chem. 2000;275:10893–10898. doi: 10.1074/jbc.275.15.10893. [DOI] [PubMed] [Google Scholar]
- 38.Tsunemi T, Saegusa H, Ishikawa K, Nagayama S, Murakoshi T, Mizusawa H, Tanabe T. Novel Cav2.1 splice variants isolated from Purkinje cells do not generate P-type Ca2+ current. J Biol Chem. 2002;277:7214–7221. doi: 10.1074/jbc.M108222200. [DOI] [PubMed] [Google Scholar]
- 39.von Gersdorff H, Borst JG. Short-term plasticity at the calyx of held. Nat Rev Neurosci. 2002;3:53–64. doi: 10.1038/nrn705. [DOI] [PubMed] [Google Scholar]
- 40.Wheeler DB, Tsien RW. Voltage-gated calcium channels. In: Carafoli E, Lee C, editors. Calcium as a cellular regulator. Oxford UP; New York: 1999. pp. 171–199. [Google Scholar]
- 41.Wielowieyski PA, Wigle JT, Salih M, Hum P, Tuana BS. Alternative splicing in intracellular loop connecting domains II and III of the alpha 1 subunit of Cav1.2 Ca2+ channels predicts two-domain polypeptides with unique C-terminal tails. J Biol Chem. 2001;276:1398–1406. doi: 10.1074/jbc.M006868200. [DOI] [PubMed] [Google Scholar]
- 42.Zamponi GW, Bourinet E, Nelson D, Nargeot J, Snutch TP. Crosstalk between G proteins and protein kinase C mediated by the calcium channel alpha1 subunit. Nature. 1997;385:442–446. doi: 10.1038/385442a0. [DOI] [PubMed] [Google Scholar]
- 43.Zhang MQ. Statistical features of human exons and their flanking regions. Hum Mol Genet. 1998;7:919–932. doi: 10.1093/hmg/7.5.919. [DOI] [PubMed] [Google Scholar]
- 44.Zhong H, Yokoyama CT, Scheuer T, Catterall WA. Reciprocal regulation of P/Q-type Ca2+ channels by SNAP-25, syntaxin and synaptotagmin. Nat Neurosci. 1999;2:939–941. doi: 10.1038/14721. [DOI] [PubMed] [Google Scholar]
- 45.Zhuchenko O, Bailey J, Bonnen P, Ashizawa T, Stockton DW, Amos C, Dobyns WB, Subramony SH, Zoghbi HY, Lee CC. Autosomal dominant cerebellar ataxia (SCA6) associated with small polyglutamine expansions in the alpha 1A-voltage-dependent calcium channel. Nat Genet. 1997;15:62–69. doi: 10.1038/ng0197-62. [DOI] [PubMed] [Google Scholar]
- 46.Zuhlke RD, Reuter H. Ca2+-sensitive inactivation of L-type Ca2+ channels depends on multiple cytoplasmic amino acid sequences of the a1C subunit. Proc Natl Acad Sci USA. 1998;95:3287–3294. doi: 10.1073/pnas.95.6.3287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zuhlke RD, Pitt GS, Deisseroth K, Tsien RW, Reuter H. Calmodulin supports both inactivation and facilitation of L-type calcium channels. Nature. 1999;399:159–162. doi: 10.1038/20200. [DOI] [PubMed] [Google Scholar]