

Abstract
Neurons of the medial nucleus of the trapezoid body, which transmit auditory information that is used to compute the location of sounds in space, are capable of firing at high frequencies with great temporal precision. We found that elimination of the Kv3.1gene in mice results in the loss of a high-threshold component of potassium current and failure of the neurons to follow high-frequency stimulation. A partial decrease in Kv3.1 current can be produced in wild-type neurons of the medial nucleus of the trapezoid body by activation of protein kinase C. Paradoxically, activation of protein kinase C increases temporal fidelity and the number of action potentials that are evoked by intermediate frequencies of stimulation. Computer simulations confirm that a partial decrease in Kv3.1 current is sufficient to increase the accuracy of response at intermediate frequencies while impairing responses at high frequencies. We further establish that, of the two isoforms of the Kv3.1 potassium channel that are expressed in these neurons, Kv3.1a and Kv3.1b, the decrease in Kv3.1 current is mediated by selective phosphorylation of the Kv3.1b isoform. Using site-directed mutagenesis, we identify a specific C-terminal phosphorylation site responsible for the observed difference in response of the two isoforms to protein kinase C activation. Our results suggest that modulation of Kv3.1 by phosphorylation allows auditory neurons to tune their responses to different patterns of sensory stimulation.
Keywords: Kv3.1, potassium channel, MNTB neurons, protein kinase C, phosphorylation, auditory timing, channel isoforms
Introduction
The Kv3.1 potassium channel is expressed at high levels in neurons that characteristically fire rapid trains of action potentials (Perney et al., 1992; Weiser et al., 1995;Rudy, 1999). Particularly high levels of this channel are found in neurons of the auditory brainstem, such as bushy cells of the cochlear nucleus and neurons of the medial nucleus of the trapezoid body (MNTB). These neurons participate in neural circuits that determine the intensity and timing of auditory stimuli and use this information to determine the location of sounds in space (Joris, 1996). To perform their function, these neurons are endowed with a number of cellular specializations that allow them to fire at rates of many hundreds of Hertz both in vivo and in vitro (Spirou et al., 1990; Banks and Smith, 1992; Wu and Kelly, 1993; Trussell, 1999;Taschenberger and von Gersdorff, 2000). Such cells lock their action potentials precisely to the phase of auditory stimuli at frequencies of up to 2000–4000 Hz or to rapid fluctuations in the amplitude of higher-frequencies sounds (Joris and Yin, 1995; Joris, 1996). At those frequencies at which an action potential cannot be generated to every single stimulus, their firing pattern is characterized by alternating action potentials and failures of action potential generation (Banks and Smith, 1992; Brew and Forsythe, 1995; Wang et al., 1998). For example, in response to a 600 Hz stimulus, an MNTB neuron may fire at 300 Hz, locking its action potentials to every other stimulus (Wang et al., 1998). The synaptic and electrophysiological specializations of bushy cells and MNTB neurons ensure that the delay from a suprathreshold stimulus to the occurrence of an action potential varies by no more than a few tens of microseconds throughout a stimulus train (Borst et al., 1995; Oertel, 1999; Trussel, 1999).
A major component of potassium current in MNTB neurons is a high-threshold voltage-dependent potassium current (IHT) that is selectively blocked by low concentrations of tetraethylammonium (TEA) and whose properties match those of the Kv3.1 potassium channel in heterologous expression systems (Perney and Kaczmarek, 1991; Brew and Forsythe, 1995; Wang et al., 1998; Grigg et al., 2000). Blockade of this current in MNTB neurons significantly impairs their ability to fire high-frequency trains of action potentials (Wang et al., 1998).
The Kv3.1 gene has retained a high degree of similarity between human and rodents throughout evolution. Alternate splicing at the 3′ end of the Kv3.1 gene results in two channel isoforms that differ exclusively at their C termini (Luneau et al., 1991). Previous work has shown that these two variants, termed Kv3.1a and Kv3.1b, share a similar expression pattern in the rat brain (Perney et al., 1992; Weiser et al., 1995). Although both variants are present in the same neurons, including MNTB neurons, levels of the Kv3.1b variant increase markedly at the time of synapse formation, and this isoform predominates in adult neurons (Perney et al., 1992; Liu and Kaczmarek, 1998). The functional significance of such alternately spliced channels that produce similar currents is unknown, although there is evidence that divergent C termini may be involved in targeting channels to different regions of the neuronal membrane (Pounce et al., 1997; Rudy, 1999).
Previous work has shown that, in heterologous expression systems, the Kv3.1 channel can be modulated by activation of protein kinase C, which produces a decrease in current amplitude (Critz at al., 1993; Kanemasa et al., 1995). We now found that regulation by this enzyme is specific to the Kv3.1b isoform and results from phosphorylation of a single site in the C terminus. We also compared the effects of the protein kinase C-induced reduction of Kv3.1 current in MNTB neurons with those of total elimination of the IHT current through homologous recombination, using both native neurons and a computer simulation of their firing patterns. We demonstrate that, whereas total elimination of the Kv3.1 current in MNTB neurons severely impairs high-frequency firing, a partial reduction of current significantly increases both the number of action potentials generated and their temporal fidelity at those intermediate frequencies at which failures to generate action potentials are first detected. Our results suggest that the transition from the Kv3.1a to the Kv3.1b isoform during development allows MNTB neurons to modulate their firing pattern to improve temporal fidelity at intermediate stimulus frequencies.
Materials and Methods
Generation of Kv3.1 mutant mice. The Kv3.1gene was mutated by homologous recombination by a replacement vector as previously described in 129/Sv mice (Ho et al., 1997). Briefly, the coding region between EcoRI and MscI (35 bp in the S2–S3 linker) was replaced with a neomycin-resistant cassette. Identification of mutant animals was confirmed by PCR using oligonucleotides directed against the 5′ coding region (primer 5′ GAA ATC GAG AAC GTT CGA AAC GG 3′) and the 35 bp recombinant sequence (primer 5′ CTA CTT CCA TTT GTC ACG TCC TG 3′).
Stable expression of Kv3.1a in a Chinese hamster ovary cell line. Chinese hamster ovary (CHO) cells with dihydrofolate reductase thymidylate (DHFR) deficiency [CHO/DHFR(−)] were maintained in Iscove's modified Dulbecco's medium (Invitrogen, San Diego, CA) supplemented with 10% fetal bovine serum, 0.1 mm hypoxanthine, and 0.01 mm thymidine and maintained in a 5% CO2 incubator at 37°C. Cells were seeded 1 d before transfection at ∼5 × 105cells/60 mm plate. Kv3.1a expression vector (pcDNA3/Kv3.1a) was added 24 hr later to transfect CHO/DHFR(−) using Lipofectamine (Invitrogen). The cells were then grown in normal medium for 48 hr to develop antibiotic resistance and subsequently exposed to geneticin (0.5 mg/ml; Invitrogen) for another 10–14 d. The geneticin-resistant cells were subjected to single-cell sorting using FACSIV (Becton Dickinson, Mountain View, CA) to generate individual stable cell lines. The stable transfection of Kv3.1b into CHO cells has been described previously (Wang et al., 1998).
Electrophysiological recordings from CHO cells. CHO/DHFR(−) cells were maintained in Iscove's modified Dulbecco's medium (Invitrogen) supplemented with 10% fetal bovine serum, 0.1 mm hypoxanthine, and 0.05 mg/ml geneticin (Invitrogen) and maintained in a 5% CO2 incubator at 37°C. CHO cells were grown on coverslips 24–48 hr preceding recordings and transferred to extracellular solution (in mm: 140 NaCl, 1.3 CaCl2, 5.4 KCl, 25 HEPES, and 33 glucose, pH 7.4) 1 hr before voltage clamping. Recordings were made as specified either with the perforated-patch technique using nystatin or in the whole-cell configuration using an Axopatch 1D amplifier (Axon Instruments, Foster City, CA). The patch electrodes had a resistance of 3–5 MΩ when filled with intracellular solution (in mm: 32.5 KCl, 97.5 K-gluconate, 5 EGTA, and 10 HEPES, pH 7.2). All data were low-pass filtered at 2 kHz and digitized using a modified digital data recorder (Instrutech, Great Neck, NY). Data were analyzed using pClamp 6.0 software. Average data are expressed as means ± SE. Conductance values were obtained by dividing current by the electrochemical driving force [IK/(Vm− Ek)]. Normalized conductance–voltage plots were obtained by normalizing conductance (G) to maximal conductance (Gmax) and fit using the Boltzmann isoform G =Gmax/[1 + exp ((V −V1/2)/k)], whereV1/2 is the voltage at half-maximal activation, and k is the slope factor.
Preparation of brainstem slices. Brains were rapidly removed from postnatal 9–14 d old mice (control 129/Sv or Kv3.1 mutant mice) or rats after decapitation and placed into ice-cold bicarbonate-buffered artificial CSF (ACSF) (in mm: 125 NaCl, 2.5 KCl, 26 NaHCO3, 1.25 NaH2PO4, 2 Na pyruvate, 3 myo-inositol, 10 glucose, 2 CaCl2, and 1 MgCl2, pH 7.4) solution gassed with 95% O2–5% CO2. The area of the brainstem containing MNTB nuclei were cut into four to six transverse slices using a vibrotome. The slices were incubated at 37°C for 1 hr and thereafter kept at room temperature (22–25°C).
Electrophysiological recordings from MNTB. Recordings were conducted on MNTB neurons as described previously (Macica and Kaczmarek, 2001). Briefly, a slice was transferred to a recording chamber that was continually perfused (1 ml/min) with gassed ACSF. Whole-cell patch-clamp recordings were made from visually identified MNTB neurons using an Axopatch 2D amplifier (Axon Instruments). The patch electrodes had a resistance of 3–5 MΩ when filled with intracellular solution (in mm: 32.5 KCl, 97.5 K-gluconate, 5 EGTA, 10 HEPES, and 1 MgCl2, pH 7.2). For voltage-clamp recordings, the extracellular calcium concentration was lowered to 0.5 mm to minimize the contribution of calcium-activated K channels. TTX (0.5 μm) was also included in ACSF to block sodium currents. The mean cell capacitance was 12.0 ± 0.4 pF, with a mean series resistance of 5.3 ± 0.4 MΩ. The compensation for series resistance was set at least 85%, with a lag of 10 μsec. Data were low-pass filtered at 5 kHz, digitized, and acquired on-line with pClamp 6.0 software (Axon Instruments). Total current was compared before and after addition of activators of PKC in the presence and absence of PKC inhibitors, and averaged data are expressed as means ± SE. Conductance and normalized conductance values were obtained as above.
Immunoprecipitation and chemiluminescence. Stably transfected CHO cells expressing Kv3.1a or Kv3.1b or untransfected cells were grown to 80% confluence. Medium was removed, and cells were washed three times with ice-cold PBS. Cells were lysed with radioimmunoprecipitation assay buffer (150 mm NaCl, 1.0% Nonidet P-40, 0.5% deoxycholate, 0.1% SDS, and 50 mm Tris, pH 8.0) containing a protease inhibitor cocktail (Boehringer Mannheim, Indianapolis, IN) and phosphatase inhibitors (100 μm NaF and 0.2 mmNaVO3) and allowed to incubate on a rocking platform for 30 min at 4°C. Lysates were spun in a microfuge for 15 min, and the supernatant was transferred to a new tube. Rat brain membranes were prepared from whole brain homogenized in 320 mm sucrose–PBS, pH 7.4, containing a protease cocktail inhibitor and centrifuged at 1000 × g for 10 min at 4°C. The supernatant was then centrifuged at 15,000 ×g for 30 min at 4°C, followed by centrifugation of the supernatant at 105,000 × g for 1 hr at 4°C. Pellets were reconstituted in sucrose–PBS. Lysates and membranes (brought up to volume with PBS) were precleared with a 50% slurry aliquot of protein A Sepharose beads, followed by incubation with the indicated anti-Kv3.1 antibody (1:1000 dilution) at 4°C overnight. Lysates and membranes were immunoprecipitated with protein A Sepharose beads for 2 hr at 4°C, spun, and washed three times in Triton X-100 buffer (0.1% Triton X-100, 0.1% SDS, 300 mm NaCl, and 50 mm Tris, pH 7.5). Samples were eluted by boiling in 1× SDS-PAGE sample buffer (62.5 mm Tris, pH 6.8, 4% SDS, 10% glycerol, 0.02% bromophenol blue, and 4% β-mercaptoethanol) for 5 min. Samples were subjected to SDS-PAGE on a 7.0% gel and transferred to a polyvinylidene difluoride (PVDF) membrane (Millipore, Bedford, MA). Peptides were visualized by chemiluminescence. Briefly, the membrane was incubated with the Kv3.1 antibody against the C terminus at a 1:4000 dilution in antibody diluent (1% casein and 0.04% Tween 20 in PBS, pH 7.4) for 1 hr, washed three times, incubated with secondary antibody (1:10,000 in diluent buffer), and washed three times. The blot was incubated with substrate (5.5 mg of luminol, 0.28 mg of p-coumaric acid, and 1% H2O2–25 ml of PBS) for 1 min and briefly exposed to film.
Metabolic labeling, immunoprecipitation, and phosphoamino acid analysis. Phosphorylation of Kv3.1 was analyzed in stably transfected CHO cells expressing either Kv3.1a or Kv3.1b that was metabolically labeled to equilibrium with [32P]orthophosphate as described previously (Macica and Kaczmarek, 2001). Cells were lysed, immunoprecipitated, and subjected to SDS-PAGE as described above. This gel was fixed as above and dried, and bands were visualized by autoradiography. Phosphoamino acid analysis was also performed on samples prepared as above and analyzed by two-dimensional electrophoresis, as described previously (Macica and Kaczmarek, 2001).
Numerical simulations. Computer modeling was performed using equations that have been used previously to model MNTB neurons (Wang et al., 1998) and other auditory neurons (Perney and Kaczmarek, 1997;Richardson and Kaczmarek, 2000). The model comprised a sodium currentINa, the Kv3.1 currentIKv3.1, a low-threshold potassium current ILT, and a leakage conductanceIL.INa andIL were given by the equationsINa =gNam3h(50 − V) and IL =gL(−63 − V), respectively. ILT andIKv3.1 were simulated by the equationsILT =gLTlr(−80 −V) and IKv3.1 =gKv3.1n3(0.8 + 0.2p)(−80 − V). Evolution of the variables m, h, l, r,n, and p were determined by Hodgkin-Huxley-like equations as described previously (Perney and Kaczmarek, 1997). Parameters for the sodium current were as follows:gNa = 0.5 μS,kαm = 76.4 msec−1, ηαm = 0.037 mV−1,kβm = 6.93 msec−1, ηβm = −0.043 mV−1, andkαh = 0.000533 msec−1, ηαh = −0.0909 mV−1,kβh = 7.87 msec−1, and ηβh = 0.0691 mV−1 . The leakage conductancegL was 0.002 μS. Parameters for potassium current were obtained from direct fits to traces recorded from MNTB neurons and Kv3.1-transfected CHO cells (Wang et al., 1998). Parameters for the low-threshold potassium current were as follows:gLT = 0.02 μS ,kα1 = 1.2 msec−1, ηα1 = 0.03512 mV−1,kβ1 = 0.2248 msec−1, ηβ1 = −0.0319 mV−1,kαr = 0.0438 msec−1, ηαr = −0.0053 mV−1,kβr = 0.0562 msec−1, and ηβr = −0.0047 mV−1. Parameters for the Kv3.1 channel were as follows: gHT = 0.15 μS (control) or 0.10 μS [phorbol 12-myristate 13-acetate (PMA) treated], kαn = 0.2719 msec−1, ηαn = 0.04 mV−1,kβn = 0.1974 msec−1, ηβn = 0 mV−1,kαp = 0.00713 msec−1, ηαp = −0.1942 mV−1,kβp = 0.0935 msec−1, and ηβp = 0.0058 mV−1. Numerical simulations of the responses of the cells to external stimulation was performed using the equation C dV/dt =INa +IKv3.1 +ILT +Iext(t), where Cis the cell capacitance (0.01 nF), and external currentsIext(t) were presented as repeated current steps (1.4 nA, 0.25 msec) applied at frequencies from 100 to 600 Hz.
Results
Contribution of Kv3.1 to the firing pattern of MNTB neurons
We first tested the role of the Kv3.1 channel in MNTB neurons by comparing the properties of MNTB neurons in 9- to 14-d-old brainstem slices from wild-type mice with those in which the Kv3.1gene had been deleted by homologous recombination (Ho et al., 1997). As described previously, wild-type mice express a high-threshold potassium current IHT whose properties closely match those of Kv3.1 in transfected cell lines (Wang et al., 1998). This current can be isolated by stepping to positive potentials from a holding potential of −40 mV, at which potential the low-threshold component of total outward current in these neurons is inactivated (Brew and Forsythe, 1995; Wang et al., 1998). This current can be substantially inhibited by extracellular application of 1 mm TEA ions, consistent with the IC50 of 250 μm for the Kv3.1 channel (Kanemasa et al., 1995; Wang et al., 1998; Rudy, 1999) (Fig.1a,b).
Fig. 1.

Representative traces comparing native high-threshold current from a 14 d wild-type MNTB neuron with the high-threshold current from a 14 d Kv3.1 mutant MNTB neuron.a, b, Outward currents evoked by stepping from a holding potential of −40 mV for 2 min (to ensure complete inactivation of low-threshold current) to test potentials from −80 to +40 mV in 20 mV increments in wild-type mice before and after treatment with 1 mm TEA or in Kv3.1 mutant mice (c). d, e, Representative recording from an MNTB neuron in response to brief current injections (2 nA, 0.3 msec) at three different test frequencies (100–300 Hz) in wild-type mice before and after treatment with 1 mm TEA or in Kv3.1 mutant mice (f). Bars denote failure to evoke an action potential in response to a stimulus. Failure was defined as a membrane depolarization to less than −10 mV in response to a current injection. Expanded traces below compare a successful action potential with failures, which showed little regenerative response.
When recordings were made in MNTB neurons from Kv3.1−/− mice, the current evoked by depolarizing commands from −40 mV current was reduced by >90%, indicating that theIHT current had been eliminated (Fig.1c). The remaining ∼10% high-threshold current was blocked by 1 mm TEA. In contrast, the low-threshold currents evoked by stepping from a holding potential of −80 mV could still readily be recorded in the Kv3.1−/− mice (data not shown). The low-threshold current, which is TEA insensitive in wild-type mouse, however, displays sensitivity to 1 mm TEA, suggesting a compensatory increase in a TEA-sensitive low-threshold current in these mice (data not shown). It is possible that the afterhyperpolarization in the Kv3.1−/− mice reflects a compensatory increase in low-threshold current in these animals. Combined with previous work demonstrating the loss of Kv3.1 mRNA and protein in the Kv3.1−/− mice (Ho et al., 1997), our findings strongly support the identification of theIHT current recorded at the somata of MNTB neurons with the Kv3.1 channel.
To test the impact of knock-out of the Kv3.1 gene on the firing properties of MNTB neurons, neurons were stimulated with trains of brief current pulses (0.3 msec, 2 nA) at frequencies ranging from 100 to 300 Hz (Fig. 1d). As described previously, wild-type MNTB neurons faithfully follow stimulus frequencies up to 300 Hz (Wang et al., 1998). In the presence of extracellular TEA (1 mm) a normal pattern of action potentials is evoked at the lower frequencies (100–200 Hz), but a progressive loss of full action potentials occurs during a train at 300 Hz and at higher frequencies (Fig. 1e). A similar, but more severe, deficit occurs in MNTB neurons from Kv3.1−/− mice. These can follow stimulation at 100 Hz, but attenuation of action potentials is already evident even at 200 Hz. At 300 Hz and at all higher frequencies, only a single action potential is evoked at the start of the train (Fig.1f). This complete loss of response at higher frequencies exactly matches that predicted in numerical simulations for elimination of the IHT current (Wang et al., 1998).
The Kv3.1 isoforms differ in their response to activation of protein kinase C
In transfected cells, the amplitude of Kv3.1 current is known to be regulated by activators of protein kinase C, attributed to a decrease in single-channel open probability (Critz et al., 1993;Kanemasa et al., 1995). There exist two isoforms of the Kv3.1 channel, Kv3.1a and Kv3.1b, which arise by alternate splicing of theKv3.1 gene. These differ in the number of consensus sites for phosphorylation by protein kinase C. The longer Kv3.1b isoform has two additional consensus sites in the C terminus that are absent in Kv3.1a. Both isoforms are coexpressed in the same neurons, with Kv3.1b predominating in the mature nervous system (Perney et al., 1992; Weiser et al., 1995; Rudy, 1999). To determine the physiological consequences of modulation by protein kinase C, we compared the electrophysiological properties of the two isoforms in transfected cells and of the nativeIHT current in MNTB neurons.
We first compared the basal electrophysiological properties of the two isoforms. Currents were evoked in CHO cells stably transfected with either Kv3.1a or Kv3.1b by depolarizations to test potentials between −80 and 60 mV (Fig. 2a). In some but not all experiments, a transient peak that decreased slightly to a steady-state value was seen at positive potentials at the beginning of the pulse for both Kv3.1a and Kv3.1b, a finding reported previously for Kv3.1b (Rettig et al., 1992; Critz et al., 1993;Kanemasa et al., 1995; Rudy, 1999). The conductance–voltage relationships were fit by Boltzmann isotherms with a midpoint of activation of 17.3 ± 1.26 mV (n = 9) versus 20.8 ± 1.60 mV (n = 9) for Kv3.1a and Kv3.1b, respectively (Fig. 2b). The 10–90% rise times for maximal activation at 60 mV were 1.25 ± 0.16 msec (n = 9) versus 1.22 ± 0.09 msec, (n = 9) respectively. Deactivation kinetics were obtained from tail currents recorded after a 100 msec depolarizing pulse to +40 mV, followed by membrane potential repolarization to −40mV. These were fit by a single exponential and yielded time constants of 2.38 ± 0.14 msec (n = 18) and 2.20 ± 0.12 msec (n = 10), respectively. In addition, both Kv3.1a and Kv3.1b were inhibited to a similar degree by 1 mm TEA (87 ± 1.5%, n= 3 vs 82 ± 5.1%, n = 12) (Fig.2c).
Fig. 2.

Comparison of Kv3.1 current recorded from CHO cells stably transfected with Kv3.1a or Kv3.1b. a, Whole-cell current was evoked by depolarizing the membrane from a holding potential of −80 to +60 mV in 20 mV increments.b, Normalized conductance–voltage relationship for Kv3.1a versus Kv3.1b. Conductance values (G) were obtained as described in Materials and Methods. c, Sensitivity of Kv3.1a or Kv3.1b to 1 mm TEA. Currents were evoked as described ina.
Although the biophysical properties of Kv3.1a and Kv3.1b appear indistinguishable under basal conditions, their response to 100 nm PMA, an activator of protein kinase C, was very different. Kv3.1a or Kv3.1b currents were recorded from nystatin-perforated patches before and after exposure to 100 nm PMA (n = 9) (Fig.3a,b). PMA produced a significant reduction of Kv3.1b current (32.4 ± 4.3% inhibition), which was maximal by 10–15 min (Fig.4). 4α-PMA, an analog of PMA that does not activate protein kinase C, did not significantly affect Kv3.1b current (Fig. 4). In contrast, Kv3.1a current amplitude was not significantly affected by PMA (2.26 ± 1.7% inhibition) (Figs.3a, 4). No effect of PMA was detected on the kinetics (data not shown) or the voltage dependence of activation of either isoform (midpoint of activation, 20.3 ± 1.3 mV during control period vs 19.1 ± 1.6 mV after PMA treatment in nystatin-perforated patches).
Fig. 3.

Effect of 100 nm PMA on Kv3.1 current.a, Whole-cell currents evoked from CHO cells stably expressing Kv3.1a from a holding potential of −70 mV to test potentials of −80 to +60 mV in 20 mV increments (left) and corresponding current–voltage relationship of evoked currents (right) before and after PMA treatment.b, Whole-cell currents evoked from CHO cells stably expressing Kv3.1b (left) and current–voltage relationship of evoked currents. c, Whole-cell currents evoked from CHO cells stably expressing Kv3.1b mutant S503A (left) and current–voltage relationship of evoked currents.
Fig. 4.

Time course of the effect of 100 nmPMA on Kv3.1 current amplitude. Outward current evoked from a holding potential of −70 mV in Kv3.1-transfected cells or from a holding potential of −40 mV in MNTB neurons to a test potential of +40 mV was monitored in perforated patches every 1 min in response to treatment with exogenous PMA in Kv3.1a-transfected cells (filled circles), in Kv3.1b-transfected cells (filled squares), or in MNTB neurons (filled triangles). The effect of the inactive phorbol ester 4α-PMA was also tested on Kv3.1b-transfected cells (open squares) and MNTB neurons (open triangles).
Phosphorylation of Kv3.1 channel proteins
To determine whether the actions of activators of protein kinase C on Kv3.1b currents result from the direct phosphorylation of the Kv3.1b protein, CHO cells stably expressing either Kv3.1a or Kv3.1b were radiolabeled to equilibrium with [32P]orthophosphate and were then stimulated for 15 min with 100 nm PMA. Immunoprecipitation revealed substantial basal incorporation of32P into both channel proteins, as well as stimulated incorporation in the presence of PMA (Fig.5a). Incorporation could be eliminated by treatment of the immunoprecipitated phosphoprotein with alkaline phosphatase. To determine the identity of the amino acids into which 32P was incorporated, the immunoprecipitates were then subjected to phosphoamino acid analysis. Radioactivity comigrating with unlabeled phosphoserine, but not phosphothreonine or phosphotyrosine, was detected after acid hydrolysis of 32P-labeled Kv3.1 (Fig. 5b). These results indicate that, of the putative consensus sites for phosphorylation, only those containing serine residues are phosphorylated.
Fig. 5.

In vivo phosphorylation and phosphoamino acid analysis of Kv3.1 in CHO cells. a, CHO cells expressing Kv3.1a or Kv3.1b were radiolabeled with [32P]orthophosphate to equilibrium, stimulated with or without 100 nm PMA for 15 min, and lysed. Lysates were immunoprecipitated with Kv3.1 antibody (Kv3.1a, lanes 1 and 2, with and without PMA, respectively or Kv3.1b, lanes 4 and 5, with and without PMA, respectively) and resolved as outlined in Materials and Methods. An additional 32P-labeled Kv3.1a or Kv3.1b immunoprecipitate was treated with calf intestinal alkaline phosphatase for 1 hr at 37°C (lanes 3 and 6, respectively). b, Phosphoamino acid analysis of the Kv3.1a or Kv3.1b channel protein obtained from lysates and immunoprecipitated and electrophoresed as above. The gel was transferred to a PVDF membrane, and the Kv3.1 band was visualized by autoradiography. The excised protein was subjected to phosphoamino acid analysis as outlined in Materials and Methods and visualized by ninhydrin staining (top), and standard phosphoamino acids were visualized by autoradiography (bottom).
Protein kinase C acts at serine 503 of Kv3.1b
Kv3.1b has 11 consensus sites for PKC-mediated phosphorylation, two of which are absent from Kv3.1a as a result of the divergent C terminus. Only one of these (S503), however, is a serine site. The phosphoamino acid analysis suggests, therefore, that this is the only consensus protein kinase C site that differs between the two isoforms. To test the hypothesis that this unique site is responsible for response of Kv3.1b to activation of protein kinase C, a mutant Kv3.1b channel was constructed in which this serine was mutated to alanine. When this mutation (S503A) was stably expressed in CHO cells, the currents appeared identical to those of wild-type Kv3.1 but were completely insensitive to PMA treatment (n = 6) (Fig.3c).
The Kv3.1b channel subunit predominates in mature neurons
To identify native Kv3.1 protein isoforms, membranes were prepared from adult rat brain homogenates or from CHO cells stably transfected with either isoform. These were immunoprecipitated with antibodies against either the N terminus, which recognizes both isoforms, or the Kv3.1b C terminus, which recognizes only Kv3.1b (Perney and Kaczmarek, 1991) (Fig. 6a). Immunoprecipitated Kv3.1a and Kv3.1b in CHO cells had mobilities corresponding to ∼98 and 110 kDa, respectively (Fig. 6a,lanes 1–3). Immunoprecipitation in both cell lines also yielded additional bands corresponding to the predicted molecular weights of the unglycosylated forms of the channel proteins (85 and 80 kDa, respectively). In support of this interpretation, the size of thein vitro translated product of the Kv3.1 channels in the absence of membranes corresponded precisely to the Mr of the lower bands (data not shown). The predominant isoform in brain homogenates is Kv3.1b (Fig. 6a,lane 4), consistent with the dominant expression pattern of Kv3.1 mRNA transcripts in adult animals (Perney et al., 1992). Although the expression of both isoforms in the same cells (Perney et al., 1992; Weiser et al., 1995) suggests that they could form heteromultimers in vivo, Kv3.1a protein could not be detected in immunoprecipitates from rat brain membranes using the C-terminus antibody, which recognizes only the Kv3.1 b channel protein (Fig. 6a, lane 5).
Fig. 6.

Molecular identity and effect of 100 nm PMA on native Kv3.1 current. a, Coimmunoprecipitation of Kv3.1a and Kv3.1b in brain homogenates or in stably transfected CHO cells. Lane 1, Kv3.1a/CHO with N-terminal antibody; lane 2, Kv3.1b/CHO with N-terminal antibody; lane 3, Kv3.1b/CHO with C-terminal antibody; lane 4, rat brain membranes with N-terminal antibody; lane 5, rat brain membranes with C-terminal antibody. b, Whole-cell currents evoked from a 13 d MNTB neuron from a holding potential of −40 mV to test potentials of −80 to +60 mV (left) and current–voltage relationship of evoked currents before and after PMA treatment.
Effect of phorbol ester treatment on high-threshold current in MNTB neurons
Having confirmed the molecular identity of the native high-threshold current as Kv3.1, we next tested the effect of PMA on potassium currents and the firing pattern of MNTB neurons. In 9 of 12 experiments, the amplitude of the high-threshold component of current evoked from a holding potential of −40 mV was reduced by 44.8 ± 2.17% in response to 100 nm PMA (Fig. 6b). The time course of inhibition closely matched that for the inhibition of Kv3.1b by PMA in transfected cells. Moreover, the inactive phorbol ester 4α-PMA had no effect on MNTB current amplitude (Fig. 4). In 3 of the 12 experiments conducted, however, there was very little effect of PMA on IHT current (4.50 ± 0.76%). Finally, PMA had little or no effect on the low-threshold component of current, obtained by subtracting the high-threshold component of current from the total outward current at a test potential of −20 mV, in which a majority of the total outward current is of the low-threshold type (Fig. 7a). Consistent with this finding was the observation that the spike threshold is unaltered and that MNTB neurons fire a single action potential in response to a sustained current injection both before and after PMA treatment (Fig. 7b). This limited depolarization has been shown to be mediated by the activation of the low-threshold current, which results in the lowering of the input resistance and a shortening of membrane time constants in the depolarizing voltage range, thus preventing repetitive firing in response to a sustained current injection (Wu and Kelly, 1993; Brew and Forsythe, 1995; Wang et al., 1998).
Fig. 7.
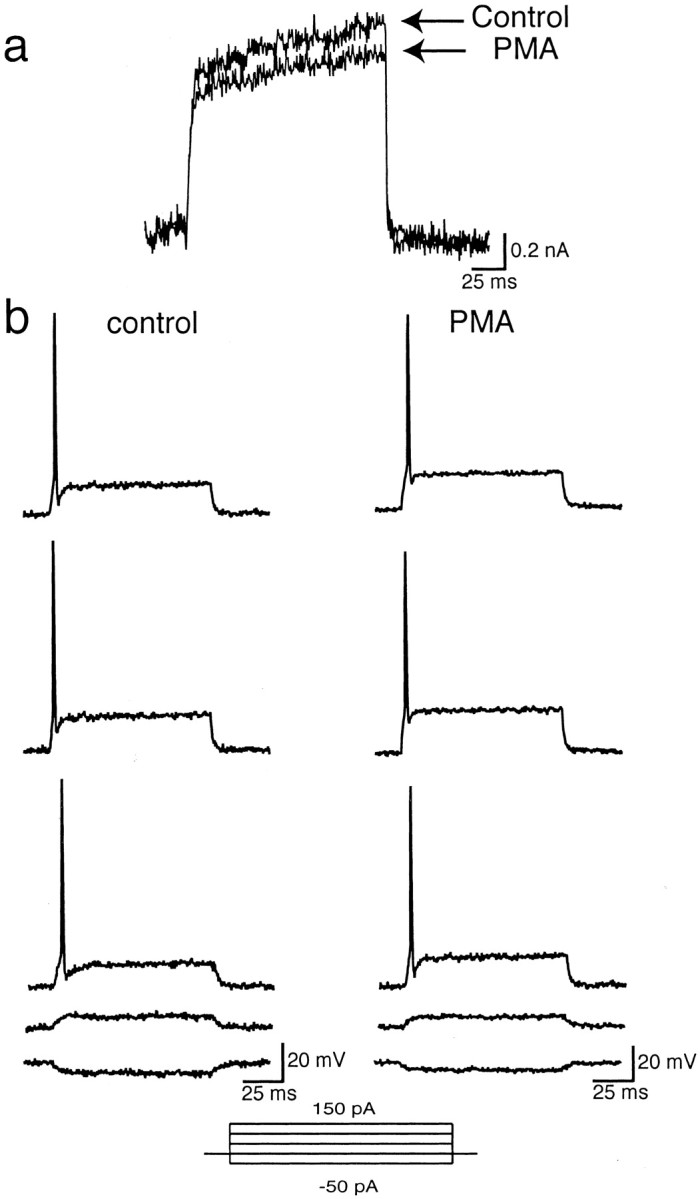
Effect of 100 nm PMA on low-threshold current from an MNTB neuron. a, Low-threshold current was obtained by subtracting the high-threshold component of outward current evoked from a holding potential of −40 mV to a test potential of −20 mV from total outward current evoked from a holding potential of −70 mV to a test potential of −20 mV (ILT =ITOT −IHT) before and after PMA treatment (n = 8). b, Current-clamp recording from an MNTB neuron in response to a series of 100 msec current injections ranging from −50 to 150 pA before and after PMA treatment (n = 4).
Additional analysis of the action of PMA on theIHT current also showed that its effects closely matched those for Kv3.1 in CHO cells and that PMA produced no change in the activation or deactivation kinetics in any experiment. Native IHT currents had 10–90% rise times for maximal activation at 60 mV of 1.41 ± 0.18 msec (n = 8) during the control period versus 1.12 ± 0.15 msec (n = 8) after 15 min PMA treatment. Deactivation kinetics of macroscopic currents were obtained by tail currents generated by a 100 msec depolarizing pulse to +40 mV, followed by membrane potential repolarization to test potentials between −100 and −20 mV. Deactivation kinetics were fit by a single exponential and yielded time constants of 2.35 ± 0.25 (n = 9) during the control period and 2.24 ± 0.28 (n = 8) at −40 mV after treatment of 15 min PMA. Like the cloned Kv3.1b channel, there was also no effect on the voltage dependence of activation of IHT(midpoint of activation, 16.6 ± 1.5 mV during control period vs 17.8 ± 1.0 mV after PMA).
Other parameters that were measured before and after PMA treatment included the mean slope of the rising phase of action potentials, which remained unchanged (before PMA, 171.8 ± 16.6 mV/msec; after PMA, 189.3 ± 13.8 mV/msec). In addition, the peak voltage reached by action potentials remained unchanged (before PMA, +28.8 ± 0.4 mV; after PMA, +30.6 ± 0.8 mV), and the mean resting potential remained unchanged (before PMA, −64.2 ± 1.12 mV; after PMA, −65.2 ± 0.87 mV). The findings argue that a change in sodium currents is unlikely to contribute to the observed effects.
Effect of phorbol ester on firing properties of MNTB neurons
In contrast to the complete loss of Kv3.1 current produced by knock-out of the Kv3.1 gene, the partial suppression of Kv3.1b current by protein kinase C might be expected to produce smaller changes in firing characteristics and contribute to the fine-tuning of auditory information processing. We therefore examined the effect of 100 nm PMA on the response of MNTB neurons to different frequencies of stimulation. To avoid the presynaptic effects of PMA at the MNTB synapse (Hori et al., 1999), we used direct current stimulation of MNTB neurons. At frequencies up to 300 Hz, at which MNTB neurons fire action potentials in response to every stimulus pulse, there was no effect of PMA on the ability of the cells to follow stimulation. At slightly higher frequencies, however, action potentials are not generated in response to every stimulus, and the firing pattern is characterized by intermittent failures separated by brief trains of action potentials. For example, in the cell shown in Figure8a, the control response at 400 Hz consists of groups of two or three consecutive action potentials separated by failures to reach threshold for spike initiation. At these intermediate frequencies at which failures are first detected, exposure of MNTB neurons to PMA reduced the number of failures. For example, in the cell shown in Figure 8a, PMA produced a small reduction in the number of failures in response to stimulation.
Fig. 8.

Effect of PMA treatment on firing properties of an MNTB neuron (13 d old) in response to different frequencies of stimulation (n = 6). a, Plots of the delay from the onset of the stimulus pulse to the peak of the action potential before (Control) and 15 min after (PMA) treatment with 100 nm PMA.Arrows denote failure to evoke an action potential in response to a stimulus. Failure was defined as a membrane depolarization to less than −10 mV in response to a current injection (one that has no detectable regenerative component). b, Superimposed action potentials in response to 100, 300, and 400 Hz stimulation. Failures were omitted from the superimposedtraces.
More striking than the effect of PMA on the number of failures was its effect on the timing of action potentials. To examine differences in the timing of the evoked action potentials, we superimposed successive action potentials locked to the stimulus (Fig. 8b). In addition, we plotted the change in the delay from the onset of the stimulus to the peak of the action potential during the stimulus trains. At frequencies up to 300 Hz at which all stimuli evoked action potentials, the latency from the onset of the stimulus pulse to the peak of the action potential was invariant, as seen by the superposition of the response to consecutive current pulses. The occurrence of failures, which occurs at higher frequencies, however, substantially disrupts the timing of the action potentials. The latency from onset of the current pulse to the peak of the action potential was found to vary by 500 μsec or more in the burst of action potentials separated by failures. Exposure of MNTB neurons to PMA, however, was found to reduce significantly the variance of the latency at 400 Hz [at 400 Hz, control delay of 0.99 ± 0.17 (SD) msec; after PMA, delay of 0.85 ± 0.11 msec;p(delay) < 0.0001;p(variance) < 0.015;n = 46 action potentials] (Fig. 8b).
Because it is certain that, in addition to their effects on Kv3.1 currents, activators of protein kinase C have a variety of other actions on MNTB neurons, we performed numerical simulations to compare the effects of a partial reduction of Kv3.1 current with those of PMA. We used a model that had been used previously to predict the firing pattern of MNTB neurons and that incorporates the amplitudes and kinetics of currents measured directly in these cells (Wang et al., 1998). The response of the model neurons to current pulses at different frequencies were tested with control conditions (150 nS Kv3.1 conductance) and in response to altering the level of Kv3.1 current to a similar degree as in PMA-treated MNTB neurons (100 nS Kv3.1 conductance) (Fig. 9). As with the native neurons, the model neuron was able to respond to each stimulus pulse up to 300 Hz, and the timing of action potentials was uniform throughout a maintained stimulus train, with either level of Kv3.1 current. However, as the stimulus frequency was increased, a critical frequency was reached at which failures began to occur (350 Hz with 150 nS Kv3.1 in Fig. 9a). At this point, the timing of the action potentials was also disrupted, with a progressive increase in the latency of the response with consecutive action potentials. At “intermediate” stimulus frequencies above this critical frequency, the latency during a train of action potentials could vary by >1 msec. When the level of Kv3.1 current was reduced by 33% at these frequencies, the failures were reduced or eliminated, and the timing of the action potentials was restored (Fig. 9b). As the stimulus frequency was increased, however, the effects of the reduction of Kv3.1 were less apparent (410 Hz in Fig. 9), and, at higher frequencies, the ability of the model neurons to respond was attenuated by the reduction in Kv3.1 current (data not shown). The simulation studies show that a reduction in Kv3.1 current alone is predicted to decrease the number of failures and to significantly reduce the variance of the timing of action potentials at intermediate frequencies of stimulation.
Fig. 9.

Model of an MNTB neuron in response to different frequencies of stimulation. a, Plots of the delay from the onset of the stimulus pulse to the peak of the action potential under control conditions (150 nS Kv3.1 conductance) and under conditions in which the level of Kv3.1 current amplitude is reduced to a similar degree as in PMA-treated neurons (100 nS Kv3.1 conductance).Arrows denote failure to evoke an action potential in response to a stimulus. Failure was defined as a membrane depolarization to less than −10 mV in response to a current injection.b, Superimposed action potentials in response to 100, 350, 360, or 410 Hz stimulation. Failures were omitted from the superimposed traces.
Discussion
Kv3.1 is critical for the preservation of timing information
Within the auditory brainstem, the Kv3.1 channel is expressed at high levels in spherical and globular bushy cells of the cochlear nucleus (Perney and Kaczmarek, 1997) and in principal neurons of the MNTB (Li et al., 2001), which receive a secure synaptic input from the globular bushy cells (Smith et al., 1991). The large calyx of Held synaptic ending from globular bushy cells onto the somata of MNTB neurons is specialized for temporally invariant transmission of excitatory inputs (Morest, 1968; Borst et al., 1995; Joris, 1996;Trussell, 1999), and evidence suggests that MNTB neurons faithfully transmit temporal information from globular bushy cells (Smith et al., 1998). Many globular bushy cells have spontaneous firing rates of over 100 Hz and, when driven by sounds, attain firing frequencies of 500–600 Hz (Rhode and Smith, 1986; Spirou et al., 1990). Our findings suggest that the Kv3.1 potassium channel is required for the normal ability of these cells to follow such high-frequency stimulation.
MNTB neurons provide an inhibitory input to the lateral superior olive and participate in a pathway that detects interaural intensity differences in high-frequency sounds (Joris and Yin, 1995, 1998; Tollin and Yin, 2002). They also project to other auditory nuclei, including the medial superior olive, which detects interaural timing differences in lower-frequency sounds. The characteristic frequencies of a population of globular bushy cells, which provide the excitatory input to the MNTB, lie within the range at which phase-locking occurs (<4000 Hz), and experiments with cats have shown that these cells have the ability to lock their action potentials very precisely to the phase of auditory stimuli at these frequencies (Smith et al., 1991). Nevertheless, the majority of these neurons, as well as their MNTB targets, have characteristic frequencies >4000 Hz (Guinan et al., 1972; Smith et al., 1991, 1998), and neurons are incapable of phase locking to such higher frequencies. Neurons with high characteristic frequencies, however, also respond to lower sound frequencies, and their ability to phase lock at these lower frequencies is even enhanced over that of cells with lower characteristic frequencies (Joris et al., 1994). Our present results suggest that direct phosphorylation of the Kv3.1 channel by protein kinase C provides a potential mechanism that can adjust the accuracy of timing of the response of MNTB neurons. Such adjustments of timing could occur either at frequencies close to the characteristic frequency or when a neuron is driven at quite different frequencies. Indeed, it has been proposed that the pathway from the globular bushy cells to the lateral superior olive via the MNTB comprises a circuit that can determine timing differences in high-frequency sounds that are amplitude modulated at lower frequencies (Joris and Yin, 1995; Joris, 1996).
Because the effects of PKC activation on the amplitude of Kv3.1 current occur over a period of several minutes, such modulation could reflect a slow adaptation to the auditory environment. Nevertheless, although the pathways that lead to the regulation of protein kinase C in MNTB neurons remains to be defined, it is likely that receptor-mediated activation is more rapid than pharmacological activation and that modulation could occur on a faster time scale.
The Kv3.1b isoform predominates in the principal neurons of the MNTB
The simplest interpretation of the finding that deletion of the Kv3.1 gene results in the near total elimination of the high-thresholdIHT current is that theIHT channel represents a homomultimer of Kv3.1 subunits. In addition to Kv3.1, MNTB neurons express mRNA for Kv3.3, another Shaw subfamily channel. In contrast to Kv3.1, Kv3.3 produces potassium currents with variable inactivation rates dependent on the heterologous expression system in which the gene is expressed (Rudy, 1999). The IHTcurrent is, however, noninactivating, and its kinetics very closely match those of Kv3.1 (Brew and Forsythe, 1995; Wang et al., 1998). Nevertheless, the small amount of residual high-threshold TEA-sensitive current remaining in Kv3.1−/− mice may also represent Kv3.3 current or some other unidentified potassium channel.
We found that Kv3.1a and Kv3.1b, the two channel subunits that are generated by the Kv3.1 gene, produce indistinguishable basal currents and that both channel proteins are substrates for phosphorylation, but that the two isoforms are differentially modulated by protein kinase C. In particular, phosphorylation of the consensus protein kinase site at serine 503 of Kv3.1b produces a decrease in current for this isoform. This is consistent with previous observations that activators of protein kinase C produce a decrease in channel open probability in Kv3.1b-transfected cells (Kanemasa et al., 1995). Nevertheless, because activation of this enzyme produces little or no change in the voltage dependence or kinetic behavior of the macroscopic currents, we cannot eliminate the possibility that a proportion of channels becomes silent during phosphorylation, as would occur if phosphorylation at serine 503 allowed them to interact with an endogenous inhibitor.
In situ hybridization and RNase protection assays indicate that the regional expression of both Kv3.1 isoforms overlaps in all brain areas (Perney et al., 1992). The Kv3.1a transcript predominates early in development and can be detected as early as embryonic day 17. There is a pronounced increase in the level of the Kv3.1b transcript from postnatal day 8 to postnatal day 14, the major period of synaptogenesis, and Kv3.1b becomes the major isoform in the mature brain, although the Kv3.1a transcript also persist into adulthood (Perney et al., 1992; Liu and Kaczmarek, 1998). In the cerebellum, in which both splice variants are expressed, it has been shown that the levels of the two variants are regulated by distinct mechanisms during development (Weiser et al., 1995; Liu and Kaczmarek, 1998). It has been suggested that the C terminus of Kv3.1a, which diverges from Kv3.1b in the last 10 amino acids, plays a role in targeting heteromultimers of Kv3.1a and Kv3.1b to axons and terminals. Although we did not find evidence of heteromultimerization between Kv3.1a and Kv3.1b by coimmunoprecipitation from whole-brain homogenates, we cannot eliminate the possibility that Kv3.1b heteromultimerizes with Kv3.1a in certain neuronal populations (Rudy, 1999).
PKC-mediated phosphorylation of Kv3.1 improves timing at intermediate frequencies
A high level of basal phosphorylation of Kv3.1 has been demonstrated previously and can be attributed primarily to the actions of casein kinase 2 (Macica and Kaczmarek, 2001). Our present data suggest that the switch from to Kv3.1a to Kv3.1b during development permits the IHT current of MNTB neurons to be modulated by protein kinase C, after synaptic transmission has been established. Activation of this enzyme influences the amplitude of the IHT current with no apparent change in voltage dependence or kinetics. Genetic knock-out, pharmacological, and computer modeling studies all indicate that a decrease in IHT current impairs the ability of the neurons to follow very-high-frequency stimulation. Nevertheless, a partial decrease inIHT current significantly improves the fidelity of firing and the timing of action potentials at those intermediate frequencies at which stimulation produces bursts of two or more action potentials interrupted by failures. Under these conditions, there is a progressive build up of inhibitory potassium conductance (both IHT andILT) during the burst, resulting in a progressive delay in the occurrence of action potentials. This delay can reach hundreds of microseconds, a condition that would preclude computation of small timing differences by the auditory brainstem circuits. A decrease in IHT current decreases the inhibitory conductance, generating additional action potentials and restoring more uniform spike latencies. At high-stimulus frequencies at which single action potentials are separated by failures, however, a decrease in IHTdoes not improve responses.
The biophysical characteristics of Kv3.1, particularly its activation only at positive potentials, make it particularly suitable for modulation of fidelity of propagation through the MNTB. In addition to simulating the effects of small changes in Kv3.1 current, we modeled the actions of decreases in the low-threshold potassium current or of increases in sodium current. Although both of these manipulations can restore one-to-one firing at intermediate frequencies of stimulation, they render the cells more likely to fire spontaneous action potentials and to generate more than one action potential in response to depolarizing currents, changes that would degrade auditory information. Nevertheless, although we did not find any modulation ofILT by PKC, it is probable that channels other than Kv3.1 are also subject to modulation in MNTB neurons.
Each individual auditory nerve fiber, neuron of the cochlear nucleus, or principal neuron of the MNTB can be assigned a characteristic frequency, which represents that frequency of sound for which the neuron has the lowest threshold. Neurons with different characteristic frequencies are organized tonotopically within their nuclei, reflecting their innervation by fibers originating in different regions of the cochlea. At higher sound intensities, however, MNTB neurons respond over a wider range of frequencies (Joris et al., 1994). Modulation of potassium conductances may provide one mechanism for adjusting the firing pattern of a neuron so that it can accurately follow a specific pattern of auditory stimulation. Although the pathways that lead to the regulation of protein kinase C in MNTB neurons remains to be defined, modulation of Kv3.1 by PKC may provide one mechanism for fine-tuning the frequency-dependent responses of MNTB neurons to auditory stimuli.
Footnotes
This work was supported by National Institutes of Health Grant DC-01919 (L.K.K.) and National Research Service Award Fellowship MH12257-02 (C.M.M.). We thank Donata Oertel for valuable comments on this manuscript.
Correspondence should be addressed to Leonard K. Kaczmarek, Department of Pharmacology, Yale University School of Medicine, 333 Cedar Street, New Haven, CT 06520. E-mail: leonard.kaczmarek@yale.edu.
References
- 1.Banks MI, Smith PH. Intracellular recordings from neurobiotin-labeled cells in brain slices of the rat medial nucleus of the trapezoid body. J Neurosci. 1992;12:2819–2837. doi: 10.1523/JNEUROSCI.12-07-02819.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Borst JG, Helmchen F, Sakmann B. Pre- and postsynaptic whole-cell recordings in medial nucleus of trapezoid body of the rat. J Physiol (Lond) 1995;489:825–840. doi: 10.1113/jphysiol.1995.sp021095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brew HM, Forsythe ID. Two voltage-dependent K+ conductances with complementary functions in postsynaptic integration at a central auditory synapse. J Neurosci. 1995;15:8011–8022. doi: 10.1523/JNEUROSCI.15-12-08011.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Critz SD, Wible BA, Lopez HS, Brown AM. Stable expression and regulation of a rat brain K channel. J Neurochem. 1993;60:1175–1178. doi: 10.1111/j.1471-4159.1993.tb03273.x. [DOI] [PubMed] [Google Scholar]
- 5.Grigg JJ, Brew HM, Tempel BL. Differential expression of voltage-gated potassium channel genes in auditory nuclei of the mouse brainstem. Hear Res. 2000;140:77–90. doi: 10.1016/s0378-5955(99)00187-2. [DOI] [PubMed] [Google Scholar]
- 6.Guinan JJ, Jr, Norris BE, Guinan SS. Single auditory units in the superior olivary complex. II. Locations of unit categories and tonotopic organization. Int J Neurosci. 1972;4:147–166. [Google Scholar]
- 7.Ho CS, Grange RW, Joho RH. Pleiotropic effects of a disrupted K+ channel gene: reduced body weight, impaired motor skill and muscle contraction, but no seizures. Proc Natl Acad Sci USA. 1997;94:1533–1538. doi: 10.1073/pnas.94.4.1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hori T, Takai Y, Takahashi T. Presynaptic mechanism for phorbol ester-induced synaptic potentiation. J Neurosci. 1999;19:7262–7267. doi: 10.1523/JNEUROSCI.19-17-07262.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Joris PX. Envelope coding in the lateral superior olive. II. Characteristic delays and comparison with responses in the medial superior olive. J Neurophysiol. 1996;76:2137–2156. doi: 10.1152/jn.1996.76.4.2137. [DOI] [PubMed] [Google Scholar]
- 10.Joris PX, Yin TC. Envelope coding in the lateral superior olive. I. Sensitivity to interaural time differences. J Neurophysiol. 1995;73:1043–1062. doi: 10.1152/jn.1995.73.3.1043. [DOI] [PubMed] [Google Scholar]
- 11.Joris PX, Yin TC. Envelope coding in the lateral superior olive. III. Comparison with afferent pathways. J Neurophysiol. 1998;79:253–269. doi: 10.1152/jn.1998.79.1.253. [DOI] [PubMed] [Google Scholar]
- 12.Joris PX, Smith PH, Yin TC. Enhancement of neural synchronization in the anteroventral cochlear nucleus. II. Responses in the tuning curve tail. J Neurophysiol. 1994;71(3):1037–1051. doi: 10.1152/jn.1994.71.3.1037. [DOI] [PubMed] [Google Scholar]
- 13.Kanemasa T, Gan L, Perney TM, Wang LY, Kaczmarek LK. Electrophysiological and pharmacological characterization of a mammalian Shaw channel expressed in NIH 3T3 fibroblasts. J Neurophysiol. 1995;74:207–217. doi: 10.1152/jn.1995.74.1.207. [DOI] [PubMed] [Google Scholar]
- 14.Li W, Kaczmarek LK, Perney TM. Localization of two high threshold potassium channel subunits in rat central auditory system. J Comp Neurol. 2001;437:196–218. doi: 10.1002/cne.1279. [DOI] [PubMed] [Google Scholar]
- 15.Liu SJ, Kaczmarek LK. The expression of two splice variants of the Kv3.1 potassium channel gene is regulated by different signaling pathways. J Neurosci. 1998;18:2881–2890. doi: 10.1523/JNEUROSCI.18-08-02881.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Luneau CJ, Williams JB, Marshal J, Levitan ES, Oliva C, Smith JS, Antanavage J, Folander K, Stein RB, Swanson R, Kaczmarek L, Buhrow SA. Alternative splicing contributes to K+ channel diversity in the mammalian central nervous system. Pro Natl Acad Sci USA. 1991;88:3932–3936. doi: 10.1073/pnas.88.9.3932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Macica CM, Kaczmarek LK. Casein kinase 2 determines the voltage dependence of the Kv3.1 channel in auditory neurons and transfected cells. J Neurosci. 2001;21:1160–1168. doi: 10.1523/JNEUROSCI.21-04-01160.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Morest DK. The collateral system of the medial nucleus of the trapezoid body of the cat, its neuronal architecture and relation to the olivo-cochlear bundle. Brain Res. 1968;9:288–311. doi: 10.1016/0006-8993(68)90235-7. [DOI] [PubMed] [Google Scholar]
- 19.Oertel D. The role of timing in the brainstem auditory nuclei of mammals. Annu Rev Physiol. 1999;61:497–519. doi: 10.1146/annurev.physiol.61.1.497. [DOI] [PubMed] [Google Scholar]
- 20.Perney TM, Kaczmarek LK. The molecular biology of K+ channels. Curr Opin Cell Biol. 1991;3:663–670. doi: 10.1016/0955-0674(91)90039-2. [DOI] [PubMed] [Google Scholar]
- 21.Perney TM, Kaczmarek LK. Localization of a high threshold potassium channel in the rat cochlear nucleus. J Comp Neurol. 1997;386:178–202. [PubMed] [Google Scholar]
- 22.Perney TM, Marshall J, Martin KA, Hockfield S, Kaczmarek LK. Expression of the mRNAs for the Kv3.1 potassium channel gene in the adult and developing brain. J Neurophysiol. 1992;68:756–766. doi: 10.1152/jn.1992.68.3.756. [DOI] [PubMed] [Google Scholar]
- 23.Pounce A, Vega-Saenz de Miera, Kentrol C, Moreno H, Thornbill B, Rudy B. K+ channel isoforms with divergent carboxy-terminal sequences carry distinct membrane targeting signals. J Membr Biol. 1997;159:149–159. doi: 10.1007/s002329900278. [DOI] [PubMed] [Google Scholar]
- 24.Rettig J, Wunder F, Stocker M, Lichtinghagen R, Mastiaux F, Beckh S, Kues W, Pedarzani P, Schroter KH, Ruppersberg JP, Veh R, Pongs O. Characterization of a Shaw-related potassium channel family in rat brain. EMBO J. 1992;11:2473–2486. doi: 10.1002/j.1460-2075.1992.tb05312.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rhode WS, Smith PH. Encoding timing and intensity in the ventral cochlear nucleus of the cat. J Neurophysiol. 1986;56:261–286. doi: 10.1152/jn.1986.56.2.261. [DOI] [PubMed] [Google Scholar]
- 26.Richardson FR, Kaczmarek LK. Modification of delayed rectfier potassium currents by the Kv9.1 potassium channel subunit. Hear Res. 2000;147:21–30. doi: 10.1016/s0378-5955(00)00117-9. [DOI] [PubMed] [Google Scholar]
- 27.Rudy B. Contributions of Kv3 channels to neuronal excitability. Ann NY Acad Sci. 1999;868:304–343. doi: 10.1111/j.1749-6632.1999.tb11295.x. [DOI] [PubMed] [Google Scholar]
- 28.Smith PH, Joris PX, Carney LH, Yin TC. Projections of physiologically characterized globular bushy cell axons from the cochlear nucleus of the cat. J Comp Neurol. 1991;304:387–407. doi: 10.1002/cne.903040305. [DOI] [PubMed] [Google Scholar]
- 29.Smith PH, Joris PX, Yin TC. Anatomy and physiology of principal cells of the medial nucleus of the trapezoid body (MNTB) of the cat. J Neurophysiol. 1998;79:3127–3142. doi: 10.1152/jn.1998.79.6.3127. [DOI] [PubMed] [Google Scholar]
- 30.Spirou GA, Brownell WE, Zidanic M. Recordings from cat trapezoid body and HRP labeling of globular bushy cell axons. J Neurophysiol. 1990;63:1169–1190. doi: 10.1152/jn.1990.63.5.1169. [DOI] [PubMed] [Google Scholar]
- 31.Taschenberger H, von Gersdorff H. Fine-tuning an auditory synapse for speed and fidelity: developmental changes in presynaptic waveform, EPSC kinetics, and synaptic plasticity. J Neurosci. 2000;20:9162–9173. doi: 10.1523/JNEUROSCI.20-24-09162.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tollin DJ, Yin TC. The coding of spatial location by single units in the lateral superior olive of the cat. II. The determinants of spatial receptive fields in azimuth. J Neurosci. 2002;22:1468–1479. doi: 10.1523/JNEUROSCI.22-04-01468.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Trussell LO. Synaptic mechanisms for coding timing in auditory neurons. Annu Rev Physiol. 1999;61:477–496. doi: 10.1146/annurev.physiol.61.1.477. [DOI] [PubMed] [Google Scholar]
- 34.Wang LY, Gan L, Forsythe ID, Kaczmarek LK. Contribution of the Kv3.1 potassium channel to high-frequency firing in mouse auditory neurons. J Physiol (Lond) 1998;509:183–194. doi: 10.1111/j.1469-7793.1998.183bo.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Weiser M, Bueno E, Sekirnjak C, Martone ME, Baker H, Hillman D, Chen S, Thornhill W, Ellisman M, Rudy B. The potassium channel subunit Kv3.1b is localized to somatic and axonal membranes of specific populations of CNS neurons. J Neurosci. 1995;15:4298–4314. doi: 10.1523/JNEUROSCI.15-06-04298.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wu SH, Kelly JB. Response of neurons in the lateral superior olive and medial nucleus of the trapezoid body to repetitive stimulation: intracellular and extracellular recordings from mouse brain slice. Hear Res. 1993;68:189–201. doi: 10.1016/0378-5955(93)90123-i. [DOI] [PubMed] [Google Scholar]