

NPR4 has been shown to be a key regulator of the salicylic acid-mediated signal perception and transduction pathways. This project aimed to obtain structural details of NPR4, thus elucidating the molecular mechanism of salicylic acid perception and its signalling role in the systemic acquired resistance pathway.
Keywords: salicylic acid, systemic acquired resistance, protein purification, regulatory mechanism, crystal diffraction, Arabidopsis thaliana
Abstract
Salicylic acid (SA) is an important phytohormone that is involved in the regulation of plant defence, growth and development. A large number of proteins have been shown to have the ability to interact with SA, and NPR4 has been demonstrated to be a receptor of SA that plays significant roles in the innate immune response of plants. In this study, Spodoptera frugiperda (Sf9) cells were used to express full-length AtNPR4 from Arabidopsis thaliana. To facilitate crystallization, T4 lysozyme (T4L) was added to the N-terminus of the AtNPR4 protein. The recombinant T4L-AtNPR4 protein was expressed, purified and crystallized using the sitting-drop and hanging-drop vapour-diffusion methods. The T4L-AtNPR4 crystals have symmetry consistent with space group C2, with unit-cell parameters a = 93.7, b = 85.8, c = 88.2 Å, β = 90° and one molecule per asymmetric unit. The best crystal diffracted to a resolution of 2.75 Å. Structure determination is in progress.
1. Introduction
Salicylic acid (SA) plays an important role in the innate immune response of plants, acting as a signalling molecule and triggering the systemic acquired resistance (SAR) response (Vlot et al., 2009 ▸; Shah, 2003 ▸). Once plants have been infected by pathogens, the amount of SA increases significantly, yet how this key hormone signals and the number of receptors to which it binds remain unknown. Moreover, how it further mediates the SAR process is still unknown, despite drawing the attention of numerous scientists in recent decades. Large numbers of proteins have been shown to interact with the SA molecule. The protein NPR4, along with its paralogues NPR1 and NPR3, has been shown to be a key regulator within the SA-mediated signal perception and transduction pathways (Liu et al., 2005 ▸). Recent studies have shown that NPR4 binds directly to SA, which regulates the amount of NPR1 through the NPR4–SA complex by degradation control (Fu et al., 2012 ▸). The foundations of the SA-mediated resistance reaction have been qualitatively outlined; however, many crucial details remain unclear and many other predictions of the underlying mechanism are also plausible. As opinions vary, understanding could be advanced by structural, quantitative analysis of NPR4. Consequently, our project aims to determine the structure of NPR4, to elucidate the molecular mechanism of SA perception and its signalling role in the SAR pathway at the atomic level, and to attempt to further study the biochemical mechanism of the SAR process.
NPR4 is 36.0% identical to NPR1, specifically in the BTB/POZ and ankyrin-repeat domains, which have been shown to be key regulators within the SA-mediated signal perception and transduction pathways (Fig. 1 ▸; Liu et al., 2005 ▸). Sequence alignments indicate that NPR4 and its homologues share four conserved cysteines in their BTB/POZ domain, and a stretch of five variable basic amino acids at the C-terminus which may be involved in nuclear localization (Fig. 1 ▸; Shi et al., 2013 ▸). The interaction of SA with NPR4 has a higher affinity than that of SA with NPR3 (Spoel et al., 2009 ▸). NPR4 serves as a cullin 3 (CUL3) E3 ubiquitin ligase adaptor to promote the proteasomal degradation of NPR1 (Fu et al., 2012 ▸). The role of NPR4 in promoting the degradation of NPR1, together with the higher affinity of SA for NPR4, explains many aspects of NPR1 homeostasis at the site of infection, in distal tissues and with respect to basal resistance. Upon infection, the concentration of SA increases at the site of infection. This very high SA concentration promotes interaction between NPR1 and SA-bound NPR3, thereby leading to the proteasomal degradation of NPR1 and thus promoting local cell death. In the distal site of infection, a moderately high concentration of SA occurs (higher than the basal concentration but not as high as that at the infection site) and the NPR1–NPR3 interaction is reduced, allowing some NPR1 to enter the nucleus to promote the transcription of PR-1 (Janda & Ruelland, 2015 ▸). In the absence of infection, the very low basal SA concentration favours the interaction of NPR1 with unliganded NPR4 while some NPR1 escapes degradation, thus allowing limited NPR1-dependent expression of defence genes to confer basal resistance (Kaltdorf & Naseem, 2013 ▸).
Figure 1.

Sequence alignment of homologues of AtNPR. All of them share four conserved cysteines in their BTB/POZ domain.
The primary sequence of Arabidopsis thaliana NPR4 (AtNPR4) shares very low sequence similarity with the structures of homologues deposited in the PDB. Therefore, we decided to perform structural characterization of the protein. In this study, we report the expression, purification and crystallization of full-length AtNPR4.
2. Materials and methods
2.1. Macromolecule production
Because the AtNPR4 protein formed inclusion bodies during expression in Escherichia coli BL21 cells, Spodoptera frugiperda (Sf9) cells were used to express the recombinant protein. Full-length AtNPR4 and a fusion with T4 lysozyme (T4L-AtNPR4) were cloned using similar protocols. The cDNA for AtNPR4 was generated by PCR using the gene library of A. thaliana. T4L-AtNPR4 was cloned into a modified pFastBac1 vector (Invitrogen) between the BamHI and XhoI restriction sites. High-titre recombinant baculovirus (>108 viral particles per millilitre) was obtained using the Bac-to-Bac Baculovirus Expression System (Invitrogen). Sf9 cells at a cell density of 2–3 × 106 cells per millilitre were infected with virus at a multiplicity of infection (MOI) of 5 (Table 1 ▸). Cells were harvested by centrifugation at 48 h post-infection.
Table 1. Macromolecule-production information.
| Source organism | A. thaliana |
| DNA source | cDNA |
| Forward primer† | CGGGATCCATGGCTGCAACTGCAATAGAG |
| Reverse primer‡ | CCGCTCGAGTCATGTTGGATTCTCTAAGGCTTCT |
| Cloning vector | pFastBac1 |
| Expression vector | pFastBac1 |
| Expression host | Sf9 cells |
| Complete amino-acid sequence of the construct produced§ | MNIFEMLRIDERLRLKIYKDTEGYYTIGIGHLLTKSPSLNAAKSELDKAIGRNTNGVITKDEAEKLFNQDVDAAVRGILRNAKLKPVYDSLDAVRRAALINMVFQMGETGVAGFTNSLRMLQQKRWDEAAVNLAKSIWYNQTPNRAKRVITTFRTGTWDAYMAATAIEPSSSISFTSSHLSNPSPVVTTYHSAANLEELSSNLEQLLTNPDCDYTDAEIIIEEEANPVSVHRCVLAARSKFFLDLFKKDKDSSEKKPKYQMKDLLPYGNVGREAFLHFLSYIYTGRLKPFPIEVSTCVDSVCAHDSCKPAIDFAVELMYASFVFQIPDLVSSFQRKLRNYVEKSLVENVLPILLVAFHCDLTQLLDQCIERVARSDLDRFCIEKELPLEVLEKIKQLRVKSVNIPEVEDKSIERTGKVLKALDSDDVELVKLLLTESDITLDQANGLHYAVAYSDPKVVTQVLDLDMADVNFRNSRGYTVLHIAAMRREPTIIIPLIQKGANASDFTFDGRSAVNICRRLTRPKDYHTKTSRKEPSKYRLCIDILEREIRRNPLVSGDTPTCSHSMPEDLQMRLLYLEKRVGLAQLFFPAEANVAMDVANVEGTSECTGLLTPPPSNDTTENLGKVDLNETPYVQTKRMLTRMKALMKTVETGRRYFPSCYEVLDKYMDQYMDEEIPDMSYPEKGTVKERRQKRMRYNELKNDVKKAYSKDKVARSCLSSSSPASSLREALENPT |
The underlined sequence denotes the BamHI restriction site.
The underlined sequence denotes the XhoI restriction site.
The sequence of T4L is shown in bold.
For purification of the protein, the cell pellets were resuspended in lysis buffer (20 mM Tris–HCl pH 8.0, 150 mM NaCl) and lysed using a high-pressure cell disruptor. After centrifugation at 18 000 rev min−1 for 30 min, the precipitate was discarded and the supernatant was loaded onto a disposable column containing nickel-affinity resin (Invitrogen) to purify the His-tagged T4L-AtNPR4 protein. The contaminants were removed using a wash buffer containing lysis buffer supplemented with 20 mM imidazole and the recombinant T4L-AtNPR4 protein was eluted with elution buffer consisting of 20 mM Tris–HCl pH 8.0, 100 mM NaCl, 300 mM imidazole. T4L-AtNPR4 was further purified using a Superdex S200 gel-filtration column equilibrated with 20 mM Tris–HCl pH 8.0, 150 mM NaCl, 2 mM DTT. The protein was greater than 95% pure after this step as judged by SDS–PAGE analysis (Fig. 2 ▸). The purified protein was concentrated to approximately 10 mg ml−1 for crystallization.
Figure 2.
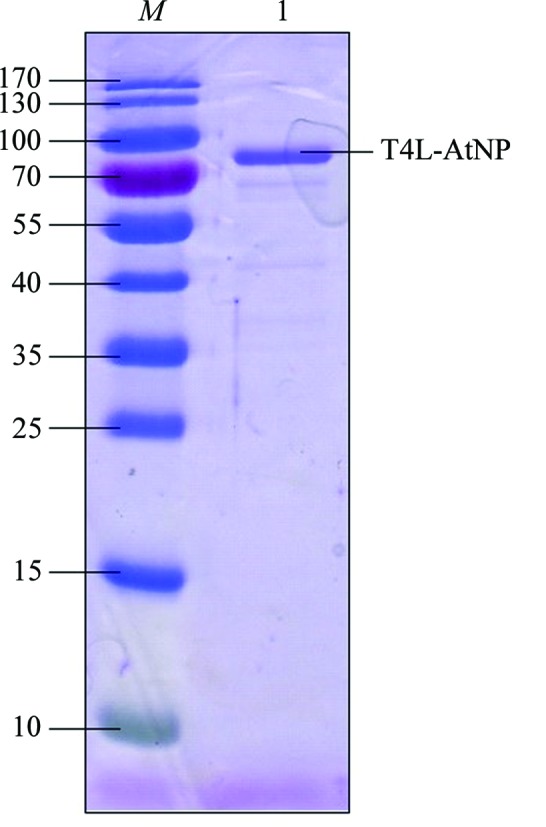
SDS–PAGE analysis of recombinant T4L-AtNPR4. Lane M, molecular-mass marker (labelled in kDa); lane 1, recombinant T4L-AtNPR4 protein with 2 mM DTT stained using Coomassie Brilliant Blue.
2.2. Crystallization
The initial crystallization experiments were performed by the sitting-drop vapour-diffusion method at 293 K with a 1:1 ratio of protein and precipitant solutions (0.5 µl of each). The crystallization conditions were screened in 96-well crystal-growth plates with a crystallization robot (Art Robbins Instruments) using the following commercial screening kits: Crystal Screen, Crystal Screen 2, Index, PEG/Ion, PEG/Ion 2 and SaltRx from Hampton Research and Wizard I, II, III and IV from Rigaku Reagents. The initial crystals appeared in several conditions after 3–5 d (Fig. 3 ▸ a). Further optimization was performed by altering the pH range, salt concentration and precipitant concentration and by screening additives using the hanging-drop vapour-diffusion method by mixing 2 µl protein solution and 2 µl reservoir solution. Optimal crystals for X-ray diffraction were obtained in a condition consisting of 0.1 M bis-Tris pH 6.5, 1.0 M ammonium sulfate, 2.0%(w/v) PEG 3350 (Fig. 3 ▸ b and Table 2 ▸). The crystals were harvested 3 d after the crystallization experiment, when the crystals had reached their largest size.
Figure 3.

Crystals of T4L-AtNPR4 before and after optimization. (a) Crystals of T4L-AtNPR4 grown in 0.1 M bis-Tris pH 5.5, 1.0 M ammonium sulfate, 1.0%(w/v) PEG 3350. (b) The optimization conditions that gave the largest T4L-AtNPR4 crystals: 0.1 M bis-Tris pH 6.5, 1.0 M ammonium sulfate, 2.0%(w/v) PEG 3350.
Table 2. Crystallization.
| Method | Hanging drop |
| Temperature (K) | 293 |
| Protein concentration (mg ml−1) | 10 |
| Buffer composition of protein solution | 20 mM Tris–HCl pH 8.0, 150 mM NaCl, 2 mM DTT |
| Composition of reservoir solution | 0.1 M bis-Tris pH 6.5, 1.0 M ammonium sulfate, 2.0%(w/v) PEG 3350 |
| Volume and ratio of drop | 2 µl, 1:1 |
| Volume of reservoir (µl) | 500 |
2.3. Data collection and processing
Crystals were soaked in a cryoprotectant solution for 5 s before being flash-cooled in liquid nitrogen. The cryoprotectant consisted of 25%(v/v) ethylene glycol in the mother liquor. Initial experiments showed that the crystals diffracted X-rays and we therefore proceeded to collect data. Data-collection parameters are listed in Table 3 ▸. The crystals showed high-quality diffraction patterns (Fig. 4 ▸). All intensity data were indexed, integrated and scaled with the HKL-3000 package (Minor et al., 2006 ▸). The R merge and completeness in the outer resolution shell were less than 48.4% and greater than 99.9%, respectively. The data set was then processed in space group C2 and evaluated using phenix.xtriage, and no twinning or pseudo-translational symmetry were detected (Adams et al., 2010 ▸). Based on the molecular weight of the protomer, the Matthews coefficient was calculated to be 2.14 Å3 Da−1 and the solvent content was 42.6%, with one molecule per asymmetric unit (Matthews, 1968 ▸). A final diffraction data set (2.75 Å resolution) was collected and the related data-collection and processing statistics are summarized in Table 3 ▸.
Table 3. Data collection and processing.
Values in parentheses are for the outer shell.
| Diffraction source | BL19U1, SSRF |
| Wavelength (Å) | 0.9778 |
| Temperature (K) | 100 |
| Detector | PILATUS 6M |
| Crystal-to-detector distance (mm) | 300 |
| Rotation range per image (°) | 1.0 |
| Total rotation range (°) | 360 |
| Exposure time per image (s) | 0.5 |
| Resolution range (Å) | 30.00–2.75 (2.80–2.75) |
| Space group | C2 |
| Unit-cell parameters (Å, °) | a = 93.66, b = 85.75, c = 88.22, α = γ = 90.00, β = 90.93 |
| Solvent content (%) | 42.60 |
| Mosaicity range (°) | 0.93–1.69 |
| Total No. of reflections | 86897 |
| No. of unique reflections | 18637 (942) |
| Completeness (%) | 99.80 (99.90) |
| Multiplicity | 5.20 (4.90) |
| 〈I/σ(I)〉 | 50.70 (4.80) |
| R merge † | 0.174 (0.485) |
| R mean | 0.193 (0.543) |
| CC1/2 | 0.973 (0.871) |
R
merge =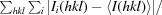
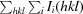 , where Ii(hkl) is the intensity of the ith observation of reflection hkl and 〈I(hkl)〉 is the average intensity.
, where Ii(hkl) is the intensity of the ith observation of reflection hkl and 〈I(hkl)〉 is the average intensity.
Figure 4.

A typical diffraction pattern of T4L-AtNPR4; the black circles correspond to the resolution shells (labelled in Å). (a) A diffraction pattern from an optimized T4L-AtNPR4 crystal obtained on the BL19U1 beamline. (b) Expanded view of the boxed region of the diffraction pattern in (a).
3. Results and discussion
The NPR4 gene from A. thaliana encodes a 574-amino-acid protein. A search for homologues using the BLASTp suite at NCBI retrieved sequences that share conserved regions. Analysis of the sequences of the AtNPRs showed that all of them share four conserved cysteines in their BTB/POZ domain (Fig. 1 ▸).
The T4L-AtNPR4 construct was expressed in Sf9 cells with a yield of 10 mg l−1. The protein was purified using nickel-affinity chromatography followed by size-exclusion chromatography. The purity of the protein was checked by SDS–PAGE with staining by Coomassie Brilliant Blue. The purity of the sample used for crystallization is shown in Fig. 2 ▸.
Although AtNPR4 was easily purified, the crystallization process was not smooth. To facilitate crystallization, T4 lysozyme (T4L) was added to the N-terminus of the AtNPR4 protein. Purified T4L-AtNPR4 was screened for crystallization. Crystals appeared in multiple conditions after initial screening (Fig. 3 ▸ a). Several optimization strategies were used to improve the crystals, including varying the salt concentration, precipitant concentration, pH range, protein concentration, crystallization temperature, time of crystal growth and cryoprotectant. The crystals were harvested 3 d after the crystallization experiment, when the crystals had reached their largest size (Fig. 3 ▸ b).
A 2.75 Å resolution data set was collected on the BL19U1 beamline at Shanghai Synchrotron Radiation Facility (SSRF; Fig. 4 ▸). Analysis of the data suggested that the crystal had high mosaicity. The crystal belonged to space group C2, with unit-cell parameters a = 93.66, b = 85.75, c = 88.22 Å, α = γ = 90.00, β = 90.93°. The unit-cell volume is consistent with the presence of one molecule in the asymmetric unit, with a solvent content of 42.6% (Table 3 ▸).
The primary sequence of AtNPR4 shares very low sequence similarity with the structures of homologues deposited in the PDB; the structure that shows the highest homology is PDB entry 2qyj (Merz et al., 2008 ▸). In the two stretches of 107 and 125 amino acids where the sequences resemble each other, the identities in the sequences were 31/107 (29%) and 35/125 (28%), the similarities in the two stretches were 56/107 (52%) and 62/125 (50%), and the gaps in the two stretches were 5/107 (5%) and 5/125 (4%), respectively. Molecular replacement was thus not successful. Moreover, in contrast to expression in Sf9 cells, the expression of AtNPR4 in E. coli is very low, so that selenomethionine substitution is difficult. Therefore, soaking of the crystals with heavy atoms will be attempted in order to solve the phase problem.
Acknowledgments
We are grateful to the staff of beamline BL19U1 at SSRL for support during data collection. We appreciate Qingya Shen (from Beijing Normal University) for helping to process the data and create some of the figures. We appreciate the help that we received from our colleagues in Dr Jia’s laboratory.
Funding Statement
This work was funded by National Natural Science Foundation of China grant 21773014. Postdoctoral Research Foundation of China grant 2124400209.
References
- Adams, P. D., Afonine, P. V., Bunkóczi, G., Chen, V. B., Davis, I. W., Echols, N., Headd, J. J., Hung, L.-W., Kapral, G. J., Grosse-Kunstleve, R. W., McCoy, A. J., Moriarty, N. W., Oeffner, R., Read, R. J., Richardson, D. C., Richardson, J. S., Terwilliger, T. C. & Zwart, P. H. (2010). Acta Cryst. D66, 213–221. [DOI] [PMC free article] [PubMed]
- Fu, Z. Q., Yan, S., Saleh, A., Wang, W., Ruble, J., Oka, N., Mohan, R., Spoel, S. H., Tada, Y., Zheng, N. & Dong, X. (2012). Nature (London), 486, 228–232. [DOI] [PMC free article] [PubMed]
- Janda, M. & Ruelland, E. (2015). Environ. Exp. Bot. 114, 117–128.
- Kaltdorf, M. & Naseem, M. (2013). Sci. Signal. 6, jc3. [DOI] [PubMed]
- Liu, G., Holub, E. B., Alonso, J. M., Ecker, J. R. & Fobert, P. R. (2005). Plant. J. 41, 304–318. [DOI] [PubMed]
- Matthews, B. W. (1968). J. Mol. Biol. 33, 491–497. [DOI] [PubMed]
- Merz, T., Wetzel, S. K., Firbank, S., Pluckthun, A., Grütter, M. G. & Mittl, P. R. E. (2008). J. Mol. Biol. 376, 232–240. [DOI] [PubMed]
- Minor, W., Cymborowski, M., Otwinowski, Z. & Chruszcz, M. (2006). Acta Cryst. D62, 859–866. [DOI] [PubMed]
- Shah, J. (2003). Curr. Opin. Plant Biol. 6, 365–371.
- Shi, Z., Maximova, S., Liu, Y., Verica, J. & Guiltinan, M. J. (2013). Mol. Plant. 6, 802–816. [DOI] [PubMed]
- Spoel, S. H., Mou, Z., Tada, Y., Spivey, N. W., Genschik, P. & Dong, X. (2009). Cell, 137, 860–872. [DOI] [PMC free article] [PubMed]
- Vlot, A. C., Dempsey, D. A. & Klessig, D. F. (2009). Annu. Rev. Phytopathol. 47, 177–206. [DOI] [PubMed]