

Abstract
Mammalian C to U RNA is mediated by APOBEC1, the catalytic deaminase, together with RNA binding cofactors (including A1CF and RBM47) whose relative physiological requirements are unresolved. Although A1CF complements APOBEC1 for in vitro RNA editing, A1cf–/– mice exhibited no change in apolipoproteinB (apoB) RNA editing, while Rbm47 mutant mice exhibited impaired intestinal RNA editing of apoB as well as other targets. Here we examined the role of A1CF and RBM47 in adult mouse liver and intestine, following deletion of either one or both gene products and also following forced (liver or intestinal) transgenic A1CF expression. There were minimal changes in hepatic and intestinal apoB RNA editing in A1cf–/– mice and no changes in either liver- or intestine-specific A1CF transgenic mice. Rbm47 liver-specific knockout (Rbm47LKO) mice demonstrated reduced editing in a subset (11 of 20) of RNA targets, including apoB. By contrast, apoB RNA editing was virtually eliminated (<6% activity) in intestine-specific (Rbm47IKO) mice with only five of 53 targets exhibiting C-to-U RNA editing. Double knockout of A1cf and Rbm47 in liver (ARLKO) eliminated apoB RNA editing and reduced editing in the majority of other targets, with no changes following adenoviral APOBEC1 administration. Intestinal double knockout mice (ARIKO) demonstrated further reduced editing (<10% activity) in four of five of the residual APOBEC1 targets identified in ARIKO mice. These data suggest that A1CF and RBM47 each function independently, yet interact in a tissue-specific manner, to regulate the activity and site selection of APOBEC1 dependent C-to-U RNA editing.
Keywords: APOBEC1, A1CF, RBM47, ApoB, intestine, liver
INTRODUCTION
Mammalian RNA editing encompasses a process in which select sequences within the original genomic template are enzymatically altered to produce a change in the corresponding RNA transcript (for review, see Gagnidze et al. 2018). By far the most prevalent form of RNA editing is adenosine to inosine deamination (A-to-I), which is mediated by members of the family of adenosine deaminases acting on RNA (ADARs) (for review, see Nishikura 2010; Keegan et al. 2017). ADAR-mediated RNA editing requires optimal configuration of sites within a double-stranded RNA substrate, most typically residing within intronic or intergenic regions enriched in Alu repeats (Nishikura 2010). The other, less prevalent form of RNA editing involves cytidine to uridine deamination (C-to-U) of single-strand RNA substrates, which is mediated by APOBEC1, and was first described 30 yr ago as the molecular mechanism underlying the tissue-specific production of two distinct isoforms of the lipid transport protein, apolipoproteinB (apoB), from the liver and small intestine (Chen et al. 1987; Powell et al. 1987). Genetic deletion and rescue experiments have demonstrated that the catalytic deaminase, APOBEC1, is absolutely required for apoB C-to-U RNA editing (Hirano et al. 1996; Nakamuta et al. 1996), but those experiments also revealed that additional factor(s) are required since APOBEC1 alone was insufficient to mediate C-to-U RNA editing on synthetic apoB RNA templates (Driscoll et al. 1993; Sowden et al. 1996b; Anant et al. 2003).
In 2000, two groups simultaneously reported the identification of a plausible complementation factor, APOBEC1 complementation factor (A1CF), demonstrating that a two-component system containing recombinant A1CF and APOBEC1 alone was sufficient for efficient C-to-U RNA editing of apoB RNA in vitro (Lellek et al. 2000; Mehta et al. 2000). However, formal evidence of the role and requisite physiological functions for A1CF in vivo, including its role in C-to-U RNA editing, has been challenging to elucidate and those efforts have yielded somewhat conflicting results. Earlier studies demonstrated early embryonic lethality in germline A1cf−/− mice with null embryos failing to implant at E3.5d (Blanc et al. 2005). However, heterozygous A1cf+/− mice were viable and exhibited defective liver regeneration following partial hepatectomy, but no change in intestinal apoB RNA editing and showed, if anything, a subtle increase in hepatic apoB RNA editing (Blanc et al. 2010). More recently, another line of A1cf−/− mice was reported, that used a different gene targeting strategy, and bypassed the early embryonic lethality reported with the first line (Snyder et al. 2017). Those A1cf−/− mice were viable as adults, with no overt growth phenotype in either liver or intestine and exhibited no change in either apoB RNA editing or in several other candidate RNAs that were identified from reports of a range of APOBEC1 targets of C-to-U editing (Rosenberg et al. 2011; Blanc et al. 2014). Those findings in adult A1cf−/− mice (Snyder et al. 2017) suggested that A1CF is dispensable for apoB RNA editing and raised the possibility that A1CF may not participate in regulating C-to-U RNA editing activity of other physiological targets.
An alternative candidate complementation factor, RBM47, was identified in a screen of genes in foregut endoderm (Loebel et al. 2011), and a role in apoB RNA editing emerged with the discovery that gene-trap Rbm47 mutant mice exhibited prenatal lethality with surviving pups exhibiting defective C-to-U editing of intestinal apoB RNA and several other targets (Fossat et al. 2014). In particular, based on the loss of C-to-U RNA editing activity in the surviving Rbm47 mutant pups, we wondered what functions might be impaired in adult Rbm47 null mice where conditional targeting approaches might bypass the severe perinatal, developmental defects?
Here we examined the role of A1CF and of RBM47 in physiological, tissue-specific regulation of C-to-U RNA editing, both individually and in combination, in order to examine the range of target specificity and activity in vivo for each of these potential complementation factors. The findings suggest a model in which both A1CF and RBM47 exhibit distinctive, tissue-specific functions that together modulate APOBEC1 dependent RNA editing site selection and activity.
RESULTS
Subtle changes in apoB RNA editing following A1CF deletion and transgenic A1CF overexpression
We replicated the recent findings (Snyder et al. 2017) that A1cf−/− mice show no A1CF protein expression in liver (Fig. 1A,B) with only minor alterations in apoB RNA editing (65% vs. 53%, Fig. 1C) and no change in the ratio of apoB100 vs. apoB48 protein products (Fig. 1D). There was a small (albeit statistically significant) increase in apoB RNA editing (84%, Fig. 1C), with no change in hepatic apoB100/48 ratio (Fig. 1E) with liver-transgenic A1CF overexpression (approximately fourfold overexpression, Fig. 1F). Since prior studies have shown that overexpression of APOBEC1 results in promiscuous editing of apoB RNA, beyond the canonical site (Sowden et al. 1996a; Yamanaka et al. 1996), we examined the impact of adenoviral APOBEC1 (Ad-A1) overexpression in either A1cf−/− mice deletion or with liver transgenic A1CF overexpression. Those findings revealed increased RNA editing at the canonical site (6666) in all genotypes but generally decreased or absent editing at downstream sites in both transgenic A1cf and A1cf−/− mice (Fig. 1G).
FIGURE 1.

Alterations in hepatic A1CF expression produces only subtle changes in apoB RNA editing. (A) Hepatic expression of A1CF in wild-type (WT) and A1cf−/− mice. Actin was used as loading control. Molecular weights (kDa) are shown to the left. (B). Immunohistochemical detection of A1CF in WT livers showing nuclear localization of ACF and loss in A1cf−/− livers. (C) Hepatic apoB RNA editing by genotype. The region of apoB sequenced spans nucleotides 6563 to 7210. The data represent the average from three to four mice per genotype. Twenty clones were sequenced for each mouse. Positions of the edited cytidines are indicated on the left. Every circle represents a sequenced clone. Solid circles indicate C to U editing at the specified cytidine base, position indicated on the left. In all three genotypes, C to U editing is detected only at apoB canonical site C6666. Editing frequency (% edited clones at cytidine 6666) is indicated to the right. The numbers in parentheses under each panel represent the number of edited clones versus the total number of sequenced clones. The P-value under each panel indicates that editing at the canonical site of ApoB is statistically significant between A1cf−/− and WT liver; between A1cf−/− and A1cfTg; between A1cfTg and WT liver. Editing frequency at canonical C6666 editing site as mean ± SE: WT: 64 ± 4.2; A1cf−/− 53 ± 1.8; A1cfTg 84.5 ± 1.8. (D) Western blot detection of ApoB100 and ApoB48 in WT and A1cf−/− livers. Detection of Actin is used as loading control (n = 5–7 per genotype). (E) Western blot analysis of apoB100:B48 ratio in WT and transgenic A1CF liver (n = 3 per genotype). Actin is used as loading control. (F) A1CF and APOBEC1 expression in WT, A1cf−/−, and AcfTg mice injected with adenovirus-Apobec-1. Actin is used as loading control. (G) Hyperediting profile of hepatic ApoB RNA following adenoviral-expression of Apobec-1 in WT, A1cf−/− and A1cfTg mice. Representative data from three to four mice per genotype. Twenty clones per mouse were sequenced. Positions of the edited cytidines are indicated on the left. Editing frequencies at specific cytidine positions are shown to the right.
We extended those findings in small intestine, comparing A1cf−/− mice to those with intestinal transgenic A1CF overexpression (approximately fourfold overexpression, Fig. 2A). We observed no alteration in canonical site editing of apoB RNA by genotype but again observed decreased editing at downstream sites in both transgenic A1CF and A1cf−/− mice (Fig. 2B). Taken together, the findings suggest that basal hepatic apoB RNA editing at the canonical site exhibits minor changes over a range of A1CF expression. Under conditions of forced APOBEC1 overexpression, all genotypes exhibited virtually 100% RNA editing at the canonical site with subtle alterations in editing activity at downstream sites in the setting of either loss of A1CF or with transgenic A1CF overexpression. By contrast, there was no change in intestinal apoB RNA editing activity (versus WT) at the canonical site in either transgenic A1CF or A1cf−/− mice.
FIGURE 2.
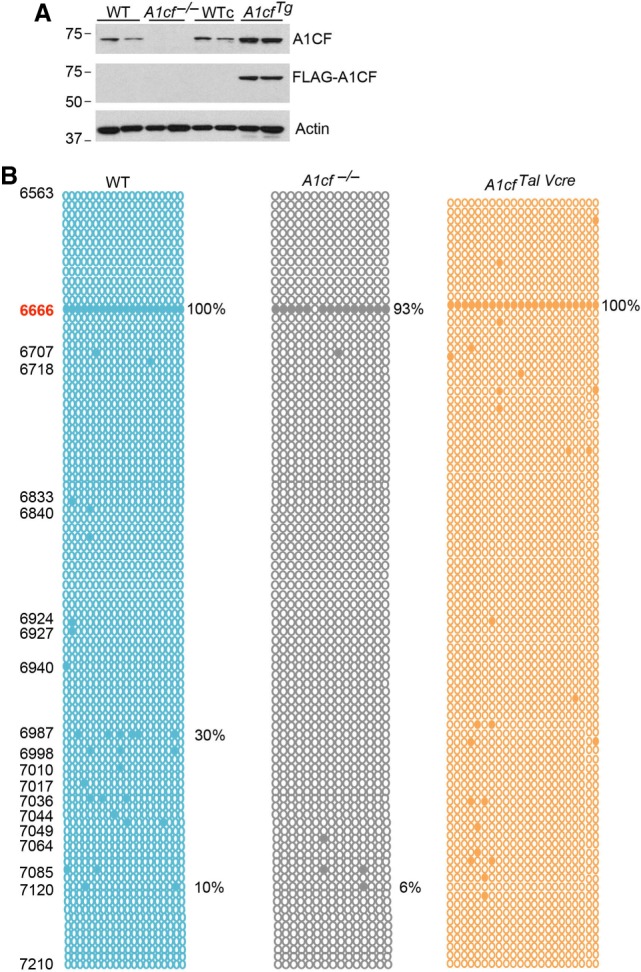
Neither A1cf deletion or overexpression in intestinal transgenic A1CF mice alter intestinal ApoB RNA editing. (A) Intestinal A1CF expression in WT, A1cf−/−, AcfTg and littermate controls (WTc). Transgenic A1CF expression detected using both anti-A1CF and anti-FLAG antibody. Detection of Actin is used as loading control. (B) Intestinal apoB RNA editing profile. The data represent the average from three mice per genotype, 15–22 clones were sequenced for each genotype. Solid circles identify clone with editing at the site indicated to the left.
Tissue-specific Rbm47 deletion differentially impairs apoB RNA editing in liver and intestine
Earlier observations in Rbm47 mutant mice demonstrated prenatal lethality (Fossat et al. 2014), making it challenging to understand the role of RBM47 in RNA editing in adult animals. This is an important consideration in examining apoB RNA editing activity, since work has demonstrated that apoB RNA editing is developmentally regulated in a tissue-specific manner in rodents and other mammals, including humans (Teng et al. 1990a,b; Inui et al. 1992). Accordingly, we generated conditional Rbm47 floxed mice (Fig. 3A and Methods) and crossed them into either Albumin-Cre (Jax) or Villin-Cre (Jax) transgenic mice to generate liver-specific (Rbm47LKO, Fig. 3B,C) and intestine-specific (Rbm47IKO, Fig. 3D) mice, respectively, both of which were viable as adults. We then examined apoB RNA editing in the respective tissues, the findings demonstrating that hepatic apoB RNA editing in Rbm47LKO mice was reduced (∼25% C-to-U editing at the canonical site) but not eliminated, while intestinal apoB RNA editing was virtually eliminated in Rbm47IKO mice (6% residual activity) at the canonical site (Fig. 3E,F). Accompanying these changes in RNA editing activity there was a corresponding increase in relative hepatic apoB100 vs. apoB48 protein expression (Fig. 3G), with no change in hepatic A1CF expression in Rbm47LKO mice (Fig. 3H), suggesting that Rbm47 deletion does not produce a compensatory change in total A1CF abundance.
FIGURE 3.

Generation and characterization of conditional Rbm47 knockout mice. (A) Endogenous mouse Rbm47 gene and targeting construct. Schematic illustration of genomic structure of Rbm47 allele containing nine exons (orange boxes). Schematic of Rbm47tm1a composition. (Numbered oranges boxes) Rbm47 exons; (blue boxes) Lac Z and neo cassettes; (green triangles) FRT sites; (purple triangles) LoxP sites. FLP recombination eliminates the LacZ and neo cassettes. Exon 6 containing transcriptional start codon and sequences encoding RNA recognition motifs was targeted for Cre-dependent homologous recombination. (B) Representative DNA electrophoresis showing genotype of WT and targeted alleles using primers P1 and P2 surrounding the 3′arm LoxP site. Analysis of the WT allele results in a 490 bp PCR product (lane 1) whereas recombination at the floxed allele generates a 530 bp product (f/f) (lane 3). Cre activation resuts in the loss of exon 6, evidenced by the absence of PCR amplification (lane 5). Molecular weights (bp) are indicated to the left. (C) Western blot detection of RBM47 in liver of floxed and Rbm47 liver-specific knockout (Rbm47LKO) mice with Actin used as loading control. (D) Western blot analysis of RBM47 in Rbm47IKO mice showing no detectable protein in intestinal nuclear extract. (E) Hepatic apoB RNA editing profile in Rbm47LKO mice, representative of three individual livers. Forty-two clones were sequenced, with each clone represented by a circle. Solid circles indicate editing at the specified position. We examined an apoB region encompassing nucleotides 6563–7210, with editing only at the canonical cytidine (6666) detected. (F) Intestinal apoB RNA editing profile in Rbm47IKO mice. Representative distribution of RNA editing sites from the same region (6563–7210) revealing only 2/19 clones exhibiting RNA editing. (G) Western blot analysis of hepatic apoB100 and apoB48 isoforms (three to five mice per genotype) with actin as loading control. (Panel to the right ) Quantitation of apoB100 isoform as a fraction of total apoB showing significant increase of apoB100 in liver of Rbm47LKO mice. (H) Western blot analysis of RBM47 and A1CF in Rbm47LKO mice at baseline and following adenoviral APOBEC1 overexpression. (I) Editing and hyperediting profile of hepatic apoB RNA following adenoviral overexpression of APOBEC1 in two Rbm47LKO mice. Representative distribution of edited sites from nucleotide 6563 to nucleotide 7210.
We then examined the response to adenoviral APOBEC1 (Ad-A1) overexpression in Rbm47LKO mice, the findings demonstrating no change in apoB RNA editing activity at the canonical site (6666) in Rbm47LKO mice (Fig. 3I, ∼30% editing with Ad-A1 vs. ∼25% editing at baseline, as in Fig. 3E), suggesting that hepatic RNA editing activity is constrained even with supplemental Apobec-1.
These findings together suggest that RBM47 is required for intestinal apoB RNA editing in adult mice, because Rbm47 deletion essentially eliminates editing activity. By contrast, the findings suggest the possibility that A1CF alone provides basal complementation for hepatic APOBEC1 dependent RNA editing activity (i.e., in the absence of RBM47) while suggesting that potential stoichiometric interactions between A1CF and RBM47 are required to establish physiologic hepatic apoB RNA editing.
Combined hepatic deletion of A1cf and of Rbm47 eliminates hepatic apoB RNA editing and is partially restored with A1CF rescue, both in vivo and in vitro
Based on the suggestion above, we crossed Rbm47LKO mice with A1cf−/− mice to generate mice with combined deletion of both genes in the liver, referred to as ARLKO mice. As expected, those ARLKO mice demonstrated loss of both A1CF and RBM47 in liver (Fig. 4A), but we observed that hepatic A1CF appeared more abundant by western blotting than RBM47, an impression confirmed at the RNA level as shown by quantitative PCR (Fig. 4B). We next examined those ARLKO mice under basal conditions (chow fed) or following Ad-A1 administration (discussed below) to supplement basal expression of APOBEC1 (Fig. 4A). The findings at baseline in adult ARLKO mice indicate no change in total apoB mRNA abundance (Fig. 4C), with elimination of apoB RNA editing (<1%, Table 1) and no detectable apoB48 in liver protein extracts (Fig. 4D). Editing activity of other targets is presented in a subsequent section below.
FIGURE 4.

Combined hepatic deletion of A1cf and Rbm47 in double knockout (ARLKO) liver eliminates apoB RNA editing. (A) Western blot analysis of A1CF, RBM47, and APOBEC1 in livers of ARLKO mice, with or without Ad-Apobec1. Actin was used as a loading control. (B) Quantitative PCR analysis of hepatic A1cf and Rbm47 RNAs demonstrating greater A1cf mRNA abundance versus Rbm47 RNA. Data represents mean ± SE of four to five mice per genotype. (**) P-value < 0.01. (C) Quantitative PCR analysis of hepatic apoB RNA, showing no differences by genotype (mean ± SE of three to four mice per genotype). (D) Western blot of hepatic apoB protein in Rbm47LKO, A1cf−/−, and ARLKO mice. Actin was used as a loading control. (E) Western blot detection of FLAG-A1CF following adenovirus A1CF administration to ARLKO mice. The calculated percent C-to-U apoB RNA editing indicates apoB editing frequency at the canonical site C6666. Parentheses indicate number of clones containing the edited site vs. the total number of sequenced clones. (F) In vitro apoB RNA editing assay using liver S100 extracts (lane 1 negative control contains primer and bovine serum albumin only) prepared from either WT mice (lane 2), or from ARLKO mice (lanes 3–8), or using recombinant (rec) APOBEC1 (lanes 9,10), supplemented where indicated with recombinant A1CF (lanes 4,5) or RBM47 (lanes 6–8). ApoB RNA editing was determined by poison primer extension (Materials and Methods). The relative mobility of the unedited (C) and the edited (U) C6666 is indicated to the right. Representative of three independent assays. (G) Baseline hepatic editing efficiency of 3′UTR APOBEC1 RNA targets and hierarchy of RNA binding complementation factors (see Table 1 for data). Wild-type (black dots), Rbm47LKO (green dots), A1cf−/− (blue dots), ARLKO (orange dots), and Apobec1−/− (white dots). RNAs preferentially edited by RBM47 show decreased editing efficiency in Rbm47LKO and ARLKO and no change in A1cf−/−. In contrast, RNAs preferentially edited by A1CF show no change in the absence of A1CF but increased editing frequency in the absence of RBM47 and almost complete loss of editing in the absence of both A1CF and RBM47.
TABLE 1.
C-to-U editing efficiency by genotype of hepatic 3′UTR APOBEC1 RNA targets

To examine the possibility that A1CF might exert an RBM47-independent role in hepatic apoB RNA editing, we administered adenoviral A1CF (Ad-A1CF) to ARLKO mice. Those ARLKO mice, injected intravenously with Ad-A1CF, expressed hepatic A1CF (Fig. 4E) and revealed partial restoration of apoB RNA editing (∼6% activity, Fig. 4E). We further examined the role of A1CF using an in vitro RNA editing assay and a synthetic apoB RNA template (Blanc et al. 2001) in the presence of S100 extracts prepared from liver of wild-type or ARLKO mice, supplemented, where indicated, with either recombinant A1CF or RBM47 as previously described (Blanc and Davidson 2010). S100 extracts prepared from wild-type mice produced robust C-to-U editing of apoB (Fig. 4E, lane 2) while extracts from ARLKO mice were inactive (Fig. 4F, lane 3). However, supplementing extracts from ARLKO mice with recombinant A1CF partially restored editing activity, albeit not to the level observed in extracts from wild-type mice (Fig. 4F, lanes 4,5 vs. lane 2). Similarly, supplementing extracts from ARLKO mice with recombinant RBM47 restored only modest editing activity (Fig. 4F, lanes 6–9). Those findings suggest that S100 extracts from the liver of ARLKO mice are devoid of C-to-U RNA editing activity, but that the addition of either recombinant A1CF or RBM47 proteins alone produces partial restoration. We confirmed observations that recombinant A1CF alone with recombinant APOBEC1 yields robust editing activity, (Fig. 4F, lane 9), and also that recombinant RBM47 with recombinant APOBEC1 yielded editing activity (Fig. 4F, lane 10). Those findings suggest that either recombinant A1CF or RBM47 provide complementation activity to recombinant APOBEC1 in editing apoB RNA in vitro and support the earlier observations regarding A1CF function (Mehta and Driscoll 2002). Previous work has suggested that there was no additional effect on apoB RNA editing activity following supplementation of in vitro reactions containing APOBEC1 with both RBM47 and A1CF (Fossat et al. 2014) but we did not independently explore this question.
Combined hepatic deletion of A1cf and of Rbm47 modifies RNA activity of a subset of targets
Based on the finding that hepatic apoB RNA editing was reduced but not eliminated in Rbm47LKO mice, we examined C-to-U RNA editing efficiency across the range of previously identified hepatic targets (Blanc et al. 2014), in the four genotypes (WT, A1cf−/−, Rbm47LKO, and ARLKO). The findings show that deletion of A1cf alone failed to alter editing activity in any of 21 targets examined (Table 1). By contrast, hepatic C-to-U RNA editing efficiency in Rbm47LKO mice was decreased (average of 64%, range 44%–84%), but not eliminated, in 11 targets (including apoB), unchanged in four targets (Table 1) and, to our surprise, increased in four targets (Table 1). Of those four targets whose C-to-U RNA editing efficiency was increased in Rbm47LKO mice, all showed decreased editing in ARLKO mice (including to <1% in Cmtm6, Sh3bgrl, and Rnf128), suggesting that these particular loss-of-function effects of hepatic Rbm47 deletion also require loss of A1CF. In addition, there was a further subset of targets (including Sep15, Mpeg, and Cybb) whose editing activity was either unchanged or only slightly decreased in ARLKO mice (Table 1). A summary figure of the proposed hierarchy for A1CF, RBM47 and the combination (A1CF plus RBM47) in hepatic C-to-U RNA editing is presented in Figure 4G.
Combined intestinal deletion of both A1cf and Rbm47 eliminates C-to-U RNA editing in the majority of RNAs and reveals tissue-specific roles in target selection
The findings above indicate a distinct tissue-specific requirement for RBM47, since hepatic apoB RNA editing was reduced but not eliminated in Rbm47LKO mice (Fig. 3E), while apoB RNA editing was virtually eliminated in Rbm47IKO mice (Fig. 3F). Accordingly, we examined the possibility that editing at other RNA target sites was altered in Rbm47IKO mice. The findings indicate that the majority (48/53) of targets exhibited >90% reduction in RNA editing activity in Rbm47IKO mice (Table 2), including four targets (B2M, Cmtm6, Rnf128 and Sh3bgrl) where (hepatic) RNA editing activity was found to be increased in Rbm47LKO mice (compare data in Table 1, vs. Table 2). While RNA editing was reduced in the majority of targets in Rbm47IKO mice, we identified five targets (Cyp4v3, Kctd12, Tmbim6, Sep15, and Bche) with significant residual editing activity (Table 2). In order to examine the impact of combined loss of A1CF and RBM47 in adult intestine, we crossed Rbm47IKO mice with A1cf−/− mice to generate mice with combined deletion referred to as ARIKO mice (Fig. 5A). As expected, ARIKO mice exhibited no apoB RNA editing at any of the previously identified sites (Fig. 5B), and no change in total intestinal apoB mRNA abundance was observed in any genotype (Fig. 5C). We then examined editing activity in ARIKO mice in the five targets identified above demonstrating residual editing activity in Rbm47IKO mice. Those findings demonstrated decreased editing activity in four of five targets with one (Kctd12) exhibiting 30% C-to-U RNA editing, despite the absence of both A1CF and RBM47. These findings strongly suggest that RBM47 plays a dominant role in regulating Apobec-1 dependent RNA editing in mouse intestine, but also support the conclusions that A1CF plays a permissive role in a subset of targets. The one exception was C-to-U RNA editing of Kctd12, which was uninfluenced by the loss of either complementation factor.
TABLE 2.
C-to-U RNA editing efficiency by genotype of intestinal 3′UTR APOBEC1 targets
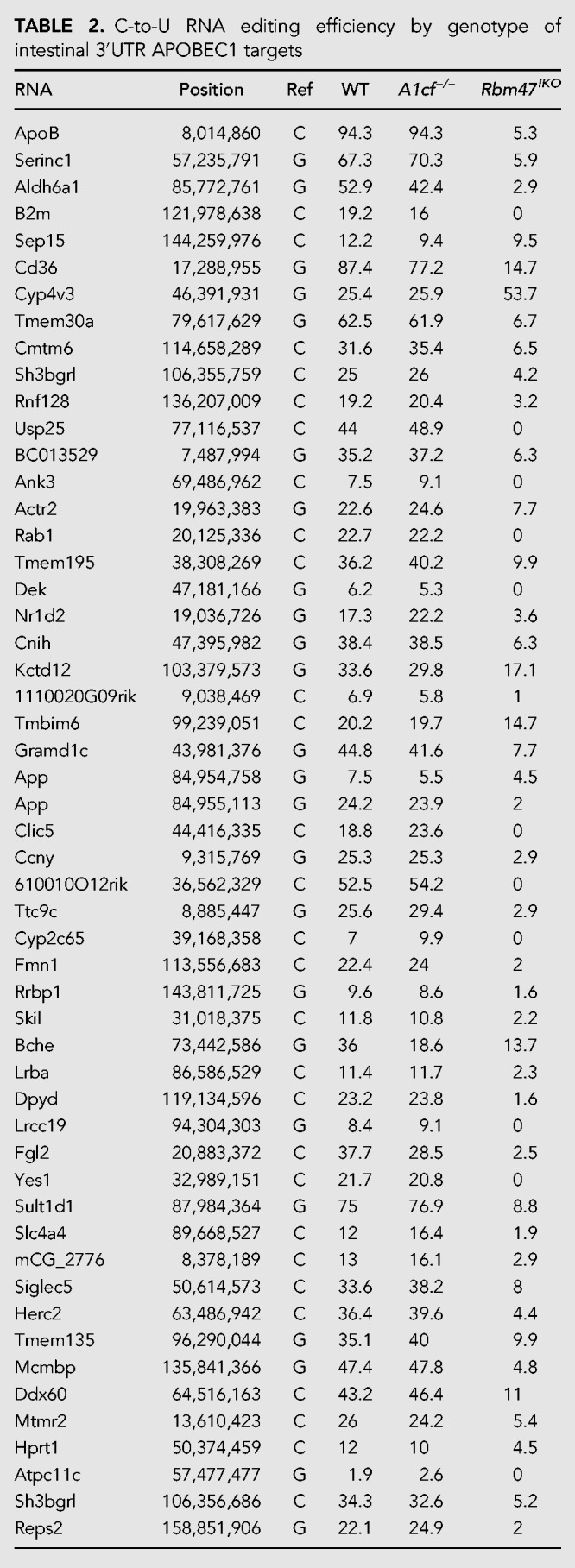
FIGURE 5.

Combined intestinal deletion of A1cf and Rbm47 in double knockout (ARIKO) mice eliminates apoB RNA editing and reduces C-to-U editing activity in four of five residual targets from Rbm47IKO mice. (A) Western blot analysis of intestinal A1CF and RBM47 in ARIKO mice. Actin was used as a loading control. (B) Intestinal apoB RNA editing is eliminated in ARIKO mice. Data are representative of four individual animals. Open circles represent individual clones (total of 32 clones sequenced) in the region spanning nucleotides 6563 to 7210. (C) Quantitative PCR analysis of intestinal apoB RNA showing no differences by genotype, from representative mice (n = 3–4 per genotype). (D) Baseline editing efficiency of the subset of intestine 3′UTR APOBEC1 RNA targets that revealed residual editing activity in Rbm47IKO mice. Data are indicated as percentage of edited clones with numbers in parentheses indicating edited versus total number of sequenced clones.
DISCUSSION
There is considerable information regarding the physiological regulation of mammalian C-to-U RNA editing, particularly for the prototype target apoB RNA, which undergoes tissue-specific regulation, modulated by developmental, hormonal, and nutritional cues in mouse and rat liver, through mechanisms proposed to involve regulation of the complementation factors (Teng et al. 1990a; Inui et al. 1992, 1994; Lau et al. 1995; Lehmann et al. 2006). In addition, intestinal apoB RNA editing is developmentally regulated in both mouse and human small intestine, findings at least partially accounted for by the temporal patterns of APOBEC1 expression but which may also reflect alterations in A1CF activity (Teng et al. 1990b; Giannoni et al. 1995). These earlier studies, coupled with findings showing that A1CF undergoes alternative splicing with differential functions assigned to each splice form (Galloway et al. 2010), supported the view that A1CF undergoes nuclear-cytoplasmic shuttling in conjunction with metabolically regulated, post-translational modifications that in turn regulate C-to-U RNA editing (Galloway and Smith 2010). The view that A1CF was a necessary cofactor for C-to-U RNA editing was radically revised with the report that Rbm47 mutant mice exhibit virtually no intestinal apoB RNA editing and also that a two-component system containing recombinant RBM47 with recombinant APOBEC1 together catalyze in vitro apoB RNA editing (Fossat et al. 2014). Those findings, coupled with the recent report that A1cf−/− mice exhibit no detectable alterations in C-to-U RNA editing (Snyder et al. 2017), led to the alternative suggestion that RBM47 acts alone as the complementation factor for APOBEC1 dependent RNA editing, and left open the function (if any) for A1CF in vivo. The findings from this report offer a more nuanced view, namely, that A1CF and RBM47 function independently, yet interact in a tissue-specific manner to regulate the activity and site selection of Apobec-1 dependent C-to-U RNA editing in adult mouse intestine and liver. Several aspects of this report merit further discussion.
A1CF was independently identified by two groups, through approaches based in part on RNA binding affinity of factors purified from either baboon or rat liver that were enriched by binding to an apoB RNA fragment flanking the C-to-U editing site, which eventually yielded a single protein that exhibited complementation activity for in vitro apoB RNA editing (Lellek et al. 2000; Mehta et al. 2000). Those findings, coupled with findings that RBM47 colocalizes and physically interacts with both APOBEC1 and with A1CF (Fossat et al. 2014) suggest that the in-vivo complementation activity originally assigned to A1CF may reflect the optimal configuration of RBM47 and APOBEC1in the context of a heteromeric, functional editosome complex, containing A1CF (Fossat et al. 2014). By contrast, the structural and functional homology of A1CF and RBM47 likely accounts for the ability of either protein to provide complementation activity in a reconstituted, two component in vitro assay (Fossat et al. 2014) and Figure 4F. These observations allow us in principle to reconcile the apparent paradox that A1CF alone with APOBEC1 is sufficient for in vitro RNA editing yet appears dispensable for physiological editing activity in vivo.
Based on the finding that Rbm47 mutant mice exhibit virtually no intestinal apoB RNA editing in the surviving animals (Fossat et al. 2014), we expected to find that hepatic apoB RNA editing would be similarly eliminated in Rbm47LKO mice. However, that was not the case. We observed instead that RNA editing was reduced but not eliminated in Rbm47LKO mice and that both isoforms of apoB were readily detectable, albeit with an altered ratio reflecting the decreased RNA editing activity. Similarly, we observed decreased activity in 11 of 20 other targets of C-to-U RNA editing in those Rbm47LKO mice, no change in a further subset and an increase in yet another subset of four targets (B2m, Cmtm6, Sh3bgrl, Rnf128, Table 1). We interpret those findings to indicate that RBM47 provides the dominant source of complementation activity for the 11 of 20 targets that exhibit decreased editing in Rbm47LKO mice, while the subset of four targets showing increased editing activity in Rbm47LKO mice may be predominantly A1CF dependent, as evidenced by the loss of editing activity in ARLKO mice. The additional subset of hepatic targets demonstrating ∼12%–14% residual editing activity in ARLKO mice (Table 1) might conceivably reflect residual expression of A1CF or RBM47 within non-parenchymal cells (Kupffer cells, stellate cells, endothelial cells, or cholangiocytes), which would not be amenable to Cre deletion with the albumin promoter used in these studies, which is reportedly hepatocyte predominant (Postic et al. 1999).
The same caveats might be considered for the findings with Rbm47IKO mice where a small subset of targets exhibited residual editing activity, most of which were further reduced in ARIKO mice. Those studies were conducted using scraped intestinal mucosa which contains >90% enterocytes but may also include submucosal and lamina propria cell types, including neuronal cells. It is worth noting that the villin promoter, used to drive intestinal Cre expression in our studies, is reported to be confined to villus enterocytes and colonocytes with some expression in renal tubular cells (el Marjou et al. 2004). This cell-restricted pattern of villin-Cre expression may be a relevant consideration for the observations regarding RNA editing of Kctd12, which demonstrated robust (30%) Apobec-1 dependent editing in ARIKO mice. Kctd12 encodes a potassium channel which is known to be widely expressed in human and mouse tissues including in intestinal neuroendocrine and colorectal tumors as well as throughout the brain (Resendes et al. 2004; Li et al. 2016; Suehara et al. 2018). Thus, it is possible that Kctd12 expression in cells other than enterocytes might account for the preserved RNA editing activity in Rbm47IKO and ARIKO mice. Nevertheless, an alternative interpretation might also be considered, namely that other (to be identified) complementation factors might be responsible. A further consideration is that Apobec-1 alone may be sufficient to mediate C-to-U RNA editing of a small subset of transcripts, as implied earlier (Chester et al. 2004). Resolution of these possibilities will require further investigation beyond the scope of the current studies.
The tissue-specific roles of A1CF and RBM47 in RNA editing was further supported by the finding that four hepatic targets (B2m, Cmtm6, Sh3bgrl Rnf128) that were edited more efficiently by A1CF in the liver (Table 1) were edited only by RBM47 in the intestine (Table 2). One possibility to explain this apparent discrepancy is that the binding site(s) in those RNAs for A1CF may not be as accessible in the intestine as in the liver, perhaps because the folding of RNAs may be different and/or there may be other RBPs that in turn modify access of target transcripts to A1CF. The identification of A1CF/RNA/protein partners will require extended investigation beyond the focus of this study.
The proposed dominance of RBM47 in hepatic apoB RNA editing is further supported by the observation that hepatic A1CF appears to be expressed at higher levels than RBM47 (Fig. 4A) while loss of A1CF alone exerts only minor if any effect on apoB RNA editing. This may imply either greater affinity or sustained occupancy with an RBM47-apoB RNA- APOBEC1 complex compared to an A1CF-apoB RNA-APOBEC1 complex. It is also conceivable that both RBM47-Apobec-1 and A1CF-APOBEC1 complexes coexist, with RBM47-APOBEC1 complex being more efficient in mediating RNA editing, but this speculation will require formal examination and a more complete understanding of the relative molar distribution of each cofactor within these complexes.
The current findings also raise the question of a broader biological role for A1CF and RBM47 in mammalian physiology. As noted above, our initial attempt to generate germline A1cf−/− mice revealed early developmental lethality with homozygous A1cf−/− blastocysts failing to implant at embryonic day 3.5 (Blanc et al. 2005). A different line of A1cf−/− mice (used in the studies reported here) employed a distinct genomic targeting strategy (Snyder et al. 2017) and also used sox2-Cre which is active at embryonic day 6.5 and which would bypass the early, preimplantation lethality observed in our studies (Blanc et al. 2005; Moore et al. 2014). Those A1cf−/− mice were viable as adults yet showed abnormalities in renal function, while the viable heterozygous A1cf+/− mice from the initial knockout attempts demonstrated a growth phenotype with defective liver regeneration following partial hepatectomy (Blanc et al. 2010). Along the same lines, the original line of mutant, gene-trap Rbm47−/− mice demonstrated perinatal lethality with the surviving animals showing a runted phenotype (Fossat et al. 2014). Our findings used a different genomic targeting strategy and conditional tissue-specific deletion which again bypassed the perinatal lethality. While we observed no gross defects in liver or intestinal development in Rbm47IKO, Rbm47LKO, or the corresponding double knockout lines (i.e., ARIKO and ARLKO mice), we are currently examining in more detail the possibilities of other growth-related and metabolic phenotypes.
As a final comment, we recognize that the current findings are limited to the tissue-specific impact of A1CF and of RBM47 within the liver and small intestine but we certainly acknowledge the importance of APOBEC1 dependent C-to-U RNA editing activity in other cell types as revealed by recent studies in microglia and monocytes derived from Apobec1−/− mice (Cole et al. 2017; Rayon-Estrada et al. 2017). We envision future studies utilizing a range of other cell-specific Cre drivers to elucidate the role of RBM47 in those contexts also.
MATERIALS AND METHODS
Animals
Rbm47 conditional knockout animals were generated by in vitro fertilization of C57BL/6 oocytes (Washington University Mouse Genetics Core) with sperm carrying the Rbm47tm1a mutation (Rbm47tm2a(EUCOMM)Wtsi) obtained from the International Mouse Phenotyping Consortium/European Mouse Mutant Archive (EMMA). Offspring were then bred with Flp-1 Tg mice (Jax# 009086) to remove the Frt-flanked LacZ-Neo cassette (Fig. 3A). Genotyping was performed using the following primers surrounding the 3′arm LoxP site: Rbm47 Fwd LoxP (P1) (5′-ACT CCT GTG ACC CCT ACA CG-3′) and Rbm47 Rev 24157 (P2) (5′-GTA ACC CAG GCT GGC CTA-3′), using the following conditions: 94°C for 5 min; 94°C for 30 sec, 60°C for 45 sec, 72°C for 50 sec (35 cycles); 72° C 10 min. The PCR reaction generates a 490 bp wild-type band and a 530 bp Rbm47 floxed (Rbm47f/f) band. Heterozygous intercrosses resulted in viable and fertile homozygous Rbm47f/f mice with a normal Mendelian frequency. To generate liver-specific knockout mice (Rbm47LKO), Rbm47tm1a mice were crossed with Albumin-Cre transgenic mice (Jax# 003570). Intestine-specific Rbm47 knockout (Rbm47IKO) were generated by crossing Rbm47tm1a mice with transgenic villin-Cre-ERT2 mice (el Marjou et al. 2004). Cre recombinase migration to the nuclear compartment was induced by intraperitoneal injection of 1 mg/100 µL tamoxifen (Sigma) 1 mg/day for 5 d (el Marjou et al. 2004). All experimental and control animals were maintained on a C57BL/6J background.
A1cftm1b (A1cf−/−) were obtained from Jax (#005650) (Snyder et al. 2017) and maintained on a C57BL/6NJ background. Liver (ARLKO) and intestine (ARIKO)-specific A1cf-Rbm47 double knockouts were generated by breeding A1cf−/− mice with Rbm47LKO or Rbm47IKO, respectively. Apobec-1−/− mice were maintained on a C57BL/6J background. For hepatic overexpression of Apobec-1, animals were injected with 6 × 108 plaque-forming units of recombinant adenovirus encoding rat Apobec1 (Kozarsky et al. 1996). Mice were 8–12 wk old and fed an ad libitum chow diet. All animals were treated according to the National Institutes of Health guidelines and all protocols were approved by the Washington University Institutional Animal Care and Use Committee.
RNA isolation, cDNA synthesis, and quantitative PCR
RNA was extracted from tissues of the indicated genotype using TRIzol (Invitrogen) following manufacturer's instructions. DNase-free RNAs (RNase-Free DNase kit, Qiagen) were used for cDNA preparation using the High Capacity cDNA Reverse Transcription kit (Applied Biosystems). Quantitative evaluation of RNA abundance was performed using Fast SYBR Green Master Mix (Applied Biosystems) in a StepOne PLus Real Time PCR system instrument (Applied Biosystems). The following primers were used: A1cf: Fwd: 5′-GCC AGA ATC CTCG CAA TCCA-3′; Rev 5′-AGC ATA CCT CTT CGC TTC ATC C-3′, Rbm47 Fwd: 5′-GCT TCG CCT TTG TGG AGT ATG-3′; Rev: 5′-ATC CGA CCTGGC ATG AG-3′, ApoB Fwd: 5′-CAC TGC CGT GGC CAA AA-3′, Rev: GCT AGA GAG TTG GTC TGA AAA ATC CT-3′. Total mRNA abundance was determined by normalization to Gapdh RNA level in each sample. Primers for Gapdh are as follows: Fwd: 5′- TGTGTCCGTCGTGGATCTGA-3′; Rev: CCTGCTTCACCACCTTCTTGA-3′.
RNA editing analysis by Sanger sequencing
Following reverse transcription, cDNA was used to amplify sequences containing Apobec-1-dependent editing sites previously identified in liver and small intestine (Blanc et al. 2014). PCR amplifications were performed using AccuPrime Pfx DNA polymerase (Thermo Fisher Scientific). Primer sequences are available (Blanc et al. 2014). PCR products were then gel purified using Qiaquick Gel Extraction kit (Qiagen) and cloned using Zero Blunt TOPO PCR Cloning kit (Invitrogen) following manufacturer's recommendations. At least 20 individual clones were sequenced at Genewiz Inc.
Amplicon sequencing
PCR products of Apobec-1 hepatic and small intestine RNA targets were concatemerized as follows. The PCR products (∼12 ng) were treated with DNA polymerase I, Large Klenow Fragment (NEB) for 30 min at 37° C following manufacturer's instructions. After purification using PCR Purification kit (Qiagen), the PCR products were ligated overnight at room temperature using a mix of T4 DNA ligase and T4 polynucleotide kinase in the presence of 50% PEG-8000. Ligated products were purified using PCR Purification kit and ligation was confirmed by running aliquot of ligation on a 1% agarose gel. Concatemerized amplicons were then submitted for automated high-throughput DNA sequencing (Washington University Genome Center). The results of sequencing hepatic and intestinal amplicons are presented in Tables 1 and 2.
Protein extraction and western blotting
Total liver and scraped intestinal mucosa were homogenized as previously described (Blanc et al. 2014). Nuclear extracts were prepared using Glass Dounce Tissue homogenizer. Fifty milligrams of tissue was homogenized in 300 µL of Buffer A (25 mM Tris pH 7.5, 50 mM KCl, 2 mM MgCl2, 1 mM EDTA, 5 mM DTT, 1× protease inhibitor [Roche], 100 mM NaF, 1 mM NaVa, 50 mM α-glycerophosphate) performing 30 strokes with pestle A followed by 20 strokes with pestle B. The nuclear pellet was collected by centrifugation at 4000 rpm at 4°C for 10 min. The nuclei were washed three times with Buffer A, resuspended in 80 µL of Buffer B (25 mM Tris pH 7.5, 420 mM NaCl, 1.5 mM MgCl2, 0.5 mM EDTA, 1 mM DTT, 20% glycerol, 1× protease inhibitor) and incubated in ice for 30 min with a regular period of 30 sec vortex. Nuclear fraction was then collected by centrifugation 15 min at 10,000 rpm at 4°C. Fifteen micrograms of nuclear protein were resolved on a SDS–PAGE, transferred to PVDF membrane and probed with rabbit anti-A1CF antibody, rabbit anti-RBM47 (Abcam ab94638), anti-APOBEC1 (Kozarsky et al. 1996). Equal loading was confirmed using a rabbit anti-actin antibody (Sigma–Aldrich).
For detection of hepatic apoB isoforms, liver tissue was homogenized using modified RIPA buffer containing 1 M urea (Smagris et al. 2016). Fifty milligrams of tissue were suspended in 300 µL RIPA/urea buffer and homogenized in a bullet blender with 200 µL of 1 mm Glass Beads (Next Advance) twice for 5 min at maximum speed (speed 10) at 4°C. An additional 400 µL of RIPA/urea buffer was added and the mixture homogenized at speed 3 for 3 min at 4°C. The Homogenates were supplemented with one tenth of the volume with 10× detergent buffer (10% SDS, 10% NP-40) and incubated in ice for 30 min, before centrifugation 15 min at 4°C 15,000 rpm. Eighty micrograms of extracts were then fractionated on a 4%–20% SDS gradient gel, transferred on PVDF membrane and probed with rabbit anti-apoB (1:4000). Equal loading was checked using rabbit anti-actin (1:2000) antibody.
In vitro RNA editing analysis by poisoned primer extension
A synthetic 360-nt apoB RNA (20 fmol) was incubated with liver extracts prepared from wild-type or A1cf-Rbm47 liver-specific double knockout (ARLKO) mice. S100 extract (100 µg) or recombinant Apobec-1 (400 ng) was supplemented where indicated with increasing amounts (50 to 200 ng) of recombinant A1CF (Blanc et al. 2001) or recombinant mouse RBM47. Recombinant RBM47 protein was generated by cloning Rbm47 cDNA from mouse liver into the NdeI-XhoI sites of pTYB1 expression vector (New England Biolabs), resulting in synthesis of a C-terminal RBM47-intein fusion protein. RBM47 was purified as previously described (Blanc et al. 2001). S100 extracts with or without recombinant proteins were incubated for 3 h at 30°C in in-vitro conversion buffer (Blanc et al. 2001; Blanc and Davidson 2011). Following incubation, RNA was phenol/chloroform extracted, precipitated and resuspended in cDNA synthesis reaction mix. Single-stranded DNA was then subjected to PCR amplification using the following apoB primers: Fwd:5′-ATCTGACTGGGAGAGACAAGTAG-3′, Rev: 5′-GCTCGCTCAGGCTATATCTGTGGGC-3′. PCR product was then used as template in poison primer extension as previously described (Blanc and Davidson 2011). Extension products were separated by electrophoresis on a 7 M urea-acrylamide gel and analyzed by autoradiography.
ACKNOWLEDGMENTS
This work was supported by grants HL-38180, DK-56260, and DK-112378 to N.O.D.; grants DK-106382 and DK-112378 to D.C.R.; grants DK094989, DK105129, DK110406, Alvin J. Siteman Cancer Center/Barnes Jewish Hospital Foundation Cancer Frontier Fund, National Institutes of Health NCI P30 CA091842, and The Barnard Trust to J.C.M.; grants DK-093885, DK-108764, DK-052574, the Siteman Cancer Center, and the Cancer Research Foundation Young Investigator Award to B.B.M.; and grant DP 1HD-075624 to J.H.N. J.D.R. was supported by F32 DK-109651. The authors acknowledge the assistance of the Murine Models and ARAC cores of the Washington University DDRCC (DK-52574) for outstanding support.
Footnotes
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.068395.118.
REFERENCES
- Anant S, Blanc V, Davidson NO. 2003. Molecular regulation, evolutionary, and functional adaptations associated with C to U editing of mammalian apolipoproteinB mRNA. Prog Nucleic Acid Res Mol Biol 75: 1–41. 10.1016/S0079-6603(03)75001-6 [DOI] [PubMed] [Google Scholar]
- Blanc V, Davidson NO. 2010. APOBEC-1-mediated RNA editing. Wiley Interdiscip Rev Syst Biol Med 2: 594–602. 10.1002/wsbm.82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanc V, Davidson NO. 2011. Mouse and other rodent models of C to U RNA editing. Methods Mol Biol 718: 121–135. 10.1007/978-1-61779-018-8_7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanc V, Henderson JO, Kennedy S, Davidson NO. 2001. Mutagenesis of apobec-1 complementation factor reveals distinct domains that modulate RNA binding, protein-protein interaction with apobec-1, and complementation of C to U RNA-editing activity. J Biol Chem 276: 46386–46393. 10.1074/jbc.M107654200 [DOI] [PubMed] [Google Scholar]
- Blanc V, Henderson JO, Newberry EP, Kennedy S, Luo J, Davidson NO. 2005. Targeted deletion of the murine apobec-1 complementation factor (acf) gene results in embryonic lethality. Mol Cell Biol 25: 7260–7269. 10.1128/MCB.25.16.7260-7269.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanc V, Sessa KJ, Kennedy S, Luo J, Davidson NO. 2010. Apobec-1 complementation factor modulates liver regeneration by post-transcriptional regulation of interleukin-6 mRNA stability. J Biol Chem 285: 19184–19192. 10.1074/jbc.M110.115147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanc V, Park E, Schaefer S, Miller M, Lin Y, Kennedy S, Billing AM, Hamidane HB, Graumann J, Mortazavi A, et al. 2014. Genome-wide identification and functional analysis of Apobec-1-mediated C-to-U RNA editing in mouse small intestine and liver. Genome Biol 15: R79 10.1186/gb-2014-15-6-r79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen SH, Habib G, Yang CY, Gu ZW, Lee BR, Weng SA, Silberman SR, Cai SJ, Deslypere JP, Rosseneu M, et al. 1987. Apolipoprotein B-48 is the product of a messenger RNA with an organ-specific in-frame stop codon. Science 238: 363–366. 10.1126/science.3659919 [DOI] [PubMed] [Google Scholar]
- Chester A, Weinreb V, Carter CW Jr, Navaratnam N. 2004. Optimization of apolipoprotein B mRNA editing by APOBEC1 apoenzyme and the role of its auxiliary factor, ACF. RNA 10: 1399–1411. 10.1261/rna.7490704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole DC, Chung Y, Gagnidze K, Hajdarovic KH, Rayon-Estrada V, Harjanto D, Bigio B, Gal-Toth J, Milner TA, McEwen BS, et al. 2017. Loss of APOBEC1 RNA-editing function in microglia exacerbates age-related CNS pathophysiology. Proc Natl Acad Sci 114: 13272–13277. 10.1073/pnas.1710493114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Driscoll DM, Lakhe-Reddy S, Oleksa LM, Martinez D. 1993. Induction of RNA editing at heterologous sites by sequences in apolipoprotein B mRNA. Mol Cell Biol 13: 7288–7294. 10.1128/MCB.13.12.7288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- el Marjou F, Janssen KP, Chang BH, Li M, Hindie V, Chan L, Louvard D, Chambon P, Metzger D, Robine S. 2004. Tissue-specific and inducible Cre-mediated recombination in the gut epithelium. Genesis 39: 186–193. 10.1002/gene.20042 [DOI] [PubMed] [Google Scholar]
- Fossat N, Tourle K, Radziewic T, Barratt K, Liebhold D, Studdert JB, Power M, Jones V, Loebel DA, Tam PP. 2014. C to U RNA editing mediated by APOBEC1 requires RNA-binding protein RBM47. EMBO Rep 15: 903–910. 10.15252/embr.201438450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gagnidze K, Rayon-Estrada V, Harroch S, Bulloch K, Papavasiliou FN. 2018. A new chapter in genetic medicine: RNA editing and its role in disease pathogenesis. Trends Mol Med 24: 294–303. 10.1016/j.molmed.2018.01.002 [DOI] [PubMed] [Google Scholar]
- Galloway CA, Smith HC. 2010. The expression of apoB mRNA editing factors is not the sole determinant for the induction of editing in differentiating Caco-2 cells. Biochem Biophys Res Commun 391: 659–663. 10.1016/j.bbrc.2009.11.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galloway CA, Kumar A, Krucinska J, Smith HC. 2010. APOBEC-1 complementation factor (ACF) forms RNA-dependent multimers. Biochem Biophys Res Commun 398: 38–43. 10.1016/j.bbrc.2010.06.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giannoni F, Chou SC, Skarosi SF, Verp MS, Field FJ, Coleman RA, Davidson NO. 1995. Developmental regulation of the catalytic subunit of the apolipoprotein B mRNA editing enzyme (APOBEC-1) in human small intestine. J Lipid Res 36: 1664–1675. [PubMed] [Google Scholar]
- Hirano K, Young SG, Farese RV Jr, Ng J, Sande E, Warburton C, Powell-Braxton LM, Davidson NO. 1996. Targeted disruption of the mouse apobec-1 gene abolishes apolipoprotein B mRNA editing and eliminates apolipoprotein B48. J Biol Chem 271: 9887–9890. 10.1074/jbc.271.17.9887 [DOI] [PubMed] [Google Scholar]
- Inui Y, Hausman AM, Nanthakumar N, Henning SJ, Davidson NO. 1992. Apolipoprotein B messenger RNA editing in rat liver: Developmental and hormonal modulation is divergent from apolipoprotein A-IV gene expression despite increased hepatic lipogenesis. J Lipid Res 33: 1843–1856. [PubMed] [Google Scholar]
- Inui Y, Giannoni F, Funahashi T, Davidson NO. 1994. REPR and complementation factor(s) interact to modulate rat apolipoprotein B mRNA editing in response to alterations in cellular cholesterol flux. J Lipid Res 35: 1477–1489. [PubMed] [Google Scholar]
- Keegan L, Khan A, Vukic D, O'Connell M. 2017. ADAR RNA editing below the backbone. RNA 23: 1317–1328. 10.1261/rna.060921.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozarsky KF, Bonen DK, Giannoni F, Funahashi T, Wilson JM, Davidson NO. 1996. Hepatic expression of the catalytic subunit of the apolipoprotein B mRNA editing enzyme (apobec-1) ameliorates hypercholesterolemia in LDL receptor-deficient rabbits. Hum Gene Ther 7: 943–957. 10.1089/hum.1996.7.8-943 [DOI] [PubMed] [Google Scholar]
- Lau PP, Cahill DJ, Zhu HJ, Chan L. 1995. Ethanol modulates apolipoprotein B mRNA editing in the rat. J Lipid Res 36: 2069–2078. [PubMed] [Google Scholar]
- Lehmann DM, Galloway CA, Sowden MP, Smith HC. 2006. Metabolic regulation of apoB mRNA editing is associated with phosphorylation of APOBEC-1 complementation factor. Nucleic Acids Res 34: 3299–3308. 10.1093/nar/gkl417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lellek H, Kirsten R, Diehl I, Apostel F, Buck F, Greeve J. 2000. Purification and molecular cloning of a novel essential component of the apolipoprotein B mRNA editing enzyme-complex. J Biol Chem 275: 19848–19856. 10.1074/jbc.M001786200 [DOI] [PubMed] [Google Scholar]
- Li L, Duan T, Wang X, Zhang RH, Zhang M, Wang S, Wang F, Wu Y, Huang H, Kang T. 2016. KCTD12 regulates colorectal cancer cell stemness through the ERK pathway. Sci Rep 6: 20460 10.1038/srep20460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loebel DA, Studdert JB, Power M, Radziewic T, Jones V, Coultas L, Jackson Y, Rao RS, Steiner K, Fossat N, et al. 2011. Rhou maintains the epithelial architecture and facilitates differentiation of the foregut endoderm. Development 138: 4511–4522. 10.1242/dev.063867 [DOI] [PubMed] [Google Scholar]
- Mehta A, Driscoll DM. 2002. Identification of domains in apobec-1 complementation factor required for RNA binding and apolipoprotein-B mRNA editing. RNA 8: 69–82. 10.1017/S1355838202015649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta A, Kinter MT, Sherman NE, Driscoll DM. 2000. Molecular cloning of apobec-1 complementation factor, a novel RNA-binding protein involved in the editing of apolipoprotein B mRNA. Mol Cell Biol 20: 1846–1854. 10.1128/MCB.20.5.1846-1854.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore R, Tao W, Smith ER, Xu XX. 2014. The primitive endoderm segregates from the epiblast in β1 integrin-deficient early mouse embryos. Mol Cell Biol 34: 560–572. 10.1128/MCB.00937-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamuta M, Chang BH, Zsigmond E, Kobayashi K, Lei H, Ishida BY, Oka K, Li E, Chan L. 1996. Complete phenotypic characterization of apobec-1 knockout mice with a wild-type genetic background and a human apolipoprotein B transgenic background, and restoration of apolipoprotein B mRNA editing by somatic gene transfer of Apobec-1. J Biol Chem 271: 25981–25988. 10.1074/jbc.271.42.25981 [DOI] [PubMed] [Google Scholar]
- Nishikura K. 2010. Functions and regulation of RNA editing by ADAR deaminases. Annu Rev Biochem 79: 321–349. 10.1146/annurev-biochem-060208-105251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Postic C, Shiota M, Niswender KD, Jetton TL, Chen Y, Moates JM, Shelton KD, Lindner J, Cherrington AD, Magnuson MA. 1999. Dual roles for glucokinase in glucose homeostasis as determined by liver and pancreatic β cell-specific gene knock-outs using Cre recombinase. J Biol Chem 274: 305–315. 10.1074/jbc.274.1.305 [DOI] [PubMed] [Google Scholar]
- Powell LM, Wallis SC, Pease RJ, Edwards YH, Knott TJ, Scott J. 1987. A novel form of tissue-specific RNA processing produces apolipoprotein-B48 in intestine. Cell 50: 831–840. 10.1016/0092-8674(87)90510-1 [DOI] [PubMed] [Google Scholar]
- Rayon-Estrada V, Harjanto D, Hamilton CE, Berchiche YA, Gantman EC, Sakmar TP, Bulloch K, Gagnidze K, Harroch S, McEwen BS, et al. 2017. Epitranscriptomic profiling across cell types reveals associations between APOBEC1-mediated RNA editing, gene expression outcomes, and cellular function. Proc Natl Acad Sci 114: 13296–13301. 10.1073/pnas.1714227114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Resendes BL, Kuo SF, Robertson NG, Giersch AB, Honrubia D, Ohara O, Adams JC, Morton CC. 2004. Isolation from cochlea of a novel human intronless gene with predominant fetal expression. J Assoc Res Otolaryngol 5: 185–202. 10.1007/s10162-003-4042-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg BR, Hamilton CE, Mwangi MM, Dewell S, Papavasiliou FN. 2011. Transcriptome-wide sequencing reveals numerous APOBEC1 mRNA-editing targets in transcript 3′ UTRs. Nat Struct Mol Biol 18: 230–236. 10.1038/nsmb.1975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smagris E, Gilyard S, BasuRay S, Cohen JC, Hobbs HH. 2016. Inactivation of Tm6sf2, a gene defective in fatty liver disease, impairs lipidation but not secretion of very low density lipoproteins. J Biol Chem 291: 10659–10676. 10.1074/jbc.M116.719955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder EM, McCarty C, Mehalow A, Svenson KL, Murray SA, Korstanje R, Braun RE. 2017. APOBEC1 complementation factor (A1CF) is dispensable for C-to-U RNA editing in vivo. RNA 23: 457–465. 10.1261/rna.058818.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sowden M, Hamm JK, Smith HC. 1996a. Overexpression of APOBEC-1 results in mooring sequence-dependent promiscuous RNA editing. J Biol Chem 271: 3011–3017. 10.1074/jbc.271.6.3011 [DOI] [PubMed] [Google Scholar]
- Sowden M, Hamm JK, Spinelli S, Smith HC. 1996b. Determinants involved in regulating the proportion of edited apolipoprotein B RNAs. RNA 2: 274–288. [PMC free article] [PubMed] [Google Scholar]
- Suehara Y, Akaike K, Mukaihara K, Kurisaki-Arakawa A, Kubota D, Okubo T, Mitomi H, Mitani K, Takahashi M, Toda-Ishii M, et al. 2018. KCTD12 is negatively regulated by Kit in gastrointestinal stromal tumors. Oncotarget 9: 27016–27026. 10.18632/oncotarget.25469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teng B, Black DD, Davidson NO. 1990a. Apolipoprotein B messenger RNA editing is developmentally regulated in pig small intestine: nucleotide comparison of apolipoprotein B editing regions in five species. Biochem Biophys Res Commun 173: 74–80. 10.1016/S0006-291X(05)81023-X [DOI] [PubMed] [Google Scholar]
- Teng B, Verp M, Salomon J, Davidson NO. 1990b. Apolipoprotein B messenger RNA editing is developmentally regulated and widely expressed in human tissues. J Biol Chem 265: 20616–20620. [PubMed] [Google Scholar]
- Yamanaka S, Poksay KS, Driscoll DM, Innerarity TL. 1996. Hyperediting of multiple cytidines of apolipoprotein B mRNA by APOBEC-1 requires auxiliary protein(s) but not a mooring sequence motif. J Biol Chem 271: 11506–11510. 10.1074/jbc.271.19.11506 [DOI] [PubMed] [Google Scholar]