

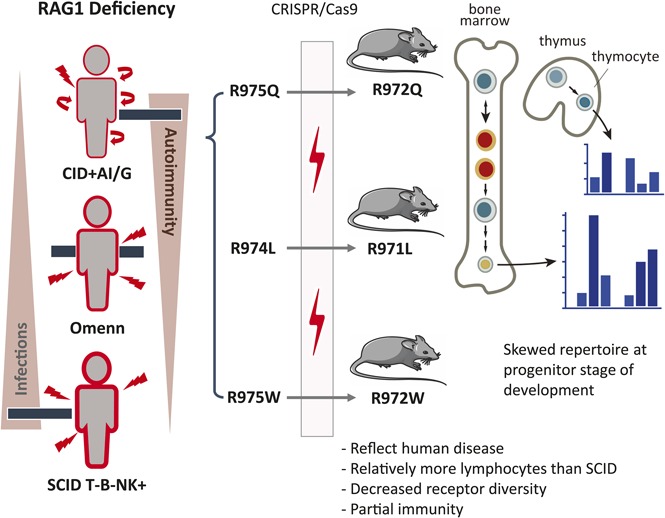
Key Points
Mice with hypomorphic mutations in the Rag1 C-terminal domain are a model of leaky combined immunodeficiency with autoantibodies.
Hypomorphic C-terminal domain Rag1 mutations cause repertoire skewing at the earliest stages of B- and T-cell development.
Abstract
Hypomorphic RAG1 mutations allowing residual T- and B-cell development have been found in patients presenting with delayed-onset combined immune deficiency with granulomas and/or autoimmunity (CID-G/AI) and abnormalities of the peripheral T- and B-cell repertoire. To examine how hypomorphic Rag1 mutations affect the earliest stages of lymphocyte development, we used CRISPR/Cas9 to generate mouse models with mutations equivalent to those found in patients with CID-G/AI. Immunological characterization showed partial development of T and B lymphocytes, with persistence of naïve cells and preserved serum immunoglobulin but impaired antibody responses and presence of autoantibodies, thereby recapitulating the phenotype seen in patients with CID-G/AI. By using high-throughput sequencing, we identified marked skewing of Igh V and Trb V gene usage in early progenitors, with a bias for productive Igh and Trb rearrangements after selection occurred and increased apoptosis of B-cell progenitors. Rearrangement at the Igk locus was impaired, and polyreactive immunoglobulin M antibodies were detected. This study provides novel insights into how hypomorphic Rag1 mutations alter the primary repertoire of T and B cells, setting the stage for immune dysregulation frequently seen in patients.
Visual Abstract
Introduction
Adaptive immunity relies on the dynamic response of lymphocytes to generate specific antigen receptors to fight pathogens. Recombination activation gene 1 (RAG1) and RAG2 are crucial for effective combinatorial joining of variable (V), diversity (D), and joining (J) genes that encode the antigen-binding regions of T-cell receptor (TCR) and B-cell immunoglobulin molecules.1 More than 200 disease-causing mutations in the RAG1 and RAG2 genes have been identified that can cause a wide spectrum of clinical and immunological phenotypes.2 In particular, functionally null RAG mutations cause a complete arrest of T- and B-cell development, resulting in T− B− severe combined immunodeficiency.3-5 Hypomorphic mutations allowing minimal residual function of RAG can lead to Omenn syndrome, with presence of a variable number of activated, oligoclonal T cells that infiltrate and damage target tissues.6 By contrast, hypomorphic RAG mutations with higher residual activity have been identified in patients with delayed-onset combined immunodeficiency associated with granulomas and/or autoimmunity (CID-G/AI).7
A significant proportion of patients with CID-G/AI carry missense mutations in the coding flank–sensitive region of the carboxy-terminal domain (CTD) of RAG1 (human amino acid 892-977; mouse amino acid 889-974; supplemental Figure 1A, available on the Blood Web site). These mutations have been postulated to favor targeting of certain coding elements.8 Although abnormalities of the peripheral T- and B-cell repertoire have been observed in patients with CID-G/AI and Rag1-mutant mice,9,10 it is unknown whether similar abnormalities are present in the preimmune repertoire of progenitor lymphocytes or whether they are secondary to antigen-mediated pressure in the periphery. We used gene editing to generate 3 mouse models carrying homozygous Rag1 mutations (F971L, R972Q, and R972W), corresponding to human mutations (F974L, R975Q, and R975W) described in patients with CID-G/AI,7,11-13 to understand how these mutations affect repertoire composition, cell selection, and survival during T- and B-cell development.
Methods
Mice
Rag1-mutant mice were generated by gene editing as previously described.14,15 For the analysis of regulatory T cells (Treg) and conventional T cells (Tconv), Rag1-mutant mice were crossed with Foxp3.EGFPCre mice. Animal work was conducted in specific pathogen–free conditions, in accordance with the US Public Health Service Policy on Humane Care and Use of Laboratory Animals, with protocols approved by Boston Children’s Hospital (protocols 13-08-2472R and 16-05-3176R) and by the National Institute of Allergy and Infectious Diseases Animal Care and Use Committee (protocol LCIM 16E).
Flow cytometry, cell isolation, and histochemistry
Detailed methods for flow cytometry and cell isolation are provided in the supplemental Methods. Immunohistochemistry of thymus and spleen was performed as previously described.16,17
Measurement of serum BAFF, immunoglobulin levels, specific antibody levels, and autoantibodies
BAFF, immunoglobulin, and specific antibody serum levels were measured by enzyme-linked immunosorbent assay as described in the supplemental Methods. Serum immunoglobulin M (IgM) autoantibodies and anticytokine antibodies were measured using microarrays, as described in the supplemental Methods.
T- and B-cell repertoire analysis
High-throughput sequencing of TCR β (Trb) and immunoglobulin heavy chain (Igh) rearrangements was performed as described in the supplemental Methods. Analysis of Vκ-Jκ rearrangements in pre-B cells was performed by polymerase chain reaction amplification as described.18
Statistical analysis
One-way analysis of variance with multiple comparisons was used when comparing wild-type (WT) with Rag1-mutant mice or comparing different Rag1-mutant mice. Two-way analysis of variance was used to compare antibody responses in WT and mutant mice. The Mann-Whitney U test was used when only 2 groups of mice were compared. Distribution of V and J gene usage was compared using the Kolmogorov-Smirnov test. Individual V and J gene usage was analyzed by the χ2 test.
Results
Generation of mice with targeted mutations in RAG1 CTD
We selected 3 mutations (F971L, R972Q, and R972W) corresponding to human mutations (F974L, R975Q, and R975W) that have been previously described in patients with CID-G/AI. All 3 fall in the coding flank–sensitive region of RAG1 CTD8 (supplemental Figure 1A). Crystallography predicted that the R972 residue located near the catalytic amino acid E962 (supplemental Figure 1B) may participate in the recognition sequence specificity of the DNA coding flank that is directly adjacent to the recombination signal sequence.19 On the basis of amino acid properties and in vitro studies,10 we predicted that the R972Q and the F971L mutations would have a moderate effect on RAG1 protein stability. To extend our analyses, we included a mutation (R972W) that protein structure and in vitro activity predicted to be highly disruptive.7
Incomplete block of T- and B-cell development in Rag1-mutant mice
All 3 Rag1-mutant mouse models showed abnormalities of thymic architecture that were most prominent in R972W mice, with a marked decrease of the thymic medulla but partial preservation of the corticomedullary demarcation (Figure 1A). The total number of thymocytes was reduced 50- to 100-fold in all 3 models (Figure 1B). Flow cytometric analysis revealed a near-complete block of thymocyte development at CD4−CD8− DN stage in R972W mice, similar to what was observed in Rag1−/− (knockout) mice, whereas a significant fraction of CD4+CD8+ DP cells and CD4+CD8− and CD4−CD8+ single-positive cells were detected in R972Q and F971L mice (Figure 1C; supplemental Figure 2A). In all mutant mice, DN cells were largely blocked at CD25+CD44− DN3 stage (Figure 1D; supplemental Figure 2B). An increase in the relative proportion of γδT cells was noted in the 2 leakiest models (R972Q and F971L; Figure 1E; supplemental Figure 2C).
Figure 1.

Thymic T-cell development in Rag1-mutant mice. (A) Tissue sections of thymus stained with hematoxylin and eosin for medulla (lighter staining) and cortex (darker staining). Original magnification ×4. (B) Live cell counts from individual thymuses of WT, hypomorphic Rag1-mutant, and Rag1 knockout (KO) mice. Thymocyte developmental stages were analyzed by flow cytometry for double-negative (DN; CD4−CD8−) cells, double-positive (DP; CD4+CD8+) cells, and single-positive (CD4+ or CD8+) cells (C); lineage-negative DN populations, DN1 (CD44+CD25−), DN2 (CD44+CD25+), DN3 (CD44−CD25+), and DN4 (CD44−CD25−) (D), and thymocytes expressing the αβ or γδ form of the TCR (E). Representative flow cytometry panels with 6 thymuses per group (open circles). Error bars represent standard error of the mean. Statistical analysis was performed with 1-way analysis of variance. *P ≤ .05, **P ≤ .01, ***P ≤ .001, ****P ≤ .0001.
In the bone marrow, a significant increase in the proportion of B220loIgM−CD43+ cells (pro-B cells and pre-BI, here collectively called pro-B) was seen in Rag1-mutant mice as compared with WT mice (Figure 2A-B). By contrast, the proportion and absolute number of B220loIgM−CD43− cells (small pre-B, including pre-BII), B220loIgM+CD43− cells (immature), and B220hiIgM+CD43− cells (mature recirculating B cells) were all decreased in Rag1-mutant mice (Figure 2A-C). This block in B-cell development was less severe in R972Q than in F971L and R972W mice. During B-cell development, RAG expression occurs at the pro-B stage for Igh rearrangement and at the pre-BII stage for light chain (LC) rearrangement.20 To characterize these specific transitions, we performed flow cytometric analysis to identify the proportions of B220loc-kit+ pro/pre-BI and of B220loCD25+ pre-BII bone marrow cells. Among B220+ IgM− B-cell precursors, a significant increase in the proportion of pro/pre-BI cells was demonstrated in all 3 hypomorphic Rag1-mutant mouse strains (Figure 2D-E), indicating a major block at the assembly of LC. Although the proportion of B220loCD25+ pre-BII cells was significantly reduced in all 3 Rag1 hypomorphic mutants compared with WT mice, this difference was less severe in R972Q mice (Figure 2E), indicating a more pronounced leakiness of defective lymphocyte development in this model.
Figure 2.

Bone marrow B-cell development in Rag1-mutant mice. (A) Gating strategy for pro-B cells (B220loIgM−CD43+), pre-B cells (B220loIgM−CD43−), immature B cells (B220loIgM+CD43−), and mature recirculating (B220hiIgM+CD43−) cells, shown as percentages of total B220+ cells (B) and absolute numbers (n = 6) (C) for WT, hypomorphic mutant, and KO mice. Gating strategy for pro-pre-BI stage (B220loc-kit+) and pre-BII stage (B220loCD25+) (D) and percentage of these cells among B220+IgM− B-cell precursors (n = 3) (E). Error bars represent standard error of the mean. (B-C,E) One-way analysis of variance: *P ≤ .05, **P ≤ .01, ***P ≤ .001, ****P ≤ .0001.
Rag1-mutant mice have a reduced number of mature T and B lymphocytes in the periphery
In the spleen, the number of CD3+, CD4+, and CD8+ cells was diminished in all 3 Rag1-mutant models, and this defect was more pronounced in F971L and R972W mice (Figure 3A-B). In T-cell lymphopenic hosts, compensatory homeostatic proliferation can result in an increased proportion of peripheral T cells with an activated phenotype and virtual absence of peripheral naïve T cells.9,21,22 Although all 3 mutants showed depletion of naïve T cells, a significant fraction of CD62L+CD44− naïve cells was present in both F971L and R972Q mice (Figure 3C-D). As compared with WT mice, the absolute number of B-cell splenocytes was reduced in all Rag1-mutant models and especially in R972W and F971L compared with R972Q mice (Figure 4A). The number and size of B-cell follicles in the spleen correlated with the degree of B-cell lymphopenia (Figure 4B). Analysis of the proportion and absolute number of follicular (CD19+CD93−CD21+CD23+) and MZ; (CD19+CD93−CD21+CD23−) B cells showed that B-cell depletion was more prominent for follicular than MZ B cells and more severe in F971L than in R972Q mice (Figure 4C).
Figure 3.

Distribution and phenotype of splenic T-cell subsets in Rag1-mutant mice. (A) Representative example of CD4+ and CD8+ T cells (top) and of CD4−CD8− cells expressing the γδ form of the TCR (bottom) among splenic CD3+ cells. Percentages (among CD3+ cells) (B) and absolute count of CD4+ and CD8+ cells (C). (C) Gating strategy for naïve (CD62L+CD44−), central memory (CD44+CD62L+), and effector memory T cells (CD44+CD62L−) among CD4+ (top) and CD8+ (bottom) T cells. (D) Distribution of naïve, central memory, and effector memory T cells among total splenic CD4+ (top) and CD8+ (bottom) cells (n = 6 per group). Error bars represent standard error of the mean. (B,D) One-way analysis of variance: *P ≤ .05, **P ≤ .01, ***P ≤ .001, ****P ≤ .0001.
Figure 4.

Distribution and phenotype of splenic B cells and humoral immunity in Rag1-mutant mice. (A) Representative examples (left) and absolute count (right) of mature CD19/IgM+ splenic B cells (error bars represent standard error of the mean [SEM]). (B) Hematoxylin and eosin–stained sections of spleen counterstained for B220 and CD3 concentrated within follicles (dark staining). Original magnification ×4. (C) Spleens were analyzed by flow cytometry for follicular (CD19+CD93−CD21+CD23+) and marginal zone (MZ) B cells (CD19+CD93−CD21+CD23−) with percentage and absolute number of CD19+ cells shown on the right. (D) Immunoglobulin serum concentration (mean ± SEM) in naïve mice (n = 6-10 per group). Mice were immunized with TNP-Ficoll, and TNP-specific IgM (E) and IgG3 (F) were measured by enzyme-linked immunosorbent assay (ELISA; n = 4). (G) Mice were immunized with T-dependent antigen (TNP-KLH), and TNP-specific IgG was measured by ELISA (n = 4). (H) Th17 responses to immunization with Pneumococcal whole-cell antigen (WCA) antigen were measured by antigen-specific total IgG. One-way (A,C-D) and 2-way (E-H) analysis of variance: *P ≤ .05, **P ≤ .01, ***P ≤ .001, ****P ≤ .0001. OD, optical density.
Dysregulation of humoral immunity in Rag1-mutant mice
In contrast to patients with Omenn syndrome, who are profoundly hypogammaglobulinemic but show increased levels of serum IgE, variable levels of serum IgG and IgM and of antigen-specific antibody responses have been reported in patients with CID-G/AI.11,13,23 R972Q mice had normal levels of IgG with increased serum IgM; increased serum IgE was detected in R972W mice (Figure 4D). Naïve Rag1-mutant mice had higher serum titers of TNP-binding IgM (Figure 4E). Upon immunization with the T-independent antigen TNP-Ficoll, equivalent amounts of anti-TNP IgM and IgG3 antibodies were detected in WT, R972Q, and F971L mice, whereas a defective response was observed in R972W mutants (Figure 4E-F). The antibody response to the T-dependent antigen TNP-KLH was impaired in all 3 Rag1-mutant mice, although it was greatest for R972W mice (Figure 4G). Furthermore, R972Q mice mounted an impaired antibody response to the Th17-dependent pneumococcal whole-cell antigen (Figure 4H). Altogether, these data indicate that antibody responses to T-independent responses, potentially stemming from the demonstrable MZ B-cell population, are preserved in R972Q and F971L mice, but their T-dependent antibody responses are compromised, despite their relative leakiness of T- and B-cell development.
One of the characteristics of patients with hypomorphic mutations in the RAG1 CTD domain is the frequent association with autoimmune disease.24 We hypothesized that increased levels of TNP-binding IgM antibodies in Rag1-mutant mice might reflect the presence of low-affinity, polyreactive antibodies. High levels of antiphosphorylcholine (PC) antibody were detected in the serum of 8- to 12-week-old R972Q-mutant mice (Figure 5A). Because B1 B cells are thought to be a principal source of anti-PC antibodies,25 we characterized the peritoneal compartment and observed a significant reduction of B1a cells in R972Q and F971L mice, whereas the B1b compartment was not significantly compromised (Figure 5B). These results suggest that elevated levels of anti-PC antibodies in R972Q mice cannot be attributed to expansion of B1a cells. When probing a proteomic microarray that contained a panel of 95 common self-antigens, IgM autoantibodies were detected in the serum of all 3 mutant strains, most abundantly in the leakiest mutant, R972Q (Figure 5C). Because the proteomic microarray was normalized for total IgM concentration, the increase seen in R972Q mice was not simply a result of the higher IgM levels in these mice. Specificity of the autoantibody repertoire was further interrogated for anticytokine antibodies, which have been seen in patients with CID-G/AI24; however, no distinctive anticytokine antibody signature was observed in Rag1 hypomorphic mutant mice (supplemental Figure 3). We then looked for mechanisms that could account for autoantibody formation both centrally and in the periphery. RAG-mediated receptor editing and revision are important checkpoints of central B-cell tolerance to prevent autoimmunity.26,27 This process results in further κ, and eventually λ, LC rearrangement. The proportion of Igλ expressing splenic B cells was significantly decreased in all 3 mutant strains, indicating impairment of receptor editing (Figure 5D). Elevated BAFF levels, which may allow rescue of self-reactive immature B cells,28,29 were found in all 3 models and correlated with the severity of B-cell lymphopenia (Figure 5E). Finally, R972Q mice had an increased proportion of splenic age-associated B cells, which express CD11c and T-bet and have been reported to contribute to autoantibody production30 (Figure 5F). Altogether, these data suggest that multiple checkpoints of B-cell tolerance are affected in Rag1-mutant mice.
Figure 5.

Serum autoantibodies and mechanisms of altered B-cell tolerance in Rag1-mutant mice. (A) Anti-PC IgM measured by enzyme-linked immunosorbent assay (ELISA). (B) Peritoneal lavage was analyzed by flow cytometry for B1 (CD19+CD11b+) and B2 (CD19+CD11b−) cells and B1a (CD19+CD11b+CD5+) and B1b (CD19+CD11b+CD5−) cells. (C) Heat map of serum IgM binding to a protein autoantigen microarray (n = 7-8 mice per strain), normalized to WT reactivity. (D) Percentage of IgM+ B cells in spleen expressing Igλ (n = 4-7 per group). (E) BAFF serum concentrations measured by ELISA (n = 4 per group). (F) Age-associated B cells (ABCs) (Tbet+CD11c+) as percentage of CD19+ cells in spleen (n = 7-8 per group). Error bars represent standard error of the mean. One-way analysis of variance (A-B,D-E) and Mann-Whitney U test (F): *P ≤ .05, **P ≤ .01, ***P ≤ .001, ****P ≤ .0001.
Abnormalities of B-cell repertoire are detected at early stages of B-cell development
To investigate how Rag1 mutations affect composition of the B-cell repertoire, we adopted a protocol using specific primers for each of the 4 J genes to detect in an unbiased manner VDJ and DJ rearrangements, as well as productive and nonproductive Igh recombination products.18,31 First, we analyzed B220+IgM−CD43+ bone marrow B cells, a population containing pro- and pre-BI cells, collectively termed pro-B cells18,31 and in which both DJ and VDJ rearrangements have occurred. At this stage, if neither allele undergoes productive VDJ rearrangement, the cell undergoes apoptosis.20 Although equal numbers of pro-B cells and DNA input were sequenced, too few Igh rearrangements were detected in R972W mice, preventing further analysis. We recovered significantly fewer DJ and VDJ rearrangement products in Rag1 R972Q and F971L than in WT mice. To take this into account, absolute numbers instead of percentages were used to test for statistical differences (supplemental Tables 2 and 3). DJ rearrangements accounted for a higher percentage of total rearrangements in pro-B cells from R972Q and F971L mice than in WT mice (Figure 6A), consistent with decreased Rag1 activity and corresponding impaired sequential recombination.
Figure 6.

Abnormalities of Igh repertoire in Rag1-mutant mice. (A) Bar graph of Igh VDJ (green) and DJ (yellow) rearrangements as a percentage of total rearrangements recovered in pro-B cells and IgM+ splenocytes. (B) Bar graph of productive (blue) vs nonproductive (red) reads of total unique Igh VDJ rearrangements in pro-B cells, pre-B cells, and IgM+ splenic B cells. (C) Fivefold dilution of input DNA was polymerase chain reaction amplified for Vκ-Jκ LC rearrangements in pre-B cells with loading control (Dlg5). (D) Apoptotic B cells through development were analyzed by flow cytometry using PI and annexin. (E) Frequency of D gene utilization of Igh DJ rearrangements in pro-B cells. (F) Frequency of V gene utilization in unique Igh VDJ rearrangements in pro-B cells. Top shows WT mice; bottom shows R972Q mice. (G) Frequency of V gene utilization in unique Igh VDJ rearrangements in pro-B cells. Top shows WT mice (data as in panel F); bottom shows F971L mice. (H) Utilization of V genes in unique Igh VDJ rearrangements in splenic IgM+ B cells. Top shows WT mice; bottom shows R972Q mice. Mann-Whitney U test (D) and χ2 test (comparing individual V gene usage) (F-G): *P ≤ .05, ****P ≤ .0001. imm, immature; mat, mature.
In the periphery, 40% of mature B cells are expected to harbor VDJ rearrangements on both alleles (only 1 productive), and 60% are anticipated to carry a productive VDJ rearrangement on 1 allele and a DJ rearrangement on the other allele.20 Consistent with this, DJ rearrangements accounted for 31% of total rearrangements in splenic B cells from WT mice. However, DJ rearrangements represented <5% of rearrangements in splenic B cells from R972Q mice (Figure 6A). The low proportion of DJ rearrangements in mutant splenic B cells and the lower absolute number of Igh DJ and VDJ rearrangements in pro-B cells from mutant mice suggest that 1 allele remains in germ line configuration. This implies that productive VDJ rearrangement must occur on the other allele for the cell to survive. To test this hypothesis, we evaluated the proportion of productive and nonproductive Igh VDJ rearrangements at different stages of B-cell development. Unique sequences from pro-B cells revealed a similar frequency of nonproductive Igh rearrangements in Rag1-mutant and in WT mice (Figure 6B). By contrast, the proportion of nonproductive rearrangements in pre-B cells was significantly lower in R972Q and F971L mice (6.5% and 6.8%, respectively) than in WT mice (27%), a pattern that was maintained in splenic B cells (Figure 6B). To further examine the efficiency of hypomorphic RAG-mediated recombination in vivo, we assayed for Igκ gene rearrangements in pre-B cells using a semiquantitative polymerase chain reaction strategy20 and observed a fivefold reduction of Vκ-Jκ products in R972Q when compared with WT mice (Figure 6C).
Reduced capacity of hypomorphic Rag1 mutations to develop productive V(D)J rearrangements on at least 1 allele for both the Igh and Igk loci and ineffective pairing of the 2 could result in increased cell death. To test this hypothesis, we stained bone marrow B220loIgM−CD43+ (pro-B cells), B220loIgM−CD43− cells (pre-B cells that have rearranged the LC and include pre-BII cells), immature B cells (B220loIgM+CD43−), and mature recirculating B cells (B220hiIgM+CD43−) from WT and R972Q mice. A significantly higher proportion of apoptotic cells was detected in pre-B and immature B cells from Rag1-mutant mice, with an increased trend seen for pro-B cells (Figure 6D).
Subsequently, we analyzed individual V and D gene usage at distinct stages of B lymphocyte development. Analysis of D gene usage in DJ rearrangements from sorted pro-B cells did not reveal any differences between WT and R972Q or F971L mice (Figure 6E). V gene usage is tightly regulated during early B-cell development and requires the interplay of transcription factors to alter accessibility of the Igh locus.32,33 The overall distribution of IghV gene usage among total unique VDJ sequences was significantly different between pro-B cells of WT and Rag1-mutant mice (P = 1.99e-12 for R972Q; P = 4.27e-22 for F971L; Figure 6F-G; supplemental Table 3). In particular, Rag1-mutant pro-B cells revealed fewer rearrangements to the normally highly used proximal VH2 and VH5 families, including the very proximal VH5-2.18,31 The 10 most proximal V genes were involved in 14.3% of total unique VDJ rearrangements in WT pro-B cells, compared with 4.6% in R972Q pro-B cells and 4.5% in F971L pro-B cells (Figure 6F-G; supplemental Table 4). Conversely, there was a trend for more frequent usage of distal IghV genes in Rag1-mutant pro-B cells (Figure 6F). These findings indicate that the V gene repertoire is significantly skewed at the pro–B-cell stage, independent of classical selection after LC rearrangement. Similar trends in proximal and distal Igh V gene usage between WT and mutant mice were observed in pre-B cells (supplemental Figure 4A-B), splenic B cells (Figure 6H), and peritoneal B1 cell (supplemental Figure 5), although less prominent than at the pro–B-cell level. Overall distribution of V genes was significantly different for all B-cell populations between WT and mutants. The difference was more significant for pro-B cells than for the more mature B-cell populations and more significant for F971L than R972Q mice (supplemental Table 3). No differences were observed in CDR3 length or P or N nucleotide modifications between WT and R972Q pro-B and mature B cells (supplemental Figure 4C).
Finally, despite altered usage of IghV genes, we observed that B-cell splenocytes from R972Q mice had a diversified repertoire, as indicated by the D50 index, a measure for clonotypic expansion corresponding to the percentage of unique CDR3 sequences that account for 50% of the total number of sequences observed10 (supplemental Figure 4D).
T-cell repertoire of Rag1 hypomorphic mutant mice is skewed at early stages of V(D)J recombination
Next-generation sequencing of the Trb repertoire revealed many similarities to what was observed at the Igh locus during B-cell development. In particular, the proportion of nonproductive Trb rearrangements was similar in DN4 cells of WT and mutant mice, but an increased proportion of productive rearrangements was detected in DP and peripheral T cells from R972Q- and F971L-mutant compared with WT mice (Figure 7A). Furthermore, important differences in Trb V and J gene usage were detected in thymocytes from Rag1-mutant and WT mice (supplemental Figure 6A-B), including DN4 thymocytes (P = 3.340e-08; Figure 7B; supplemental Tables 3 and 5), indicating that they had occurred before selection. The same pattern was documented in DP thymocytes (Figure 7C-D; supplemental Table 6), splenic CD4+ Tconv (Figure 7E-F; supplemental Tables 5 and 6), and Treg (Figure 7G-H; supplemental Tables 5 and 6). In the periphery, a mildly reduced diversity of the T-cell repertoire was observed in both Tconv and Treg from R972Q and F971L mice as shown by modestly lower Shannon entropy index (supplemental Figure 6C); however, the capacity of Treg to suppress proliferation of T effector cells in response to polyclonal mitogenic stimulation was preserved (supplemental Figure 7).
Figure 7.

Abnormalities of Trb repertoire in Rag1-mutant mice. (A) Percentage of productive and nonproductive reads of total unique Trb joins from WT and R972Q DN4 thymocytes (CD4−CD8−CD44−CD25−); WT, R972Q, and F971L CD4+CD8+ DP thymocytes; WT, R972Q, and F971L Tconv (CD4+EGFP−) cells; and WT, R972Q, and F971L Treg (CD4+EGFP+) cells. (B) Utilization of V (left) and J (right) genes in Trb rearrangements in DN4 thymocytes (CD4−CD8−CD44−CD25−). Top shows WT mice; bottom shows R972Q mice. (C) Trb unique sequences in CD4+CD8+ DP thymocytes. Top shows WT mice; bottom shows R972Q mice. (D) Trb unique sequences in CD4+CD8+ DP thymocytes. Top shows WT mice; bottom shows F971L mice. (E) Trb unique sequences in splenic Tconv (CD4+EGFP−) cells from WT (top) and R972Q (bottom) mice. (F) Trb unique sequences in splenic Tconv (CD4+EGFP−) cells from WT (top) and F971L (bottom) mice. (G) Utilization of V (left) and J (right) genes in Trb unique sequences in splenic Treg (CD4+EGFP+) cells from WT (top) and R972Q (bottom) mice. (H) Utilization of V (left) and J (right) genes in Trb unique sequences in splenic Treg (CD4+EGFP+) cells from WT (top) and F971L (bottom) mice. χ2 test: *P ≤ .05, ***P ≤ .001, ****P ≤ .0001.
Discussion
We have generated mouse models with missense mutations in the RAG1 CTD corresponding to those identified in patients with CID-G/AI and shown that 2 models (R972Q and F971L mice) mimic the human phenotype, with partial preservation of T- and B-cell development and immunoglobulin levels but abnormalities of T- and B-cell repertoire and autoantibody production. Alterations of T- and B-cell development did not affect all lymphocyte subsets equally. Generation of TCRγδ+ T cells was relatively preserved, perhaps because of the distinct developmental origin, which requires fewer RAG-dependent checkpoints.34 In the spleen, partial preservation of MZ B-cell development was observed in R972Q and F971L mice, whereas follicular B cells were severely depleted. The B-cell compartment seems most vulnerable to impaired RAG1 activity, with proportionately more depletion in F971L compared with R972Q mice. Similar findings in RAG1-deficient patients have been observed, where B-cell defects appear in greater magnitude than those in T cells.2 Additional studies of the constraints of RAG1 activity at the Igh versus the TCR locus may uncover mechanisms for this difference in severity of lymphoid development.
This study has uncovered novel effects of hypomorphic Rag1 mutations on the composition of the preimmune repertoire, cell survival, and selection at early stages of T and B lymphocytes. In particular, we observed a strong bias for productive V(D)J rearrangements in peripheral B and T cells from Rag1-mutant mice, previously recognized only in peripheral B cells of patients with more severe hypomorphic RAG mutations.35,36 Before LC rearrangement, both DJ and VDJ rearrangements are detectable in pro-B cells, but only productive VDJ rearrangements subsequently pair with the λ5/V pre–B-cell surrogate LC complex enabling selection.37 We saw no significant bias for productive versus nonproductive rearrangements at the pro–B-cell stage; however, a significantly higher proportion of productive VDJ rearrangements were detected in pre-B cells from Rag1-mutant than WT mice. Our data are consistent with a model where in Rag1-mutant mice, fewer alleles accomplish DJ rearrangement, and only those cells with inframe VDJ rearrangements subsequently progress further along B-cell development, with a high proportion of cells maintaining the second allele in germ line configuration. A similar selective pressure was observed in T-cell development, with no significant bias for productive Trb VDJ rearrangements at the DN4 stage of T-cell development, before selection and pairing with TCRα,34 but an increased proportion of productive rearrangements in DP thymocytes (where pairing with TCRα enables positive selection) and in the periphery. These findings indicate that despite the relatively leaky block in lymphoid development observed in R972Q and F971L mice, a significant impairment of gene rearrangement at the Igh, Igκ, and Trb loci limits antigen receptor diversity from a very early stage in development.
Signaling at the pre-BI and pre-BII stages represents an important checkpoint during B-cell development. Failure to correctly signal via the newly generated antigen receptor results in cell death, usually at the immature B-cell stage of development, where LC rearrangement can potentially rescue mispaired or autoimmune-prone B cells.1,27 Correspondingly, Rag1-mutant mice manifested increased apoptosis that was more prominent in immature B cells, with a reduced proportion of Igλ+ B cells in the spleen. However, enhanced apoptosis was also observed earlier in development (pre-B cells), potentially because of failed surrogate LC pairing or failed LC rearrangement. These findings suggest that RAG activity unexpectedly dictates survival very early in lymphocyte development, possibly by constraining the timeframe that is responsive to a productive recombination.
Pro-B cells from Rag1-mutant mice displayed a significantly decreased usage of proximal IghV genes. This is in marked contrast to what has been previously described in normal development, where proximal V genes are preferentially rearranged.31,38 Targeting of the Igh locus is regulated by several transcription factors that regulate pro– to pre–B-cell transition and recombination of the distal IghV genes and by cohesin and CTCF factors that affect V gene usage by controlling proximal to distal interactions.18,33,39-42 Impaired RAG activity may alter the timing dynamics and magnify the regulation of recombination by CTCF versus the transcription factors.40,43
Using next-generation sequencing, we demonstrated that the skewing of Trb repertoire previously reported in the periphery of R972Q mice9 is already present at the DN4 stage. Remarkably, similar abnormalities of Trb V and J gene usage were observed in F971L and R972Q mice. One of the proposed functions of the CTD of RAG1 is to ensure recognition of specific antigen receptor genes, with in vitro data suggesting that this region interacts with the terminal 2 base pair coding ends of V, D, and J genes.8,44,45 However, no difference in D gene usage in DJ rearrangements was observed in pro-B cells of mutant mice, and no differences in the composition of the 2 base pair coding ends were detected when comparing IghV and TrbV genes that were differentially used in mutant mice versus WT mice (supplemental Tables 7 and 8).
Finally, Rag1-mutant mice displayed signs of immune dysregulation, with the presence of broad-spectrum autoantibodies, recapitulating similar features observed in patients with CID-G/AI.24 This phenotype was especially evident in the leakiest model (R972Q mouse), where anti-PC antibodies were detected at higher titer. An important difference between patients with CID-G/AI and the mouse models described here is that the former have a characteristic anticytokine antibody signature (predominantly directed against type 1 interferon),24 especially in patients with a history of severe viral infections, whereas such an anticytokine signature was not observed in Rag1-mutant mice. Future studies may help address whether infectious triggers elicit anticytokine antibodies in Rag1-mutant mice.
Various mechanisms may contribute to immune dysregulation both in patients and in mice with hypomorphic RAG mutations. In particular, we have confirmed previous findings of altered receptor editing.17,46 Abnormalities in the composition of the preimmune repertoire may affect subsequent survival and selection of T and B cells. In the periphery, relative expansion of age-associated B cells and increased serum BAFF levels may also lead to breakage of B-cell tolerance. Furthermore, although Treg function was apparently preserved, as measured by suppression of proliferation of T effector cells in response to polyclonal stimulation via CD3/CD28/CD2, abnormalities of Treg repertoire in Rag1-mutant mice and patients47 may limit their capacity to effectively suppress immune responses against defined sets of self-antigens.
Supplementary Material
The online version of this article contains a data supplement.
Acknowledgments
The authors thank K. Musunuru for his guidance in designing the gRNA to generate the Rag1-mutant mice and W. Yang and M. Gellert for the structural prediction model of the RAG mutants (supplemental Figure 1); F. W. Alt for providing the HTGTS-Rep-seq platform and L. Charbonnier for help with T regulatory studies; and M. L. Boes for reading the manuscript and discussing the study.
This work was supported by the National Institutes of Health, National Institute of Allergy and Infectious Diseases grant #R01 AI100887 (J.P.M.) and by the Division of Intramural Research, National Institute of Allergy and Infectious Diseases, National Institutes of Health.
The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does the mention of trade names, commercial products, or organizations imply endorsement by the US Government.
Footnotes
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: L.M.O.d.B. generated the mice, performed the experiments, analyzed the data, and wrote the manuscript; M.B. and A.B. performed phenotypic characterization of mice; M.B., S.G.L., J.H.R., and K.C. contributed to the high-throughput sequencing of T- and B-cell repertoire; P.L.P. performed immunohistochemistry of thymus and spleen; D.E., N.L., and O.K. performed analysis of anticytokine antibodies; and J.P.M. and L.D.N. supervised the project and the writing of the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: L. D. Notarangelo, Laboratory of Clinical Immunology and Microbiology, National Institute of Allergy and Infectious Diseases, National Institutes of Health, Building 10 CRC–Room 5-3950, 10 Center Dr, Bethesda, MD 20892; e-mail: luigi.notarangelo2@nih.gov; and J. P. Manis, Harvard Medical School, Joint Program in Transfusion Medicine, Department of Laboratory Medicine, Boston Children’s Hospital, 320 Longwood Ave, Enders Research Laboratories 809, Boston, MA 02115; e-mail: manis@enders.tch.harvard.edu.
References
- 1.Schatz DG, Swanson PC. V(D)J recombination: mechanisms of initiation. Annu Rev Genet. 2011;45:167-202. [DOI] [PubMed] [Google Scholar]
- 2.Notarangelo LD, Kim MS, Walter JE, Lee YN. Human RAG mutations: biochemistry and clinical implications. Nat Rev Immunol. 2016;16(4):234-246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mombaerts P, Iacomini J, Johnson RS, Herrup K, Tonegawa S, Papaioannou VE. RAG-1-deficient mice have no mature B and T lymphocytes. Cell. 1992;68(5):869-877. [DOI] [PubMed] [Google Scholar]
- 4.Schwarz K, Gauss GH, Ludwig L, et al. RAG mutations in human B cell-negative SCID. Science. 1996;274(5284):97-99. [DOI] [PubMed] [Google Scholar]
- 5.Shinkai Y, Rathbun G, Lam KP, et al. RAG-2-deficient mice lack mature lymphocytes owing to inability to initiate V(D)J rearrangement. Cell. 1992;68(5):855-867. [DOI] [PubMed] [Google Scholar]
- 6.Villa A, Santagata S, Bozzi F, et al. Partial V(D)J recombination activity leads to Omenn syndrome. Cell. 1998;93(5):885-896. [DOI] [PubMed] [Google Scholar]
- 7.Schuetz C, Huck K, Gudowius S, et al. An immunodeficiency disease with RAG mutations and granulomas. N Engl J Med. 2008;358(19):2030-2038. [DOI] [PubMed] [Google Scholar]
- 8.Wong SY, Lu CP, Roth DBA. A RAG1 mutation found in Omenn syndrome causes coding flank hypersensitivity: a novel mechanism for antigen receptor repertoire restriction. J Immunol. 2008;181(6):4124-4130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Khiong K, Murakami M, Kitabayashi C, et al. Homeostatically proliferating CD4 T cells are involved in the pathogenesis of an Omenn syndrome murine model. J Clin Invest. 2007;117(5):1270-1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lee YN, Frugoni F, Dobbs K, et al. Characterization of T and B cell repertoire diversity in patients with RAG deficiency. Sci Immunol. 2016;1(6):eaah6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Henderson LA, Frugoni F, Hopkins G, et al. Expanding the spectrum of recombination-activating gene 1 deficiency: a family with early-onset autoimmunity. J Allergy Clin Immunol. 2013;132(4):969-971.e1-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Villa A, Sobacchi C, Notarangelo LD, et al. V(D)J recombination defects in lymphocytes due to RAG mutations: severe immunodeficiency with a spectrum of clinical presentations. Blood. 2001;97(1):81-88. [DOI] [PubMed] [Google Scholar]
- 13.Avila EM, Uzel G, Hsu A, et al. Highly variable clinical phenotypes of hypomorphic RAG1 mutations. Pediatrics. 2010;126(5):e1248-e1252. [DOI] [PubMed] [Google Scholar]
- 14.Ott de Bruin L, Yang W, Capuder K, et al. Rapid generation of novel models of RAG1 deficiency by CRISPR/Cas9-induced mutagenesis in murine zygotes. Oncotarget. 2016;7(11):12962-12974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yang H, Wang H, Jaenisch R. Generating genetically modified mice using CRISPR/Cas-mediated genome engineering. Nat Protoc. 2014;9(8):1956-1968. [DOI] [PubMed] [Google Scholar]
- 16.Rucci F, Poliani PL, Caraffi S, et al. Abnormalities of thymic stroma may contribute to immune dysregulation in murine models of leaky severe combined immunodeficiency. Front Immunol. 2011;2(15):00015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cassani B, Poliani PL, Marrella V, et al. Homeostatic expansion of autoreactive immunoglobulin-secreting cells in the Rag2 mouse model of Omenn syndrome. J Exp Med. 2010;207(7):1525-1540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Guo C, Yoon HS, Franklin A, et al. CTCF-binding elements mediate control of V(D)J recombination. Nature. 2011;477(7365):424-430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kim MS, Lapkouski M, Yang W, Gellert M. Crystal structure of the V(D)J recombinase RAG1-RAG2. Nature. 2015;518(7540):507-511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jung D, Giallourakis C, Mostoslavsky R, Alt FW. Mechanism and control of V(D)J recombination at the immunoglobulin heavy chain locus. Annu Rev Immunol. 2006;24:541-570. [DOI] [PubMed] [Google Scholar]
- 21.Marrella V, Poliani PL, Casati A, et al. A hypomorphic R229Q Rag2 mouse mutant recapitulates human Omenn syndrome. J Clin Invest. 2007;117(5):1260-1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Giblin W, Chatterji M, Westfield G, et al. Leaky severe combined immunodeficiency and aberrant DNA rearrangements due to a hypomorphic RAG1 mutation. Blood. 2009;113(13):2965-2975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.De Ravin SS, Cowen EW, Zarember KA, et al. Hypomorphic Rag mutations can cause destructive midline granulomatous disease. Blood. 2010;116(8):1263-1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Walter JE, Rosen LB, Csomos K, et al. Broad-spectrum antibodies against self-antigens and cytokines in RAG deficiency. J Clin Invest. 2015;125(11):4135-4148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Baumgarth N. The double life of a B-1 cell: self-reactivity selects for protective effector functions. Nat Rev Immunol. 2011;11(1):34-46. [DOI] [PubMed] [Google Scholar]
- 26.Nemazee D. Receptor editing in lymphocyte development and central tolerance. Nat Rev Immunol. 2006;6(10):728-740. [DOI] [PubMed] [Google Scholar]
- 27.Nemazee D. Mechanisms of central tolerance for B cells. Nat Rev Immunol. 2017;17(5):281-294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lesley R, Xu Y, Kalled SL, et al. Reduced competitiveness of autoantigen-engaged B cells due to increased dependence on BAFF. Immunity. 2004;20(4):441-453. [DOI] [PubMed] [Google Scholar]
- 29.Mackay F, Woodcock SA, Lawton P, et al. Mice transgenic for BAFF develop lymphocytic disorders along with autoimmune manifestations. J Exp Med. 1999;190(11):1697-1710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rubtsov AV, Rubtsova K, Fischer A, et al. Toll-like receptor 7 (TLR7)-driven accumulation of a novel CD11c+ B-cell population is important for the development of autoimmunity. Blood. 2011;118(5):1305-1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lin SG, Ba Z, Du Z, Zhang Y, Hu J, Alt FW. Highly sensitive and unbiased approach for elucidating antibody repertoires. Proc Natl Acad Sci USA. 2016;113(28):7846-7851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Teng G, Schatz DG. Regulation and evolution of the RAG recombinase. Adv Immunol. 2015;128:1-39. [DOI] [PubMed] [Google Scholar]
- 33.Proudhon C, Hao B, Raviram R, Chaumeil J, Skok JA. Long-range regulation of V(D)J recombination. Adv Immunol. 2015;128:123-182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Krangel MS. Mechanics of T cell receptor gene rearrangement. Curr Opin Immunol. 2009;21(2):133-139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ohm-Laursen L, Nielsen C, Fisker N, Lillevang ST, Barington T. Lack of nonfunctional B-cell receptor rearrangements in a patient with normal B cell numbers despite partial RAG1 deficiency and atypical SCID/Omenn syndrome. J Clin Immunol. 2008;28(5):588-592. [DOI] [PubMed] [Google Scholar]
- 36.IJspeert H, Driessen GJ, Moorhouse MJ, et al. Similar recombination-activating gene (RAG) mutations result in similar immunobiological effects but in different clinical phenotypes. J Allergy Clin Immunol. 2014;133(4):1124-1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Melchers F. The pre-B-cell receptor: selector of fitting immunoglobulin heavy chains for the B-cell repertoire. Nat Rev Immunol. 2005;5(7):578-584. [DOI] [PubMed] [Google Scholar]
- 38.Yancopoulos GD, Desiderio SV, Paskind M, Kearney JF, Baltimore D, Alt FW. Preferential utilization of the most JH-proximal VH gene segments in pre-B-cell lines. Nature. 1984;311(5988):727-733. [DOI] [PubMed] [Google Scholar]
- 39.Bertolino E, Reddy K, Medina KL, Parganas E, Ihle J, Singh H. Regulation of interleukin 7-dependent immunoglobulin heavy-chain variable gene rearrangements by transcription factor STAT5. Nat Immunol. 2005;6(8):836-843. [DOI] [PubMed] [Google Scholar]
- 40.Ebert A, Hill L, Busslinger M. Spatial regulation of V-(D)J recombination at antigen receptor loci. Adv Immunol. 2015;128:93-121. [DOI] [PubMed] [Google Scholar]
- 41.Busslinger M. Transcriptional control of early B cell development. Annu Rev Immunol. 2004;22:55-79. [DOI] [PubMed] [Google Scholar]
- 42.Reynaud D, Demarco IA, Reddy KL, et al. Regulation of B cell fate commitment and immunoglobulin heavy-chain gene rearrangements by Ikaros. Nat Immunol. 2008;9(8):927-936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kumari G, Sen R. Chromatin interactions in the control of immunoglobulin heavy chain gene assembly. Adv Immunol. 2015;128:41-92. [DOI] [PubMed] [Google Scholar]
- 44.Gerstein RM, Lieber MR. Coding end sequence can markedly affect the initiation of V(D)J recombination. Genes Dev. 1993;7(7B):1459-1469. [DOI] [PubMed] [Google Scholar]
- 45.Mo X, Bailin T, Sadofsky MJ. A C-terminal region of RAG1 contacts the coding DNA during V(D)J recombination. Mol Cell Biol. 2001;21(6):2038-2047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Walter JE, Rucci F, Patrizi L, et al. Expansion of immunoglobulin-secreting cells and defects in B cell tolerance in Rag-dependent immunodeficiency. J Exp Med. 2010;207(7):1541-1554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rowe JH, Stadinski BD, Henderson LA, et al. Abnormalities of T-cell receptor repertoire in CD4+ regulatory and conventional T cells in patients with RAG mutations: implications for autoimmunity. J Allergy Clin Immunol. 2017;140(6):1739-1743.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.