

Abstract
Aims
Cardiac β-adrenergic receptor (βAR) signalling is susceptible to heterologous desensitization by different neurohormonal stimuli in clinical conditions associated with heart failure. We aim to examine the underlying mechanism of cross talk between βARs and a set of G-protein coupled receptors (GPCRs) activated by hormones/agonists.
Methods and results
Rat ventricular cardiomyocytes were used to determine heterologous phosphorylation of βARs under a series of GPCR agonists. Activation of Gs-coupled dopamine receptor, adenosine receptor, relaxin receptor and prostaglandin E2 receptor, and Gq-coupled α1 adrenergic receptor and angiotensin II type 1 receptor promotes phosphorylation of β1AR and β2AR at putative protein kinase A (PKA) phosphorylation sites; but activation of Gi-coupled α2 adrenergic receptor and activation of protease-activated receptor does not. The GPCR agonists that promote β2AR phosphorylation effectively inhibit βAR agonist isoproterenol-induced PKA phosphorylation of phospholamban and contractile function in ventricular cardiomyocytes. Heterologous GPCR stimuli have minimal to small effect on isoproterenol-induced β2AR activation and G-protein coupling for cyclic adenosine monophosphate (cAMP) production. However, these GPCR stimuli significantly promote phosphorylation of phosphodiesterase 4D (PDE4D), and recruit PDE4D to the phosphorylated β2AR in a β-arrestin 2 dependent manner without promoting β2AR endocytosis. The increased binding between β2AR and PDE4D effectively hydrolyzes cAMP signal generated by subsequent stimulation with isoproterenol. Mutation of PKA phosphorylation sites in β2AR, inhibition of PDE4, or genetic ablation of PDE4D or β-arrestin 2 abolishes this heterologous inhibitory effect. Ablation of β-arrestin 2 or PDE4D gene also rescues β-adrenergic stimuli-induced myocyte contractile function.
Conclusions
These data reveal essential roles of β-arrestin 2 and PDE4D in a common mechanism for heterologous desensitization of cardiac βARs under hormonal stimulation, which is associated with impaired cardiac function during the development of pathophysiological conditions.
Keywords: Heterologous desensitization, PKA, PKC, β-Arrestin 2, Phosphodiesterase 4
1. Introduction
β-adrenergic receptor (βAR) signal acts as a linchpin of cardiac regulation under stress. Over the last decades, homologous desensitization of βARs has been extensively studied, which has revealed essential roles of G protein receptor kinases (GRKs) and β-arrestins in inhibiting the coupling of βARs to their G proteins as well as removing βARs from the cell surface for termination of cyclic adenosine monophosphate (cAMP) generation. This paradigm has served as a cornerstone to understand βAR turn-off mechanism, which protects the heart from excessive or repetitive stimuli.1 Meanwhile, in clinical conditions, a variety of elevated neurohormonal stimuli can impair heart contractile function via interaction with cardiac βAR signal. This phenomenon is mainly attributed to heterologous desensitization of βARs, in which βARs are phosphorylated through other pathways [such as activation of protein kinase A (PKA), and protein kinase C (PKC)] in a βAR agonist-independent manner.1,2 Yet, the mechanisms underlying cross talk between cardiac βAR signal and these neurohormonal stimuli are not completely understood.
GPCRs usually possess multiple phosphorylation sites (serine, threonine, and tyrosine) on their intracellular domains. In the classical paradigm, the second messenger cAMP and diacylglycerol induced by other GPCRs leads to activation of PKA and PKC, respectively, which phosphorylate serine and threonine residues on the GPCR intracellular domains. The phosphorylation also contributes to a process known as heterologous desensitization or ‘non-agonist-specific’ desensitization.3–5 The phosphorylation event leads to a reduction in receptor-G-protein coupling,6 thus attenuating the effector-mediated production of secondary messengers for downstream cellular responses. Meanwhile, other mechanism such as enhancing cAMP degradation by PKA- or CaMKII-mediated activation or recruitment of phosphodiesterase (PDE) is lately discovered and added into the portrait of receptor heterologous desensitization system.7–11
β-arrestins play two important roles in GPCR homologous desensitization (‘agonist-specific’ desensitization). First, β-arrestins bind to GRK phosphorylated receptors to interfere with G protein coupling and initiate endocytosis through clathrin-coated vesicles12; Second, β-arrestins act as a scaffold protein for various other signalling molecules such as PDE to further diminish βAR-induced cAMP signal.13 Several studies have reported that β-arrestins are also involved in heterologous desensitization of certain types of cell surface receptors, such as α2 adrenergic receptor, metabotropic glutamate receptor, and lysophosphatidic acid receptor.14–16 Yet, compared to homologous desensitization of βAR,9 the roles of β-arrestins in heterologous desensitization of cardiac βAR signal, especially those triggered by activation of other GPCRs, are still poorly understood. Moreover, it is not clear how the heterologous desensitization of cardiac βAR induced by other GPCRs affects excitation-contraction coupling in cardiomyocytes.
In our previous study, we have shown that PDE4 is crucial in prostaglandin E2 (PGE2)-induced heterologous desensitization of cardiac βAR signalling.17 Notably, the cross talk between EP-R and βAR is β2AR dependent, as the inhibitory effect of PGE2 on βAR signalling is absent in β2AR knockout cardiomyocytes, implying the important role of β2AR in coordination and integration of diversified signal pathways.17 In the current study, we focused on understanding molecular events on β2AR that are involved in the heterologous cross talk. Meanwhile, we extended our exploration to the cross talk between several GPCRs and cardiac βARs. Our results reveal that some Gs- and Gq-coupled receptors, but not Gi-coupled receptors promote phosphorylation of β2ARs via PKA and PKC, respectively. Heterologous phosphorylation of β2AR has minimal to small effect on βAR agonist-induced receptor active conformation for Gs-coupling and receptor trafficking, but significantly promotes recruitment of PDE4D to phosphorylated β2AR in a β-arrestin 2-dependent manner. The β2AR/PDE4D complex effectively suppresses subsequent βAR agonist-induced cAMP/PKA signal. Consequently, it efficiently blocks βAR agonist-induced PKA phosphorylation of phospholamban (PLB), a critical protein involved in the regulation of myocyte calcium cycling, as well as impairs myocyte contractile response to β-adrenergic stimulation. Our data offer a general mechanism to understand how other GPCR signals intercept β-adrenergic regulation in cardiomyocytes, which may compromise cardiac function under pathophysiological conditions.
2. Methods
2.1 Cardiomyocytes isolation and contractility and calcium transient assay
All animal experiments conform to the NIH guide for the care and use of laboratory animals and were approved by the Institutional Animal Care and Use Committees at the University of California, Davis. Neonatal cardiomyocytes from wild-type (WT), β1AR knockout (KO), β2AR KO, β-arrestin 1 KO, or β-arrestin 2 KO pups were isolated and cultured as described previously.18 Ventricular cardiomyocytes from adult male mice or rats (8–10 weeks old) were isolated and myocyte contractility was measured as previously described.17,19 For neonatal ventricular cardiomyocytes isolation, rapid decapitation with sharp scissors was performed to ensure viability of cardiomyocytes. For adult ventricular cardiomyocytes isolation, mice and rat were anesthetized with 3–5% isoflurane. Hearts were excised quickly, then mounted on Langendorff perfusion apparatus and perfused with collagenase and protease solution (0.5 mg/mL collagenase and 0.1 mg/mL protease for mouse; 1.0 mg/mL collagenase, and 0.2 mg/mL protease for rat). Freshly isolated adult cardiomyocytes were loaded with Fluo-4 AM (5 µM; Molecular Probes, Eugene, OR) for 10 min along with different drugs in a dark environment before placed in the middle of a glass-bottomed petri dish filled with beating buffer (NaCl 120 mM, KCl 5.4 mM, NaH2PO4 1.2 mM, MgSO4 1.2 mM, HEPES 20 mM, Glucose 5.5 mM, CaCl2 1mM, pH 7.1). Electrical stimulation was carried out by immersing platinum electrodes in the beating buffer and paced at 1 Hz with voltage of 30 V using the SD9 stimulator (Grass Technology, Warwick, RI, Oberkochen, Germany). The calcium transient and cell length were recorded on an inverted microscope (Zeiss AX10) at 20 × magnification lens using the MetaMorph® software (Molecular Devices, Sunnyvale, CA). The percent sarcomere length shortening (%ΔL/L0) was analysed using MetaMorph® software. The calcium transient analysis was performed using ImageJ software and homemade routines in IDL (Interactive Data Language, ITT) as previously described.20
All experiments using PDE4D KO mice were carried out according to the European Community guiding principles in the Care and Use of Animals (2010/63/UE, 22 September 2010), the local Ethics Committee (CREEA Ile-de-France Sud) guidelines, and the French decree no. 2013-118, 1 February 2013 on the protection of animals used for scientific purposes (JORF no. 0032, 7 February 2013, p2199, text no. 24). Authorizations to perform animal experiments according to this decree were obtained from the Ministère français de l'Agriculture, de l'Agroalimentaire et de la Forêt (agreement no. B 92-019-01). Generation of Pde4d homozygous null mice has been described previously.21 Ventricular myocytes were obtained from 8- to 10-week-old males as previously described.22 Animals were anesthetized by intraperitoneal injection of pentothal (150 mg/kg), and the heart was quickly removed and placed into a cold Ca2+-free Tyrode’s solution containing 113 mM NaCl, 4.7 mM KCl, 4 mM MgSO4, 0.6 mM KH2PO4, 0.6 mM NaH2PO4, 10 mM butanedione monoxime (BDM), 1.6 mM NaHCO3, 10 mM HEPES, 30 mM Taurine, and 20 mM D-glucose, adjusted to pH 7.4. The ascending aorta was cannulated and the heart was perfused with oxygenated Ca2+-free Tyrode’s solution at 37 °C for 4 min using retrograde Langendorff perfusion. For enzymatic dissociation, the heart was perfused with Ca2+-free Tyrode’s solution containing Liberase TM Research Grade (Roche Diagnostics) for 10 min at 37 °C. Then the heart was removed and placed into a dish containing Tyrode’s solution supplemented with 0.2 mM CaCl2 and 5 mg/mL BSA (Sigma-Aldrich). The ventricles were separated from the atria, cut into small pieces, and triturated with a pipette to disperse the myocytes. Ventricular myocytes were filtered on gauze and allowed to sediment by gravity for 10 min. The supernatant was removed and cells were suspended in Tyrode’s solution supplemented with 0.5 mM CaCl2 and 5 mg/mL BSA. The procedure was repeated once and cells were suspended in Tyrode’s solution with 1 mM CaCl2. Freshly isolated ventricular myocytes were plated in 35-mm culture dishes coated with laminin (10 μg/mL) and stored at room temperature until use. All experiments were performed at room temperature. Isolated cardiomyocytes were loaded with 1 μM Fura-2 AM (Thermo Fischer Scientific) at room temperature for 10 min. The loaded cells were field stimulated (5 V, 4 ms) at a frequency of 1 Hz. Sarcomere length and Fura-2 ratio (measured at 512 nm upon excitation at 340 nm and 380 nm) were simultaneously recorded using an IonOptix System (IonOptix). Cell contractility was assessed by the percentage of sarcomere shortening, which is the ratio of twitch amplitude (difference of end-diastolic and peak systolic sarcomere length) to end-diastolic sarcomere length. Ca2+ transients were assessed by the percentage of variation of the Fura-2 ratio by dividing the twitch amplitude (difference of end-diastolic and peak systolic ratios) to end-diastolic ratio. The t1/2off (τ) was used as an index of relaxation and Ca2+ transient decay kinetics. All parameters were calculated offline using a dedicated software (IonWizard).
2.2 Adenovirus infection
Adult ventricular cardiomyocytes were cultured in 6-well plates in minimum essential medium with 10% 2,3-BDM. Neonatal ventricular cardiomyocytes were cultured in 24-well plates with coated coverslips in Dulbecco’s modified Eagle’s media containing 10% bovine fetal serum. The recombinant adenovirus encoding flag-β2AR, flag-β2AR-PKA4A mutant (flag-β2AR with point mutations of serine residues at 261, 262, 345, 346 to alanine), HA-tagged mouse β1AR, GFP-N-terminal-PDE4D5 and mCherry-tagged PDE4D5 were generated with the pAdEasy system (Qbiogene, Carlsbad, CA). PM-ICUE3 and ICUE3 and SR-AKAR fluorescent resonance energy transfer (FRET) biosensor adenoviruses are generated in our lab.23 Cells were infected for 2 h for neonatal cardiomyocytes and 24 h for adult cardiomyocytes, media were then removed and replaced with fresh media. All experiments were carried out after 48 h after virus infection.
2.3 Immunoprecipitation, co-immunoprecipitation, and western blotting
Ventricular cardiac cardiomyocytes were cultured in 6-well plates for 48 h. After addition of different agonists, cells were then harvested in lysis buffer (25 mM Hepes, pH 7.4, 5 mM EDTA, 150 mM NaCl, 0.5% Triton X-100, and protease inhibitors containing 2 mM Na3VO4, 1 mM PMSF, 10 mM NaF, 10 µg/mL Aprotinin, 5 mM Bestatin, 10 µg/mL Leupeptin, and 2 µg/mL Pepstain A). The crude cell lysate was centrifuged at 13 200 × g for 30 min. For immunoprecipitation and co-immunoprecipitation, the supernatant was then incubated with anti-FLAG M2 affinity gel (Sigma, St. Louis, MO) for 4 h at 4 °C. The immune complexes were washed three times with lysis buffer. The bound proteins were eluted by 2 × sodium dodecyl sulfate (SDS)-loading buffer with 5% beta-mercaptoethanol and separated on 8% SDS-PAGE gels and probed with corresponding primary antibodies; for western blotting, the protein amount in the supernatant was quantified by standard BCA assay and equal amount of proteins were resolved on SDS-PAGE gels. Anti-flag antibody (mouse monoclonal IgG2b, Sigma, St. Louis, MO); Phospho-(Ser/Thr) PKA substrate antibody (Rabbit polyclonal IgG, Cell Signaling Technology, Danvers, MA); Phospho-Troponin I (Cardiac) (Ser23/24) antibody (Rabbit polyclonal IgG, Cell Signaling Technology, Danvers, MA); anti-phospho-PDE4D (serine 190) antibody (Rabbit polyclonal IgG, Abcam, Cambridge, United Kingdom); anti-GFP antibody (Rabbit polyclonal IgG, Convance, Princeton, NJ); anti-RFP antibody (Rabbit polyclonal IgG, Rockland, Limerick, PA). Specially, for phosphorylation of PLB (serine 16) detection, isolated cardiomyocytes pretreated with different GPCR agonists were incubated at 37 °C water bath in Eppendorf tubes, then stimulated with isoproterenol for 5 min. After stimulation, cardiomyocytes were spun down using bench-top centrifuge machine at top speed for 30 s, supernatant was quickly removed and cell pellets were lysed using lysis buffer described above. Same amount of protein (20 µg) was resolved on SDS-PAGE gels and later detected with anti-phospho-PLB (serine 16) antibody (Badrilla, Leeds, United Kingdom). All primary antibodies were revealed with IRDye 800 CW goat secondary antibodies using Odyssey detection system (Li-cor Biosciences, Lincoln, Nebraska). The optical density of the bands was analysed with NIH Image J software.
2.4 Confocal imaging
The confocal imaging was carried out on Zeiss LSM 700 Axio Observer confocal microscope (Oberkochen, Germany) using a Zeiss Plan-Apochromat 63 ×/1.4 oil objective. Neonatal cardiomyocytes were cultured in 24-well plates with coverslips and infected with flag-β2AR and Nb80-GFP adenovirus. After stimulation, cardiomyocytes were fixed with phosphate-buffered saline (PBS) containing 4% paraformaldehyde at room temperature and then blocked with PBS containing 0.2% Nonidet P-40 and 2% goat serum followed by staining with anti-flag antibody (mouse monoclonal IgG2b, Sigma, St. Louis, MO). The primary antibody was labelled with Alexa Fluor 594 labelled goat anti-mouse IgG antibody (Life Technologies, Carlsbad, CA) and images were collected using 488 nm and 555 nm laser lines for excitation, respectively. Images were processed and analysed with Zen Software from Zeiss (Oberkochen, Germany).
2.5 FRET assay
Neonatal cardiomyocytes infected with PM-ICUE3, ICUE3 or SR-AKAR3 biosensor were maintained in PBS during FRET recording as described previously.24 Images were acquired with a Leica DMI 3000B microscope with a 40 ×/1.3 NA oil-immersion objective lens. FRET images acquisition setting and FRET intensity measurement were carried out as previously described.17
2.6 Data analysis
All bar graphs with scatter plots were show as mean ± standard error of the mean of at least three independent experiments. One-way analysis of variance (ANOVA) with Dunnett’s multiple comparison test or student’s unpaired t-test was performed using the GraphPad Prism 7 software (GraphPad Inc., San Diego, CA) to determine statistical significance as follows: n.s, not significant; *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001; or #P < 0.05; ##P < 0.01; ###P < 0.001; ####P < 0.0001.
3. Results
3.1 Different hormones/GPCR agonists promote PKA/PKC phosphorylation of βAR in adult rat ventricular cardiomyocytes
We first examined the effect of several neurohormonal stimuli/GPCR agonists on phosphorylation of β2AR at putative PKA phosphorylation sites, a well-known β-adrenergic stimuli-induced post-translational modification that promotes desensitization of β2AR.25 Adult rat ventricular cardiomyocytes expressing flag-β2AR were stimulated with the βAR agonist isoproterenol (ISO, 1 µM) or a set of different GPCR agonists, including Gs-coupled receptors: prostaglandin E2 receptor (EP-R) agonist prostaglandin E2 (PGE2), β1AR agonist dobutamine, dopamine receptor agonist dopamine, corticotropin releasing factor type 2 receptor agonist urocortin, relaxin receptor agonist relaxin and adenosine receptor agonist adenosine; Gq-coupled receptors: α adrenergic receptor (α1AR) agonist phenylephrine, angiotensin receptor type I (AT1R) agonist angiotensin II (Ang II) and protease-activated receptors agonist thrombin; and Gi-coupled α2 adrenergic receptor (α2AR) agonist clonidine. Stimulation with PGE2, dopamine, urocortin, relaxin, adenosine, phenylephrine, or Ang II significantly enhances the phosphorylation of β2AR at putative PKA phosphorylation sites (Figure 1A), which is similar to βAR agonist ISO. In contrast, stimulation with dobutamine, thrombin, or clonidine has either no effect or minimal effect. In addition, we show that β1AR is subjected to a similar influence by the tested GPCR agonists, except that urocortin has no significant effect on β1AR phosphorylation, but β1AR selective agonist dobutamine does (see Supplementary material online, FigureS1). Besides PKA, the putative PKA phosphorylation sites can also be phosphorylated by other kinases including PKC.25 Application of PKA selective peptide inhibitor PKI effectively diminishes ISO-, PGE2-, phenylephrine-, and dopamine-induced phosphorylation of β2AR (Figure 1C). In contrast, the PKC inhibitor GO6976 selectively inhibits dopamine-and phenylephrine-induced β2AR phosphorylation (Figure 1B). The inhibitory effect of PKI on phenylephrine-induced phosphorylation of β2AR is probably due to non-selective inhibition of PKC by high concentration of PKI (10 µM) or activation of the βAR-PKA cascade by phenylephrine at high concentrations.26,27
Figure 1.

Activation of different GPCRs promotes phosphorylation of β2AR in adult ventricular cardiomyocytes. Adult rat ventricular cardiomyocytes expressing flag-β2AR were cultured for 48 h before stimulation with different GPCR agonists as indicated (1 µM for 5 min, respectively). The flag-β2AR was immunoprecipitated using anti-FLAG beads. The elution was subjected to western blot to detect β2AR phosphorylation. The phosphorylation of β2AR at putative PKA phosphorylation sites is quantified and expressed as fold over non-stimulated group (Control). Data are obtained from 3–7 independent experiments and shown as mean ± S.E.M. n.s, not significant; *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001 (vs. Control group) by student’s unpaired t-test. ISO, isoproterenol; PGE2, Prostaglandin E2; Dobu, Dobutamine; Dopa, Dopamine; Phe, Phenylephrine; Ang II, Angiotensin II. Adult rat ventricular cardiomyocytes expressing flag-β2AR were pretreated with 1 µM of PKC inhibitor GO6976 (B) or 10 µM of PKA peptide inhibitor PKI (C) for 30 min followed by stimulation with different GPCR agonists as indicated (1 µM for 5 min, respectively). Flag-β2AR was immunoprecipitated using anti-FLAG beads. Phosphorylated β2AR at putative PKA phosphorylation sites and total β2AR were detected by western blot. Data represent 3–5 independent experiments.
3.2 Different hormones/GPCR agonists inhibit β-adrenergic stimulation induced adult cardiomyocyte contractile response
We then examined whether heterologous phosphorylation of βAR leads to desensitization of cardiac β-adrenergic signal in native rat ventricular cardiomyocytes. Stimulation of βAR agonist ISO (100 nM) promotes robust increases in myocyte contractile shortening (Figure 2A and L). While pretreatment with PGE2, Ang II, dopamine, or phenylephrine alone has no effect on resting sarcomere lengths (see Supplementary material online, FigureS2A), it significantly inhibits β-adrenergic stimulation-induced contractile shortening responses (Figure 2B–E and L). In parallel, these GPCR stimuli significantly diminishes calcium transient amplitudes and prolongs recovery tau in response to β-adrenergic stimulation (Figure 2F–J,M, and N), indicating impaired calcium handling for contractile function after pretreatment with these GPCR agonists. Consistently, pretreatment of cardiomyocytes with PGE2, Ang II, dopamine, or phenylephrine significantly inhibits β-adrenergic-induced PKA phosphorylation of PLB at serine 16 site (Figure 2K), an essential event involved in regulating myocyte calcium cycling and contractile function. In addition, phosphorylation of other PKA targets in response to ISO stimulation, such as cardiac Troponin I, is also inhibited by pretreatment with Ang II, dopamine or phenylephrine, but not by PGE2 (see Supplementary material online, FigureS2C), suggesting divergent inhibition of local PKA activity in response to β-adrenergic stimulation after various GPCR agonists pretreatment.
Figure 2.

Activation of different GPCRs inhibits β-adrenergic stimulation induced PLB phosphorylation, calcium handling and myocyte contractility. (A–E) Representative traces of myocyte sarcomere length fractional shortening and (F–J) representative traces of intracellular calcium release (calcium transient) from adult rat ventricular cardiomyocytes treated with or without 100 nM isoproterenol (ISO, A and F); 1 µM PGE2 before 100 nM ISO (B and G); 1 µM angiotensin II (Ang II) before 100 nM ISO (C and H); 1 µM dopamine (Dopa) before 100 nM ISO (D and I); 1 µM phenylephrine (Phe) before 100 nM ISO (E and J). (K) Adult rat ventricular cardiomyocytes were pretreated with indicated GPCR agonists (1 µM for 5 min, respectively), then stimulated by 100 nM ISO for 5 min. Phosphorylation of PLB at Serine 16 site was detected by western blot and quantified as fold over non-stimulated group (Control group, without ISO stimulation condition), and shown in bar graph with scatter plots. Data are collected from 7 to 9 independent experiments. The percent shortening in adult rat ventricular myocyte sarcomere length (L), the amplitude of calcium release (calcium transient) (M), and the rate of calcium decay (τ) are shown in bar graph with scatter plots. Data are obtained from four independent experiments and shown as mean ± S.E.M. In (K)—(N), *P ≤ 0.05; **P ≤ 0.01; ****P ≤ 0.0001 (vs. ISO alone group) by one-way ANOVA with Dunnett’s multiple comparison test. Between paired groups, n.s, not significant; #P ≤ 0.05; ###P ≤ 0.001; ####P ≤ 0.0001 by student’s unpaired t- test.
3.3 Phosphorylation of β2AR at PKA sites does not affect βAR agonist-induced receptor activation
Phosphorylation of βARs at PKA sites can affect receptor signalling via modulation of either receptor-G protein coupling to inhibit adenylyl cyclase-dependent cAMP synthesis28 or receptor-associated phosphodiesterase 4 (PDE4)-dependent cAMP hydrolysis.29,30 Notably, the cross talk between EP-R and βARs is β2AR dependent.17 Thus we used PGE2 and β2AR as an example to further explore the underlying mechanism of heterologous desensitization of β-adrenergic signal and impairment of contractile function. We first applied a set of conformation-specific camelid single-domain nanobodies (Nbs): Nb80 and Nb37 to examine activation of β2AR and Gs protein, respectively.31–33 Nb80, an indicator of active receptor conformation, selectively binds agonist-occupied β2AR. In cardiomyocytes, while βAR agonist ISO stimulation significantly induces recruitment of Nb80 to β2AR on the plasma membrane, PGE2 fails to do so (Figure 3). However, PGE2 pretreatment does not prevent recruitment of Nb80 to β2AR on the plasma membrane upon subsequent stimulation with ISO (Figure 3). This phenomenon is also preserved in HEK293 cells (see Supplementary material online, FigureS3A). The association of Nb80 to β2AR is confirmed by co-immunoprecipitation (see Supplementary material online, FigureS3B and D), suggesting that PKA phosphorylation of β2AR does not influence agonist-induced receptor activation. Meanwhile, Nb37 specifically recognizes the guanine-nucleotide-free form of Gαs representing the catalytic intermediate of G protein activation.34 While PGE2 is incapable of recruit Nb37 to β2AR, after PGE2 pretreatment, ISO still effectively promotes β2AR and Nb37 association with a small reduction when compared to those induced by ISO directly (see Supplementary material online, FigureS3C and E). In comparison, the myocyte contractile response to ISO stimulation is completely abolished after PGE2 pretreatment (Figure 2L), indicating the existence of additional regulation components besides reduction of β2AR coupling to Gs.
Figure 3.

Heterologous phosphorylation of β2AR does not affect β-adrenergic stimulation-induced receptor active conformation change. WT neonatal cardiomyocytes expressing flag-β2AR and Nb80-GFP were stimulated with or without isoproterenol (ISO, 1 μM) for 5 min or in the presence of PGE2 (1 μM) for 5 min then stimulated with or without ISO (1 μM) for 5 min. The cells were then fixed and labelled with anti-flag antibody, followed by a red fluorescent secondary antibody. Data represents five independent experiments. The representative dual colour confocal immunofluorescence images show Nb80-GFP (green) and flag-β2AR (red) distribution and their localization (shown as merge) (Scale bar = 10 µm). The relative fluorescence intensity of Nb80-GFP and flag-β2AR along the white solid line are measured and plotted in the graphs below each individual image, respectively. The arrowheads indicate the positions where fluorescence peaks of Nb80-GFP and flag-β2AR are overlapped.
3.4 PKA phosphorylation of β2AR promotes assembly of β2AR/PDE4D complex at the plasma membrane to promote desensitization of β adrenergic signalling
We then examined the potential role of PDE in hormone-induced inhibitory effect on βAR signal. In isolated adult rat ventricular cardiomyocytes, PGE2 promotes significant association of β2AR with PDE4D5 (Figure 4B and C), in a fashion similar to the β2AR/PDE4D5 complex formation induced by βAR agonist ISO.29,30 However, unlike βAR agonist ISO, which could induce receptor endocytosis, PGE2 stimulation fails to induce β2AR internalization (see Supplementary material online, FigureS3F and G). Accordingly, PGE2 stimulation promotes significant recruitment of PDE4D5 to β2AR in the plasma membrane fraction (see Supplementary material online, FigureS3I), suggesting that PDE4D and β2AR form a complex on the cell surface. Moreover, mutation of PKA phosphorylation sites on the β2AR abolishes the PGE2-induced binding of β2AR with PDE4D5 (Figure 4C). In addition, PGE2, phenylephrine, and Ang II, but not adenosine promote PDE4D phosphorylation at putative PKA phosphorylation sites (Figure 4A and see Supplementary material online, FigureS3H), which increases enzymatic activity to attenuate βAR signalling.35
Figure 4.

Heterologous phosphorylation of β2AR promotes β-arrestin 2 dependent recruitment and activation of PDE4D to β2AR. (A) Adult rat ventricular cardiomyocytes were stimulated with different GPCR agonists as indicated (1 µM for 5 min, respectively). Cells were then lysed, and phosphorylation of PDE4D at Serine 190 site was detected in total lysate by western blot using anti-phospho-specific PDE4D antibody. The levels of phosphorylation of PDE4D are quantified and expressed as fold over non-stimulated group (Control). Data are obtained from three independent experiments and shown as mean ± S.E.M. *P < 0.05; ***P < 0.001; ****P < 0.0001 by one-way ANOVA with Dunnett’s multiple comparison test. (B) WT or β-arrestin-2 KO neonatal cardiomyocytes were infected with flag-β2AR together GFP-N-terminal-PDE4D5 adenoviruses. After stimulation with 1 µM PGE2 for 5 min, flag-β2AR was immunoprecipitated using anti-FLAG M2 beads. The elution was subjected to western blot to detect β2AR and PDE4D5, respectively. The PDE4D levels are quantified and expressed as fold over β2AR levels in corresponding non-stimulated groups. (C) Adult rat ventricular cardiomyocytes were infected with mCherry-PDE4D5 together with flag-β2AR or flag-β2AR-PKA4A (lacking four PKA phosphorylation sites) adenovirus and cultured for 48 h. After stimulation with 1 µM PGE2 or 1 µM Angiotensin II (Ang II) for 5 min, cardiomyocytes were lysed and β2AR was immunoprecipitated using anti-FLAG M2 beads. The elution was subjected to western blot to detect β2AR and PDE4D5, respectively. The PDE4D5 levels are quantified and expressed as fold over β2AR levels in corresponding non-stimulated groups. In (B) and (C), data are obtained from four independent experiments and shown as mean ± S.E.M. Between paired groups, n.s, not significant; *P < 0.05 by student’s unpaired t-test.
We then assessed whether inhibition of PDE4D is able to rescue the desensitized β-adrenergic signal on the plasma membrane. We took advantage of a FRET-based cAMP biosensor, which is selectively targeted to the plasma membrane (PM-ICUE3).36 In WT neonatal cardiomyocytes, βAR agonist ISO induces robust cAMP signal across the plasma membrane (Figure 5A and G). Pretreatment with PGE2 blunts ISO (1 μM)-induced cAMP signal (Figure 5B and G). At a concentration when applying alone has no effect on cAMP and PKA signal (data not shown and ref.17), PDE4 inhibitor rolipram significantly rescues β-adrenergic stimulation-induced cAMP signal dampened by PGE2 pretreatment (Figure 5D–G). To avoid the potential saturation of FRET biosensor induced by 1 μM of ISO, a submaximal concentration of ISO (10 nM) was used. Stimulation with ISO (10 nM) generates a moderate cAMP signal that can be further enhanced by addition of forskolin (50 µM) and IBMX (100 µM); and the modest increase in cAMP induced by ISO is also blunted by PGE2 pretreatment (Figure 5H). However, addition of a submaximal concentration (100 nM) of rolipram significantly restores ISO-induced cAMP signal in PGE2 pretreatment group (Figure 5H). At both saturated (1 μM) and submaximal (10 nM) concentration of ISO, the rolipram-induced recoveries in cAMP signal in the PGE2-pretreated groups are still slightly below the control groups without PGE2 treatment, respectively (Figure 5G and H). These data indicate that PGE2 induces a significant yet small impairment of the β2AR-Gs coupling. We also measured global cAMP levels in cardiomyocytes with another FRET biosensor ICUE3. Similar to those observed on the plasma membrane, ISO-induced cAMP signal in the whole cells is inhibited by PGE2 pretreatment, which is also rescued by rolipram (see Supplementary material online, FigureS4). Together, these data suggest that PGE2-induced of activation and recruitment of PDE4D to the plasma membrane effectively metabolizes the βAR-induced cAMP at both the plasma membrane and in the cytoplasm.
Figure 5.

Inhibition of PDE4 or deletion of PDE4D rescues desensitized β-adrenergic signal after PGE2 treatment. WT mouse neonatal cardiomyocytes expressing PM-ICUE3 FRET biosensor were stimulated with 1 µM ISO without (A) or with 1 µM selective PDE4 inhibitor rolipram (Roli) (E); or cardiomyocytes were pretreated with 1 µM PGE2 for 5 min followed by stimulation with 1 µM ISO in the absence (C) or presence of 1 µM Roli addition at different times (B, D, and F). Representative traces of normalized YFP/CFP (FRET) ratio changes with different stimulations as indicated are shown. (G) The normalized FRET ratio changes corresponding to (A–F) are shown in bar graph with scatter plots. Data are obtained from five independent experiments and shown as mean ± S.E.M. ****P ≤ 0.0001 (vs. ISO + PGE2 group); Between paired groups, n.s, not significant; #P ≤ 0.05; ##P ≤ 0.05; ####P ≤ 0.0001 by one-way ANOVA with Dunnett’s multiple comparison test. (H) WT mouse neonatal cardiomyocytes expressing PM- ICUE3 FRET biosensor were stimulated with 10 nM ISO followed by Forskolin (50 µM) and IBMX (100 µM); or 10 nM ISO together with 100 nM selective PDE4 inhibitor rolipram (Roli). Alternatively cardiomyocytes were pretreated with 1 µM PGE2 for 5 min followed by stimulation with 10 nM ISO in the absence or presence of 100 nM Roli. The normalized FRET ratio changes are shown in bar graph with scatter plots. Data are obtained from three experiments and shown as mean ± S.E.M. ****P ≤ 0.0001 (6 vs. ISO + PGE2 group); Between paired groups, n.s, not significant; #P ≤ 0.05; ##P ≤ 0.05 by one-way ANOVA with Dunnett’s multiple comparison test. The percent shortening in adult WT or PDE4D KO mouse ventricular cardiomyocyte sarcomere length (I), the amplitude of calcium release (calcium transient) (J) and the rate of calcium decay (τ) (K) are shown in bar graph with scatter plots. In (I–K), data are obtained from three independent experiments and shown as mean ± S.E.M. Between paired groups, n.s, not significant; *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001; ****P ≤ 0.0001 by student’s unpaired t-test.
We further validated the functional involvement of PDE4D in GPCR signal cross talk using cardiomyocyte contractile shortening assay. In WT cardiomyocytes, stimulation with βAR agonist ISO greatly increases myocyte contractile shortening (Figure 5I and see Supplementary material online, FigureS5A), and pretreatment with PGE2 significantly inhibits ISO-induced contractile shortening response (Figure 5I and see Supplementary material online, FigureS5B). In parallel, PGE2 pretreatment reduces calcium transient amplitude and prolongs calcium decay tau in response to β-adrenergic stimulation, indicating impaired calcium handling for contractile function (Figure 5J, K, and see Supplementary material online, FigureS5C and D). Compared to general βAR stimulation, stimulation of β2AR (revealed by in the presence of β1AR selective antagonist CGP20712A) induces small but significant increases in the contractility and intracellular calcium transient, respectively (see Supplementary material online, FigureS6A and B and refs.37,38). PGE2 pretreatment inhibits β2AR stimulation-induced increases in contractile shortening and calcium transient. Addition of rolipram (100 nM) rescues both contractile shortening and calcium transient amplitude in PGE2 pretreatment groups (see Supplementary material online, FigureS6A and B). In contrast, deletion of PDE4D rescues ISO-induced contractile response in cardiomyocytes pretreated with PGE2 (Figure 5I and see Supplementary material online, FigureS5E and F). Ablation of PDE4D, to a large extent, also restores calcium transient amplitude and decay tau in response to β-adrenergic stimulation (Figure 5J, K, and see Supplementary material online, FigureS5G and H). Together, these results suggest that PDE4 negates βAR responses during heterologous cross talks.
3.5 β-arrestin 2 is required for PDE4-mediated desensitization of β-adrenergic signal
We have recently shown that PGE2 is able to attenuate βAR stimulated cardiac contractility in mouse hearts by preventing cAMP propagation into the sarcoplasmic reticulum (SR), a critical organelle for calcium cycling and EC coupling.17 Thus, we further assessed the role of GPCR cross talk on β adrenergic stimuli-induced PKA activity at the SR by applying a FRET-based biosensor, SR-AKAR3.23 In WT neonatal cardiomyocytes, PGE2 significantly attenuates ISO-induced PKA activity at the SR (Figure 6A, B, G, and see Supplementary material online, FigureS7A, B, and E). Mutation of PKA phosphorylation sites of β2AR abolishes this inhibitory effect (see Supplementary material online, FigureS7C–E). Meanwhile, studies show that ISO induced association of PDE4D and β2AR is dependent on β-arrestin 2.13,30 Here, the PGE2-induced association between β2AR and PDE4D5 is absent in the cardiomyocytes lacking β-arrestin 2 (Figure 4B), confirming a scaffold role of β-arrestin 2 in connecting PDE4D to the β2AR after PGE2 stimulation. We thus examined the roles of β-arrestin 2 in the observed cross talk between the tested GPCRs and β2AR in cardiomyocytes. Deletion of β-arrestin 2, but not β-arrestin 1 abolishes the PGE2 inhibitory effect on ISO-induced PKA activity at the SR (Figure 6C–G). While PKA-mediated phosphorylation of β2AR is involved in promoting receptor coupling to Gi,39 pretreatment with Gi inhibitor pertussis toxin does not affect the inhibitory effect of PGE2 on ISO-induced PKA activity at the SR (see Supplementary material online, FigureS7F).
Figure 6.

Deletion of β-arrestin 2 rescues desensitized β-adrenergic signal after PGE2 treatment. WT (A, B), β-arrestin 1 KO (C, D) or β-arrestin 2 KO (E, F) neonatal cardiomyocytes expressing SR-AKAR3 FRET biosensor were stimulated with 1 µM ISO directly or pretreated with 1 µM PGE2 for 5 min followed by stimulation with 1 µM ISO. Representative traces of normalized YFP/CFP (FRET) ratio changes with different stimulations as indicated are shown. (G) The normalized FRET ratio changes corresponding to (A–F) are shown in bar graph with scatter plots. Data are obtained from four independent experiments and shown as mean ± S.E.M. Between paired groups, n.s, not significant; ***P ≤ 0.001; ****P ≤ 0.0001 by student’s unpaired t-test. The percent shortening in adult WT or β-arrestin 2 KO mouse ventricular cardiomyocyte sarcomere length (H), the amplitude of calcium release (calcium transient) (I) and the rate of calcium decay (τ) ( J) are shown in bar graph with scatter plots. In (H–J), data are obtained from three independent experiments and shown as mean ± S.E.M. Between paired groups, *P ≤ 0.05; **P ≤ 0.01; ****P ≤ 0.0001 by student’s unpaired t-test.
We then further assessed the role of β-arrestin 2 in receptor cross talk-dependent modulation of cardiomyocytes contractile function. In WT mouse ventricular cardiomyocytes, βAR agonist ISO stimulation significantly enhances myocyte contractile shortening (Figure 6H and see Supplementary material online, FigureS8A), pretreatment with PGE2 blunts the contractile response (Figure 6H and see Supplementary material online, FigureS8B). In parallel, PGE2 pretreatment decreases calcium transient amplitudes and prolongs calcium decay tau in response to β-adrenergic stimulation (Figure 6I, J and see Supplementary material online, FigureS8F and G). In contrast, deletion of β-arrestin 2 rescues ISO-induced contractile response as well as calcium transient amplitude and recovery tau in cardiomyocytes pretreated with PGE2 (Figure 6H–J and see Supplementary material online, FigureS9A, B, F, and G). Together, our data suggest that the PGE2-induced binding of β-arrestin 2 and PDE4D to β2AR effectively hydrolyzes cAMP produced by adenylyl cyclases in response β-adrenergic stimulation, contributing to desensitization of the βAR-PKA signalling in cardiomyocytes.
3.6 β-arrestin 2 serves as a nexus to coordinate PDE4-dependent desensitization of β-adrenergic signal by different GPCRs stimulations
We further examined whether other GPCRs utilize the same machinery to cross talk to β2AR. Ang II or phenylephrine alone does not affect PKA activity at the SR, but significantly blunts ISO-induced PKA activity at the SR (see Supplementary material online, FigureS10A and B). While Ang II promotes phosphorylation of PDE4D and recruitment of PDE4D to the phosphorylated β2AR, deletion of the β2AR PKA sites abolishes the recruitment of PDE4D to receptors (Figure 4A and C). Accordingly, PDE4 inhibitor rolipram to a great extent rescues the β-adrenergic-stimulation-induced PKA activity after pretreatment with Ang II or phenylephrine (see Supplementary material online, FigureS10A). In addition, pretreatment with PKC inhibitor GO6976 abolishes the inhibitory effects of Ang II and phenylephrine on β-adrenergic-stimulation induced PKA activity at the SR (see Supplementary material online, FigureS10B), confirming the involvement of PKC in the cross talk between these Gq-coupled receptors and β2AR in cardiomyocytes.
Similar to PGE2, Ang II, dopamine or phenylephrine significantly blunts ISO-induced PKA phosphorylation of PLB at serine 16 site in WT mouse cardiomyocytes (Figure 7A). These GPCR stimuli also inhibit myocyte contractile response and calcium transient amplitudes and decay tau to β-adrenergic stimulation (Figure 7B–D and see Supplementary material online, FigureS8C–E and H–J). However, deletion of β-arrestin 2 rescues ISO-induced PKA phosphorylation of PLB and myocyte contractile response in cardiomyocytes pretreated with dopamine or phenylephrine, and to a lesser extent recovers the responses in the cells pretreated with Ang II (Figure 7E, F, and see Supplementary material online, FigureS9C–E). Similarly, deletion of β-arrestin 2 also rescues calcium transient amplitude and decay tau in response to β-adrenergic stimulation in cardiomyocytes pretreated with dopamine or phenylephrine, yet lesser effect on those treated with Ang II (Figure 7G, H, and see Supplementary material online, FigureS9H–J). Together, these data suggest a general mechanism of cardiac βAR desensitization promoted by other GPCRs via recruitment of PDE4D to PKA/PKC phosphorylated βARs in a β-arrestin 2-dependent manner, which inhibits adrenergic signalling by increasing cAMP hydrolysis (Figure 8).
Figure 7.

Deletion of β-arrestin 2 abolishes the inhibitory effect of different GPCR agonists on β-adrenergic signal in cardiomyocytes. Adult mouse ventricular cardiomyocytes were pretreated with indicated GPCR agonists (1 µM for 5 min, respectively), then stimulated by 100 nM ISO for 5 min. Phosphorylation of PLB at Serine 16 site was detected by western blot. PLB Phosphorylation levels in WT (A) or β-arrestin 2 KO (E) cardiomyocytes are quantified and expressed as fold over non-stimulated group (Control group, without ISO stimulation condition). Data are obtained from 3 to 4 independent experiments and shown as mean ± S.E.M. *P ≤ 0.05; ***P ≤ 0.001; ****P ≤ 0.0001 (vs. ISO alone group) by one-way ANOVA with Dunnett’s multiple comparison test. The percent shortening in adult WT (B) or β-arrestin 2 KO (F) mouse ventricular cardiomyocyte sarcomere length, the amplitude of calcium release (calcium transient) in WT (C) or β-arrestin 2 KO myocytes (G), and the rate of calcium decay (τ) in WT (D) or β-arrestin 2 KO myocytes (H) are shown in bar graph with scatter plots. Data are obtained from three independent experiments and shown as mean ± S.E.M. **P ≤ 0.01; ****P ≤ 0.0001 (vs. ISO alone group) by one-way ANOVA with Dunnett’s multiple comparison test.
Figure 8.
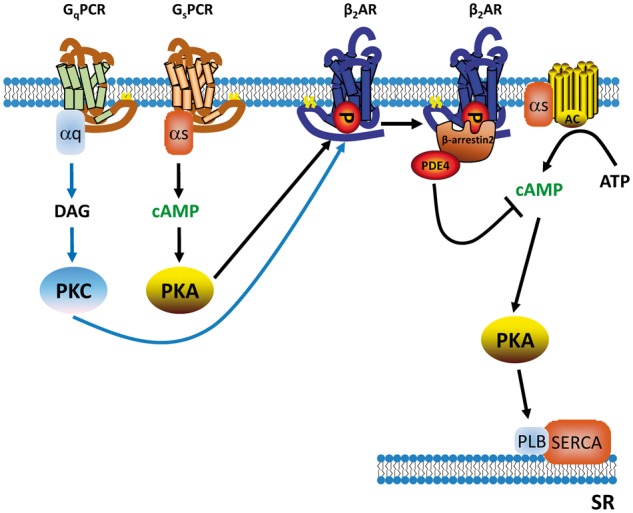
Working model of cross talk between different GPCRs and β2AR resulting in decreased cardiac contractile response. Schematic diagram depicts key signalling components responsible for cross talk between Gs- or Gq-coupled receptors and β2AR. Gs- and Gq-coupled receptors promote PKA- and PKC-mediated phosphorylation of β2AR. The phosphorylated β2AR recruits PDE4D to the cell surface in a β-arrestin 2-dependent manner, which enhances hydrolysis of cAMP induced by β-adrenergic stimulation, effectively blocking β-adrenergic signal transduction to the inner cell organelle such as the SR.
4. Discussion
It is well known that βAR signalling undergoes sophisticated modulation by an increasing number of factors, including homologous desensitization1,40 and heterologous desensitization.28 Here, we reveal that some Gs- and Gq-coupled receptors promote βAR phosphorylation at putative PKA phosphorylation sites through activation of PKA and PKC, respectively (Figure 1 and see Supplementary material online, FigureS1). The phosphorylation of β2AR had small impact on βAR agonist ISO-induced receptor-G-protein coupling without influence on receptor internalization. Further evidence shows that phosphorylated β2AR promotes β-arrestin 2-dependent recruitment of active PDE4 isoform to the receptors at the plasma membrane, which effectively hydrolyzes cAMP generated by subsequent application of βAR agonist ISO. Genetic deletion of PDE4D or β-arrestin 2 and pharmacological inhibition of PDE4D abolish the inhibitory effects of other GPCRs on βAR-induced contractile responses. Together, this study reveals a general mechanism for desensitization of cardiac βAR signalling by other Gs- and Gq-coupled receptors, which is in contrast to the inhibitory effect of Gi-coupled muscarinic receptors on βAR signal via inhibition of adenylyl cyclase for cAMP production.41
Traditionally, GPCR desensitization is attributed to the diminished coupling between phosphorylated receptors to G proteins. In agreement, using novel biosensors, the PKA-phosphorylated β2AR induced by PGE2 pretreatment displays a significant reduction in binding to Gs proteins in response to ISO stimulation, but without alteration in binding to Nb80, a nanobody selectively binding to ISO-induced receptor active conformation (see Supplementary material online, FigureS3B and C). This reduced receptor-Gs coupling has a small impact on βAR agonist-induced cAMP signal (Figure 5G and H). In comparison, the inhibitory effect of PGE2 on βAR signal appears to be due primarily to an enhanced hydrolysis of cAMP mediated by receptor-associated PDE4Ds. Disruption of β2AR-PDE4D complex or inhibition of PDE4 abolishes the inhibition of PGE2 on ISO-induced cAMP signal in cardiomyocytes. Classic literature and crystal structure studies suggest that G protein and arrestin potentially share a common GPCR-binding interface,42–44 which means their binding to β2AR would be mutually exclusive. The observations in this study suggest that PKA phosphorylation of β2AR unlikely switch receptor from Gs-coupling to arrestin binding completely, therefore resulting into a reduced binding to Gs while gaining increase in arrestin binding. Alternatively, there could be existence of a tertiary complex between receptor, Gs, and arrestin, as suggested by a recent co-crystallography study.45 Moreover, there may be different pools of β2ARs that favour different binding partners, particularly under certain post-translational modification such as PKA phosphorylation.
Among cardiac βARs, β1AR is the primary subtype that mediates cardiac contractile response whereas stimulation of β2AR promotes small increases in cardiac contractility. However, β2AR appears to be essential to mediate cross talk between EP-R and βAR since deletion of β2AR completely abolishes the inhibition of PGE2 on β1AR signal in cardiac myocytes.17 In contrast, deletion of β1AR does not affect the inhibitory effect of PGE2 on β2AR signal.17 This apparent paradox is further explored in the current study. While PGE2 stimulation promotes PKA phosphorylation on both β1AR and β2AR, they seem to induce opposite modulation of the receptor association with PDE4D in cardiac myocytes. Agonist stimulation of β2AR promotes arrestin-dependent binding of PDE4Ds to the activated receptor.46 In agreement, we show that PGE2 promotes PKA phosphorylation-dependent and arrestin-mediated recruitment of PDE4D to the β2AR on the plasma membrane (Figure 4B, C, and see Supplementary material online, FigureS3I). This preassembled β2AR-PDE4D complex effectively attenuates signal induced by βAR agonist ISO (see Supplementary material online, FigureS6 and ref.46). In contrast, PDE4D binds to β1AR at the baseline,47 which undergoes dissociation upon stimulation with βAR agonist ISO.46 Pretreatment with PGE2 however abolishes ISO-induced dissociation of PDE4 from β1AR, contributing to inhibition of β1AR in cardiomyocytes.17 Interestingly, this inhibitory effect of PGE2 on β1AR signal is absent in β2AR-KO myocytes, supporting a necessary role of β2AR in the process.17 Together, the cross talk between PGE2 and β2AR is sufficient to inhibit cAMP signalling and attenuate contractile responses induced by general βAR stimulation WT myocytes. The mechanism underlying the inhibitory effect of PGE2 on dissociation of β1AR-PDE4 complex is currently unclear; a possible role of β2AR and/or PGE2-induced PKA phosphorylation of β1AR in this process remain to be examined.
Agonist-induced homologous desensitization of β2AR is primarily dependent on GRK-mediated receptor phosphorylation and GRK phosphorylation-dependent β-arrestin binding.48,49 GPCRs usually possess multiple GRK phosphorylation sites that can be phosphorylated in a tissue-, cell-, or agonist-dependent manner, which has recently been postulated to create a ‘phosphorylation barcode’ that offers a way to regulate divergent signalling and trafficking of GPCRs.50 This notion could explain how the pattern of receptor phosphorylation determines the receptor conformation of binding to β-arrestins and its functional capabilities.50,51 Here, we observe an increased association between β2AR, β-arrestin 2, and PDE4D in a PKA phosphorylation-dependent manner after stimulation of EP-R by PGE2 or AT1R by Ang II. However, the PGE2 stimulation does not promote receptor internalization, which is different from βAR agonist ISO-induced β-arrestin 2-mediated receptor endocytosis in a GRK phosphorylation-dependent manner48,49. This observation is in line with previous reports showing that recruitment of β-arrestin 2 alone is not sufficient for initiating receptor internalization,52 and additional regulators such as GRK and PI3K are necessary for promoting receptor endocytosis.53 Meanwhile, GRKs can also participate in heterologous desensitization of cardiac βARs by activation of tumour necrosis factor receptors,54 insulin receptors19 or vasopressin receptors.55 Together, agonist occupancy in combination with receptor phosphorylation by PKA or GRK may offer different structural interface to interact signalling molecules. These observations essentially expand the ‘barcode’ regulation from GRKs to a broad range of kinases including PKA and PKC, revealing the complexity in the regulation of GPCR activity and trafficking under homologous and heterologous stimuli.
In the current study, why stimulation with some Gs- and Gq-coupled receptor agonists but not others promotes phosphorylation of β2AR is currently unclear; the subcellular distribution of the receptors likely plays a key role. In the case of Gs-coupled receptors, both relaxin and urocortin effectively promote cardiac contraction,56,57 but PGE2 fails to do so. In contrast, PGE2-induced cAMP diffuses into lipid rafts,36 a membrane microdomain containing β2AR. These observations argue that relaxin and urocortin induce segregated intracellular signals from those induced by PGE2. On the other hand, stimulation of either α1 adrenergic receptor by phenylephrine or angiotensin II type 1 receptor by Ang II are known to promote myocyte contractility58,59; these stimuli also promote β2AR phosphorylation (Figure 1). In comparison, thrombin is not known to promote myocyte contractility; and it fails to promote β2AR phosphorylation. Therefore, the actions of α1 adrenergic receptor and angiotensin II type I receptor activation on myocyte contractility may be converged with or relevant to β-adrenergic signal. Meanwhile, angiotensin II type I receptor may use a β-arrestin-dependent mechanism to promote contractility independent of β-adrenergic signal.60 Together, these observations suggest that different GPCRs are spatially and functionally segregated to fine-tune their functional roles in cardiomyocytes. In addition, despite that both β-arrestin 2 and β-arrestin 1 are capable of binding to βARs and PDE4,13 we observe a selective role of β-arrestin 2 in the desensitization of βAR after heterologous GPCR activation. This selectivity could fine-tune cross talk between GPCRs and βARs for local subcellular signalling, which may be further modulated by biased ligands and/or hetero-oligomeric status of GPCRs in cardiomyocytes.61,62
Our data indicate that PKC is involved in phenylephrine/Ang II induced-β2AR phosphorylation (Figure 1B and see Supplementary material online, FigureS10B). In line with these observations, phenylephrine/Ang II also promotes PDE4D phosphorylation (Figure 4A). The antibody that detects putative PKA phosphorylation sites of PDE4D (pPDE4D S190) has been used extensively in the field to detect phosphorylation of PDE4D.17,63–66 Here, we further validated the antibody specificity with tissues from WT and PDE4D-/- mouse hearts by western blots. The result shows that PDE4D-/- heart tissue display significantly reduced phosphorylation in the pPDE4DS190 antibody detection (see Supplementary material online, FigureS3H). The remaining signal in PDE4D-/- tissues is probably due to the expression of PDE4A and PDE4B proteins, which share high-sequence homologue to the PDE4D. Together, these data in Figure 4A indicate that PKC is capable to induce phosphorylation of PDE4D in animal hearts.
Under clinical conditions such as congestive heart failure or hypertrophy, many neuronal hormones and cytokines such as PGE267,68 and Ang II69,70 are evaluated. Our study reveals a general mechanism that β-arrestin 2 and PDE4D act as essential mediators in heterologous desensitization of βARs by other GPCR stimuli, which effectively block β-adrenergic-induced PKA phosphorylation of PLB and myocyte contractility. This happens in a manner depending on the phosphorylation of β2AR at putative PKA phosphorylation sites. Meanwhile, the expression, activity, and subcellular distribution of PDE4/PDE4D are depending on the stage of pathological development,71–73 which could further contribute to divergent modulation of cardiac adrenergic signalling. Together, this study expands the complexity on the regulation of cardiac βAR signal under elevated neurohormonal stimuli, which may contribute to the development of pathophysiological conditions such as heart failure.
Supplementary material
Supplementary material is available at Cardiovascular Research online.
Conflict of interest: none declared.
Funding
Studies were supported by China NSFC grants [81473212 to Q.F.] and [81428022 to Y.K.X.], and by NIH grants [S10 OD10389, HL127764, and HL112413 to Y.K.X.], and by French grant [ANR-16-CE14-0014 to D.M.]. Y.K.X. is an established investigator of the American Heart Association and a Shanghai Eastern Scholar. Q.S. and A.S. are supported by a post-doctoral fellowship from the American Heart Association. D.M. is supported by the Fondation Lefoulon-Delalande post-doctoral fellowship.
Supplementary Material
References
- 1. Gainetdinov RR, Premont RT, Bohn LM, Lefkowitz RJ, Caron MG.. Desensitization of G protein-coupled receptors and neuronal functions. Annu Rev Neurosci 2004;27:107–144. [DOI] [PubMed] [Google Scholar]
- 2. Chuang TT, Iacovelli L, Sallese M, De Blasi A. . G protein-coupled receptors: heterologous regulation of homologous desensitization and its implications. Trends Pharmacol Sci 1996;17:416–421. [DOI] [PubMed] [Google Scholar]
- 3. Rockman HA, Koch WJ, Lefkowitz RJ.. Seven-transmembrane-spanning receptors and heart function. Nature 2002;415:206–212. [DOI] [PubMed] [Google Scholar]
- 4. Moulédous L, Froment C, Dauvillier S, Burlet-Schiltz O, Zajac J-M, Mollereau C.. GRK2 protein-mediated transphosphorylation contributes to loss of function of μ-opioid receptors induced by neuropeptide FF (NPFF2) receptors. J Biol Chem 2012;287:12736–12749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cheng Y, Tao Y, Sun J, Wang Y, Xu X, Chen J, Chi Z, Liu J.. Adenosine A(1) receptor agonist N(6)-cyclohexyl-adenosine induced phosphorylation of delta opioid receptor and desensitization of its signaling. Acta Pharmacol Sin 2010;31:784–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Freedman NJ, Lefkowitz RJ.. Desensitization of G protein-coupled receptors. Recent Prog Horm Res 1996;51:319–351. [PubMed] [Google Scholar]
- 7. Willoughby D, Wong W, Schaack J, Scott JD, Cooper DMF.. An anchored PKA and PDE4 complex regulates subplasmalemmal cAMP dynamics. EMBO J 2006;25:2051–2061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Terrin A, Di Benedetto G, Pertegato V, Cheung Y-F, Baillie G, Lynch MJ, Elvassore N, Prinz A, Herberg FW, Houslay MD, Zaccolo M.. PGE(1) stimulation of HEK293 cells generates multiple contiguous domains with different [cAMP]: role of compartmentalized phosphodiesterases. J Cell Biol 2006;175:441–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Willoughby D, Baillie GS, Lynch MJ, Ciruela A, Houslay MD, Cooper DMF.. Dynamic regulation, desensitization, and cross-talk in discrete subcellular microdomains during beta2-adrenoceptor and prostanoid receptor cAMP signaling. J Biol Chem 2007;282:34235–34249. [DOI] [PubMed] [Google Scholar]
- 10. Leroy J, Abi-Gerges A, Nikolaev VO, Richter W, Lechêne P, Mazet J-L, Conti M, Fischmeister R, Vandecasteele G.. Spatiotemporal dynamics of beta-adrenergic cAMP signals and L-type Ca2+ channel regulation in adult rat ventricular myocytes: role of phosphodiesterases. Circ Res 2008;102:1091–1100. [DOI] [PubMed] [Google Scholar]
- 11. Mika D, Richter W, Conti M.. A CaMKII/PDE4D negative feedback regulates cAMP signaling. Proc Natl Acad Sci USA 2015;112:2023–2028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Perry SJ, Lefkowitz RJ.. Arresting developments in heptahelical receptor signaling and regulation. Trends Cell Biol 2002;12:130–138. [DOI] [PubMed] [Google Scholar]
- 13. Perry SJ, Baillie GS, Kohout TA, McPhee I, Magiera MM, Ang KL, Miller WE, McLean AJ, Conti M, Houslay MD, Lefkowitz RJ.. Targeting of cyclic AMP degradation to beta 2-adrenergic receptors by beta-arrestins. Science 2002;298:834–836. [DOI] [PubMed] [Google Scholar]
- 14. Dang VC, Chieng BC, Christie MJ.. Prolonged stimulation of μ-opioid receptors produces β-arrestin-2-mediated heterologous desensitization of α(2)-adrenoceptor function in locus ceruleus neurons. Mol Pharmacol 2012;82:473–480. [DOI] [PubMed] [Google Scholar]
- 15. Mundell SJ, Pula G, McIlhinney RAJ, Roberts PJ, Kelly E.. Desensitization and internalization of metabotropic glutamate receptor 1a following activation of heterologous Gq/11-coupled receptors. Biochemistry 2004;43:7541–7551. [DOI] [PubMed] [Google Scholar]
- 16. Dalle S, Imamura T, Rose DW, Worrall DS, Ugi S, Hupfeld CJ, Olefsky JM.. Insulin induces heterologous desensitization of G-protein-coupled receptor and insulin-like growth factor I signaling by downregulating beta-arrestin-1. Mol Cell Biol 2002;22:6272–6285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Liu S, Li Y, Kim S, Fu Q, Parikh D, Sridhar B, Shi Q, Zhang X, Guan Y, Chen X, Xiang YK.. Phosphodiesterases coordinate cAMP propagation induced by two stimulatory G protein-coupled receptors in hearts. Proc Natl Acad Sci USA 2012;109:6578–6583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Devic E, Xiang Y, Gould D, Kobilka B.. Beta-adrenergic receptor subtype-specific signaling in cardiac myocytes from beta(1) and beta(2) adrenoceptor knockout mice. Mol Pharmacol 2001;60:577–583. [PubMed] [Google Scholar]
- 19. Fu Q, Xu B, Liu Y, Parikh D, Li J, Li Y, Zhang Y, Riehle C, Zhu Y, Rawlings T, Shi Q, Clark RB, Chen X, Abel ED, Xiang YK.. Insulin inhibits cardiac contractility by inducing a Gi-biased β2-adrenergic signaling in hearts. Diabetes 2014;63:2676–2689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Pereira L, Cheng H, Lao DH, Na L, Oort RJ van, Brown JH, Wehrens XHT, Chen J, Bers DM.. Epac2 mediates cardiac β1-adrenergic-dependent sarcoplasmic reticulum Ca2+ leak and arrhythmia. Circulation 2013;127:913–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Jin S-LC, Latour AM, Conti M.. Generation of PDE4 knockout mice by gene targeting. Methods Mol Biol 2005;307:191–210. [DOI] [PubMed] [Google Scholar]
- 22. Mika D, Bobin P, Pomérance M, Lechêne P, Westenbroek RE, Catterall WA, Vandecasteele G, Leroy J, Fischmeister R.. Differential regulation of cardiac excitation-contraction coupling by cAMP phosphodiesterase subtypes. Cardiovasc Res 2013;100:336–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Liu S, Zhang J, Xiang YK.. FRET-based direct detection of dynamic protein kinase A activity on the sarcoplasmic reticulum in cardiomyocytes. Biochem Biophys Res Commun 2011;404:581–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Soto D, De Arcangelis V, Zhang J, Xiang Y.. Dynamic protein kinase a activities induced by beta-adrenoceptors dictate signaling propagation for substrate phosphorylation and myocyte contraction. Circ Res 2009;104:770–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Yuan N, Friedman J, Whaley BS, Clark RB.. cAMP-dependent protein kinase and protein kinase C consensus site mutations of the beta-adrenergic receptor. Effect on desensitization and stimulation of adenylylcyclase. J Biol Chem 1994;269:23032–23038. [PubMed] [Google Scholar]
- 26. Valks DM, Cook SA, Pham FH, Morrison PR, Clerk A, Sugden PH.. Phenylephrine promotes phosphorylation of Bad in cardiac myocytes through the extracellular signal-regulated kinases 1/2 and protein kinase A. J Mol Cell Cardiol 2002;34:749–763. [DOI] [PubMed] [Google Scholar]
- 27. Markou T, Hadzopoulou-Cladaras M, Lazou A.. Phenylephrine induces activation of CREB in adult rat cardiac myocytes through MSK1 and PKA signaling pathways. J Mol Cell Cardiol 2004;37:1001–1011. [DOI] [PubMed] [Google Scholar]
- 28. Benovic JL, Pike LJ, Cerione R a, Staniszewskis C, Yoshimasa T, Codina J, Caron MG, Lefkowitz RJ.. Phosphorylation of the Mammalian beta-Adrenergic Receptor by Cyclic AMP-dependent Protein Kinase. J Biol Chem 1985;260:7094–7101. [PubMed] [Google Scholar]
- 29. Baillie GS, Sood A, McPhee I, Gall I, Perry SJ, Lefkowitz RJ, Houslay MD.. beta-Arrestin-mediated PDE4 cAMP phosphodiesterase recruitment regulates beta-adrenoceptor switching from Gs to Gi. Proc Natl Acad Sci USA 2003;100:940–945. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 30. De Arcangelis V, Liu R, Soto D, Xiang Y.. Differential association of phosphodiesterase 4D isoforms with beta2-adrenoceptor in cardiac myocytes. J Biol Chem 2009;284:33824–33832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Steyaert J, Kobilka BK.. Nanobody stabilization of G protein-coupled receptor conformational states. Curr Opin Struct Biol 2011;21:567–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Rasmussen SGF, Choi H-J, Fung JJ, Pardon E, Casarosa P, Chae PS, Devree BT, Rosenbaum DM, Thian FS, Kobilka TS, Schnapp A, Konetzki I, Sunahara RK, Gellman SH, Pautsch A, Steyaert J, Weis WI, Kobilka BK.. Structure of a nanobody-stabilized active state of the β(2) adrenoceptor. Nature 2011;469:175–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Irannejad R, Tomshine JC, Tomshine JR, Chevalier M, Mahoney JP, Steyaert J, Rasmussen SGF, Sunahara RK, El-Samad H, Huang B, Zastrow M von.. Conformational biosensors reveal GPCR signalling from endosomes. Nature 2013;495:534–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Westfield GH, Rasmussen SGF, Su M, Dutta S, DeVree BT, Chung KY, Calinski D, Velez-Ruiz G, Oleskie AN, Pardon E, Chae PS, Liu T, Li S, Woods VL, Steyaert J, Kobilka BK, Sunahara RK, Skiniotis G.. Structural flexibility of the G alpha s alpha-helical domain in the beta2-adrenoceptor Gs complex. Proc Natl Acad Sci USA 2011;108:16086–16091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Baillie G, MacKenzie SJ, Houslay MD.. Phorbol 12-myristate 13-acetate triggers the protein kinase A-mediated phosphorylation and activation of the PDE4D5 cAMP phosphodiesterase in human aortic smooth muscle cells through a route involving extracellular signal regulated kinase (ERK). Mol Pharmacol 2001;60:1100–1111. [DOI] [PubMed] [Google Scholar]
- 36. Agarwal SR, Yang P-CC, Rice M, Singer C a, Nikolaev VO, Lohse MJ, Clancy CE, Harvey RD.. Role of membrane microdomains in compartmentation of cAMP signaling. PLoS One 2014;9:e95835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zhou YY, Cheng H, Bogdanov KY, Hohl C, Altschuld R, Lakatta EG, Xiao RP.. Localized cAMP-dependent signaling mediates beta 2-adrenergic modulation of cardiac excitation-contraction coupling. AmJPhysiol 1997;273:H1611–H1618. [DOI] [PubMed] [Google Scholar]
- 38. Xiao RP, Lakatta EG.. Beta 1-adrenoceptor stimulation and beta 2-adrenoceptor stimulation differ in their effects on contraction, cytosolic Ca2+, and Ca2+ current in single rat ventricular cells. Circ Res 1993;73:286–300. [DOI] [PubMed] [Google Scholar]
- 39. Zamah AM, Delahunty M, Luttrell LM, Lefkowitz RJ.. Protein kinase A-mediated phosphorylation of the beta 2-adrenergic receptor regulates its coupling to Gs and Gi. Demonstration in a reconstituted system. J Biol Chem 2002;277:31249–31256. [DOI] [PubMed] [Google Scholar]
- 40. Lefkowitz RJ, Hausdorff WP, Caron MG.. Role of phosphorylation in desensitization of the beta-adrenoceptor. Trends Pharmacol Sci 1990;11:190–194. [DOI] [PubMed] [Google Scholar]
- 41. Warrier S, Belevych AE, Ruse M, Eckert RL, Zaccolo M, Pozzan T, Harvey RD.. Beta-adrenergic- and muscarinic receptor-induced changes in cAMP activity in adult cardiac myocytes detected with FRET-based biosensor. Am J Physiol Cell Physiol 2005;289:C455–C461. [DOI] [PubMed] [Google Scholar]
- 42. Rasmussen SGF, Choi H-J, Rosenbaum DM, Kobilka TS, Thian FS, Edwards PC, Burghammer M, Ratnala VRP, Sanishvili R, Fischetti RF, Schertler GFX, Weis WI, Kobilka BK.. Crystal structure of the human β2 adrenergic G-protein-coupled receptor. Nature 2007;450:383–387. [DOI] [PubMed] [Google Scholar]
- 43. Shukla AK, Westfield GH, Xiao K, Reis RI, Huang L-Y, Tripathi-Shukla P, Qian J, Li S, Blanc A, Oleskie AN, Dosey AM, Su M, Liang C-R, Gu L-L, Shan J-M, Chen X, Hanna R, Choi M, Yao XJ, Klink BU, Kahsai AW, Sidhu SS, Koide S, Penczek PA, Kossiakoff AA, Woods VL, Kobilka BK, Skiniotis G, Lefkowitz RJ.. Visualization of arrestin recruitment by a G-protein-coupled receptor. Nature 2014;512:218–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Szczepek M, Beyrière F, Hofmann KP, Elgeti M, Kazmin R, Rose A, Bartl FJ, Stetten D von, Heck M, Sommer ME, Hildebrand PW, Scheerer P.. Crystal structure of a common GPCR-binding interface for G protein and arrestin. Nat Commun 2014;5:4801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Thomsen ARB, Plouffe B, Iii TJC, Skiniotis G, Bouvier M, Lefkowitz RJ, Thomsen ARB, Plouffe B, Iii TJC, Shukla AK, Tarrasch JT, Dosey AM.. GPCR-G protein-beta-arrestin super-complex mediates sustained G protein signaling. Cell 2016;166:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Richter W, Day P, Agrawal R, Bruss MD, Granier S, Wang YL, Rasmussen SGF, Horner K, Wang P, Lei T, Patterson AJ, Kobilka B, Conti M.. Signaling from beta1- and beta2-adrenergic receptors is defined by differential interactions with PDE4. EMBO J 2008;27:384–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. De Arcangelis V, Soto D, Xiang Y, De Arcangelis V, Soto D, Xiang Y.. Phosphodiesterase 4 and phosphatase 2A differentially regulate cAMP/protein kinase A signaling for cardiac myocyte contraction under stimulation of beta1 adrenergic receptor. Mol Pharmacol 2008;74:1453–1462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Lohse MJ, Andexinger S, Pitcher J, Trukawinski S, Codina J, Faure JP, Caron MG, Lefkowitz RJ.. Receptor-specific desensitization with purified proteins. Kinase dependence and receptor specificity of beta-arrestin and arrestin in the beta 2-adrenergic receptor and rhodopsin systems. J Biol Chem 1992;267:8558–8564. [PubMed] [Google Scholar]
- 49. Pitcher J, Lohse MJ, Codina J, Caron MG, Lefkowitz RJ.. Desensitization of the isolated beta 2-adrenergic receptor by beta-adrenergic receptor kinase, cAMP-dependent protein kinase, and protein kinase C occurs via distinct molecular mechanisms. Biochemistry 1992;31:3193–3197. [DOI] [PubMed] [Google Scholar]
- 50. Nobles KN, Xiao K, Ahn S, Shukla AK, Lam CM, Rajagopal S, Strachan RT, Huang T, Bressler EA, Hara MR, Shenoy SK, Gygi SP, Lefkowitz RJ.. Distinct phosphorylation sites on the β(2)-adrenergic receptor establish a barcode that encodes differential functions of β-arrestin. Sci Signal 2011;4:ra51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Yang F, Yu X, Liu C, Qu C-X, Gong Z, Liu H-D, Li F-H, Wang H-M, He D-F, Yi F, Song C, Tian C-L, Xiao K-H, Wang J-Y, Sun J-P.. Phospho-selective mechanisms of arrestin conformations and functions revealed by unnatural amino acid incorporation and (19)F-NMR. Nat Commun 2015;6:8202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Krasel C, Zabel U, Lorenz K, Reiner S, Al-Sabah S, Lohse MJ.. Dual role of the beta2-adrenergic receptor C terminus for the binding of beta-arrestin and receptor internalization. J Biol Chem 2008;283:31840–31848. [DOI] [PubMed] [Google Scholar]
- 53. Naga Prasad S V., Laporte SA, Chamberlain D, Caron MG, Barak L, Rockman HA.. Phosphoinositide 3-kinase regulates beta2-adrenergic receptor endocytosis by AP-2 recruitment to the receptor/beta-arrestin complex. J Cell Biol 2002;158:563–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Vasudevan NT, Mohan ML, Gupta MK, Martelli EE, Hussain AK, Qin Y, Chandrasekharan UM, Young D, Feldman AM, Sen S, Dorn GW, Dicorleto PE, Naga Prasad S V.. Gβγ-independent recruitment of G-protein coupled receptor kinase 2 drives tumor necrosis factor α-induced cardiac β-adrenergic receptor dysfunction. Circulation 2013;128:377–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Khan AR, Moukarbel G V, Tilley DG, Zhu W, Myers VD, Barr LA, Gao E, Li X, Song J, Carter RL, Makarewich CA, Yu D, Troupes CD, Grisanti LA, Coleman RC, Koch WJ, Houser SR, Cheung JY, Feldman AM.. β-adrenergic receptor-mediated cardiac contractility is inhibited via vasopressin type 1A-receptor-dependent signaling. Circulation 2014;130:e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Kakouris H, Eddie LW, Summers RJ.. Cardiac effects of relaxin in rats. Lancet 1992;339:1076–1078. [DOI] [PubMed] [Google Scholar]
- 57. Calderón-Sanchez E, Delgado C, Ruiz-Hurtado G, Domínguez-Rodríguez A, Cachofeiro V, Rodríguez-Moyano M, Gomez AM, Ordóñez A, Smani T.. Urocortin induces positive inotropic effect in rat heart. Cardiovasc Res 2009;83:717–725. [DOI] [PubMed] [Google Scholar]
- 58. Benfey BG, Carolin T.. Effect of phenylephrine on cardiac contractility and adenyl cyclase activity. Can J Physiol Pharmacol 1971;49:508–512. [DOI] [PubMed] [Google Scholar]
- 59. Ishihata A, Endoh M.. Pharmacological characteristics of the positive inotropic effect of angiotensin II in the rabbit ventricular myocardium. Br J Pharmacol 1993;108:999–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Rajagopal K, Whalen EJ, Violin JD, Stiber J a, Rosenberg PB, Premont RT, Coffman TM, Rockman H a, Lefkowitz RJ.. Beta-arrestin2-mediated inotropic effects of the angiotensin II type 1A receptor in isolated cardiac myocytes. Proc Natl Acad Sci USA 2006;103:16284–16289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Violin JD, Lefkowitz RJ.. Beta-arrestin-biased ligands at seven-transmembrane receptors. Trends Pharmacol Sci 2007;28:416–422. [DOI] [PubMed] [Google Scholar]
- 62. Maggio R, Novi F, Scarselli M, Corsini GU.. The impact of G-protein-coupled receptor hetero-oligomerization on function and pharmacology. FEBS J 2005;272:2939–2946. [DOI] [PubMed] [Google Scholar]
- 63. Zhu B, Kelly J, Vemavarapu L, Thompson WJ, Strada SJ.. Activation and induction of cyclic AMP phosphodiesterase (PDE4) in rat pulmonary microvascular endothelial cells. Biochem Pharmacol 2004;68:479–491. [DOI] [PubMed] [Google Scholar]
- 64. Fu Q, Kim S, Soto D, De Arcangelis V, DiPilato L, Liu S, Xu B, Shi Q, Zhang J, Xiang YK.. A long lasting β1 adrenergic receptor stimulation of cAMP/protein kinase A (PKA) signal in cardiac myocytes. J Biol Chem 2014;289:14771–14781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Mahavadi S, Nalli AD, Kumar DP, Hu W, Kuemmerle JF, Grider JR, Murthy KS.. Cytokine-induced iNOS and ERK1/2 inhibit AC5/6 activity and stimulate PDE4D5 activity in intestinal longitudinal smooth muscle. Am J Physiol Cell Physiol 2014;6:402–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Murthy KS, Sriwai W.. Stimulatory phosphorylation of cAMP-specific PDE4D5 by contractile agonists is mediated by PKC-dependent inactivation of protein phosphatase 2A. Am J Physiol Gastrointest Liver Physiol 2008;294:G327–G335. [DOI] [PubMed] [Google Scholar]
- 67. Dzau VJ, Packer M, Lilly LS, Swartz SL, Hollenberg NK, Williams GH.. Prostaglandins in severe congestive heart failure: relation to activation of the renin-angiotensin system and hyponatremia. N Engl J Med 1984;310:347–352. [DOI] [PubMed] [Google Scholar]
- 68. Pang L, Cai Y, Tang EHC, Irwin MG, Ma H, Xia Z.. Prostaglandin E Receptor Subtype 4 Signaling in the Heart: Role in Ischemia/Reperfusion Injury and Cardiac Hypertrophy. J Diabetes Res 2016;2016:1324347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Roig E, Perez-Villa F, Morales M.. Clinical implications of increased plasma angiotensin II despite ACE inhibitor therapy in patients with congestive heart failure. Eur Heart J 2000;21:53–57. [DOI] [PubMed] [Google Scholar]
- 70. Mazzolai L, Pedrazzini T, Nicoud F, Gabbiani G, Brunner HR, Nussberger J.. Increased cardiac angiotensin II levels induce right and left ventricular hypertrophy in normotensive mice. Hypertension 2000;35:985–991. [DOI] [PubMed] [Google Scholar]
- 71. Abi-Gerges A, Richter W, Lefebvre F, Mateo P, Varin A, Heymes C, Samuel J, Lugnier C, Conti M, Fischmeister R, Vandecasteele G.. Decreased expression and activity of cAMP phosphodiesterases in cardiac hypertrophy and its impact on beta-adrenergic cAMP signals. Circ Res 2009;105:784–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Molina CE, Leroy J, Richter W, Xie M, Scheitrum C, Lee I-O, Maack C, Rucker-Martin C, Donzeau-Gouge P, Verde I, Llach A, Hove-Madsen L, Conti M, Vandecasteele G, Fischmeister R.. Cyclic adenosine monophosphate phosphodiesterase type 4 protects against atrial arrhythmias. J Am Coll Cardiol 2012;59:2182–2190. [DOI] [PubMed] [Google Scholar]
- 73. Barbagallo F, Xu B, Reddy GR, West T, Wang Q, Fu Q, Li M, Shi Q, Ginsburg KS, Ferrier W, Isidori AM, Naro F, Patel HH, Bossuyt J, Bers D, Xiang YK.. Genetically Encoded Biosensors Reveal PKA Hyperphosphorylation on the Myofilaments in Rabbit Heart Failure. Circ Res 2016;119:931–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.