

Abstract
Neuroblastoma is the most common extracranial solid tumor in childhood. The vast majority of metastatic (M) stage patients present with disseminated tumor cells (DTCs) in the bone marrow (BM) at diagnosis and relapse. Although these cells represent a major obstacle in the treatment of neuroblastoma patients, insights into their expression profile remained elusive. The present RNA‐Seq study of stage 4/M primary tumors, enriched BM‐derived diagnostic and relapse DTCs, as well as the corresponding BM‐derived mononuclear cells (MNCs) from 53 patients revealed 322 differentially expressed genes in DTCs as compared to the tumors (q < 0.001, |log2FC|>2). Particularly, the levels of transcripts encoded by mitochondrial DNA were elevated in DTCs, whereas, for example, genes involved in angiogenesis were downregulated. Furthermore, 224 genes were highly expressed in DTCs and only slightly, if at all, in MNCs (q < 8 × 10−75 log2FC > 6). Interestingly, we found the transcriptome of relapse DTCs largely resembling those of diagnostic DTCs with only 113 differentially expressed genes under relaxed cut‐offs (q < 0.01, |log2FC|>0.5). Notably, relapse DTCs showed a positional enrichment of 31 downregulated genes on chromosome 19, including five tumor suppressor genes: SIRT6, BBC3/PUMA, STK11, CADM4 and GLTSCR2. This first RNA‐Seq analysis of neuroblastoma DTCs revealed their unique expression profile in comparison to the tumors and MNCs, and less pronounced differences between diagnostic and relapse DTCs. The latter preferentially affected downregulation of genes encoded by chromosome 19. As these alterations might be associated with treatment failure and disease relapse, further functional studies on DTCs should be considered.
Keywords: neuroblastoma, disseminated tumor cells, RNA‐Seq, bone marrow
Short abstract
What's new?
More than 90% of patients diagnosed with stage 4 metastatic (4/M) neuroblastoma present with disseminated tumor cells (DTCs) in the bone marrow (BM). Despite treatment, a substantial fraction of these patients experience disease relapse. Here, sequencing analysis of tumor tissue, BM‐derived mononuclear cells (MNCs), and DTCs from stage 4/M neuroblastoma patients indicates that numerous genes are differentially expressed in DTCs but are not or are only slightly altered in tumors and MNCs. Moreover, DTCs exhibited significant downregulation of tumor suppressor genes specifically on chromosome 19. Further studies are needed to determine whether DTC transcriptomic alterations are associated with neuroblastoma relapse.
Abbreviations
- BM
bone marrow
- cnLOH
copy‐neutral loss of heterozygosity
- DTC
disseminated tumor cell
- IF
immunofluorescence
- MNA
MYCN amplification
- MNC
mononuclear cell (in the context of this study: DTC‐depleted MNC fraction of the BM)
- MRD
minimal residual disease
- OXPHOS
oxidative phosphorylation
Introduction
Neuroblastoma is the most common solid tumor diagnosed in the first year of life. It is characterized by a remarkably diverse clinical behavior ranging from spontaneous regression or maturation to malignant disease. This diversity is mainly due to the complex biology and genetics of the tumor itself.1, 2 While tumors with a favorable clinical outcome frequently show numerical chromosomal aberrations and only rarely structural ones, neuroblastomas with unfavorable clinical outcome are commonly characterized by segmental chromosomal aberrations and MYCN amplifications (MNAs).1, 3, 4 Deep‐sequencing studies have provided additional insights into the genomic landscape of the tumor, revealing that chromothripsis, as well as mutations and deletions of particular genes are associated with high‐risk disease at various frequencies.5, 6 In addition, efforts have been made to improve our understanding of the transcriptomic landscape of neuroblastomas. These studies mainly analyzed the prognostic value of gene expression‐based classifiers for neuroblastoma, concluding an improved clinical endpoint prediction.7, 8, 9
Although all these genomic and transcriptomic studies have advanced our understanding of the disease, they have mainly focused on primary tumors. However, a common feature in neuroblastoma is the presence of disseminated tumor cells (DTCs) in the bone marrow (BM) of metastatic (M) stage patients. More than 90% of stage M patients present with DTC infiltration in the BM at diagnosis.10 Detection of DTCs in the BM of patients who are older than 18 months is of high prognostic importance, as these patients frequently suffer from disease recurrence and poor outcome.11 Importantly, BM can be used for the molecular quantification of minimal residual disease (MRD) and outcome prediction.12
In our recent studies, we have shown that BM‐derived DTCs are suitable for genomic and transcriptomic analyses.13, 14 Furthermore, it has been demonstrated that DTCs can be highly informative regarding the identification of the relapse‐seeding clone.15 However, little is known about gene expression changes occurring in DTCs upon dissemination and tumor progression. So far, only a single group has performed gene expression profiling of BM‐derived DTCs from 11 neuroblastoma patients by microarray analysis. In this study, Morandi et al. mainly focused on the identification of genes differentially expressed between diagnostic DTCs and primary tumors to identify genes which could serve as prognostic markers for high‐risk neuroblastoma patients.16
With our study, we aim to shed more light on the transcriptomic landscape of DTCs, which are frequently considered to lead to relapse in various cancers.17 To our knowledge, we present the first RNA‐Seq study of disseminated neuroblastoma cells in which their expression profile was characterized by sequencing the mRNA and comparing the expression profiles of primary tumors, BM‐derived DTCs and the corresponding BM‐derived mononuclear cells (MNCs). Our findings may stimulate further functional research projects to decipher the DTCs that frequently cause relapse in neuroblastoma patients.
Materials and Methods
Overview of patient samples
Primary tumor (n = 16), DTC (n = 42), and corresponding MNC (n = 28) samples of 53 stage M neuroblastoma patients were sequenced and analyzed. For three patients (n = 3), matching DTC and tumor samples were available at diagnosis. For five patients (n = 5), matching diagnostic and relapse DTCs were available. One DTC sample was sequenced twice, before enrichment (D07r2) and after enrichment (D07r). An overview of patients and samples is shown in Figure 1 and the Supporting Information Table S1. RNA‐Seq data are available on the gene expression omnibus (GEO) repository (accession number GSE94035). This study was approved by the St. Anna Kinderspital Ethics Commission in Vienna, Austria.
Figure 1.
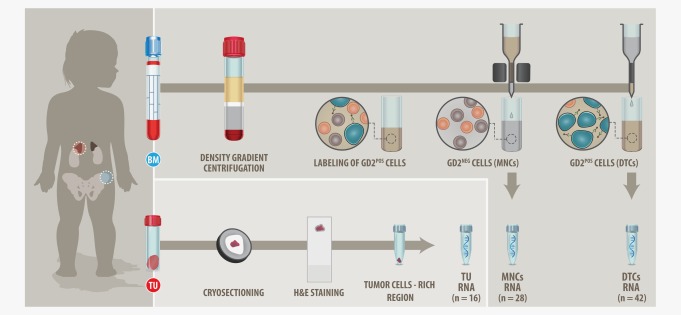
Study design. mRNA of BM cells (DTCs and MNCs) and primary tumors of stage M neuroblastoma patients was sequenced. The MNC fraction of BM aspirates was isolated by density gradient separation, DMSO frozen and stored in liquid nitrogen. Upon thawing, dead cells were removed by an additional density gradient separation and the MNCs were used for a magnetic bead‐based enrichment of DTCs. mRNA of DTCs (n = 42) was isolated and sequenced. mRNA of corresponding tumor cells‐depleted BM samples (MNCs, n = 28) was sequenced as well (MNC fractions with total RNA <30 ng or detectable DTCs were excluded from sequencing). Primary tumors were cryosectioned and H&E stained. Regions rich in tumor cells were determined by light microscopy and the corresponding regions were cut out of the frozen tumor pieces and used for RNA isolation and sequencing (n = 16). [Color figure can be viewed at wileyonlinelibrary.com]
Sample preparation for RNA‐seq
The primary tumor and BM samples were stored in liquid nitrogen. Cryosection slides of tumors were prepared and H&E stainings were performed. Identified tumor cells‐rich regions were cut out from the respective tumor pieces and homogenized in QIAzol (Qiagen) with the gentleMACS Dissociator (Miltenyi). The dimethyl sulfoxide (DMSO) frozen MNC fraction of BM aspirates was thawed and density gradient centrifugation (LymphoprepTM, AXIS‐SHIELD PoC AS) was performed. Following the density gradient centrifugation, DTCs were labeled and enriched at 4°C as described earlier.13 In brief, the MNC fraction containing DTCs was collected after density gradient separation and washed with phosphate‐buffered saline (PBS) at 300g for 10 min at 4°C. The supernatant was discarded and the cells were resuspended in 2 ml ice‐cold magnetic‐activated cell sorting (MACS) buffer (PBS pH 7.2, 0.5% bovine serum albumin, 2 mM ethylene diamine tetraacetic acid). Thereafter, the cell suspension was incubated at 4°C for 20 min with 2.5 µl of FITC‐labeled anti‐GD2 antibodies (14.18 delta CH2 clone), followed by a 15‐min incubation with 75 µl anti‐FITC magnetic beads (Miltenyi) at 4°C. The MACS sorting was continued at 4°C and the enriched DTC fraction and the corresponding tumor cells‐depleted MNC fraction were separately collected and homogenized in QIAzol.
RNA extraction
Total RNA was extracted with the miRNeasy Micro Kit (Qiagen) following the manufacturer's protocol. Quantity and integrity of extracted RNA was assessed by the Qubit RNA HS Assay Kit (Life Technologies) and the Experion RNA StdSens Assay Kit (BioRad), respectively.
Library preparation and RNA‐seq
A 30‐ng of total RNA was used for cDNA synthesis following the NEBNext Ultra RNA Library Prep Kit for Illumina protocol (New England BioLabs) with the Poly(A) mRNA Magnetic Isolation Module (New England BioLabs). After cDNA synthesis, the library was completed in an automated way at the EMBL Genomics Core Facility (Heidelberg, Germany). RNA‐Seq was performed at the Illumina HiSeq 2000 platform. Six samples were pooled per lane and 50 bp‐single‐end reads were generated.
RNA‐seq gene expression and gene set enrichment analysis
Average raw RNA sequencing yield was 36 million (M) reads per sample (range 12–172 M), of which 16 M reads (44%) mapped uniquely to protein‐coding exons (range 5–74 M) (Supporting Information Fig. S1; Table S2). Reads were mapped with GSNAP v2014‐12–2818 (“–maxsearch = 100 –npaths = 1 –max‐mismatches = 1 –novelsplicing = 0”) to human reference GRCh37 and then assigned to Ensembl gene models (build 75) using HTSeq19 (“htseq‐count ‐f bam ‐t exon ‐s no”). rRNA genes were removed from the Ensembl gene set before read counting and thus excluded from all subsequent analyses. Known single nucleotide polymorphisms (SNPs) and splice sites for GSNAP were extracted from the database SNP (dbSNP) build 138 and Ensembl GRCh37 build 75, respectively. After read mapping and counting, DESeq220 was used to call differentially expressed genes (nbinomWaldTest, minReplicates = 5, cooksCutoff = 0.7, trim = 0.4). In addition, we used DESeq2 to generate a normalized (function “fpm”, robust = TRUE) and variance‐stabilized (function “vst”) gene expression matrix for import into and further analysis in Qlucore v3.2. All samples used in this study passed internal quality control (QC) checks, including base qualities, mapping rates, duplication rates, and 5′–3′ coverage, which were performed on the basis of FastQC and RSeQC reports.21
Gene set enrichment analysis (GSEA) was performed with the xtools.gsea.GseaPreranked module implemented in Broad's javaGSEA stand‐alone desktop application (v2.0.13).22, 23 Gene sets for GSEA were downloaded from MSigDB 5.0.22 Input genes were ranked by DESeq2 p values from most to least significant, considering the directionality of change. False discovery rates were empirically assessed by permutation analysis with 1,000 iterations. GSEA command line options included “‐collapse false –mode Max_probe –norm meandiv –scoring_scheme weighted –include_only_symbols true –make_sets true –rnd_seed 149 –gui false –nperm 1000 –set_max 5000 –set_min 5” (all other options default).
The complete RNA‐Seq analysis pipeline (including alignment, QC, differential gene expression analysis, and GSEA) was implemented and run on the workflow management platform Anduril (http://www.anduril.org).24
Positional enrichment of differentially expressed genes
To find chromosomal regions enriched for differentially expressed genes, we used the Perl script “pge.pl” provided by De Preter et al.25 This method implements a hypergeometric test to test for significant co‐localization of a given query gene set among a given background (or reference) gene set. In Figure 6, the query gene set comprised all genes significantly differentially expressed between diagnosis and relapse DTC samples (q ≤ 0.2, |log2FC| ≤ 0.5) and the background gene set contained all genes from Ensembl GCRh37 build 75 except rRNA genes.
Sample preparation and DNA extraction for qPCR
DNA was extracted from samples of 10 patients (n = 10) with the high salt extraction method. For four patients (n = 4), the DNA was extracted from the primary tumor, the BM‐derived DTCs and a tumor cell‐free BM or peripheral blood sample. For another four patients (n = 4), the DNA was extracted from the primary tumor and the BM‐derived DTCs. For the remaining two patients (n = 2), the DNA was extracted from DTCs and tumor cells‐free BM.
Real‐time qPCR
For each quantitative polymerase chain reaction (qPCR) reaction, 6.25 ng DNA was used, and for each sample, three technical replicates were performed. Primers for the nuclear ALB gene (located on chromosome 4) and for a 344‐bp fragment of the mitochondrial genome (nucleotides m.8151–8494), encompassing the MT‐TK gene were used: ALB exon 12 (GenBank NM_000477.6) forward: 5′‐AAT GCT GCA CAG AAT CCT TGGT‐3′, reverse: 5′‐TCA TCG ACT TCC AGA GCT GAAA‐3′; MT‐TK forward: 5′‐CGG GGG TAT ACT ACG GTC AA‐3′, reverse: 5′‐TTT TAT GGG CTT TGG TGA GG‐3′). Template DNA was amplified in the presence of each primer (0.5 µM) and reagents of the SensiMix SYBRHi‐ROX Kit (Bioline) in 12.5 µl‐reactions using the Stratagene Mx3005P qPCR system (Agilent Technologies). Carboxy‐X‐rhodamine (ROX) was used as passive reference dye. Cycling conditions: 10 min 95°C, 40 × [15 sec 95°C, 30 sec 60°C, 20 sec 72°C], followed by a dissociation segment for melting curve analysis. SYBR Green fluorescence data were analyzed using the MxPro 4.10 software (Agilent Technologies). The relative mitochondrial DNA (mtDNA) amount for each patient sample was obtained by normalization of the MT‐TK Ct values with the Ct values of the ALB gene. Ct values were calibrated to the DNA of peripheral blood from a healthy individual.
Genomic copy‐number analysis
For 34 enriched DTCs, we used one aliquot of the samples for genome analysis that was performed earlier.13, 15 The data were analyzed with the ChAS software (Affymetrix). SNP array data analyzed for these studies are available on the GEO repository under the accession number GSE84291, and the corresponding GEO identifier can be found in the Supporting Information Table S3.
Results
Magnetic bead‐based enrichment led to a three‐fold increase in DTC purity
DTCs are usually underrepresented in the BM, which necessitates their enrichment before gene expression analysis. The median infiltration of GD2POS tumor cells in the BM aspirates (n = 42) was determined by immunofluorescence (IF) microscopy to be 20% before enrichment (range 1%–80%) and 65% (range 19%–96%) after magnetic bead‐based enrichment (Fig. 2 a). The DTC content after enrichment was sample‐dependent (Fig. 2 b) with 27% of samples containing <50% DTCs, 37% of samples containing 50%–75% DTCs, and 37% of samples with >75% DTCs after enrichment. Overall, enrichment resulted in a threefold increase in tumor cell content within DTC samples. Negative fractions (MNCs) were examined by IF (Fig. 2 c) and all samples with detectable GD2POS DTCs were excluded from further analysis.
Figure 2.
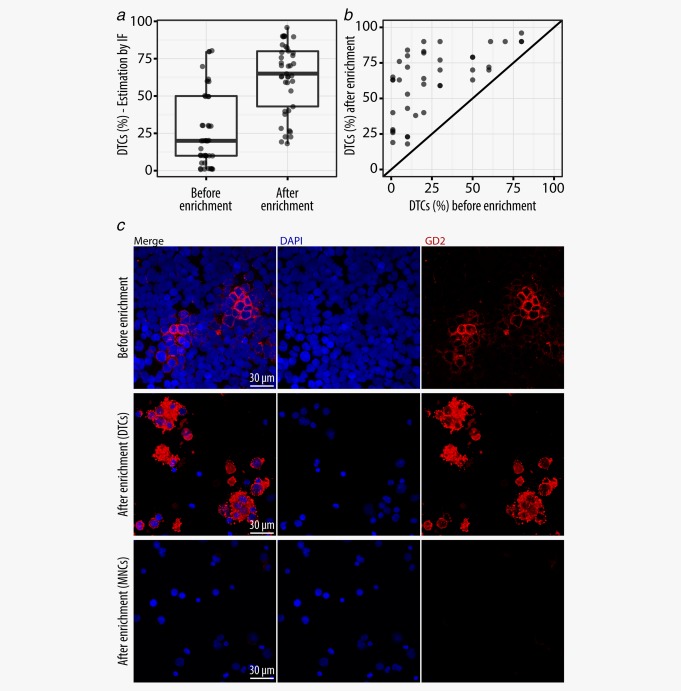
Efficiency of DTC enrichment. (a) Tumor cell content of DTC samples before and after enrichment determined by GD2‐IF. (b) Enrichment efficiency for each individual DTC sample determined by IF. (c) IF staining of BM aspirates before enrichment and corresponding positive (enriched DTCs) and negative (DTC‐depleted MNCs) fractions after enrichment. [Color figure can be viewed at wileyonlinelibrary.com]
Gene expression signature dominated by cell type and MYCN expression
We expected differences in global gene expression patterns to be largely driven by cell type (for convenience we label tumor, DTC and MNC samples as distinct cell types in this article). Indeed, principal component analysis of all genes transcribed in tumors and MNCs unambiguously separated samples by cell type along the first principal component (x‐axis), with tumor samples (16 diagnostic samples) clustering to the right and MNC samples (n = 28: 14 diagnostic samples and 14 relapse samples) to the left (Fig. 3 a). In‐between these two clusters and along the same axis, DTCs (n = 42: 22 diagnostic and 20 relapse samples) scattered according to their tumor cell content. MYCN expression status explained most of the remaining gene expression variability in tumor and DTC samples (second principal component, y‐axis) (Fig. 3 b), revealing a strong effect of MYCN upregulation on the global gene expression of tumors (n = 6 with MYCN‐low and n = 10 with MYCN‐high) and DTCs (n = 26 with MYCN‐low and n = 16 with MYCN‐high). GSEA for DTC MYCN‐high and MYCN‐low samples revealed several previously described MYCN‐associated gene sets (Supporting Information Table S4), including a gene set containing genes coregulated with MYCN upregulation in primary neuroblastomas26 (Fig. 3 c). Furthermore, unsupervised clustering with the top‐100 differentially expressed genes between MYCN‐high and MYCN‐low DTC samples (Supporting Information Table S5) separated tumors and DTCs by MYCN expression rather than cell type or tumor cell content (Fig. 3 d). Seventy six genes were upregulated in DTC MYCN‐high samples, with MYCN itself being the most significantly upregulated gene (q = 3.7 × 10−51, log2FC = 4.9). Except for one case (D04d, sample with 82% tumor cells), MYCN expression levels were in line with the MNA status as determined by fluorescence in situ hybridization (FISH) and/or SNP array (Supplementary Information Table S1, with additional information for D04d). D04d and the matching tumor sample contained approximately 25–30 copies of MYCN as determined by SNP array (Supporting Information Fig. S2). Unfortunately, no expression information on the corresponding tumor was available due to degraded RNA in this sample. The remaining 24 genes were downregulated in the DTC MYCN‐high samples. Although not among the top‐100 regulated genes, MYC was downregulated in DTC MYCN‐high samples (q = 0.006, log2FC = 1).
Figure 3.
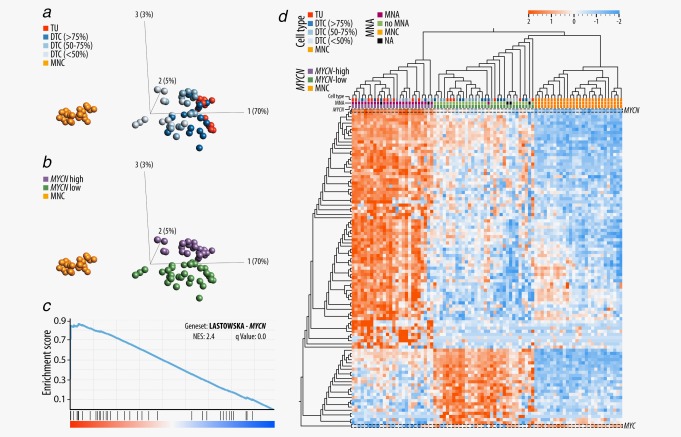
Cell type and MYCN driven expression signatures of tumor, DTC and MNC samples. (a) Principle component analysis of TU, DTC and MNC samples with cell type specific annotations. DTC samples were annotated in three different groups, according to the tumor cell content determined by IF. (b) Principle component analysis of TU, DTC and MNC samples with a MYCN expression level specific annotation. (c) A representative gene set (LASTOWSKA_COAMPLIFIED_WITH_MYCN) that was enriched in the comparison of DTC MYCN‐high and DTC MYCN‐low samples. (d) Unsupervised clustering of TU, DTC and MNC samples based on the expression of 101 genes: the top 100 differentially transcribed genes between DTC MYCN‐high and DTC MYCN‐low samples (sorted by q value, |log2FC|>1) and in addition MYC. MYCN is highlighted in the heat map as the most significantly upregulated gene in the DTC MYCN‐high samples (q = 3.7 × 10−51, log2FC = 4.9). MYC is highlighted, but is not among the top 100 differentially expressed genes (q = 0.047, Log2FC = −1). Abbreviation: NES: normalized enrichment score. [Color figure can be viewed at wileyonlinelibrary.com]
Candidate markers for MRD diagnosis in stage M neuroblastoma patients
The identification of robust MRD markers is a crucial step in the exact diagnosis of tumor cells in the BM. By comparing the gene expression of DTCs (n = 42) and MNCs (n = 28), we found that eight markers for MRD diagnosis in neuroblastoma proposed by Cheung et al.27 were upregulated in tumor and DTC samples, and only marginally or not at all transcribed in the corresponding MNC samples (Fig. 4 a). Among these eight genes, PHOX2B was the most significantly upregulated gene in our data set (q = 3 × 10−234, log2FC = 8.7). Noteworthy, in one DTC sample (D27d), PHOX2B was not transcribed. GABRB3, CRMP1, KIF1A, CCND1, TACC2, ISL1 and DDC were all significantly upregulated in DTCs as compared to MNCs (q < 8.5 × 10−74, log2FC > 6). However, it is notable that CCND1 was also transcribed by non‐tumor cells of the BM. Furthermore, we found a higher variation in the DDC expression among tumor and DTC samples as compared to the other genes. This variance was associated with MYCN expression as DDC was downregulated in DTC MYCN‐high (n = 16) samples as compared to MYCN‐low samples (n = 26) (q = 0.03, log2FC = 1). In addition to the eight published genes, we identified 224 genes that were significantly upregulated in DTCs as compared to corresponding MNCs (q < 8 × 10−75 log2FC > 6) (Supporting Information Table S6). The eight most significantly upregulated genes (q < 1 × 10−232) were ELAVL3, MAST1, SCG3, MAP1B, MAPK8IP2, DPYSL5, REEP2 and SOX11 (Fig. 4 b). The differential expression between DTCs and MNCs of the top seven identified genes was more significant (q < 1.3 × 10−243) than the differential expression of PHOX2B (q = 3 × 10−234).
Figure 4.
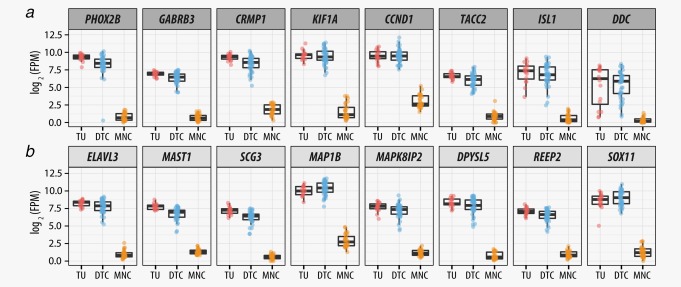
Differentially expressed genes in tumor, DTC and MNC samples. (a) Expression of MRD marker in the BM of neuroblastoma patients that are known from the literature.27 (b) The top eight upregulated genes in DTCs as compared to the corresponding MNCs. Abbreviation: FPM: fragments per million. [Color figure can be viewed at wileyonlinelibrary.com]
DTCs overexpress genes encoded by mitochondrial DNA
The tumor microenvironment is of critical importance for dissemination, arrest and progression of DTCs, and therefore, we were keen to characterize the DTCs which were infiltrating the BM. We compared the expression signature of tumors (total number = 16: 10 with MYCN‐high and 6 with MYCN‐low) to DTCs (total number = 42: 16 with MYCN‐high and 26 with MYCN‐low) in a non‐paired manner and found that the expression levels of neuroblastoma hallmark genes MYCN and PHOX2B were retained in DTCs. However, we identified 322 genes with an altered gene expression (q < 0.001, |log2FC|>2). We confirmed our observations by comparing the tumor samples with a subset of highly enriched DTC samples (tumor cells > 90%, n = 3, Supporting Information Table S7). Among the top differentially expressed genes between tumor and DTC samples (Fig. 5 a), we found genes involved in metastasis initiation and angiogenesis, for example, MGP, BGN, POSTN and ANGPT2, to be upregulated in tumors. The differentially expressed genes were enriched in several gene sets (Supporting Information Table S8) including the Hallmark Angiogenesis gene set (Fig. 5 b) and other gene sets containing genes involved in metastasis initiation. When comparing the gene expression signature of DTC and tumor samples under consideration of the MYCN expression level (Supporting Information Table S9), the expression profile largely resembled the above mentioned gene sets.
Figure 5.
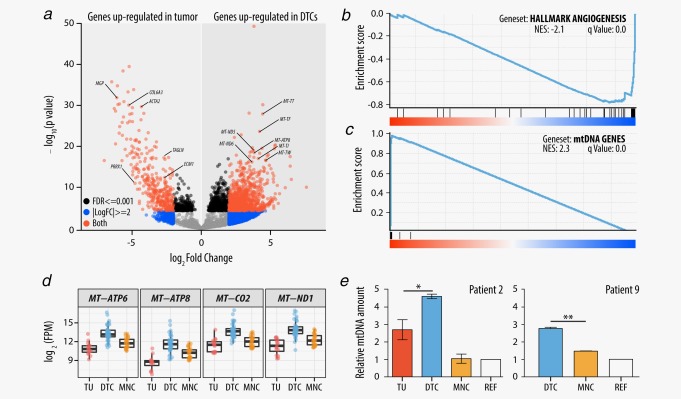
Differentially expressed genes between tumor and DTC samples. (a) Volcano plot of log2FC versus p values comparing gene expression of tumor and DTC samples. p values were capped at 1 × 10−200. Selected upregulated genes in the tumor are known drivers of metastasis initiation and or angiogenesis. A selection of genes that are encoded by the mtDNA and overexpressed in DTCs is labeled. (b) The HALLMARK_ANGIOGENESIS gene set which was enriched in the gene set enrichment analysis for tumor and DTC samples. (c) Gene set enrichment analysis for tumor and DTC samples using the complete set of genes encoded by the mitochondrial genome. (d) Expression levels of representative genes encoded by the mtDNA in tumor, DTC and MNC samples. (e) Relative amount of mtDNA in two representative patients. The mitochondrial DNA amount was determined by qPCR for each patient and with three technical replicates for each sample. Ct values were normalized with Ct values of the ALB gene (chromosome 4). All values were calibrated with the mitochondrial DNA quantity of a peripheral blood sample of a healthy individual. REF refers to BM‐derived MNCs of the particular patient at a time point when the BM was tumor cell‐free. Abbreviations: FPM: fragments per million; NES: normalized enrichment score. [Color figure can be viewed at wileyonlinelibrary.com]
Interestingly, transcript levels of genes encoded by the mtDNA were found to be significantly higher in DTCs as compared to the analyzed tumors and MNCs (Figs. 5 a, 5 c and 5 d). In a cohort of eight patients, we performed qPCR experiments to quantify the mtDNA levels. In six of these eight patients, the quantity of mtDNA was significantly higher in the DTC samples as compared to the matching tumors (Fig. 5 e; Supporting Information Table S10). However, genes encoded by the nuclear DNA (nDNA), which are essential for mitochondrial function such as the oxidative phosphorylation (OXPHOS) and which are part of the same protein complexes as those encoded by the mtDNA, were not upregulated in DTCs (Supporting Information Fig. S3).
Relapse DTCs downregulate genes on chromosome 19
By comparing the gene expression signature of diagnostic (total number = 22: 10 with MYCN‐high and 12 with MYCN‐low) and relapse DTCs (total number = 20: 6 with MYCN‐high and 14 with MYCN‐low) in a non‐paired manner, we found the gene expression to be fairly stable over time, with only 113 differentially expressed genes under relaxed cut‐offs (q < 0.01, |log2FC| > 0.5; Fig. 6 a; Supporting Information Table S11). Interestingly, among the downregulated genes in relapse DTCs, we found a strong positional enrichment on chromosome 19 (31 of 113 genes; positional enrichment q value = 9 × 10−38; Fig. 6 b). Notably, we identified five tumor suppressors among the 31 downregulated genes; STK11, SIRT6, CADM4, BBC3 (also known as PUMA) and GLTSCR2. Genomic analysis of 34 DTC samples revealed clonal deletions affecting a portion of the 31 downregulated genes encoded by chromosome 19. Although these deletions occurred at higher frequency in relapse patients (9 of 17 samples) as compared to the diagnostic samples (4 of 17 samples), the positional distribution of downregulated and deleted genes did not coincide. While chromosomal aberrations in relapse samples were mainly found on 19q (8 of 20 samples), most of the downregulated genes were encoded by 19p (Fig. 6 b; Supporting Information Table S3) which rarely contained deletions (3 of 20 samples). The three tumor suppressor genes encoded by 19q (CADM4, BBC3/PUMA and GLTSCR2) were more frequently deleted in relapse DTCs (29%) as compared to the diagnostic DTCs (12%), whereas the two tumor suppressor genes encoded by 19p were deleted at lower frequencies. By correlating the gene expression of the identified tumor suppressor genes with the overall survival and event free survival (EFS) in publically available datasets5, 28, 29, 30 (R2: Genomics Analysis and Visualization Platform http://r2.amc.nl), we found low expression levels of CADM4 and BBC3/PUMA to correlate with worse EFS (Supporting Information Fig. S4; Table S12).
Figure 6.
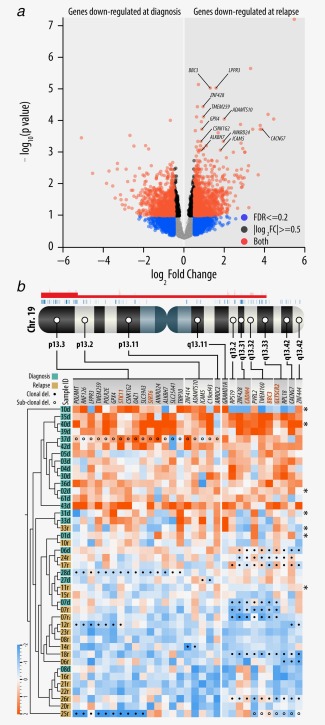
Positional enrichment of genes encoded by chromosome 19. (a) Volcano plot of log2FC versus p values comparing gene expression of diagnostic and relapse DTCs. A selection of genes which are upregulated in relapse DTCs and which are encoded by chromosome 19 is highlighted. (b) Ideogram showing the genomic location of 31 differentially expressed genes encoded by chromosome 19. Significantly enriched chromosomal regions (p < 0.01) are shown as red segments above the chromosome. Clonal deletions of particular genes are highlighted with solid circles in the heat map. Sub‐clonal deletions are labeled with non‐solid circles. Names of tumor suppressor genes are highlighted. Samples with no available SNP array data are highlighted with (*). [Color figure can be viewed at wileyonlinelibrary.com]
Discussion
BM is a frequent homing organ for DTCs of various cancer types. It can serve as a metastatic niche for DTCs, which finally can cause disease recurrence after treatment.31 Thus, the presence of DTCs is considered as an important prognostic marker for poor outcome in various cancer types, such as breast cancer,32 Ewing sarcoma,33 and neuroblastoma—with the exception of stage MS tumors which frequently show a minor BM involvement despite mostly favorable disease outcome.3, 34 Therefore, the characterization of DTCs may help to improve our knowledge about treatment failure and disease relapse.
We searched for expression changes occurring during the course of disease by analyzing the gene expression profiles of diagnostic and relapse DTCs. Unexpectedly, the transcriptional landscape of diagnostic DTCs resembled that of relapse DTCs to a large extent, although these patients had experienced multimodal treatment. This observation may be explained by the protective BM niche, just as it has been shown that the BM microenvironment promotes their survival, dormancy, growth and drug resistance.35 Remarkably, we found that the genes which were differentially expressed between diagnostic and relapse DTCs show a positional enrichment on chromosome 19, with five tumor suppressor genes being downregulated in relapse DTCs. Although chromosome 19 deletions are only rarely observed in neuroblastoma,36 several studies reported deletions or copy neutral loss of heterozygosity (cnLOH) in primary tumors at low frequency.37, 38 Interestingly, our recent study revealed a higher frequency of partial chromosome 19 deletions in DTCs of relapse patients as compared to diagnostic DTCs.15 Although most reported chromosome 19 deletions in our and other studies affected the q‐arm, we found most of the differentially expressed genes to be encoded by the p‐arm. While the downregulation of genes located on the q‐arm can be explained, at least partially, by deletions of the particular genes, the rare gene deletions in the p‐arm suggest other, trans‐regulatory mechanisms of gene expression.1 Thus, epigenetic studies focusing on DTCs seem reasonable and could shed more light on the transcriptomic alterations occurring in these cells.
Chromosome 19 encodes several well described tumor suppressor genes whose downregulation is associated with progression in various cancers. We identified five of these tumor suppressor genes to be downregulated in relapse DTCs. BBC3/PUMA is a critical apoptosis inducer whose expression ablation is oncogenic and can lead to therapeutic resistance.39 A recent study demonstrated that BBC3/PUMA is the critical determinant of the therapeutic response to p53 activation and subsequent apoptosis induced by Nutlin3a, a cancer therapeutics that is in clinical trial.40 A screening study with flubendazole, another compound that exerts anti‐cancer activity, identified neuroblastoma as a potential flubendazole‐sensitive cancer entity. The flubendazole efficacy was increased by nutlin‐3, but the flubendazole‐induced anti‐neuroblastoma effect was significantly impaired upon BBC3/PUMA depletion in the analyzed neuroblastoma cell lines.41 SIRT6 is another tumor suppressor that we found to be downregulated in relapse DTCs and whose loss was associated with poor outcome in several cancers.42 Analysis of data from the Cancer Genome Atlas database revealed that SIRT6 was deleted in 20% of analyzed cancers.43 The loss and/or downregulation of the remaining three suppressor genes in DTCs of relapse samples, namely STK11, CADM4 and GLTSCR2, was also reported to be associated with tumor progression in various cancers. Thus, we assume that the downregulation of the five tumor suppressor genes encoded by chromosome 19 in relapse DTCs represents a possible survival advantage for DTCs.
Furthermore, we found that genes involved in metastasis initiation and angiogenesis were upregulated in the primary tumors in contrast to DTCs and MNCs, which reside in the highly vascularized BM.44 Besides the downregulation of these genes, DTCs were particularly characterized by an increased amount of mtDNA and the corresponding transcripts that are encoded by this small genome. Notably, also BM‐derived MNCs had elevated levels of mtDNA transcripts as compared to primary tumors, although significantly less than BM‐derived DTCs. A numerical increase in mitochondria is a characteristic feature of various cancers, and it is well known that mitochondria impart considerable flexibility for cancer cells in harsh environments, such as hypoxic BM.45 However, the elevated level of transcripts encoded by mtDNA in DTCs is rather surprising, as these genes code only for a minority of subunits that are necessary for functional mitochondria. The vast majority of subunits, for example, for the OXPHOS complex, is encoded by the nDNA. However, unexpectedly, these genes were not upregulated in DTCs. A possible explanation for this phenomenon is the uptake of functional mitochondria by tumor cells from the BM microenvironment.46, 47 However, functional studies will be necessary to understand the exact mechanism and causes for elevated mtDNA and increased levels of transcripts encoded by it. Apart from these and other differences, DTCs also retained oncogenic features of the tumor. Particularly, the transcription of MYCN was comparable in tumor and DTC samples, as well as the transcription of MYC, which was earlier shown to be anti‐correlated with MYCN in neuroblastoma.48
As the BM is a common homing organ for disseminated neuroblastoma cells, it is widely used for MRD testing during or after cytotoxic treatment.34 Besides the standardized immunocytological detection of MRD,10 real‐time quantitative polymerase chain reaction approaches are also used for MRD detection.12 Our three‐fold enrichment of BM‐derived DTCs and their separation from the corresponding BM‐derived MNCs enabled us to characterize their gene expression profiles and to extend the variety of potential marker for MRD detection in stage M neuroblastoma. Our data indicate that the detection of one or two MRD markers can lead to unreliable results, as the expression of tumor‐specific genes varies between samples and, moreover, a number of genes can also be expressed by non‐tumor cells of the BM. Finally, not all supposed NB‐specific markers are expressed by all DTCs, as we could, for example, show for PHOX2B, the widely used MRD marker in neuroblastoma. Thus, it is necessary to consider testing and applying MRD marker panels as suggested by Stutterheim et al..49 For this purpose, for example, 2 to 3 of the 224 most highly and specifically expressed genes in BM‐derived DTCs might be considered for the design of highly specific and sensitive MRD detection markers. Furthermore, feature selection methods50 could be applied to our data to determine a minimal discriminant subset of genes being sufficient to classify presence versus absence of DTCs in the BM, serving as the basis to create a diagnostic test with a predefined sensitivity and specificity.
In conclusion, our study demonstrated that disseminated neuroblastoma cells retain some of their tumor's oncogenic features, but in addition undergo transcriptomic changes upon dissemination and cancer progression, particularly, the downregulation of tumor suppressor genes encoded by chromosome 19. These insights can stimulate further functional, and especially epigenetic, studies to decipher those cells that are assumed to be the main cause for treatment failure and disease relapse in high‐risk neuroblastoma patients.
Supporting information
Supporting Information Figure 1
Supporting Information Figure 2
Supporting Information Figure 3
Supporting Information Figure 4
Supporting Information Table 1
Supporting Information Table 2
Supporting Information Table 3
Supporting Information Table 4
Supporting Information Table 5
Supporting Information Table 6
Supporting Information Table 7
Supporting Information Table 8
Supporting Information Table 9
Supporting Information Table 10
Supporting Information Table 11
Supporting Information Table 12
Supporting Information Legends
Supporting Information Letter
Acknowledgements
The authors thank the use of the Molecular Signature Database (MSigDB) (http://www.broad.mit.edu/gsea/) and Marion Zavadil for proofreading the manuscript and Florian Kromp for his intellectual input.
Conflict of interest: The authors declare that they have no potential conflicts of interest.
Contributor Information
Fikret Rifatbegovic, Email: fikret.rifatbegovic@ccri.at.
Peter F. Ambros, Email: peter.ambros@ccri.at
References
- 1. Brodeur GM, Bagatell R. Mechanisms of neuroblastoma regression. Nat Rev Clin Oncol 2014;11:704–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Matthay KK, Maris JM, Schleiermacher G, et al. Neuroblastoma. Nat Rev Dis Primers 2016;2:16078 [DOI] [PubMed] [Google Scholar]
- 3. Cohn SL, Pearson ADJ, London WB, et al. The International Neuroblastoma Risk Group (INRG) classification system: an INRG task force report. J Clin Oncol 2009;27:289–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ambros IM, Zellner A, Roald B, et al. Role of ploidy, chromosome 1p, and Schwann cells in the maturation of neuroblastoma. N Engl J Med 1996;334:1505–11. [DOI] [PubMed] [Google Scholar]
- 5. Molenaar JJ, Koster J, Zwijnenburg DA, et al. Sequencing of neuroblastoma identifies chromothripsis and defects in neuritogenesis genes. Nature 2012;483:589–93. [DOI] [PubMed] [Google Scholar]
- 6. Pugh TJ, Morozova O, Attiyeh EF, et al. The genetic landscape of high‐risk neuroblastoma. Nat Genet 2013;45:279–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Asgharzadeh S, Pique‐Regi R, Sposto R, et al. Prognostic significance of gene expression profiles of metastatic neuroblastomas lacking MYCN gene amplification. J Natl Cancer Inst 2006;98:1193–203. [DOI] [PubMed] [Google Scholar]
- 8. Oberthuer A, Hero B, Berthold F, et al. Prognostic impact of gene expression‐based classification for neuroblastoma. J Clin Oncol 2010;28:3506–15. [DOI] [PubMed] [Google Scholar]
- 9. Vermeulen J, De Preter K, Naranjo A, et al. Predicting outcomes for children with neuroblastoma using a multigene‐expression signature: a retrospective SIOPEN/COG/GPOH study. Lancet Oncol 2009;10:663–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Méhes G, Luegmayr A, Ambros IM, et al. Combined automatic immunological and molecular cytogenetic analysis allows exact identification and quantification of tumor cells in the bone marrow. Clin Cancer Res 2001;7:1969–75. [PubMed] [Google Scholar]
- 11. Seeger RC, Patrick Reynolds C, Gallego R, et al. Quantitative tumor cell content of bone marrow and blood as a predictor of outcome in stage IV neuroblastoma: a Children's Cancer Group study. J Clin Oncol 2000;18:4067–76. [DOI] [PubMed] [Google Scholar]
- 12. Burchill SA, Beiske K, Shimada H, et al. Recommendations for the standardization of bone marrow disease assessment and reporting in children with neuroblastoma on behalf of the International Neuroblastoma Response Criteria Bone Marrow Working Group. Cancer 2017;123:1095–105. [DOI] [PubMed] [Google Scholar]
- 13. Abbasi MR, Rifatbegovic F, Brunner C, et al. Bone marrows from neuroblastoma patients: an excellent source for tumor genome analyses. Mol Oncol 2015;9:545–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Rifatbegovic F, Abbasi MR, Taschner‐Mandl S, et al. Enriched bone marrow derived disseminated neuroblastoma cells can be a reliable source for gene expression studies—a validation study. PLoS One 2015;10:e0137995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Abbasi MR, Rifatbegovic F, Brunner C, et al. Impact of disseminated neuroblastoma cells on the identification of the relapse‐seeding clone. Clin Cancer Res 2017;23:4224–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Morandi F, Scaruffi P, Gallo F, et al. Bone marrow‐infiltrating human neuroblastoma cells express high levels of calprotectin and HLA‐g proteins. PLoS One 2012;7:e29922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Klein CA. Parallel progression of primary tumours and metastases. Nat Rev Cancer 2009;9:302–12. [DOI] [PubMed] [Google Scholar]
- 18. Wu TD, Nacu S. Fast and SNP‐tolerant detection of complex variants and splicing in short reads. Bioinformatics 2010;26:873–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Anders S, Pyl PT, Huber W. HTSeq‐A Python framework to work with high‐throughput sequencing data. Bioinformatics 2015;31:166–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA‐seq data with DESeq2. Genome Biol 2014;15:550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wang L, Wang S, Li W. RSeQC: quality control of RNA‐seq experiments. Bioinformatics 2012;28:2184–5. [DOI] [PubMed] [Google Scholar]
- 22. Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: a knowledge‐based approach for interpreting genome‐wide expression profiles. Proc Natl Acad Sci USA 2005;102:15545–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mootha VK, Lindgren CM, Eriksson K‐F, et al. PGC‐1alpha‐responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet 2003;34:267–73. [DOI] [PubMed] [Google Scholar]
- 24. Ovaska K, Laakso M, Haapa‐Paananen S, et al. Large‐scale data integration framework provides a comprehensive view on glioblastoma multiforme. Genome Med 2010;2:65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. De Preter K, Barriot R, Speleman F, et al. Positional gene enrichment analysis of gene sets for high‐resolution identification of overrepresented chromosomal regions. Nucleic Acids Res 2008;36:e43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Łastowska M, Viprey V, Santibanez‐Koref M, et al. Identification of candidate genes involved in neuroblastoma progression by combining genomic and expression microarrays with survival data. Oncogene 2007;26:7432–44. [DOI] [PubMed] [Google Scholar]
- 27. Cheung IY, Feng Y, Gerald W, et al. Exploiting gene expression profiling to identify novel minimal residual disease markers of neuroblastoma. Clin Cancer Res 2008;14:7020–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Oberthuer A, Berthold F, Warnat P, et al. Customized oligonucleotide microarray gene expression‐based classification of neuroblastoma patients outperforms current clinical risk stratification. J Clin Oncol 2006;24:5070–8. [DOI] [PubMed] [Google Scholar]
- 29. Kocak H, Ackermann S, Hero B, et al. Hox‐C9 activates the intrinsic pathway of apoptosis and is associated with spontaneous regression in neuroblastoma. Cell Death Dis 2013;4:e586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Formicola D, Petrosino G, Lasorsa VA, et al. An 18 gene expression‐based score classifier predicts the clinical outcome in stage 4 neuroblastoma. J Transl Med 2016;14:142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Morgan TM, Lange PH, Porter MP, et al. Disseminated tumor cells in prostate cancer patients after radical prostatectomy and without evidence of disease predicts biochemical recurrence. Clin Cancer Res 2009;15:677–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Braun S, Vogl FD, Naume B, et al. A pooled analysis of bone marrow micrometastasis in breast cancer. N Engl J Med 2005;353:793–802. [DOI] [PubMed] [Google Scholar]
- 33. Thiel U, Wawer A, von Luettichau I, et al. Bone marrow involvement identifies a subgroup of advanced Ewing sarcoma patients with fatal outcome irrespective of therapy in contrast to curable patients with multiple bone metastases but unaffected marrow. Oncotarget 2015;7:70959–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Stutterheim J, Zappeij‐Kannegieter L, Versteeg R, et al. The prognostic value of fast molecular response of marrow disease in patients aged over 1 year with stage 4 neuroblastoma. Eur J Cancer 2011;47:1193–202. [DOI] [PubMed] [Google Scholar]
- 35. Shiozawa Y, Eber MR, Berry JE, et al. Bone marrow as a metastatic niche for disseminated tumor cells from solid tumors. Bonekey Rep 2015;4:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Schleiermacher G, Janoueix‐Lerosey I, Delattre O. Recent insights into the biology of neuroblastoma. Int J Cancer 2014;135:2249–61. [DOI] [PubMed] [Google Scholar]
- 37. Bilke S, Chen QR, Westerman F, et al. Inferring a tumor progression model for neuroblastoma from genomic data. J Clin Oncol 2005;23:7322–31. [DOI] [PubMed] [Google Scholar]
- 38. Mosse YP, Diskin SJ, Wasserman N, et al. Neuroblastomas have distinct genomic DNA profiles that predict clinical phenotype and regional gene expression. Genes Chromosom Cancer 2007;46:936–49. [DOI] [PubMed] [Google Scholar]
- 39. Nakano K, Vousden KH. PUMA, a novel proapoptotic gene, is induced by p53. Mol Cell 2001;7:683–94. [DOI] [PubMed] [Google Scholar]
- 40. Valente LJ, Aubrey BJ, Herold MJ, et al. Therapeutic response to non‐genotoxic activation of p53 by nutlin3a is driven by PUMA‐mediated apoptosis in lymphoma cells. Cell Rep 2016;14:1858–66. [DOI] [PubMed] [Google Scholar]
- 41. Michaelis M, Agha B, Rothweiler F, et al. Identification of flubendazole as potential anti‐neuroblastoma compound in a large cell line screen. Sci Rep 2015;5:8202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Sebastián C, Zwaans BMM, Silberman DM, et al. The histone deacetylase SIRT6 is a tumor suppressor that controls cancer metabolism. Cell 2012;151:1185–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lerrer B, Gertler AA, Cohen HY. The complex role of SIRT6 in carcinogenesis. Carcinogenesis 2015;37:108–18. [DOI] [PubMed] [Google Scholar]
- 44. Schipani E, Wu C, Rankin EB, et al. Regulation of bone marrow angiogenesis by osteoblasts during bone development and homeostasis. Front Endocrinol 2013;4:85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Vyas S, Zaganjor E, Haigis MC. Leading edge mitochondria and cancer. Cell 2016;166:555–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Spees JL, Olson SD, Whitney MJ, et al. Mitochondrial transfer between cells can rescue aerobic respiration. Proc Natl Acad Sci USA 2006;103:1283–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Moschoi R, Imbert V, Nebout M, et al. Protective mitochondrial transfer from bone marrow stromal cells to acute myeloid leukemic cells during chemotherapy. Blood 2016;128:253–64. [DOI] [PubMed] [Google Scholar]
- 48. Westermann F, Muth D, Benner A, et al. Distinct transcriptional MYCN/c‐MYC activities are associated with spontaneous regression or malignant progression in neuroblastomas. Genome Biol 2008;9:R150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Stutterheim J, Gerritsen A, Zappeij‐Kannegieter L, et al. Detecting minimal residual disease in neuroblastoma: the superiority of a panel of real‐time quantitative PCR markers. Clin Chem 2009;55:1316–26. [DOI] [PubMed] [Google Scholar]
- 50. Saeys Y, Inza I, Larranaga P. A review of feature selection techniques in bioinformatics. Bioinformatics 2007;23:2507–17. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information Figure 1
Supporting Information Figure 2
Supporting Information Figure 3
Supporting Information Figure 4
Supporting Information Table 1
Supporting Information Table 2
Supporting Information Table 3
Supporting Information Table 4
Supporting Information Table 5
Supporting Information Table 6
Supporting Information Table 7
Supporting Information Table 8
Supporting Information Table 9
Supporting Information Table 10
Supporting Information Table 11
Supporting Information Table 12
Supporting Information Legends
Supporting Information Letter