

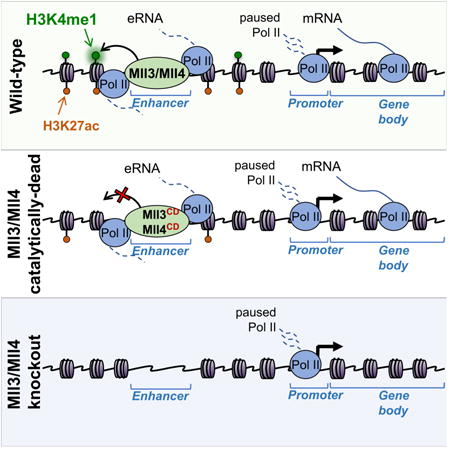
Summary
Monomethylation of histone H3 at lysine 4 (H3K4me1) and acetylation of histone H3 at lysine 27 (H3K27ac) are correlated with transcriptionally engaged enhancer elements, but the functional impact of these modifications on enhancer activity is not well understood. Here we used CRISPR/Cas9 genome editing to separate catalytic activity-dependent and independent functions of Mll3 (Kmt2c) and Mll4 (Kmt2d, Mll2), the major enhancer H3K4 monomethyltransferases. Loss of H3K4me1 from enhancers in Mll3/4 catalytically-deficient cells causes partial reduction of H3K27ac, but has surprisingly minor effects on transcription from either enhancers or promoters. In contrast, loss of Mll3/4 proteins leads to strong depletion of enhancer Pol II occupancy and eRNA synthesis, concomitant with downregulation of target genes. Interestingly, downregulated genes exhibit reduced polymerase levels in gene bodies, but not at promoters, suggestive of pause-release defects. Altogether our results suggest that enhancer H3K4me1 provides only a minor contribution to the long-range coactivator function of Mll3/4.
Keywords: Enhancers, Pol II, eRNA, H3K4me1, H2K27ac, Mll3, Mll4, Mll2, Kmt2c, Kmt2d, pausing, elongation
Graphical abstract
Introduction
Complex multicellular organisms rely on the combinatorial binding of transcription factors to distally located cis-regulatory elements, termed enhancers, to facilitate spatio-temporally precise gene expression programs during development (Levine, 2010; Long, Prescott and Wysocka, 2016). A major breakthrough in the ability to systematically annotate enhancers came through the realization that when active, they harbor stereotypical patterns of chromatin organization and modification, which are shared across cell types and conserved across species (reviewed in Zentner and Scacheri, 2012; Calo and Wysocka, 2013; Shlyueva, Stampfel and Stark, 2014). This pattern consists of a nucleosome depleted region, bound by transcription factors and coactivator complexes, and flanked by nucleosomes harboring specific histone modifications such as the monomethylation of histone H3 on lysine 4 (H3K4me1) and acetylation of histone H3 on lysine 27 (H3K27ac). Furthermore, H3K4me1 in the absence of H3K27ac (and, in some instances in the presence of the repressive mark H3K27me3) has been associated with enhancer states that are repressed or poised/primed for activation (Creyghton et al., 2010; Rada-Iglesias et al., 2011; Zentner, Tesar and Scacheri, 2011; Bonn et al., 2012; Koenecke et al., 2016). Importantly, a combination of H3K4me1/H3K27ac marks has been broadly utilized for epigenomic annotation of active enhancers in a myriad of biological contexts, facilitating systematic discovery and functional understanding of this important class of cis-regulatory elements (reviewed in Hardison and Taylor, 2012; Shlyueva, Stampfel and Stark, 2014).
Although these histone modification signatures are well correlated with (Creyghton et al., 2010; Zentner, Tesar and Scacheri, 2011) and predictive of (Karlić et al., 2010; Bonn et al., 2012) the enhancer states, their functional impact on enhancer activity is much less well understood (Hödl and Basler, 2012; Pengelly et al., 2013). However, the enzymatic activities responsible for writing the canonical enhancer marks have been identified. Two related and partially redundant members of the MLL/COMPASS family, Mll3 (a.k.a. Kmt2c, HALR) and Mll4 (a.k.a. Mll2, Kmt2d, ALR) have been recognized as major enhancer H3K4 monomethyltransferases (Herz et al., 2012; Hu et al., 2013; Lee et al., 2013). Both Mll3 and Mll4 are large nuclear proteins, containing a C-terminal SET domain responsible for H3K4me1 catalysis (Zhang et al., 2015; Li et al., 2016). Previous work demonstrated that Mll3 and Mll4 (hereafter referred to as Mll3/4) bind at enhancers and facilitate their activation, at least in part by promoting recruitment of another coactivator, p300, which acetylates H3K27 (Hu et al., 2013; Lee et al., 2013; Wang et al., 2016). Based on observations that H3K4me1 correlates with active and poised enhancer states and that loss of Mll3/4 impairs enhancer activation, it has been widely assumed that H3K4me1 plays a major role in enhancer function and gene regulation. Nonetheless, the functional impact of Mll3/4 catalytic activity has never been rigorously put to test, largely due to a lack of appropriate tools.
Here we developed an allelic series of mouse embryonic stem cell (mESC) Mll3/4 mutant lines allowing us to separate catalytically-dependent and independent functions of Mll3/4. Using these reagents, we demonstrate that loss of H3K4me1 leads to a partial depletion of H3K27ac from enhancers, but has only a minor effect on transcription originating either from enhancers or from enhancer-adjacent promoters. In contrast, loss of Mll3/4 proteins themselves strongly reduces transcription activity, both at enhancers and target genes. Interestingly, Mll3/4-dependent loss of RNA Polymerase II (Pol II) and nascent transcription from enhancers is not associated with changes in Pol II occupancy at nearby promoters, but instead impacts Pol II density across gene bodies, suggestive of pause-release defects. Taken together, our observations indicate that enhancer H3K4me1 is largely dispensable for maintenance of gene expression programs in mESCs and that the central long-range coactivator function of Mll3/4 lies outside of its methyltransferase activity.
Results
Generation and validation of a series of Mll3/4 mutant mESC lines
To study the impact of Mll3 and Mll4 functional perturbations on enhancer activity and gene regulation we used CRISPR/Cas9 genome editing in mESCs to generate lines in which both alleles of the Mll3 and Mll4 genes were altered: (i) to delete early exons, resulting in frameshifts and loss of both Mll3 and Mll4 proteins (dKO), or (ii) to introduce a point mutation in the catalytic SET domain (Y4792A in Mll3 and Y5477A in Mll4, which converts a key tyrosine involved in catalysis to an alanine) (dCD) (Figure 1A, Figures S1A,B). Immunoblot analysis of extracts from wild-type (WT), dCD and dKO mESCs with a custom antibody (see Methods) detected polypeptides larger than 460kD (in agreement with the estimated molecular weight (MW) of 540kD and 600kD of Mll3 and Mll4, respectively) in WT and dCD cells, but not in dKO cells (Figure 1B). Analysis of additional single Mll3 or Mll4 gene knockout mESC lines confirmed that the higher MW polypeptide is lost upon Mll4 knockout, whereas lower MW polypeptide is lost upon Mll3 knockout (Figure S1C). We also generated mESC lines containing mutations resulting in truncation of Mll3 or Mll4 prior to the C-terminal SET domain (dSET, Figure 1A). However, we observed that such mutations give rise to unstable protein products, which cannot be detected by immunoblotting (an example of Mll4-dSET is shown in Figure S1D), in agreement with a recent report that extensive SET domain mutations can destabilize these methyltransferases (Jang et al., 2016). In contrast, Mll3 Y4792A and Mll4 Y5477A substitutions do not appreciably affect Mll3/4 steady state levels or an interaction with known complex components UTX and Rbbp5 (Figures 1B,C).
Figure 1.
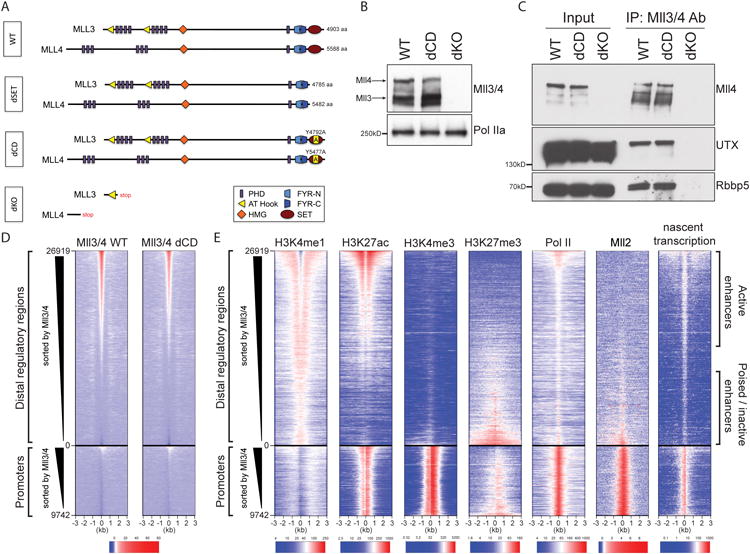
Generation and validation of the catalytically-deficient and loss-of-function Mll3 and Mll4 alleles.
(A) Schematic representation of Mll3 and Mll4 proteins in genome-engineered homozygous mESC lines. Depicted are protein domains and expected length.
(B) Western blot of protein extracts from WT, dCD or dKO Mll3/4 mESCs probed with an antibody that recognizes both Mll3 and Mll4, and Pol II as a loading control.
(C) Co-immunoprecipitation of known Mll3/4 complex components, UTX and Rbbp5, from mESC protein extracts with an antibody recognizing Mll3/4.
(D) Heatmap showing signal enrichment over 26919 enhancers and 9742 transcribed promoters (+/- 3kb from the center) following ChIP with Mll3/4 antibody in WT and dCD mESCs. Heatmap is sorted by strength of Mll3/4 signal in WT cells and centered on the mean shift clustering of p300, Oct4, Otx2 (Buecker et al., 2014) and Mll3/4 ChIP-Seq summits. A color scale indicating the relative signal intensity plotted on each heatmap is shown at the bottom.
(E) Heatmaps showing signal enrichment in WT mESCs over distal regulatory elements and promoters sorted by Mll3/4 WT signal strength as in (D) following ChIP with antibodies as indicated (Mll2 ChIP-seq from (Hu et al., 2017)) or nascent transcription as measured by Gro-seq (last column). A color scale indicating the relative signal intensity plotted on each heatmap is shown at the bottom.
See also Figure S1.
To characterize the impact of Mll3/4 mutations on bulk levels of H3K4 methylation in an antibody-independent manner, we performed quantitative mass spectrometry on acid extracted histones from WT, dKO and dCD Mll3/4 mESCs. H3K4me1 is the most abundant of the three methylation states, and occurs at nearly 30% of all histone H3 molecules in WT cells; in contrast, H3K4me2 and H3K4me3 are only detected at ∼1.4% and ∼0.35% of H3 molecules, respectively (Figure S1E). In dKO and dCD cells we observed a comparable two-fold reduction of bulk H3K4me1 levels, to about 15% of the total histone H3, with the concomitant increase in the levels of unmodified H3, but no significant changes in the H3K4me2 and H3K4me3 (Figure S1E). Altogether, these results demonstrate that mutant Mll3/4 in dCD cells are stable, retain association with known partners and show defects in global H3K4me1 deposition comparable to those seen in the dKO cells. Thus, dCD and dKO mESC lines are suitable reagents allowing us to separate the methyltransferase activity-dependent and independent functions of Mll3/4.
Association of wild type and catalytically deficient Mll3/4 with active enhancers
To characterize genomic regions bound by WT and dCD forms of Mll3/4 we performed ChIP-seq on WT, dCD and dKO mESCs using our Mll3/4 custom antibody. [Of note, all experiments described here were performed in mESC grown under 2i+LIF conditions (Silva et al., 2008; Buecker et al., 2014), which maintain them in the so-called naïve pluripotent state and minimize heterogeneity]. To ensure specificity of the observed enrichments, we subtracted any residual background signal retained in the Mll3/4 dKO ChIPs from the signals detected in WT and dCD cells. We then compared Mll3/4 binding pattern with annotation of all putative regulatory regions (using epigenomic mapping data reported in (Buecker et al., 2014) and this study), and visualized our results using heatmaps sorted by the strength of the Mll3/4 ChIP signal and organized by the cis-regulatory region class (e.g. distal regulatory regions vs promoters) (Figures 1D,E). Additionally, we used GRO-seq (Core, Waterfall and Lis, 2008) to identify nascent transcripts, including bidirectional transcripts arising from enhancers (eRNAs).
We found that, consistent with previous reports (Hu et al., 2013; Lee et al., 2013; Wang et al., 2016), Mll3/4 preferentially bound at distal elements, whereas only a very small subset of promoters had detectable binding of either WT or mutant Mll3/4 (Figure 1D). Importantly, WT and dCD Mll3/4 showed highly similar genomic occupancy patterns (Figure 1D, compare heatmaps, and examples in Figure 2A), indicating that loss of methyltransferase activity does not substantially alter the association of these factors with their genomic targets. Further comparisons with histone modification patterns in WT mESCs revealed that Mll3/4 occupancy distinguishes distal elements bearing stereotypical signatures of active (e.g. H3K4me1, H3K27ac) vs poised/repressed (e.g. H3K4me1, H3K27me3) enhancers (Rada-Iglesias et al., 2011; Zentner, Tesar and Scacheri, 2011; Cruz-Molina et al., 2017), with the former but not the latter being bound by Mll3/4 (Figures 1D,E). In contrast, another member of the MLL family of H3K4 methyltransferases, Mll2, occupies poised/repressed enhancers (albeit weakly, compared to its binding at promoters), but is largely absent from active enhancers, suggesting a division of labor among the family members at distinct classes of regulatory elements. Finally, in agreement with their active status, Mll3/4 occupied enhancers have relatively high levels of Pol II and eRNA (Figures 1D,E).
Figure 2. Loss of Mll3/4 catalytic activity impacts deposition of histone modifications at active but not poised/inactive enhancers.
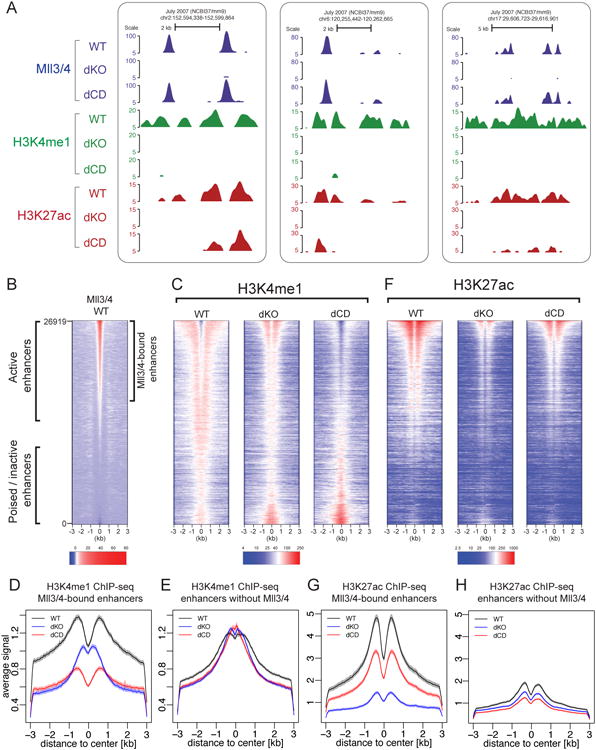
(A) UCSC genome browser tracks shown for three representative Mll3/4-bound enhancer regions. ChIPs for Mll3/4 (blue), H3K4me1 (green) and H3K27ac (red) in Mll3/4 WT, dKO and dCD mESC are shown.
(B) Heatmap of Mll3/4 ChIP signal in WT over 26919 enhancer regions, with regions classified as Mll3/4-bound illustrated to the right. Heatmap sorted by strength of Mll3/4 WT signal.
(C) Heatmap of H3K4me1 ChIP signal over same enhancer regions shown in (B) from in Mll3/4 WT, dKO and dCD mESCs.
(D-E) Aggregate plot comparing the average H3K4me1 ChIP signal over Mll3/4-bound (D) or unbound (E) enhancers in Mll3/4 WT, dKO and dCD mESCs. Shading indicates the 99% confidence interval for the mean.
(F) Heatmap of H3K27ac ChIP signal over same enhancer regions shown in (B) from Mll3/4 WT, dKO and dCD mESCs.
(G-H) Aggregate plot comparing the average H3K27ac ChIP signal over Mll3/4-bound enhancers (G) or unbound enhancers (H) in Mll3/4 WT, dKO and dCD mESCs. Shading indicates the 99% confidence interval for the mean.
See also Figure S2.
Effects of Mll3/4 mutations on the canonical enhancer histone modifications
Our quantitative mass spectrometry results demonstrated that globally, half of all H3K4me1 is depleted in dKO and dCD cells, and thus ∼15% of all histone H3 molecules within chromatin lose this methyl mark (Figure S1E). To investigate whether H3K4me1 depletion in Mll3/4 mutant cells occurs at a specific subset of genomic regions, we compared H3K4me1 ChIP-seq profiles obtained from WT, dCD and dKO mESCs. We observed a strong depletion of H3K4me1 from Mll3/4 bound active enhancers in the dKO cells, and this loss was even more pronounced in the dCD cells, whereas Mll3/4-unbound poised enhancers were not affected (Figures 2A-C). These observations suggest that in the absence of Mll3/4, another methyltransferase may partially substitute for their activity, but this does not happen when inactive Mll3/4 occupy their proper sites in the dCD cells.
Interestingly, the residual H3K4me1 signal observed at active enhancers in dKO cells was distributed distinctly from that observed in WT cells, and was enriched at a position closer to the enhancer center instead of the flanking region (Figure 2C, average H3K4me1 profiles at Mll3/4-bound and unbound enhancers are shown in Figure 2D-E, note narrow 99% confidence interval). We hypothesized that this may be due to either lower processivity of the substituting methyltransferase and/or due to the diminished nucleosomal depletion associated with Mll3/4 loss at enhancers. To explore this latter possibility, we performed ATAC-seq analysis on WT and dKO cells to map the nucleosomal depletion at Mll3/4 occupied enhancers. We observed both a reduction and narrowing of the open chromatin profile at Mll3/4 bound, but not at unbound enhancers in the absence of Mll3/4 proteins, indicating a reduction in open chromatin (Figures S2A,B).
Previous studies have shown that loss of Mll3/4 results in diminished H3K27ac at active enhancers (Herz et al., 2012; Lee et al., 2013; Wang et al., 2016). Our mutant lines afforded us an opportunity to ask to what extent this effect is mediated by the loss of Mll3/4 catalytic activity. We compared H3K27ac ChIP-seq profiles obtained from WT, dCD and dKO mESCs and found that while Mll3/4 knockout led to a strong depletion of enhancer H3K27ac, loss of catalytic activity resulted in an intermediate phenotype, with partially diminished H3K27ac (Figures 2F,G). Thus, Mll3/4 facilitates H3K27ac deposition by both catalytic activity dependent- and independent mechanisms. Notably, enhancers lacking Mll3/4 had generally low levels of H3K27ac, consistent with Mll3/4 occupying highly active enhancers (Figures 2F,H).
To ensure that observed effects are not an artifact of ChIP-seq data normalization, we validated our results using ChIP-qPCR with sets of primers amplifying representative active or poised enhancers and control negative regions. Pairwise comparisons of signals across the three genotypes confirmed our conclusions that loss of either Mll3/4 or their catalytic activity leads to diminished H3K4me1 at active enhancers, whereas H3K27ac shows strong depletion in dKO, and a more modest effect in dCD cells (Figures S2C,D).
Enhancer loss of Mll3/4 but not H3K4me1 reduces transcription from adjacent promoters
We next performed RNA-seq on Mll3/4 WT, dKO and dCD mESCs to investigate effects of Mll3/4 mutations on gene expression. Mll3/4 knockout was associated both with down- and up-regulation of a subset of genes (Figure 3A). However, genes directly targeted by Mll3/4 [that is, bound at the promoter, highlighted in red, or containing a nearby (1-5 kb from TSS) Mll3/4-bound enhancer(s), highlighted in blue] were significantly (p<10-10) biased towards downregulation (Figure 3A). These results are consistent with: (i) Mll3/4 function as a coactivator at enhancers/subset of promoters, and (ii) gene upregulation in the dKO cells being caused by indirect effects, such as downregulation of repressors. In contrast, similar analysis of gene expression changes in dCD cells revealed only minor effects on transcription, suggesting that the major coactivator function of Mll3/4 is independent of their histone methyltransferase capacity, at least in mESCs (Figure 3B).
Figure 3. Mll3/4 promote transcription of target genes and enhancer Pol II loading largely independent of H3K4me1-catalysis.
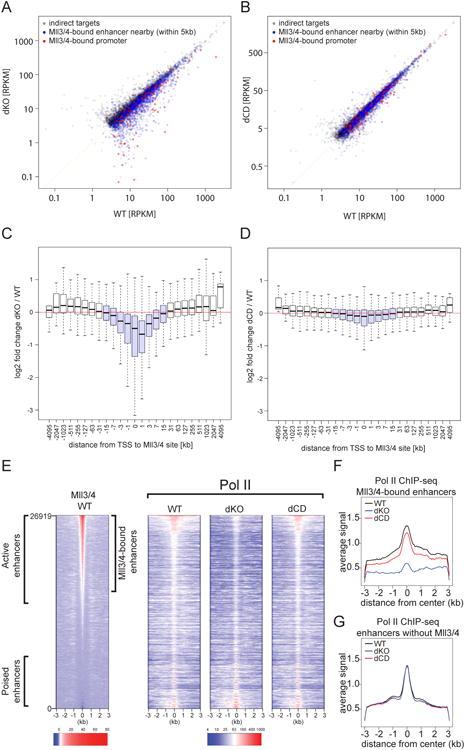
(A-B) Change in gene expression between WT (x-axis) and (A) Mll3/4 dKO or (B) Mll3/4 dCD mESCs (y-axis) plotted as RPKMs for expressed genes (RPKM > 3). Red points indicate genes with a Mll3/4 binding site overlapping the transcription start site (TSS), blue points indicate genes with a Mll3/4-bound enhancer within 1-5 kb of the promoter and grey points indicate all other genes.
(C-D) The average log 2 fold change in gene expression between Mll3/4 dKO and WT (dKO/WT) (C) or between Mll3/4 dCD and WT (dCD/WT) plotted on y-axis, relative to the distance from the TSS to the closest Mll3/4-bound region, binned as indicated (x-axis) with +/- 15kb highlighted in light blue.
(E) Heatmap of Pol II ChIP signal over 26919 enhancers in WT, dKO and dCD mESCs, sorted by strength of Mll3/4 WT ChIP signal, included on the left for reference.
(F-G) Aggregate plot showing the average Pol II ChIP signal over Mll3/4-bound (F) or unbound (G) enhancers in Mll3/4 WT, dKO and dCD mESCs.
To further corroborate these results, we analyzed changes in gene expression in relation to distance between the TSS and the nearest Mll3/4 binding site. We observed that genes containing a Mll3/4 binding site within +/- 15 kb from TSS (highlighted in light blue) were downregulated in dKO vs WT cells (Figure 3C), again in agreement with the short- and long-range coactivator function of Mll3/4. However, only minor changes in gene expression were observed in dCD cells in the vicinity of the Mll3/4 occupied sites (Figure 3D). Altogether, these results argue that loss of H3K4me1 and partial depletion of H3K27ac from active enhancers seen in dCD mutants do not substantially affect maintenance of mESC transcriptional programs.
Catalytic and loss-of-function mutations separate effects of Mll3/4 on histone methylation and polymerase loading at enhancers
We sought to identify elements of Mll3/4 function that contribute to the coactivator capacity irrespective of methyltransferase activity. Given that Mll3/4 preferentially bind enhancers that have relatively high levels of Pol II and eRNA transcription (Figures 1D,E) and that they have been previously shown to promote Pol II occupancy (Lee et al., 2013; Wang et al., 2016), we hypothesized that facilitating loading of Pol II at enhancers may provide such a catalytically-independent function of Mll3/4. To investigate this, we performed Pol II ChIP-seq (with an N-terminal antibody recognizing both initiating and elongating forms of the Pol II) from WT, dKO and dCD cells. Indeed, we found that dKO cells show a strong reduction of Pol II binding at Mll3/4-bound (but not at unbound) enhancers (Figures 3E-G). By contrast, Pol II is primarily retained at Mll3/4 bound enhancers in dCD cells (Figures 3E-G), indicating that the catalytic and loss-of-function mutations generated in our study can separate effects of Mll3/4 on histone methylation and polymerase loading at enhancers.
Mll3/4 control eRNA transcription
We next sought to address whether Mll3/4 are required for the synthesis of enhancer-originating bidirectional transcripts (eRNAs), whose impact on enhancer function and regulation of target promoters is beginning to emerge (Lam et al., 2014). To this end, we used Start-seq (a technique that allows for isolation, annotation and quantification of TSS-associated RNAs (Nechaev et al., 2010; Henriques et al., 2013)) to measure enhancer-originating nascent transcripts in WT, dCD and dKO cell lines. In the dCD cells, we observed no reduction in Start-seq signal at Mll3/4 bound enhancers on either the sense or antisense strand (Figure 4A). In contrast, eRNA transcription in Mll3/4 dKO cells was strongly (>80%) reduced, and this reduction was restricted to Mll3/4 bound enhancers (Figures 4A,B). These observations identify Mll3/4 as upstream regulators of eRNA biogenesis and show that Pol II loading and enhancer transcription can be sustained even upon loss of a canonical enhancer histone modification, H3K4me1.
Figure 4.
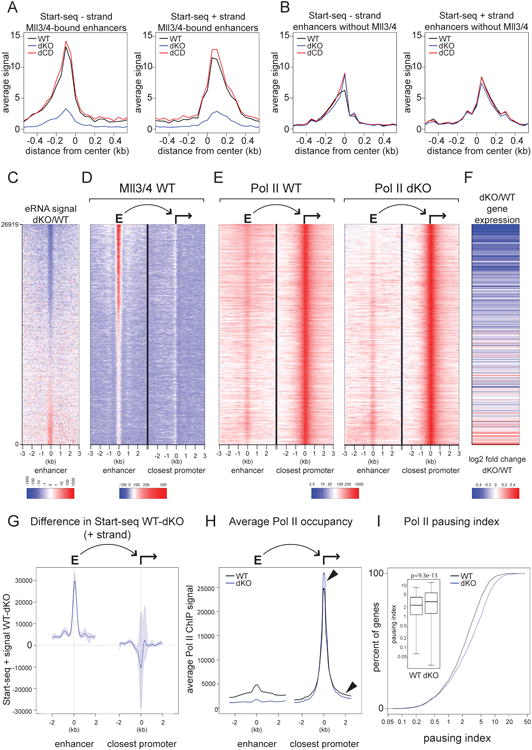
Mll3/4 is required for eRNA synthesis and Pol II gene body density at adjacent promoters.
(A-B) Average Start-seq signal over Mll3/4-bound (A) or unbound (B) enhancers (- strand shown in left panel, + strand in right panel) in Mll3/4 WT, dKO and dCD mESCs.
(C) Heatmap showing the ratio of Start-seq signal over enhancers in Mll3/4 knockout (dKO) relative to wild-type (WT) and sorted by the strength of that signal.
(D) Heatmap of Mll3/4 ChIP signal over enhancer and promoter pairs. Enhancers are same regions as in (C) and paired promoter is the closest active promoter to each enhancer.
(E) Heatmap of Pol II ChIP signal over enhancer and promoter pairs as described in (C,D) from WT and Mll3/4 dKO cells.
(F) Heatmap showing the log2 fold change in gene expression between knockout (dKO) and wild-type (WT) Mll3/4 of genes corresponding to the promoter indicated in (D,E); lines represent an average of approximately 50 genes and values are indicated in the color scale at the bottom.
(G) Change in Start-seq signal (+ strand) at top 3000 enhancer-promoter pairs ranked by the ratio of Start-seq signal (WT/dKO) over enhancers as in (C). Average signal at enhancers is shown on the left, and at linked promoters on the right; blue shading indicates the 95% confidence interval.
(H) Aggregate plot showing average Pol II ChIP signal over enhancer-promoter pairs indicated in (G). Arrows highlight elevated Pol II ChIP signal in the dKO at the TSS and lower Pol II signal over the flanking regions.
(I) Pausing Index (PI) of Pol II over genes linked to the promoter-enhancer pairs indicated in (G). Cumulative distribution function and box plot are shown, significance of the difference in PI between WT and dKO was measured by Mann-Whitney-Wilcoxon test (p-value = 9.3e-13).
See also Figure S3.
Mll3/4-dependent loss of eRNA transcription and downregulation of nearby genes are correlated
To explore the association between transcriptional changes that occur at enhancers and their adjacent promoters upon loss of Mll3/4, we quantified the Start-seq signal at all putative mESC enhancers and sorted these elements by the signal ratio in dKO/WT cells (e.g. ranked by decreasing effect of Mll3/4 loss on eRNA synthesis) (Figure 4C, Figure S3A). We then associated each enhancer to the nearest transcribed promoter and examined specific features of these enhancer-promoter pairs by heatmap visualization. Mll3/4 heatmaps (Figure 4D) indicated that enhancers that lost the greatest fraction of eRNA signal in the dKO cells also had the highest Mll3/4 enhancer occupancy in the WT cells, consistent with Mll3/4-dependent regulation of eRNA synthesis. In agreement with our previous observations, adjacent promoters were generally not occupied by Mll3/4 (Figure 4D, right panel). As expected, Pol II signal in the dKO cells was preferentially diminished at enhancers with the strongest loss of eRNA (i.e. at the top of the heatmaps, Figure 4E), as was the H3K27ac signal (Figure S3C). Notably, mRNA expression from the adjacent promoters (as measured by RNA-seq) was also downregulated in dKO cells and correlated with the loss of eRNA signal at enhancers (Figure 4F).
Mll3/4 loss from enhancers affects pausing index at adjacent promoters
Despite gene downregulation, Pol II occupancy at promoters adjacent to Mll3/4 regulated enhancers did not appear to be affected in the dKO cells (Figure 4E). To quantify this, we measured differences in the Start-seq signal in WT vs dKO cells at Mll3/4 bound enhancers and their adjacent promoters, selecting the top 3000 regions defined as in Figures 4C,D for analysis. While enhancer-originating transcripts were much lower in dKO than WT cells (in agreement with Figures 4A and C), Start-RNAs from adjacent, downregulated promoters were not decreased (Figure 4G). In fact, on average, these promoters had an increased rather than decreased Start-seq signal in the dKO as compared to WT cells, however the statistical confidence of this change is low (Figure 4G, 95% confidence interval is shown in shaded blue). The average Pol II occupancy at these same loci, though dramatically reduced at enhancers in dKO, was associated with a slight increase at adjacent promoters and decrease within gene bodies (Figure 4H, see arrows). To explore this further, we used the same gene set to calculate and compare the pausing index (PI, a.k.a. Traveling Ratio), defined as the relative ratio of Pol II density in the promoter-proximal region to that at the gene body – an increase in Pausing Index is indicative of elevated levels of paused Pol II and/or reduced levels of Pol II release and gene body density (Muse et al., 2007; Zeitlinger et al., 2007). We observed a significant (p<10-12) increase in the Pausing Index in the dKO as compared to WT cells (Figure 4I), but no significant change in PI was detected between WT and dCD cells at these loci (Figure S3D, p>0.01). Altogether, our data support a model whereby enhancer-bound Mll3/4 do not significantly impact loading of Pol II at promoters, but instead facilitate its release into elongation.
Discussion
The major question we set out to address in this work was whether H3K4me1, which constitutes a canonical component of the enhancer epigenetic landscape, is critical for enhancer function and relatedly, whether Mll3/4-catalytic activity is central to their role as transcriptional coactivators. Although we found that H3K4me1 has only a minor effect on the maintenance of enhancer activity and gene expression programs in mESCs, our results do not preclude a role for H3K4me1 in other functions or cellular contexts – reagents developed here will help address these possibilities in the future. Furthermore, H3K4me1 at poised enhancers appears to be independent of Mll3/4, and thus the possibility remains that this mark plays a role in establishment of enhancer activity. Regardless, our data illustrate the need for carefully separating enzymatic and non-enzymatic functions of chromatin modifiers in drawing conclusions about the function of their respective modifications.
Our data suggest that Mll3/4 coactivator activity is to a large extent independent of their methyltransferase function, and instead is more aligned with the catalytically-independent role in enhancer Pol II binding and/or eRNA transcription. However, it remains to be established if Mll3/4 affects enhancer Pol II loading directly or indirectly, and whether the presence of Pol II machinery at enhancers, the act of transcription or the eRNA itself are important for promoting gene expression, since loss of Mll3/4 affects all three. The influence of Mll3/4 on gene transcription typically occurs in the absence of their binding at the promoter, but is associated with the presence of a nearby Mll3/4 occupied enhancer, suggesting a long-range mechanism of action (except in rare instances when the promoter-bound Mll3/4 can also elicit effects on expression). Furthermore, the correlated Mll3/4-dependent changes in eRNA synthesis and mRNA production from adjacent promoters are consistent with a potential coordination between the two acts of transcription. Interestingly though, the disruption of Pol II binding at enhancers in Mll3/4 dKO cells does not affect Pol II binding at promoters, and instead results in diminished Pol II gene body density and increase in pausing index, in agreement with enhancers regulating Pol II pause release (Liu et al., 2013; Ghavi-Helm et al., 2014). It is also interesting to consider our data in the light of recent observations that enhancers influence Pol II bursting behavior at promoters, with weakened or lost enhancer function resulting in diminished burst frequency (Bartman et al., 2016; Fukaya, Lim and Levine, 2016). Diminished burst frequency or size in the context of a stably paused Pol II at the promoter would likely manifest as overall diminished levels of Pol II across gene bodies in population level measurements such as ChIP-seq. Thus, effects on pausing index observed upon loss of Mll3/4 may stem from enhancer dysfunction, suggesting that many other cis- and trans- perturbations affecting enhancer strength could have similar consequences.
Contact for Reagent and Resource Sharing
Further information and requests for resources and regents shold be directed to and will be fulfilled by the Lead Contact, Joanna Wysocka (wysocka@stanford.edu).
Experimental Model and Subject Details
Cell lines
Male mouse embryonic stem cells (R1) were grown as described in Buecker et al., 2014. In brief, cells were grown at 37°C in 2i+LIF media, a serum free N2B27 based medium (DMEM-F12 with glutamax and pyruvate, N2 Neuroplex, B27 without retinoic acid, non-essential amino acids, 5mg/ml BSA AlbumaxII, and penicilin/streptomycin, beta-mercaptoethanol) supplemented with MEK inhibitor PD0325901 (0.8 μM) and GSK3β inhibitor CHIR99021 (3.3μM), on tissue culture dishes coated with polyL-ornithine (7.5μg/ml) and laminin (5μg/ml). Cells were plated onto culture dishes coated with fibronectin (5μg/ml) in 2i+LIF media for 24-48 hours prior to collection.
Method Details
Generation of an Mll3 and Mll4 specific antibody
Antibody was raised against a partially degenerate repetitive peptide motif present both in Mll3 and Mll4. Peptide of the sequence TDPYSQPPGTPRPTT-C was synthesized, cross linked to maleimide activated KLH and rabbits were immunized by Covance. Antibody fraction reactive to the peptide was purified from rabbit serum by affinity chromatography with immobilized epitope peptide.
CRISPR/Cas9 genome engineering
Mouse embryonic stem cells were transfected with Lipofectamine2000 (ThermoFisher) and guideRNAs (see Table S1 for guideRNA sequences used in this study) cloned into either pSpCas9(BB)-2A-GFP (PX458)(Addgene plasmid # 48138, (Ran et al., 2013)) or pX330-U6-Chimeric_BB-CBh-hSpCas9 (Addgene plasmid # 42230, (Cong et al., 2013)) plus a GFP expressing plasmid. For generation of the point mutant lines, a 200bp single stranded oligonucleotide donor template harboring the desired mutation was included in the transfection. After 48 hours, GFP+ cells were single cell sorted into 96 well plates coated with fibronectin (5mg/ml). Cell colonies were split for DNA isolation (DirectPCR Lysis Reagent, Viagen) and propagation or freezing. A PCR product spanning the cleavage site was amplified and digested with a restriction enzyme predicted to cut the wild-type PCR product. Loss of a digestion product indicated the presence of a deletion and candidate PCR products were subsequently screened by DNA sequencing to identify the molecular lesions and by western blot to confirm absence of a protein product or stability of the mutated protein.
Chromatin immunoprecipitation
Approximately 5 million cells were fixed in 1% formaldehyde for 5 min and quenched with glycine to a final concentration of 0.125M. Chromatin was sheared to 0.5-2kb using a Covaris sonicator and incubated overnight at 4°C with 5μg antibody. 50μl Protein G Dynal magnetic beads were added and incubated for an additional 4-6 hours. Beads were washed and DNA purified following reversal of crosslinks. Antibodies used in this study are: H3K4me1 (Abcam, ab8895), H3K27ac (Active Motif, 39133), RNA Pol II (Santa Cruz Biotechnology, N-20 sc-899), custom Mll3/4 antibody (this study), H3K4me3 (Active Motif, 39159) and H3K27me3 (Active Motif, 39155).
Quantitative PCR
Primers used for qPCR in this study are shown in Table S2. qPCR analysis was performed on a Light Cycler 480II machine (Roche), using biological and technical triplicates. ChIP-qPCR signals were calculated as percentage recovery (primer efficiency(Ct Input [adjusted] - Ct ChIP)).
RNA-sequencing
Three independent biological replicates of mouse ESCs in culture were extracted with Trizol according to manufacturer's instructions (Invitrogen). 10 μg of total RNA were subjected to oligo dT purification using Dynaloligo-dT beads. 100ng mRNA were fragmented and used for first strand cDNA synthesis with Superscript II and random hexamers, followed by second strand cDNA with RNAseH and DNA Pol I.
Library preparation and Next-generation sequencing
ChIP DNA, input DNA and cDNA were subject to end repair, A-tailing, adaptor ligation and cleavage with USER enzyme, followed by size selection of 250-500 bp and amplification with NEBNext sequencing primers. Libraries were purified, quantified, multiplexed and sequenced with 1×75bp single-end reads on an Illumina NEXT-seq (Stanford Functional Genomics Facility). Genome wide sequencing data sets are deposited in NCBI Gene Expression Omnibus (GEO accession number GSE98063).
Immunoprecipitation and western blotting
Whole cell nuclear extracts were prepared by lysing cells for 30 min at 4°C in protein extraction buffer (300 mM NaCl, 100 mM Tris pH 8, 0.2 mM EDTA, 0.1% NP40, 10% glycerol) with protease inhibitors and recovery of the supernatant following centrifugation. For immunoprecipitation, 3-5 μg antibody was added to protein extract and rotated for 1-2 hours at 4°C followed by addition of 20μl protein G Dynabeads, incubation for an additional 1 hour and washing in extraction buffer without glycerol. For western blotting, protein concentration was estimated with Bradford reagent (Biorad) and equal amounts of extract were run on a 4% SDS-PAGE gel and transferred to nitrocellulose membranes. Antibodies used in this study were: UTX (Bethyl, A302-374A), Rbbp5 (Bethyl, A300-109A), RNA Pol II 8WG16 (BioLegend, 920101), Mll3/4 (this study), Mll4 (Diagenode, C15310100).
Histone Preparation and Mass Spectrometry Analysis
Histones were extracted from isolated nuclei as described previously (Sidoli et al., 2016). Briefly, nuclei were isolated, histones were extracted in 0.2 M H2SO4 for 2 hours and precipitated with 33% trichloroacetic acid (TCA) for 2 hours. Histones were resuspended in 50 mM NH4HCO3, pH 8.0, derivatized in a mixture of propionic anhydride with propanol (1:3 (v/v)) to propionylate free lysines and digested overnight with trypsin (1ug enzyme/20 ug histones). This was followed by two rounds of derivatization to propionylate peptide N-termini. Subsequently, samples were desalted using C18 Stage-tips. For LC-MS analysis, samples were then separated using a 75 μm ID × 17 cm Reprosil-Pur C18-AQ (3 μm; Dr. Maisch GmbH, Germany) nano-column mounted on an EASY-nLC nanoHPLC (Thermo Scientific, San Jose, Ca, USA) in a HPLC gradient of 2% to 28% solvent B (A = 0.1% formic acid; B = 95% MeCN, 0.1% formic acid) over 45 minutes, followed by 28% to 80% solvent B in 5 minutes and 80% B for 10 more minutes at a flow-rate of 300 nL/min. nLC was coupled online to an LTQ-Orbitrap Elite mass spectrometer (Thermo Scientific) and data were acquired using data-independent acquisition (DIA) (Sidoli et al., 2015). Briefly, full scan MS (m/z 300-1100) was acquired in the Orbitrap with a resolution of 120,000 (at 200 m/z) and an AGC target of 5×10e5. MS/MS was performed in the ion trap with sequential isolation windows of 50 m/z with an AGC target of 3×10e4, a CID collision energy of 35 and a maximum injection time of 50 msec. MS/MS data were collected in centroid mode. Data were searched using EpiProfile (Yuan et al., 2015). The peptide relative ratio was calculated using the total area under the extracted ion chromatograms of all peptides with the same amino acid sequence (including all of its modified forms) as 100%. For isobaric peptides, the relative ratio of two isobaric forms was estimated by averaging the ratio for each fragment ion with different mass between the two species.
ATAC-seq
ATAC-seq libraries were generated from 50,000 mESCs using the Nextera DNA Library Preparation Kit (Illumina) and according to published protocols (Buenrostro et al., 2013). In brief, cells were lysed in cold lysis buffer (10mM Tris-Cl pH 7.4, 10mM NaCl, 3mM MgCl, 0.1% Igepal CA-630) and transposed for 30 minutes at 37°C followed by DNA extraction and amplification with indexing primers. Multiplexed libraries were sequenced with 2×75bp paired-end reads.
GRO-seq
GRO-seq experiments were performed as described in (Williams et al., 2015). Nuclei from WT mESCs were harvested by swelling trypsinized cells for 10 minutes in 50 mL cold lysis buffer (10 mM Tris pH 7.5, 10 mM NaCl, 3 mM CaCl2, 2 mM MgCl2, 0.5% IGEPAL CA-630, 300 mM sucrose, 1 mM PMSF, 2 U/ml SUPERase-IN RNase Inhibitor (Ambion), and 1 × Protease Complete inhibitor mixture (Roche). Nuclei were resuspended in freezing buffer as in (Core, Waterfall and Lis, 2008) at the concentration of 5×106 nuclei per 100 μL, and stored at -80°C until use.
To perform nuclear run-on, the final concentration of CTP was increased to 20 μM as described in (Williams et al., 2015). To permit a quick, reproducible stop to the run-on reaction, the reaction was terminated after 5 minutes by the addition of Trizol (Invitrogen). Base hydrolysis was performed by incubating RNA on ice with 200 mM NaOH for 30 min. To normalize across samples, 3 synthetic Br-U labeled RNAs were spiked into each sample at a specific quantity per 106 cells prior to enrichment of Br-U labeled RNA. Following primary IP of Br-U RNAs, end repair was initiated by incubating RNA with 15 U RppH (NEB) and 20 U SUPERase-IN in 1× Thermopol buffer (NEB) in a final volume of 30 μL at 37°C for 1 hour. 1 μL of Polynucleotide kinase (NEB), and 0.5 μL of 5 mM MgCl2 was then added and the reaction continued for 30 min. End repair was completed as described in (Core, Waterfall and Lis, 2008). Libraries were made using the Illumina small RNA TruSeq kit.
Start-seq
For Start-seq, WT, Mll3/4 KO and Mll3/4 dCD mES cells were grown as described above. Start-RNAs were prepared from three biological replicates as described (Williams et al., 2015). In brief, approximately 15×106 cells per replicate were collected by centrifugation. After washing with ice-cold 1× PBS, cells were swelled in 10 ml of Swelling Buffer (10 mM Tris pH 7.5, 10 mM NaCl, 2 mM MgCl2, 3 mM CaCl, 300 mM sucrose, 0.5% Igepal, 5 mM dithiothreitol, 1 mM PMSF, protease inhibitors and SUPERase-IN RNAse inhibitor (Ambion)) by incubating for 15 minutes on ice followed by 14 strokes with a loose pestle. The dounced cells were spun for 5 minutes at 500g, the supernatant (cytoplasm) was discarded, the pellet resuspended in 30 ml of Swelling Buffer and spun as above. The supernatant was discarded and the nuclei pellet was resuspended in 1 ml of Swelling Buffer, aliquoted and stored at -80C. Libraries were prepared using the TruSeq Small RNA Kit (Illumina) as described in (Williams et al., 2015), with the exception that cap removal was performed using 10U of RNA 5′ pyrophosphohydrolase (NEB) in 1× ThermoPol buffer (NEB) in a final volume of 50uL at 37°C for 1 hour. To normalize samples, 10 synthetic capped RNAs were spiked into the Trizol preparation at a specific quantity per 106 cells.
Quantification and Statistical Analysis
ChIP-seq, ATAC-seq and RNA-seq data analysis
ChIP-seq and ATAC-seq reads were trimmed with cutadapt and aligned with bowtie2 to mm9 reference genome as previously described (Buecker et al., 2014). In brief, aligned reads were stripped of duplicates and 10 base-pair resolution kernel density estimates (KDE) were calculated with a R convolve() function modified to utilize FFTW library. Peak KDE values over the regions of interest were used for subsequent analysis. The read coverage was normalized to total aligned reads in a given library, and average signals from two biological replicates were plotted. Normalized density wig tracks were calculated with QuEST and visualized on the UCSC Genome Browser. Normalized heat maps were calculated with custom scripts, by tabulating read positions relative to centers regions of interest and plotted with custom R script. Centers of enhancers were obtained by mean shift clustering of summits of p300, Oct4, Otx2 (Buecker et al. 2014) and MLL3/4 ChIP-seqs. Mll3/4 peaks were called with macs2 callpeak routine against reads from Mll3/4 dKO cells to account for non-specific background. To obtain a statistically robust set of Mll3/4 occupied sites, read counts over macs2 called peaks together with large set of random decoy sites was calculated with bedtools coverage and differential count analysis was performed with DESseq2 platform. RNAseq reads were aligned with hisat2 and tabulated with featureCounts against genocode gene models (subread package), differential analysis from three biological replicates was performed with DESeq2.
GRO-seq Data Analysis
Paired-end libraries were sequenced on an Illumina NextSeq. GRO-seq reads were trimmed for quality and adapter sequence using Cutadapt 1.2.1, discarding pairs where either mate was trimmed shorter than 15 nt (-m 15 –q 10 --match-read-wildcards). Read pairs originating from rRNA and tRNA were filtered by aligning against an index consisting of the 45S, 5S, and tRNA transcripts using Bowtie 0.12.8, allowing two mismatches (-m1 -v2 -X1000 -un --max). BedGraph files were generated from strand-specific 5′-end mapping locations based on spike-normalized reads.
Start-RNA data analysis
Paired-end reads for all samples were trimmed for adapter sequence and low quality 3′ ends using cutadapt 1.2.1, discarding those containing reads shorter than 20 nt (-m 20 -q 10), and removing a single nucleotide from the 3′ end of all trimmed reads to allow successful alignment with bowtie 1.1.1. Counts of read-pairs mapping uniquely to each spike-in RNA were determined for each sample, using the same parameters. Mean counts per spike-in for each sample was determined, and least squares linear regression performed against a selected WT sample. The resulting slopes agreed well between replicates and were close to 1, thus reads were normalized solely based on sequencing depth (i.e. per million read pairs uniquely aligned to the mouse genome), by scaling the sample with the higher uniquely mappable read pair count to match the lower. Combined bedGraphs were generated by summing counts per nucleotide of both replicates for each condition. Transcription start sites for annotated genes and unannotated loci were defined as described in (Scruggs et al., 2015).
Pausing index calculation
PI was calculated as: promoter Pol II signal (-200/+300 bp from TSS)/gene body Pol II signal (+1000-3000 bp from TSS) and compared between WT and dKO cells.
Data and Software Availability
All software used in this manuscript is listed in the Key Resoures Table. Additional dedicated scripts developed for this work are available upon request. The accession number for the sequences reported in this paper is GEO: GSE98063.
Key Resources Table.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-Mll3/4 | This paper | N/A |
| Rabbit polyclonal anti-H3K4me1 Rabbit polyclonal anti-H3K27ac | Abcam Active Motif | Cat#ab8895 Cat#39133 |
| Rabbit polyclonal anti-RNA Pol II | Santa Cruz Biotechnology | Cat#sc-899, N-20 |
| Rabbit polyclonal anti-H3K4me3 Rabbit polyclonal anti-H3K27me3 | Active Motif Active Motif | Cat#39159 Cat#39155 |
| Rabbit polyclonal anti-UTX | Bethyl | Cat#A302-374A |
| Rabbit polyclonal anti-Rbbp5 | Bethyl | Cat#A300-109A |
| Rabbit polyclonal anti-Mll4 | Diagenode | Cat#C15310100 |
| Mouse monoclonal anti-PolII 8WG16 | BioLegend | Cat#920101 |
| Bacterial and Virus Strains | ||
| Biological Samples | ||
| Chemicals, Peptides, and Recombinant Proteins | ||
| MEK inhibitor PD0325901 | Selleck Chemicals | Cat#S1036 |
| GSK3b inhibitor CHIR99021 | Selleck Chemicals | Cat#S2924 |
| polyL-ornithine | Sigma-Aldrich | Cat#P4638-100MG |
| Laminin | Life Technologies | Cat#23017015 |
| Fibronectin | ThermoFishser | Cat#FC01010MG |
| Critical Commercial Assays | ||
| Lipofectamine2000 | ThermoFisher | Cat#11668019 |
| DirectPCR Lysis Reagent | Viagen | Cat#301-C |
| Dynabeads Protein G | ThermoFisher | Cat#10004D |
| Trizol | Invitrogen | Cat#15596026 |
| Dynabeads oligo(dT)25 | ThermoFisher | Cat#61002 |
| NEBNext Multiplex Oligos for Illumina | New England Biolabs | Cat#E7335S |
| Nextera DNA Library Preparation Kit | Illumina | Cat#FC-121-1030 |
| TruSeq Small RNA Kit | Illumina | Cat#RS-200-0012 |
| Deposited Data | ||
| Raw and analyzed sequencing data | This paper | GEO: GSE98063 |
| Experimental Models: Cell Lines | ||
| WT R1 mESC | ATCC | Cat#SCRC-1036 |
| dKO Mll3/4 mESC | This paper | N/A |
| dCT Mll3/4 mESC | This paper | N/A |
| Experimental Models: Organisms/Strains | ||
| Oligonucleotides | ||
| Primers for ChIP-qPCR, see Table S2. | This paper | N/A |
| Guide RNA sequences for CRISPR/Cas9, see Table S1. | This paper | N/A |
| Recombinant DNA | ||
| pX330-U6-Chimeric_BB-CBh-hSpCas9 | (Cong et al., 2013) | Addgene plasmid # 42230 |
| pX458 pSpCas9(BB)-2A-GFP | (Ran et al., 2013) | Addgene plasmid # 48138 |
| Software and Algorithms | ||
| Bowtie2 | (Langmead and Salzberg, 2012) | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| HISAT2 | (Kim, Langmead and Salzberg, 2015) | https://ccb.jhu.edu/software/hisat/index.shtml |
| DESeq2 | (Love, Huber and Anders, 2014) | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| QuEST | (Valouev et al., 2008) | http://www-hsc.usc.edu/∼valouev/QuEST/QuEST.html |
| bedtools | (Quinlan, 2014) | http://bedtools.readthedocs.io/en/latest/ |
| Other | ||
Supplementary Material
Acknowledgments
We thank Wysocka lab members for comments on the manuscript. We thank Elijah Montgomery for excellent timing and patience during revision. This research was supported in part by Howard Hughes Medical Institute, NIH R01 GM112720-01 and March of Dimes Foundation (J.W.), NIGMS IRACDA K12 award to K.M.D., the Intramural Research Program of the NIH, National Institute of Environmental Health Sciences to K.A. (Z01 ES101987), and NIH grants GM110174, CA196539 and a Leukemia and Lymphoma Society Robert Arceci Scholar award to B.A.G.
Footnotes
Author Contributions: K.M.D and J.W. conceived experiments and wrote the manuscript. K.M.D., T.S., T.H., N.V.B, B.S.S., N.N. and C.D.S. performed experiments. K.M.D., T.S., N.V.B., B.A.G., T.H. and K.A. peformed data analysis. K.M.D. and T.S. performed data visualization. J.W., B.A.G and K.A. obtained funding, provided supervision and edited the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bartman CR, Hsu SC, Hsiung CCS, Raj A, Blobel GA. Enhancer Regulation of Transcriptional Bursting Parameters Revealed by Forced Chromatin Looping. Molecular Cell. 2016;62(2):237–247. doi: 10.1016/j.molcel.2016.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonn S, Zinzen RP, Girardot C, Gustafson EH, Perez-Gonzalez A, Delhomme N, Ghavi-Helm Y, Wilczyński B, Riddell A, Furlong EEM. Tissue-specific analysis of chromatin state identifies temporal signatures of enhancer activity during embryonic development. Nature Genetics. 2012;44(2):148–156. doi: 10.1038/ng.1064. [DOI] [PubMed] [Google Scholar]
- Buecker C, Srinivasan R, Wu Z, Calo E, Acampora D, Faial T, Simeone A, Tan M, Swigut T, Wysocka J. Reorganization of enhancer patterns in transition from naive to primed pluripotency. Cell Stem Cell. 2014;14(6):838–853. doi: 10.1016/j.stem.2014.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buenrostro JD, Giresi PG, Zaba LC, Chang HY, Greenleaf WJ. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nature methods. 2013;10(12):1213–8. doi: 10.1038/nmeth.2688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calo E, Wysocka J. Molecular Cell. 5. Vol. 49. Elsevier Inc; 2013. Modification of Enhancer Chromatin: What, How, and Why? pp. 825–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, Zhang F. Multiplex Genome Engineering Using CRISPR/Cas System. Science. 2013 Feb;339:819–824. doi: 10.1126/science.1231143. doi:10.1126/science.1231143 RNA-Guided. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Core LJ, Waterfall JJ, Lis JT. Nascent RNA sequencing reveals widespread pausing and divergent initiation at human promoters. Science (New York, NY) 2008;322(5909):1845–8. doi: 10.1126/science.1162228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creyghton MP, Cheng AW, Welstead GG, Kooistra T, Carey BW, Steine EJ, Hanna J, Lodato Ma, Frampton GM, Sharp Pa, Boyer La, Young Ra, Jaenisch R. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(50):21931–21936. doi: 10.1073/pnas.1016071107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruz-Molina S, Respuela P, Tebartz C, Kolovos P, Nikolic M, Fueyo R, van Ijcken WFJ, Grosveld F, Frommolt P, Bazzi H, Rada-Iglesias A. PRC2 Facilitates the Regulatory Topology Required for Poised Enhancer Function during Pluripotent Stem Cell Differentiation. Cell Stem Cell. 2017 doi: 10.1016/j.stem.2017.02.004. [DOI] [PubMed] [Google Scholar]
- Fukaya T, Lim B, Levine M. Enhancer Control of Transcriptional Bursting. Cell. 2016;166(2):358–368. doi: 10.1016/j.cell.2016.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghavi-Helm Y, Klein FA, Pakozdi T, Ciglar L, Noordermeer D, Huber W, Furlong EEM. Enhancer loops appear stable during development and are associated with paused polymerase. Nature. 2014;512(7512):96–100. doi: 10.1038/nature13417. [DOI] [PubMed] [Google Scholar]
- Hardison RC, Taylor J. Genomic approaches towards finding cis-regulatory modules in animals. Nature Reviews Genetics. 2012;13(7):469–483. doi: 10.1038/nrg3242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henriques T, Gilchrist D, Nechaev S, Bern M, Muse G, Burkholder A, Fargo D, Adelman K. Stable pausing by rna polymerase II provides an opportunity to target and integrate regulatory signals. Molecular Cell. 2013;52(4):517–528. doi: 10.1016/j.molcel.2013.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herz HM, Mohan M, Garruss AS, Liang K, Takahashi Yhei, Mickey K, Voets O, Verrijzer CP, Shilatifard A. Enhancer-associated H3K4 monomethylation by trithorax-related, the drosophila homolog of mammalian MLL3/MLL4. Genes and Development. 2012;26(23):2604–2620. doi: 10.1101/gad.201327.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hödl M, Basler K. Transcription in the absence of histone H3.2 and H3K4 methylation. Current Biology. 2012;22(23):2253–2257. doi: 10.1016/j.cub.2012.10.008. [DOI] [PubMed] [Google Scholar]
- Hu D, Gao X, Cao K, Morgan MA, Mas G, Smith ER, Volk AG, Bartom ET, Crispino JD, Di Croce L, Shilatifard A. Molecular cell. 3. Vol. 65. Elsevier; 2017. Not All H3K4 Methylations Are Created Equal: Mll2/COMPASS Dependency in Primordial Germ Cell Specification; pp. 460–475.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu D, Gao X, Morgan Ma, Herz HM, Smith ER, Shilatifard A. The MLL3/MLL4 branches of the COMPASS family function as major histone H3K4 monomethylases at enhancers. Molecular and cellular biology. 2013;33(23):4745–54. doi: 10.1128/MCB.01181-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang Y, Wang C, Zhuang L, Liu C, Ge K. Journal of Molecular Biology. Elsevier Ltd; 2016. H3K4 methyltransferase activity is required for MLL4 protein stability. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlić R, Chung HR, Lasserre J, Vlahovicek K, Vingron M. Histone modification levels are predictive for gene expression. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(7):2926–2931. doi: 10.1073/pnas.0909344107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Langmead B, Salzberg SL. HISAT: a fast spliced aligner with low memory requirements. Nature methods. 2015;12(4):357–60. doi: 10.1038/nmeth.3317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koenecke N, Johnston J, He Q, Meier S, Zeitlinger J. Drosophila poised enhancers are generated during tissue patterning with the help of repression. Genome Research. 2016:gr.209486.116. doi: 10.1101/gr.209486.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam MTY, Li W, Rosenfeld MG, Glass CK. Enhancer RNAs and regulated transcriptional programs. Trends in Biochemical Sciences. 2014:170–182. doi: 10.1016/j.tibs.2014.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9(4):357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JE, Wang C, Xu S, Cho YW, Wang L, Feng X, Baldridge A, Sartorelli V, Zhuang L, Peng W, Ge K. H3K4 mono- And di-methyltransferase MLL4 is required for enhancer activation during cell differentiation. eLife. 2013;2013(2) doi: 10.7554/eLife.01503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine M. Transcriptional enhancers in animal development and evolution. Current Biology. 2010 doi: 10.1016/j.cub.2010.06.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Han J, Zhang Y, Cao F, Liu Z, Li S, Wu J, Hu C, Wang Y, Shuai J, Chen J, Cao L, Li D, Shi P, Tian C, Zhang J, Dou Y, Li G, Chen Y, Lei M. Structural basis for activity regulation of MLL family methyltransferases. Nature. 2016;530(7591):447–452. doi: 10.1038/nature16952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W, Ma Q, Wong K, Li W, Ohgi K, Zhang J, Aggarwal AK, Rosenfeld MG. Brd4 and JMJD6-associated anti-pause enhancers in regulation of transcriptional pause release. Cell. 2013;155(7):1581–1595. doi: 10.1016/j.cell.2013.10.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long HK, Prescott SL, Wysocka J. Ever-Changing Landscapes: Transcriptional Enhancers in Development and Evolution. Cell. 2016:1170–1187. doi: 10.1016/j.cell.2016.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome biology. 2014;15(12):550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muse GW, Gilchrist DA, Nechaev S, Shah R, Parker JS, Grissom SF, Zeitlinger J, Adelman K. RNA polymerase is poised for activation across the genome. Nature genetics. 2007;39(12):1507–11. doi: 10.1038/ng.2007.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nechaev S, Fargo DC, dos Santos G, Liu L, Gao Y, Adelman K. Global analysis of short RNAs reveals widespread promoter-proximal stalling and arrest of Pol II in Drosophila. Science (New York, NY) 2010;327(5963):335–338. doi: 10.1126/science.1181421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pengelly AR, Copur Ö, Jäckle H, Herzig A, Müller J. A histone mutant reproduces the phenotype caused by loss of histone-modifying factor Polycomb. Science. 2013;339(6120):698–699. doi: 10.1017/CBO9781107415324.004. [DOI] [PubMed] [Google Scholar]
- Quinlan AR. BEDTools: The Swiss-Army tool for genome feature analysis. Current Protocols in Bioinformatics. 2014;2014:11.12.1, 11.12.34. doi: 10.1002/0471250953.bi1112s47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rada-Iglesias A, Bajpai R, Swigut T, Brugmann S, Flynn R, Wysocka J. Nature. 7333. Vol. 470. Nature Publishing Group; 2011. A unique chromatin signature uncovers early developmental enhancers in humans; pp. 279–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat Protocols. 2013;8(11):2281–2308. doi: 10.1038/nprot.2013.143\r. http://www.nature.com/nprot/journal/v8/n11/abs/nprot.2013.143.html#supplementary-information. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scruggs BS, Gilchrist DA, Nechaev S, Muse GW, Burkholder A, Fargo DC, Adelman K. Bidirectional Transcription Arises from Two Distinct Hubs of Transcription Factor Binding and Active Chromatin. Molecular Cell. 2015;58(6):1101–1112. doi: 10.1016/j.molcel.2015.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shlyueva D, Stampfel G, Stark A. Transcriptional enhancers: from properties to genome-wide predictions. Nature reviews Genetics. 2014;15(4):272–86. doi: 10.1038/nrg3682. [DOI] [PubMed] [Google Scholar]
- Sidoli S, Bhanu NV, Karch KR, Wang X, Garcia BA. Complete Workflow for Analysis of Histone Post-translational Modifications Using Bottom-up Mass Spectrometry: From Histone Extraction to Data Analysis. Journal of visualized experiments : JoVE. 2016;111:2–3. doi: 10.3791/54112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidoli S, Lin S, Xiong L, Bhanu NV, Karch KR, Johansen E, Hunter C, Mollah S, Garcia BA. Sequential Window Acquisition of all Theoretical Mass Spectra (SWATH) Analysis for Characterization and Quantification of Histone Post-translational Modifications. Molecular cell Proteomics. 2015;14(9):2420–2428. doi: 10.1074/mcp.O114.046102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva J, Barrandon O, Nichols J, Kawaguchi J, Theunissen TW, Smith A. Promotion of reprogramming to ground state pluripotency by signal inhibition. PLoS Biology. 2008;6(10):2237–2247. doi: 10.1371/journal.pbio.0060253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valouev A, Johnson DS, Sundquist A, Medina C, Anton E, Batzoglou S, Myers RM, Sidow A. Nat Meth. 9. Vol. 5. Nature Publishing Group; 2008. Genome-wide analysis of transcription factor binding sites based on ChIP-Seq data; pp. 829–834. Available at: http://dx.doi.org/10.1038/nmeth.1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Lee JE, Lai B, Macfarlan TS, Xu S, Zhuang L, Liu C, Peng W, Ge K. Enhancer priming by H3K4 methyltransferase MLL4 controls cell fate transition. Proceedings of the National Academy of Sciences of the United States of America. 2016;113(42):11871–11876. doi: 10.1073/pnas.1606857113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams LH, Fromm G, Gokey NG, Henriques T, Muse GW, Burkholder A, Fargo DC, Hu G, Adelman K. Molecular Cell. 2. Vol. 58. Elsevier Inc; 2015. Pausing of RNA Polymerase II Regulates Mammalian Developmental Potential through Control of Signaling Networks; pp. 311–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan ZF, Lin S, Molden RC, Cao XJ, Bhanu NV, Wang X, Sidoli S, Liu S, Garcia BA. EpiProfile Quantifies Histone Peptides With Modifications by Extracting Retention Time and Intensity in High-resolution Mass Spectra. Molecular & Cellular Proteomics. 2015;14(6):1696–1707. doi: 10.1074/mcp.M114.046011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeitlinger J, Stark A, Kellis M, Hong JW, Nechaev S, Adelman K, Levine M, Young RA. RNA polymerase stalling at developmental control genes in the Drosophila melanogaster embryo. Nature genetics. 2007;39(12):1512–6. doi: 10.1038/ng.2007.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zentner GE, Scacheri PC. The chromatin fingerprint of gene enhancer elements. Journal of Biological Chemistry. 2012:30888–30896. doi: 10.1074/jbc.R111.296491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zentner GE, Tesar PJ, Scacheri PC. Epigenetic signatures distinguish multiple classes of enhancers with distinct cellular functions. Genome Research. 2011;21(8):1273–1283. doi: 10.1101/gr.122382.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Mittal A, Reid J, Reich S, Gamblin SJ, Wilson JR. Evolving catalytic properties of the MLL family SET domain. Structure. 2015;23(10):1921–1933. doi: 10.1016/j.str.2015.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.