

Abstract
Most spontaneous DNA double-strand breaks (DSBs) result from replication-fork breakage. Break-induced replication (BIR), a genome rearrangement-prone repair mechanism that requires the Pol32/POLD3 subunit of eukaryotic DNA Polδ, was proposed to repair broken forks, but how genome destabilization is avoided was unknown. We show that broken fork repair initially uses error-prone Pol32-dependent synthesis, but that mutagenic synthesis is limited to within a few kilobases from the break by Mus81 endonuclease and a converging fork. Mus81 suppresses template switches between both homologous sequences and diverged human Alu repetitive elements, highlighting its importance for stability of highly repetitive genomes. We propose that lack of a timely converging fork or Mus81 may propel genome instability observed in cancer.
Template switches and mutation clusters of <1 to tens of kilobases occur at double-strand breaks (DSBs) repaired by homologous recombination (HR) and are implicated in rapid evolution, adaptation, and tumorigenesis (1–4). We uncovered a mechanism that restricts error-prone synthesis during HR at the most common type of DSB, broken replication forks. The break-induced replication (BIR) pathway has been proposed for repair of single-ended DSBs (seDSBs), but its contribution to broken fork repair is unknown. In BIR, a displacement loop (D-loop), the initial recombination intermediate, is extended by Polδ with Pol32 and Pif1 helicase (5–7). BIR is highly mutagenic and prone to template switches (2,3), likely because of long single-strand DNA intermediates and instability of the D-loop (6, 8). A similar pathway exists in humans (9, 10). Here, we show that Mus81 and converging replication forks arriving from the opposite direction alleviate much of the genome-destabilizing consequences of BIR.
To understand the contribution of mutagenic Pol32-mediated BIR to broken fork repair, we used a galactose-inducible, nick-mediated, recombination assay (11). In this system, Flp1H305L, a step arrest mutant expressed from an inducible GAL10 promoter, creates a long-lived single-stranded break at an FRT (flippase recognition target) site, which is converted to a seDSB when encountered by a replication fork. To ascertain the potential contribution of converging forks limiting the extent of repair-specific DNA synthesis, the FRT site was inserted either between two efficient origins of replication on ChrVI or at subtelomeric regions on ChrII or ChrIV, where only inefficient or dormant origins are present distal to the FRT (12, 13). We confirmed that cells predominantly induce seDSBs originating from one side of the nick (Fig. 1A and fig. S1).
Fig. 1. Mus81 and Yen1 promote repair of broken replication forks.
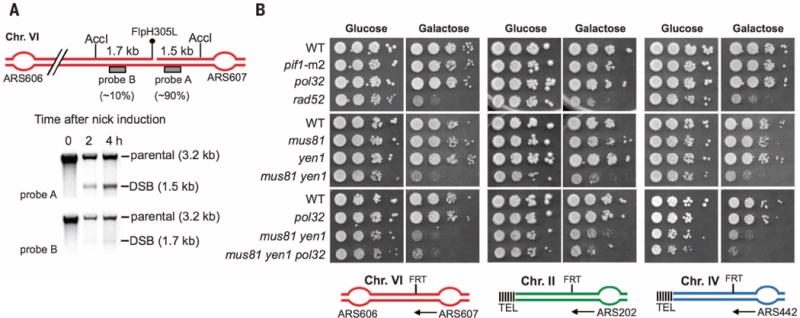
(A) Southern blot showing seDSBs formed on ChrVI after Flp-nickase induction. Approximate relative proportions of each DSB band are indicated below the location of the corresponding probe. (B) Serial 10-fold dilutions plated on YP-glucose and YP-galactose (Flp induction) to assay for repair defects in mutant strains. Arrows indicate the predominant direction of replication forks reaching the FRT.
We first determined whether proteins important for BIR—Pif1 and Pol32—are required for efficient repair of broken replication forks. Wild-type and mutant cells were plated on media containing either glucose or galactose. Both pif1-m2 and pol32Δ cells grow equivalently to wild type, even when fork breakage occurs between an efficient origin of replication and the telomere (Fig. 1B). In agreement with the nonessential role of Pol32 and Pif1 in repair of broken forks, pif11-m2 and pol32Δ cells are not sensitive to camptothecin (CPT), a topoisomerase I inhibitor, which induces DNA nicks that can be converted to broken forks during replication (fig. S2).
In BIR, studied outside the context of replication, D-loop migration results in conservative inheritance of newly synthesized strands (6,14). Consequently, structure-specific endonucleases Mus81, which can convert a D-loop to a replication fork (15, 16), and Yen1 are mostly dispensable (7,17). In contrast, elimination of Mus81 and Yen1 leads to a dramatic defect in broken fork repair (Fig. 1B), which is consistent with previous observations in budding and fission yeast (18,19). However, in those assays, an unprocessed D-loop could merge with a converging replication fork to form a single Holiday junction (HJ), which cannot be removed by Sgs1-Top3-Rmi1, likely requiring resolution by Mus81 or Yen1. In our assays with fork breakage at subtelomeric positions (ChrII and ChrIV), where only inefficient or dormant origins are present (fig. S1), the unprocessed D-loop can migrate to the end of the chromosome. In this context, mus81Δ yen1Δ cells are more resistant to fork breakage, and this improvement depends on Pol32 (Fig. 1B and fig. S3). A likely explanation for the mild repair defect in mus81Δ yen1Δ cells at subtelomeric positions, with Pol32-BIR remaining insufficient for repair, is the firing of normally inefficient or dormant origins (20,21), resulting in the merging of a converging fork with the extending D-loop. Last, key recombination proteins Rad52 and Rad51 are needed for broken fork repair (Fig. 1B and fig. S4). Together, the genetic requirements suggest that Pol32-mediated BIR is neither an efficient nor the primary pathway of broken fork repair and is suppressed by converging forks.
The other defining characteristic of BIR is a high level of template switches and point mutations at least 10 or 30 kb from the strand invasion site, respectively (2,3). We therefore determined whether broken fork repair is marked by the same events using URA3 reporters at different distances from the FRT sites (Fig. 2A). URA3 disruption confers resistance to 5-fluoroorotic acid (5-FOA); therefore, we determined the rate of 5-FOAR-conferring mutations after repair. Two versions of these assays were made, either with or without the ura3-1 point mutant allele at its natural locus on ChrV, which allows for observation of recombination template switches. Conversion to ura3-1 requires initial DNA synthesis at the sister chromatid template to reach URA3, followed by a switch to ura3-1 and either mismatch correction or DNA synthesis copying the base pair change. To recover these events, a second switch back to the original sister chromatid is required because single-switch products followed by DNA synthesis are inviable for nearly all reporter positions. The exception is the position 15 kb from the FRT on ChrII, where a single template switch to ura3-1 followed by synthesis to the end of ChrV yields a viable repair product (Fig. 2B).
Fig. 2. Mutagenesis in wild-type cells is restricted to the vicinity of the broken replication fork.
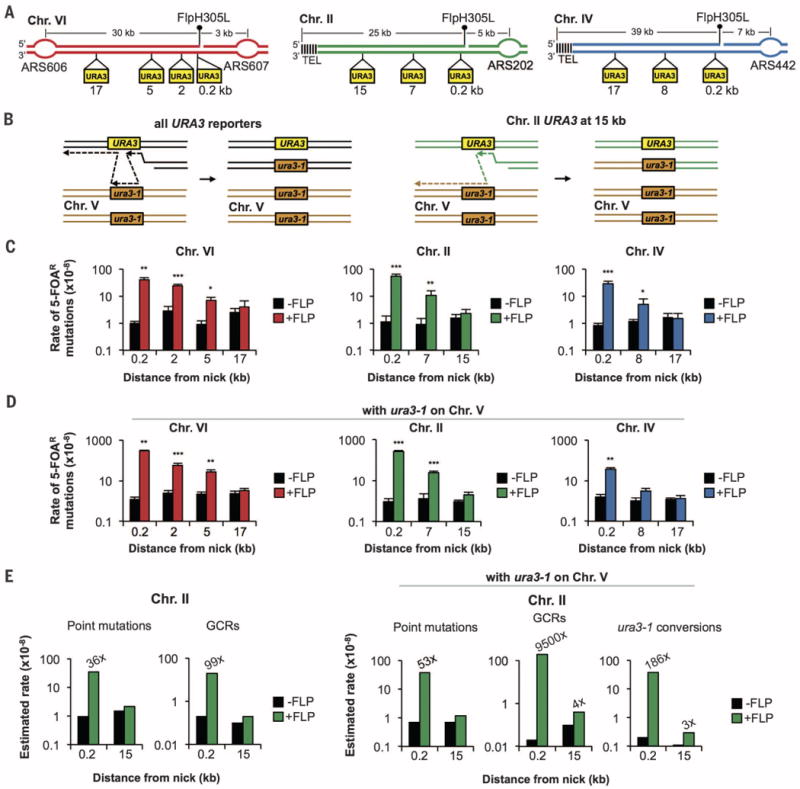
(A) Positions of URA3 reporter genes with respect to broken forks. (B) Diagram of possible recombination template switches leading to gene conversion. (C and D) Median 5-FOAR break–induced (+FLP) and spontaneous (−FLP) mutation rates in wild-type cells (C) without or (D) with ura3-1 on ChrV. Error bars show 1 SD, and significant increases over spontaneous are indicated. *P < 0.05, **P < 0.01, ***P < 0.001. (E) Estimated rates of point mutations, GCRs, and ura3-1 conversions. Fold changes above spontaneous are indicated.
In wild-type cells, mutation rates are significantly increased next to the broken fork but drop to spontaneous levels at distances beyond 10 kb from the DSB. The same pattern is observed for all break locations (Fig. 2C), indicating a shift in the fidelity of synthesis during repair. When the ura3-1 allele is present, mutation rates are further elevated, but these increases remain in proximity to the break (Fig. 2D).
To characterize mutations, we sequenced reporters positioned 0.2 or 15 kb from the broken fork on ChrII from 40 to 60 independent 5-FOAR colonies. Three primary categories of variants were scored: point mutations, conversions to ura3-1, and gross chromosomal rearrangements/deletions/duplications (called hereafter GCRs), where the reporter gene could not be amplified or had a different size. We also analyzed spontaneous mutagenesis using cells without the Flp recombinase. We observed a 36- to 53-fold increase in point mutations and a 186-fold increase in ura3-1 conversions next to the break but no increase 15 kb from the broken fork (Fig. 2E). Frameshift mutations are increased threefold more (95×) than base substitutions (35×) (table S1), which is indicative of compromised mismatch repair (22). GCRs are increased 100-fold without ura3-1 and an additional 100-fold in the presence of ura3-1, indicating that ura3-1 is used as a recombination template to generate GCRs (Fig. 2E).
To establish whether cleavage of recombination intermediates is required for the fidelity of repair, we measured mutation rates in mus81Δ and yen1Δ mutants. In both, mutation rates remain similar to that of wild-type close to the fork breakage but increase significantly, by 6- to 95-fold, in mus81Δ cells farther from the broken fork at subtelomeric locations (Fig. 3B). In the presence of the ura3-1 allele, the increase of mutations is dramatic even close to the break in mus81Δ cells, regardless of FRT positions. Farther from the break, an increase of up to 150-fold is observed, but only at subtelomeric positions (Fig. 3C). These results are consistent with a major role of Mus81 in limiting mutagenic repair, which is particularly important when there is no efficient converging fork.
Fig. 3. Mus81 suppresses error-prone Pol32-dependent synthesis at broken replication fork.
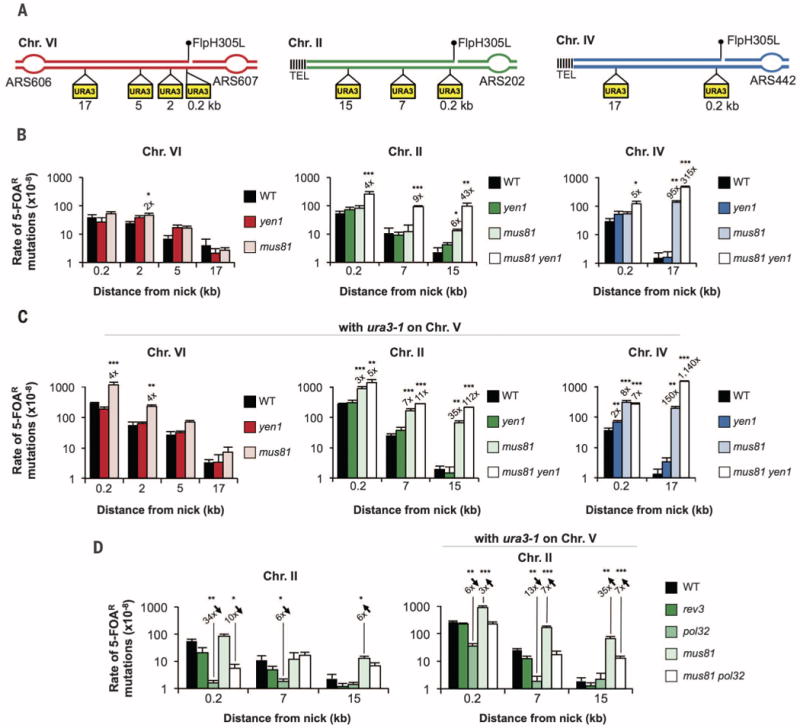
(A) Location of URA3 reporters used for mutation analysis. (B to D) Median 5-FOAR mutation rates in wild-type (WT) cells or indicated mutants. Significant fold changes compared with wild type are indicated, and error bars represent 1 SD. *P < 0.05, **P < 0.01, ***P < 0.001.
Sequencing of reporters from 5-FOAR cells at positions next to broken forks (ChrII, ChrVI, and ChrIV) or 15 to 17 kb away (ChrII and ChrVI) revealed that the most prominent change in the absence of Mus81 is an increased usage of the ura3-1 allele during repair. Resultant conversion events increase fivefold over that of wild type close to the break, with an efficient converging fork. At subtelomeric positions, conversions increase 18-or 96-fold near the break and up to 115-fold 15 kb from the break (Fig. 4, A and B, and fig. S5). Neither GCRs nor point mutations are increased more than twofold in mus81Δ cells next to the break site. In contrast, both GCRs and point mutations are increased above wild-type levels farther from the break at subtelomeric positions (Fig. 4, A and B, and fig. S5). Thus, an efficient converging fork dictates the distance of inaccurate synthesis in mus81Δ cells. Most GCRs (10 out of 10 tested) in mus81Δ cells at the URA3 reporter 15 kb from the broken fork on ChrII result from nonreciprocal translocations with ura3-1, as shown through analysis of rearrangement junctions (fig. S6), further documenting switches to nonallelic templates in mus81Δ cells, even far from the break.
Fig. 4. Mus81 prevents excessive donor site–switching during repair.
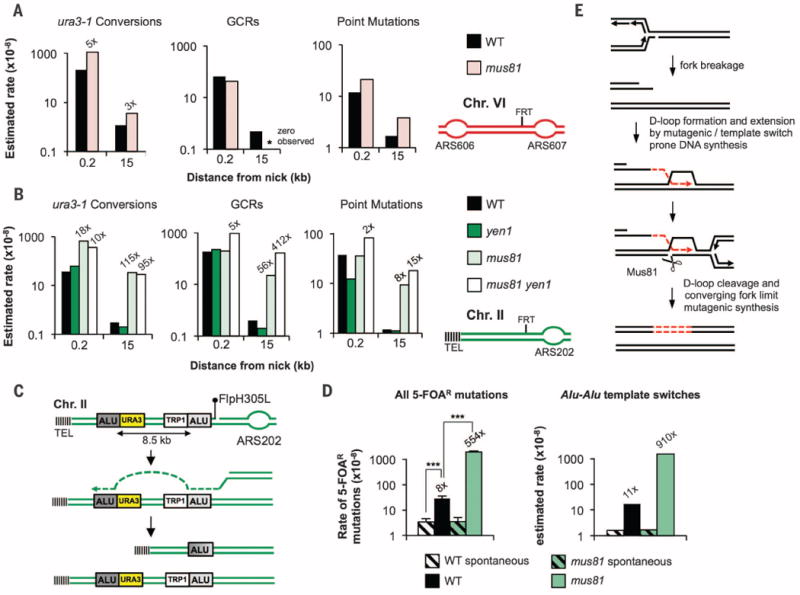
(A and B) Estimated rates of mutation categories at broken replication forks on (A) ChrVI and (B) ChrII. Fold changes over wild type are indicated. (C) Scheme of Alu-mediated template switches resulting in deletion. (D) Total 5-FOAR rates and estimated rates of Alu-mediated template switches. (E) Model for the control of repair fidelity at broken replication forks.
Although yen1Δ cells have no notable change in any type of event, elimination of Yen1 in a mus81Δ background, tested only at ChrII, leads to a further increase of GCRs but no increase in template switches and only a mild increase in point mutations (Fig. 4B and fig. S5). These results support the view that Yen1 plays a backup role in the absence of Mus81 (23), likely processing structures that arise in the absence of Mus81, which are outcomes of the D-loop merging with a converging fork. This is consistent with the late M-phase activation of Yen1 (24, 25).
BIR is mediated by Rad51 and Pol32, and indeed the majority of mutations at broken forks in wild-type and mus81Δ cells arise in a Rad51- and Pol32-dependent manner (Fig. 3D and fig. S4). This indicates that Pol32 mediates initial DNA synthesis at broken forks and that Mus81 decreases the distance it travels. Consistent with high Pol32-dependent mutation rates in mus81Δ, Pol32 plays a role in resistance to single-fork breakage induced at subtelomeric locations or by high doses of CPT (fig. S2), suggesting that repair proceeds via Pol32-BIR in the absence of Mus81 and an efficient converging fork. Pol32 also works with Polς, encoded by REV3, but this function of Pol32 is not important at broken forks, as rev3Δ does not show a significant decrease in mutations (Fig. 3D and fig. S7). Sequencing analysis revealed that nearly all break-induced point mutations in wild-type and mus81Δ cells depend on Pol32. Also, most events involving recombination template switches to ura3-1 depend on Pol32, although some GCRs clearly are Pol32-independent (fig. S7).
The presence of ura3-1 greatly increases GCRs next to the FRT on both ChrII and ChrVI in wild-type cells. Rearrangements in both cases involve deletion of sequences between the URA3 reporter and the closest Ty long terminal repeat (LTR) on the opposite sideof the FRT. The deleted regions are replaced by sequences between ura3-1 and a Ty LTR located on ChrV (figs. S8 and S9). Because these HR events use substrates on both sides of the FRT and, unlike recombination template switches, are not substantially affected by MUS81 deletion, it is likely that they correspond to repair of rare two-ended breaks by Pol32-dependent gap repair (26).
More than half of the human genome is composed of interspersed repeats, with Alu repetitive elements being the most abundant class (~1.1 million copies). Alus are often found at breakpoints of copy number variants as well as complex genomic rearrangements associated with disease (27). To address whether broken fork repair stimulates template switches between diverged Alu elements, we inserted two Alus (88.2% identical) that were previously observed to mediate human disease-associated deletions (28) onto ChrII, 1.2 and 9.7 kb from the FRT site, flanking a URA3 reporter (Fig. 4C). Alu-Alu template switches lead to deletion of the 8.5-kb sequence separating them. In wild-type cells, induction of broken forks increases 5-FOAR mutation rates by ~10-fold, the majority of which are Alu-Alu template switches. Deletion of MUS81 further increases the rate of Alu-Alu template switches 83-fold above that of wild type (Fig. 4D), underscoring the role of Mus81 in suppressing switches between highly diverged templates. Switches between Alus occurred at 4 to 21 nucleotides of microhomology (fig. S10). These results suggest that template switch mechanisms could account for a substantial proportion of Alu-Alu structural variation in humans, rather than unequal crossing over.
We demonstrate that fidelity of repair at broken replication forks depends on two partially compensatory mechanisms: cleavage by Mus81 and arrival of a converging fork (Fig. 4E and fig. S11). Converging forks limit the need to reestablish fully functional forks, illustrating an advantage of the multi-origin nature of eukaryotic chromosomes. We propose that deficiencies in Mus81 or timely converging forks may underlie the increased usage of POLD3/Pol32-mediated BIR in cancer cells (9) and consequently provide higher adaptation potential to cancer cells and promote tumor progression.
Supplementary Material
Acknowledgments
We thank P. Hastings, A. Malkova, S. Rosenberg, and D. Bates for critical comments on the manuscript. This work was supported by National Institutes of Health (NIH) grants GM080600 to G.I., NS058529 and HG006542 to J.R.L., and NS083159 to I.M.C. C.R.B is a Howard Hughes Medical Institute fellow of the Damon Runyon Cancer Research Foundation (DRG 2155-13). J.R.L. is a paid consultant for Regeneron Pharmaceuticals and has stock options in Lasergen.
Footnotes
SUPPLEMENTARY MATERIALS
www.sciencemag.org/content/349/6249/742/suppl/DC1
Materials and Methods
References (29–31)
REFERENCES AND NOTES
- 1.Shee C, Gibson JL, Rosenberg SM. Cell Rep. 2012;2:714–721. doi: 10.1016/j.celrep.2012.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Deem A, et al. PLOS Biol. 2011;9:e1000594. doi: 10.1371/journal.pbio.1000594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Smith CE, Llorente B, Symington LS. Nature. 2007;447:102–105. doi: 10.1038/nature05723. [DOI] [PubMed] [Google Scholar]
- 4.Pratto F, et al. Science. 2014;346:1256442. doi: 10.1126/science.1256442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lydeard JR, Jain S, Yamaguchi M, Haber JE. Nature. 2007;448:820–823. doi: 10.1038/nature06047. [DOI] [PubMed] [Google Scholar]
- 6.Saini N, et al. Nature. 2013;502:389–392. doi: 10.1038/nature12584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wilson MA, et al. Nature. 2013;502:393–396. doi: 10.1038/nature12585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sakofsky CJ, et al. Cell Rep. 2014;7:1640–1648. doi: 10.1016/j.celrep.2014.04.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Costantino L, et al. Science. 2014;343:88–91. doi: 10.1126/science.1243211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Carvalho CM, et al. Nat Genet. 2013;45:1319–1326. doi: 10.1038/ng.2768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nielsen I, et al. Nat Methods. 2009;6:753–757. doi: 10.1038/nmeth.1372. [DOI] [PubMed] [Google Scholar]
- 12.McGuffee SR, Smith DJ, Whitehouse I. Mol Cell. 2013;50:123–135. doi: 10.1016/j.molcel.2013.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Clausen AR, et al. Nat Struct Mol Biol. 2015;22:185–191. doi: 10.1038/nsmb.2957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Donnianni RA, Symington LS. Proc Natl Acad Sci USA. 2013;110:13475–13480. doi: 10.1073/pnas.1309800110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ehmsen KT, Heyer WD. Nucleic Acids Res. 2008;36:2182–2195. doi: 10.1093/nar/gkm1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pepe A, West SC. Cell Rep. 2014;7:1048–1055. doi: 10.1016/j.celrep.2014.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ho CK, Mazón G, Lam AF, Symington LS. Mol Cell. 2010;40:988–1000. doi: 10.1016/j.molcel.2010.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Muñoz-Galván S, et al. Mol Cell Biol. 2012;32:1592–1603. doi: 10.1128/MCB.00111-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Roseaulin L, et al. EMBO J. 2008;27:1378–1387. doi: 10.1038/emboj.2008.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Doksani Y, Bermejo R, Fiorani S, Haber JE, Foiani M. Cell. 2009;137:247–258. doi: 10.1016/j.cell.2009.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Müller CA, et al. Nucleic Acids Res. 2014;42:e3. doi: 10.1093/nar/gkt878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Malkova A, Haber JE. Annu Rev Genet. 2012;46:455–473. doi: 10.1146/annurev-genet-110711-155547. [DOI] [PubMed] [Google Scholar]
- 23.Blanco MG, Matos J, Rass U, Ip SC, West SC. DNA Repair (Amst) 2010;9:394–402. doi: 10.1016/j.dnarep.2009.12.017. [DOI] [PubMed] [Google Scholar]
- 24.Blanco MG, Matos J, West SC. Mol Cell. 2014;54:94–106. doi: 10.1016/j.molcel.2014.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Eissler CL, et al. Mol Cell. 2014;54:80–93. doi: 10.1016/j.molcel.2014.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jain S, et al. Genes Dev. 2009;23:291–303. doi: 10.1101/gad.1751209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gu S, et al. Hum Mol Genet. 2015;24:4061–4077. doi: 10.1093/hmg/ddv146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Boone PM, et al. Am J Hum Genet. 2014;95:143–161. doi: 10.1016/j.ajhg.2014.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.