

Background: Integrins regulate insulin signaling, but how they affect hepatic metabolism in vivo is unknown.
Results: Integrin α1-null mice on a high fat diet display severe hepatic insulin resistance and decreased hepatic fat accumulation.
Conclusion: Integrin α1β1 protects against hepatic insulin resistance while promoting fatty liver upon nutrient overload.
Significance: Integrin α1β1 regulates hepatic insulin action and lipid accumulation.
Keywords: Extracellular Matrix, Insulin Resistance, Integrin, Lipid Metabolism, Liver Metabolism
Abstract
Hepatic insulin resistance is associated with increased collagen. Integrin α1β1 is a collagen-binding receptor expressed on hepatocytes. Here, we show that expression of the α1 subunit is increased in hepatocytes isolated from high fat (HF)-fed mice. To determine whether the integrin α1 subunit protects against impairments in hepatic glucose metabolism, we analyzed glucose tolerance and insulin sensitivity in HF-fed integrin α1-null (itga1−/−) and wild-type (itga1+/+) littermates. Using the insulin clamp, we found that insulin-stimulated hepatic glucose production was suppressed by ∼50% in HF-fed itga1+/+ mice. In contrast, it was not suppressed in HF-fed itga1−/− mice, indicating severe hepatic insulin resistance. This was associated with decreased hepatic insulin signaling in HF-fed itga1−/− mice. Interestingly, hepatic triglyceride and diglyceride contents were normalized to chow-fed levels in HF-fed itga1−/− mice. This indicates that hepatic steatosis is dissociated from insulin resistance in HF-fed itga1−/− mice. The decrease in hepatic lipid accumulation in HF-fed itga1−/− mice was associated with altered free fatty acid metabolism. These studies establish a role for integrin signaling in facilitating hepatic insulin action while promoting lipid accumulation in mice challenged with a HF diet.
Introduction
The prevalence of type 2 diabetes has increased dramatically in parallel with excess caloric intake and sedentary lifestyles. Insulin resistance precedes the development of type 2 diabetes (1, 2). As one of the major insulin-responsive organs, the liver contributes to the pathogenesis of insulin resistance (3, 4). Hepatic insulin resistance is strongly correlated with hepatic lipid accumulation. In rats, 3 days of high fat (HF)2 feeding leads to a 3-fold increase in hepatic triglyceride (TG) accumulation and decreased suppression of hepatic glucose production during an insulin clamp (5). In addition, hepatic lipid accumulation is associated with liver damage and increases in extracellular matrix collagen proteins (3, 4, 6, 7). Liver biopsies from humans with and without type 2 diabetes demonstrate that livers from diabetic patients exhibit severe steatosis associated with increased collagen IV protein expression (8).
Extracellular matrix production and degradation are controlled by integrins, transmembrane receptors for extracellular matrix proteins that consist of an α and a β subunit (9). Upon ligand binding, integrins transduce signals across the plasma membrane to facilitate outside-in cell signaling (10). Several studies have suggested a role for integrins in the promotion of insulin action (9, 11–14). Integrin α5β1 binding to its ligand fibronectin enhances insulin-stimulated tyrosine phosphorylation of both the insulin receptor and IRS1 (13). Integrin β1 activation through its engagement with an anti-integrin β1 antibody or fibronectin leads to IRS1, PI3K, and subsequent Akt activation in isolated rat adipocytes (11). The integrin signaling molecule focal adhesion kinase (FAK) has been shown to directly bind to IRS1 and lead to IRS1 tyrosine phosphorylation in intact cells (14). Finally, the loss of the integrin β1 subunit in striated muscle leads to insulin resistance and decreased muscle glucose uptake (12).
Integrin α1β1 is a major collagen receptor expressed on various cell types, including hepatocytes (15). The expression of integrin α1β1 is up-regulated in the course of hepatic injury and extracellular matrix remodeling (16). We showed previously that global deletion of the integrin α1 subunit leads to severe impairments in insulin-induced suppression of glucose production in HF-fed mice (17). However, the mechanism for this effect and the broader role of integrin α1 in the pathogenesis of HF diet-induced hepatic insulin resistance and lipid accumulation are unknown.
In this study, we show that integrin α1 protein expression is up-regulated with HF feeding in hepatocytes. Thus, we assessed glucose tolerance and insulin sensitivity in HF-fed integrin α1-null (itga1−/−) and wild-type (itga1+/+) littermates to determine whether this response belies a protective effect against hepatic metabolic impairments in C57BL/6J mice. We show that deletion of integrin α1 results in severe hepatic insulin resistance as evidenced by complete alleviation of the insulin-mediated suppression of hepatic glucose production and decreased hepatic insulin signaling. The severe hepatic insulin resistance in HF-fed itga1−/− mice was present despite an ∼50% decrease in hepatic TG and diglyceride (diacylglycerol (DAG)) accumulation. The reduction in hepatic lipids was associated with a combination of decreased free fatty acid (FFA) availability upon insulin stimulation, decreased gene expression of the fatty acid transporter Cd36, and increased mitochondrial fatty acid utilization. This indicates that the decrease in hepatic lipid accumulation in HF-fed itga1−/− mice may be attributed to altered fatty acid metabolism.
EXPERIMENTAL PROCEDURES
Mouse Models
All animal protocols were approved by the Vanderbilt University Institutional Animal Care and Use Committee and conducted in Association for Assessment and Accreditation of Laboratory Animal Care-accredited facilities. Mice were housed in a temperature- and humidity-controlled environment with a 12/12-h light/dark cycle. Littermate wild-type (itga1+/+) and integrin α1-null (itga1−/−) male mice on a C57BL/6J background (18) were fed a standard chow diet (5.5% fat by weight; Purina Laboratory Rodent Diet 5001) or HF diet (60% kcal from fat; Bio-Serv F3282) for 16 weeks. Studies were performed between 19 and 24 weeks of age. Body composition was assessed using a mq10 nuclear magnetic resonance analyzer (Bruker Optics).
Hepatocyte Isolation
Mice were anesthetized, and their livers were perfused via the portal vein with prewarmed liver perfusion medium (Invitrogen) for 10–15 min at a rate of 4–5 ml/min. The livers were then perfused with liver digest medium containing collagenase (Invitrogen) for 15–20 min at a rate of 4–5 ml/min. After digestion, the livers were removed from the mice and placed in a dish containing liver digest medium, and the hepatic capsule was torn and shaken to free the dissociated cells. The cell suspension was filtered through a sterile 150-mesh nylon screen into a 50-ml sterile Falcon tube and centrifuged at 4 °C to collect the cells. The cell pellet was resuspended and washed three times in Hanks's balanced salt solution prior to a final resuspension in lysis buffer. Proteins were extracted, and the cellular protein content was measured using the Bradford protein assay (Bio-Rad).
Glucose Tolerance Tests (GTTs)
Indwelling carotid artery and gastric catheters were surgically implanted for sampling and glucose administration, respectively. The gastric catheter allows mice to absorb glucose via physiological mechanisms and avoids a stress response from intraperitoneal injection or gavage. Mice were fasted for 5 h, and baseline arterial glucose and insulin measurements were obtained through the arterial catheter to prevent handling the mice. Glucose was administered through the gastric catheter at a dose of 2 g/kg of body weight. Arterial glucose was measured at 5, 10, 15, 20, 30, 45, 60, 90, and 120 min after glucose administration. Arterial insulin levels were assessed during the GTT at 10, 30, 60, and 120 min. Plasma insulin was determined by radioimmunoassay.
Hyperinsulinemic-Euglycemic Clamp (Insulin Clamp)
Indwelling carotid artery and jugular vein catheters were surgically implanted for sampling and infusion 5–6 days before the insulin clamp (19). Mice were fasted for 5 h before the start of the study. [3-3H]Glucose was primed (2.4 μCi) and continuously infused for a 90-min equilibration period (0.04 μCi/min). Baseline measurements were determined in blood samples collected at −15 and −5 min for analysis of glucose, [3-3H]glucose, FFAs, and insulin. At t = 0, insulin (4 milliunits/kg/min) was infused at a fixed rate, and glucose (50% dextrose mixed with [3-3H]glucose) was infused at a variable rate to maintain euglycemia. The mixing of D50 with [3-3H]glucose prevents deviations in specific activity during the insulin clamp (20). Blood glucose levels were clamped at 150–160 mg/dl. Heparinized saline-washed erythrocytes were infused to prevent a fall in hematocrit. Blood was taken at 80–120 min for the determination of [3-3H]glucose.
Processing of Insulin Clamp Plasma Samples
The radioactivity of [3-3H]glucose was determined by liquid scintillation counting (19). Glucose appearance and disappearance rates were calculated using non-steady-state equations (21). Arterial insulin was determined by ELISA (ALPCO). FFAs were assessed using an enzymatic assay (NEFA C kit, Wako Chemicals). Basal steady-state FFA levels were calculated as an average of samples taken at t = −15 and −5 min. The levels during the insulin clamp were calculated as an average between the samples taken at t = 80 and 120 min.
Hepatic TG and DAG Content
Liver TGs were quantified using the Triglycerides-GPO reagent set (Pointe Scientific, Inc.) according to the manufacturer's protocol. Neutral lipids were stained with Oil Red O. Liver DAGs were extracted using the method of Folch et al. (22). Phospholipids, DAGs, TGs, and cholesteryl esters were scraped from the plates and methylated using BF3/methanol as described by Morrison and Smith (23). The methylated fatty acids were extracted and analyzed by gas chromatography. Gas chromatography analyses were carried out on an Agilent 7890A gas chromatograph equipped with flame ionization detectors and a capillary column (SP2380, 0.25 mm × 30 m, 0.25-μm film; Supelco Inc., Bellefonte, PA). Helium was used as a carrier gas. The oven temperature was programmed from 160 to 230 °C at 4 °C/min. Inclusion of lipid standards with odd-chain fatty acids permitted quantitation of the amount of lipid in the sample. Dipentadecanoylphosphatidylcholine (C15:0), diheptadecanoin (C17:0), trieicosenoin (C20:1), and cholesteryl eicosenoate (C20:1) were used as standards.
Hepatic TG Secretion and Measurement of Plasma TGs
Mice with indwelling carotid artery and jugular vein catheters were fasted for 8 h. Baseline arterial plasma TGs were determined in samples collected at -30 and 0 min. After the 0-min sample, VLDL-TG clearance was blocked by an intravenous injection of tyloxapol (500 mg/kg; Sigma). Blood samples were collected at 45-min intervals (0, 45, 90, 135, and 180 min post-injection). Plasma TGs were assessed using Triglycerides-GPO reagent (Raichem Reagents).
Mitochondrial Oxygen Consumption and Enzymatic Activity
Mice were fasted for 5 h prior to cervical dislocation. Fresh liver samples were mechanically permeabilized, and oxygen consumption was measured in air-saturated MiR05 medium (pH 7.4, 30 °C) with a Clark-type oxygen electrode (Oroboros Instruments GmbH, Innsbruck, Austria) (24). State 2 respiration was measured in the presence of either 10 mm glutamate and 2 mm malate or 2 mm malate and 50 μm palmitoylcarnitine prior to the addition of ADP. State 3 respiration was measured upon the addition of 0.5 mm ADP. Cytochrome c (10 μm) was added at the end of each measurement to ensure that the outer mitochondrial membrane was intact. The following criteria were applied for the inclusion of each respiration measurement: a respiratory control ratio at or above 4 and <10% response to cytochrome c addition. The respiratory control ratio was calculated as state 3/state 2. Citrate synthase activity was measured in liver homogenates according to the method of Hepple et al. (25). Respiration measurements were normalized to citrate synthase activity.
Immunoprecipitation and Immunoblotting
Frozen liver tissue was homogenized in buffer containing 25 mm Tris-HCl (pH 7.4), 10 mm EDTA, 10% glycerol, 1% Triton X-100, 50 mm sodium pyrophosphate, 100 mm sodium fluoride, 1 mm PMSF, and Halt protease and phosphatase inhibitor mixture (Thermo Scientific). For immunoprecipitation of the insulin receptor, 1 mg of protein was incubated with antibodies against the insulin receptor (Cell Signaling). Samples were incubated overnight with Protein A/G PLUS-agarose immunoprecipitation reagent (Santa Cruz Biotechnology). The immunoprecipitates were collected and applied to SDS-polyacrylamide gels and transferred to PVDF membranes. Immunoblots were incubated with primary antibodies against the insulin receptor Cell Signaling) and phosphotyrosine (Santa Cruz Biotechnology). For traditional immunoblots, the following primary antibodies were used: phospho-Akt (Ser473 and Thr308; Cell Signaling), total Akt (Cell Signaling), phospho-IRS1 (Tyr612; Invitrogen), total IRS1 (Cell Signaling), phospho-ERK1/2 (Thr202/Tyr204; Cell Signaling), total ERK 1/2 (Cell Signaling), phospho-JNK1/2 (Thr183/Tyr185; Cell Signaling), phospho-FAK (Tyr397; Abcam), total FAK (Santa Cruz Biotechnology), phospho-Foxo1 (Ser253; Upstate), total Foxo1 (Santa Cruz Biotechnology), integrin α1 (Chemicon), and β-actin (Cell Signaling). Imaging and densitometry were performed using the ImageJ program and the Odyssey imaging system (LI-COR Biosciences).
Immunohistochemistry
Collagen I was assessed by immunohistochemistry in paraffin-embedded tissue sections. Sections (5 μm) were incubated with anti-collagen I antibody for 60 min prior to staining with hematoxylin. EnVision+ System-HRP/DAB (DakoCytomation) was used to produce staining. Images were obtained using a QImaging Micropublisher camera mounted on an Olympus upright microscope.
Real-time PCR
RNA was extracted using an RNeasy mini kit (Qiagen). RNA was reverse-transcribed using an iScript cDNA synthesis kit (Bio-Rad). Quantitative PCR (qPCR) was performed using TaqMan Universal PCR Master Mix and commercially available TaqMan assays (Applied Biosystems) on a CFX real-time PCR instrument (Bio-Rad) for all genes except for Dgat1 and Dgat2. Data were normalized to the 18S ribosomal protein. The gene expression of Dgat1 and Dgat2 was measured using SYBR Green as described previously (26). Data were analyzed using the 2−ΔΔCt method (27).
Statistical Analysis
Data are presented as means ± S.E. Statistical analyses were performed using Student's t test or two-way analysis of variance, followed by Tukey's post hoc tests as appropriate. The significance level was p ≤ 0.05.
RESULTS
Integrin α1 Protein Expression Is Increased in Hepatocytes Isolated from HF-fed C57BL/6J Mice
Up-regulation of the integrin α1 subunit has been described in several states of chronic liver disease associated with inflammation and fibrosis (16). To determine the effect of HF feeding on hepatocyte integrin α1 expression, hepatocytes were isolated from both chow- and HF-fed C57BL/6J wild-type mice. Integrin α1 protein expression was increased by ∼2-fold in isolated hepatocytes from HF-fed mice (Fig. 1).
FIGURE 1.
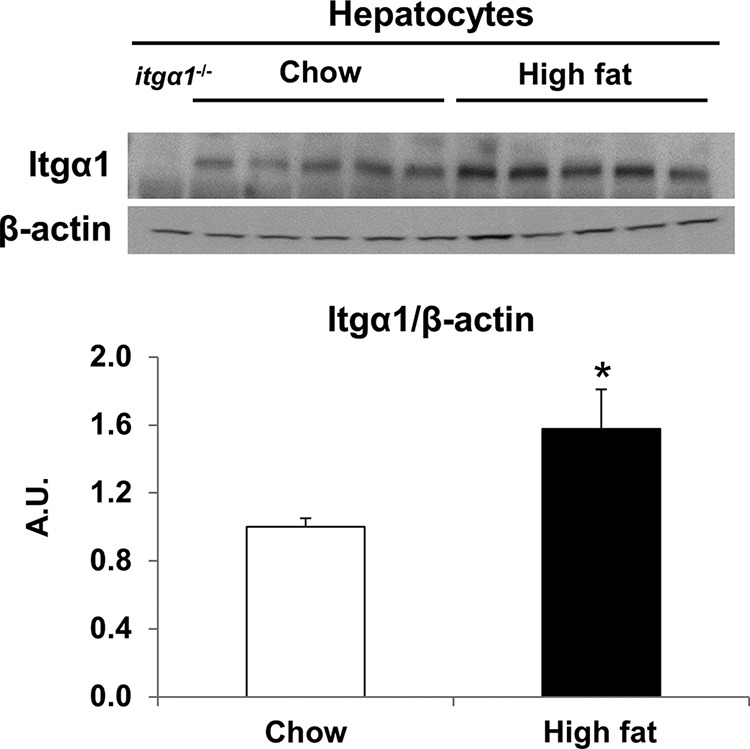
Integrin α1 protein expression is increased in hepatocytes isolated from HF-fed mice. Shown are the results of Western blot analysis for integrin α1 protein expression in isolated hepatocytes from chow- and HF-fed mice (n = 5/group). Integrin α1 protein expression was normalized to β-actin. Data are presented as means ± S.E. (n = 4–5). *, p < 0.05, chow-fed versus HF-fed. A.U., arbitrary units.
Adaptations to the Genetic Deletion of Integrin α1 in Both Chow- and HF-fed Mice
To determine the role of integrin α1 in livers from HF-fed mice, itga1+/+ and itga1−/− mice were fed a HF diet for 16 weeks. HF feeding increased body weight, percent fat mass, and fasting blood glucose levels (Table 1). Body weight did not differ between genotypes on the respective diets. Fat mass was decreased in HF-fed itga1−/− mice compared with HF-fed itga1+/+ mice. HF feeding increased fasting glucose and insulin levels in wild-type mice. Fasting glucose levels were not different between the genotypes on the respective diets. Fasting insulin levels were not different between the genotypes on a chow diet; however, they were elevated in HF-fed itga1−/− mice compared with HF-fed itga1+/+ mice, indicating insulin resistance.
TABLE 1.
Characteristics of wild-type (itga1+/+) and integrin α1-null (itga1−/−) mice on both chow and HF diets
Body composition and fasting glucose and insulin levels were determined in basal 5-h fasted mice (n = 6–8/group). Data are represented as means ± S.E.
| Chow |
HF |
|||
|---|---|---|---|---|
| itga1+/+ | itga1−/− | itga1+/+ | itga1−/− | |
| Weight (g) | 26.1 ± 0.5 | 27.0 ± 0.5 | 48.4 ± 0.9a | 48.9 ± 1.3b |
| Fat mass (%) | 10.4 ± 0.5 | 11.5 ± 2.0 | 41.6 ± 0.7a | 39.0 ± 1.8c |
| Fasting glucose (mg/dl) | 118 ± 3 | 111 ± 7 | 147 ± 12a | 151 ± 5b |
| Fasting insulin (ng/ml) | 0.80 ± 0.2 | 0.82 ± 0.1 | 2.12 ± 0.21a | 3.44 ± 0.72c |
a p < 0.05, chow-fed itga1+/+ mice versus HF-fed itga1+/+ mice.
b p < 0.05, chow-fed itga1−/− mice versus HF-fed itga1−/− mice.
c p < 0.05, HF-fed itga1+/+ mice versus. HF-fed itga1−/− mice.
HF feeding leads to excessive accumulation of collagen in the liver (7, 28). Integrin α1β1 down-regulates collagen synthesis; however, its contribution to hepatic collagen gene expression in the HF-fed state has not been investigated (29). Therefore, we measured collagen gene expression in itga1+/+ and itga1−/− mice on both chow- and HF-fed diets (Table 2). The gene expression of collagens α1(I), α2(I), and α1(III) was increased in chow-fed itga1−/− mice compared with chow-fed itga1+/+ mice. The gene expression of collagens α1(I) and α2(I) was increased by HF feeding in itga1+/+ mice. There was no difference in collagen gene expression between chow- and HF-fed itga1−/− mice. Next, we investigated whether the changes in collagen I gene expression resulted in differences in protein expression by Western blot analysis and immunohistochemistry (Fig. 2). Consistent with the collagen I gene expression results, we found that collagen I protein expression was also increased in itga1−/− mice compared with itga1+/+ mice regardless of diet by Western blot analysis (Fig. 2A). Collagen I immunohistochemistry in the liver was used to determine the localization of the collagen deposition (Fig. 2B). Collagen I staining was most prevalent around the vessels and within the sinusoidal spaces under all conditions. Thus, deletion of integrin α1 leads to increased hepatic collagen gene and protein expression independent of diet.
TABLE 2.
Collagen gene expression: interaction of gene and diet
Total RNA was extracted from the livers of 5-h fasted mice and reverse-transcribed into cDNA. qPCR was used to determine gene expression of several collagen chains (n = 5–7/group). Data are represented as means ± S.E.
| Collagen | Chow |
High fat |
||
|---|---|---|---|---|
| itga1+/+ | itga1−/− | itga1+/+ | itga1−/− | |
| α1(I) | 1.1 ± 0.2 | 3.0 ± 0.6a | 1.5 ± 0.1a | 4.2 ± 1.2b |
| α2(I) | 1.1 ± 0.3 | 9.6 ± 3.0a | 2.6 ± 0.6a | 13.6 ± 5.1b |
| α1(III) | 1.2 ± 0.2 | 4.0 ± 0.6a | 1.1 ± 0.3 | 4.5 ± 1.6b |
| α1(IV) | 1.0 ± 0.1 | 2.3 ± 0.4a | 1.8 ± 0.5 | 3.4 ± 1.0 |
a p < 0.05 compared with chow-fed itga1+/+ mice.
b p < 0.05 compared with HF fed itga1+/+ mice.
FIGURE 2.
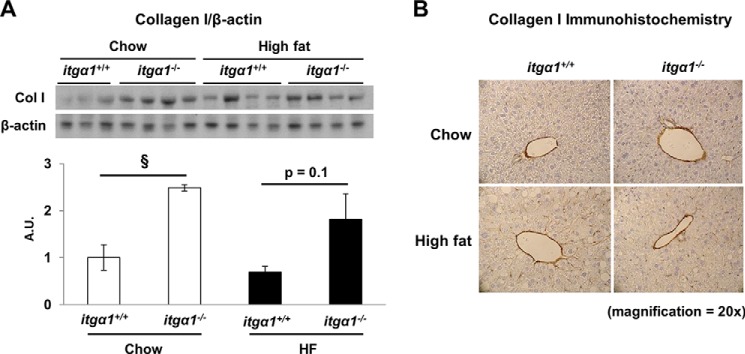
Collagen I protein expression is increased in integrin α1-null mice. A, Western blot analysis of collagen I (Col I) performed on liver homogenates from basal 5-h fasted mice (n = 4/group). Integrated intensities were obtained using ImageJ software, and collagen I protein expression was normalized to β-actin. A.U., arbitrary units. Data are presented as means ± S.E. §, p < 0.05 compared with chow-fed itga1+/+ mice. B, representative images from immunohistochemical staining of collagen I in livers from basal 5-h fasted mice (n = 5–7/group).
Integrin α1-null Mice on a HF Diet Exhibit Severe Hepatic Insulin Resistance
Fasting insulin levels were increased in HF-fed itga1−/− mice compared with HF-fed itga1+/+ mice, indicating impaired glucose metabolism. Therefore, we examined glucose tolerance in both chow- and HF-fed itga1+/+ and itga1−/− mice using gastric catheters to deliver glucose directly into the stomach. The results from the GTTs indicate that there was no difference in glucose tolerance or insulin response to a glucose challenge (insulin excursion) between the genotypes on the respective diets (Fig. 3, A–D). Considering that whole-body glucose tolerance can appear normal despite impaired insulin action (30), we next assessed insulin action in HF-fed itga1+/+ and itga1−/− mice using a hyperinsulinemic-euglycemic (insulin) clamp. During the insulin clamp, blood glucose levels were maintained between 150 and 160 mg/dl (Fig. 3E). The steady-state glucose infusion rate was lower in HF-fed itga1−/− mice (Fig. 3F). There was no difference in fasting endogenous glucose production (Fig. 3G). Endogenous glucose production during the insulin clamp was suppressed by ∼50% in HF-fed itga1+/+ mice. In contrast, endogenous glucose production was not suppressed in HF-fed itga1−/− mice (Fig. 3G), indicating complete resistance to insulin suppression of endogenous glucose production. There was no difference in basal or insulin-clamped whole-body glucose disappearance rates (Fig. 3H). Plasma insulin during the insulin clamp was not different between the groups (9.65 ± 1.22 versus 9.35 ± 1.20 ng/ml). The severe hepatic insulin resistance in HF-fed itga1−/− mice is independent of its effects on the transcriptional regulation of collagen, as there was no difference in collagen gene expression between the chow- and HF-fed itga1−/− mice (Table 2).
FIGURE 3.

Integrin α1β1 protects against diet-induced hepatic insulin resistance. GTTs were performed on chow- and HF-fed itga1+/+ and itga1−/− mice. Arterial glucose (A and C) and insulin (B and D) levels were measured during the GTTs (n = 5–7/group). Arterial glucose levels (E) and glucose infusion rates (F) were measured during the hyperinsulinemic-euglycemic (insulin) clamp (n = 5–6/group). Mice were fasted for 5 h prior to the start of the insulin clamp. Blood glucose levels were maintained between 150 and 160 mg/dl during steady state (80–120 min). Glucose (50%) was infused to maintain euglycemia. Endogenous glucose production (EndoRa; G) and whole-body disappearance rates (Rd; H) were determined during the steady-state period of the insulin clamp. Data are presented as means ± S.E. *, p < 0.05 compared with the basal measurement in HF-fed itga1+/+ mice; §, p < 0.05 compared with HF-fed itga1+/+ mice during the insulin clamp.
Integrins, including integrin α1β1, can regulate the phosphorylation and activation state of receptor tyrosine kinases and their downstream modulators (29, 31–34). To determine whether genetic deletion of the integrin α1 subunit affects insulin receptor phosphorylation, we assessed insulin receptor phosphorylation in insulin-clamped livers from HF-fed itga1+/+ and itga1−/− mice. There was no difference in insulin receptor phosphorylation (Fig. 4A), suggesting that integrin α1β1 does not affect insulin-mediated activation of its receptor. To determine whether integrin α1β1 affects insulin signaling downstream of the insulin receptor, we assessed several markers of insulin signaling in insulin-stimulated livers from HF-fed itga1+/+ and itga1−/− mice. Consistent with the hepatic glucose flux measurements, phosphorylation of IRS1 at Tyr612 and Akt at Ser473 were decreased in HF-fed itga1−/− mice (Fig. 4B), indicating decreased insulin action. There was no difference in the insulin-mediated phosphorylation of Akt at Thr308, Foxo1 at Ser253, or FAK at Ser397. To determine the effect of integrin α1 on hepatic gluconeogenic gene expression in the HF-fed state, we used qPCR to determine the gene expression of G6pc (glucose 6-phosphatase) and Pepck (phosphoenolpyruvate carboxylase) in livers from basal 5-h fasted and insulin-clamped mice (Fig. 4C). Consistent with the severe hepatic insulin resistance in the HF-fed itga1−/− mice, insulin failed to suppress G6pc gene expression, and there was a striking insulin-mediated increase in Pepck gene expression. In contrast, insulin-mediated G6pc gene expression was significantly decreased in the HF-fed itga1+/+mice. Consistent with other reports (35, 36), we found that deletion of integrin α1 reduced activation of the MAPK signaling pathway as evidenced by a slight but non-significant decrease in ERK1/2 activation and a significant decrease in JNK1/2 activation (Fig. 4B).
FIGURE 4.

Integrin α1β1 facilitates hepatic insulin action in HF-fed mice. Western blot analysis was performed on liver homogenates from basal 5-h fasted and 5-h fasted insulin-clamped mice. A, immunoprecipitation (IP) of the insulin receptor (IR) was performed on liver homogenates after the insulin clamp prior to an immunoblot (IB) for phosphotyrosine (pY; n = 4/group). Insulin receptor phosphorylation was determined as the ratio of phosphotyrosine to total insulin receptor. B, representative blots of liver insulin signaling after the insulin clamp (n = 4–5/group). C, mRNA was extracted from both basal 5-h fasted and 5-h fasted insulin-clamped livers. qPCR was performed to determine the gene expression of gluconeogenic genes G6pc and Pepck. D, FAK and FAK phosphorylation was determined in liver homogenates from basal 5-h fasted mice (n = 4/group). FAK phosphorylation was quantified as the ratio of phospho (p)-FAK to FAK. The percent increase in FAK phosphorylation was calculated relative to wild-type mice. Integrated intensities were obtained using Odyssey and ImageJ software. Data are presented as means ± S.E. §, p < 0.05 compared with HF-fed itga1+/+ mice; *, p < 0.05 compared with chow-fed itga1+/+ mice; ▵, p < 0.05 compared with chow-fed itga1−/− mice; ‡, p < 0.05 compared with basal 5-h fasted mice. A.U., arbitrary units.
Integrin α1β1 signals to activate several downstream targets, including FAK (37), and the integrin α1 subunit has been shown to bind FAK (38). In addition, FAK has been shown to interact with IRS1 (14). Thus, we sought to determine whether decreased insulin signaling in the HF-fed itga1−/− mice is associated with decreased FAK phosphorylation in both insulin-clamped and basal 5-h fasted livers. We found that there was no difference in insulin-stimulated FAK phosphorylation between the HF-fed itga1+/+ and itga1−/− mice (Fig. 4B). In the basal state, there was a significant decrease in FAK phosphorylation in HF-fed mice compared with chow-fed mice independent of genotype (Fig. 4D). However, there was no difference in the percent increase in phospho-FAK/FAK relative to the wild type in either the chow- or HF-fed mice in the basal 5-h fasted state. Thus, integrin α1β1 appears to mediate insulin signaling in a FAK-independent manner.
Genetic Deletion of Integrin α1 Improves Fatty Liver through Changes in Fatty Acid Metabolism
HF feeding leads to increased hepatic lipid accumulation and severe hepatic insulin resistance associated with decreased insulin-stimulated IRS1 tyrosine phosphorylation (5). Thus, we wanted to assess whether the severe hepatic insulin resistance in HF-fed itga1−/− mice could be attributed to increased lipid accumulation. To determine the role of integrin α1 in hepatic lipid accumulation, we assessed hepatic TG and DAG accumulation in the livers from both chow- and HF-fed itga1+/+ and itga1−/− mice. HF feeding increased liver TG and DAG content by ∼5-fold in itga1+/+ mice compared with chow-fed itga1+/+ mice (Fig. 4, A–C). Liver TG and DAG content was decreased by ∼50% in HF-fed itga1−/− mice compared with HF-fed itga1+/+ mice (Fig. 5, A–C). This was supported by the finding that lipid droplets were smaller in HF-fed itga1−/− mice compared with HF-fed itga1+/+ mice (Fig. 5A).
FIGURE 5.
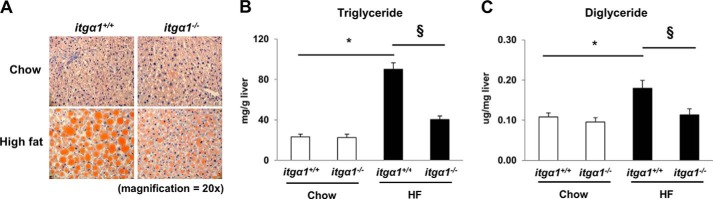
Integrin α1β1 promotes hepatic lipid accumulation. A, Oil Red O staining of liver neutral lipid droplets. Liver TG (B) and DAG (C) content was quantified in 5-h fasted mice (n = 5/group). Data are presented as means ± S.E. *, p < 0.05 compared with chow-fed itga1+/+ mice; §, p < 0.05 compared with HF-fed itga1+/+ mice.
To examine the potential mechanisms whereby HF-fed itga1−/− mice exhibit decreased TG and DAG accumulation, we first investigated several parameters that regulate hepatic TG metabolism. Circulating plasma TGs were increased in HF-fed itga1−/− mice (Fig. 6A). Thus, we tested the hypothesis that the reduction in liver TGs was due to increased secretion from the liver. However, this hypothesis was not supported by the results, as no difference in TG secretion was observed between the HF-fed itga1+/+ and itga1−/− mice (Fig. 6, B and C). Next, we examined the expression of several genes associated with hepatic lipogenesis. Despite decreased lipid accumulation, expression of the lipogenic genes Srebp-1c, Fas, Acc1, Scd1, and Elovl6 was increased in HF-fed itga1−/− mice. In contrast, the gene expression of Dgat1 and Dgat2, the genes responsible for catalyzing the final step in TG biosynthesis, was decreased in HF-fed itga1−/− mice (Fig. 6D).
FIGURE 6.

Effect of integrin α1 deletion on TG metabolism. A, circulating plasma TGs in basal 5-h fasted mice. B, hepatic TG secretion was determined using tyloxapol to block VLDL-TG clearance from the circulation. C, quantification of TG secretion rates. D, mRNA was extracted from basal 5-h fasted livers, and qPCR was used to determine lipogenic gene expression. Data are presented as means ± S.E. *, p < 0.05 compared with chow-fed itga1+/+ mice; §, p < 0.05 compared with HF-fed itga1+/+ mice (n = 5–8/group).
Circulating FFAs are typically the major substrate for the accumulation of hepatic TGs (39). Thus, we measured several markers of FFA metabolism in each mouse model. Basal 5-h fasting plasma FFAs were equal between the two genotypes (Fig. 7A). Insulin suppressed plasma FFAs to a greater extent in HF-fed itga1−/− mice. Next, we analyzed the expression of several genes involved in the regulation of fatty acid metabolism, including Cd36, Ppara, Pparg, and Cpt-1a (Fig. 7B). Consistent with the finding that hepatic lipid accumulation was decreased in HF-fed itga1−/− mice, we found that the gene expression of Cd36 and Pparg was decreased in HF-fed itga1−/− mice.
FIGURE 7.

Effect of integrin α1β1 on FFA metabolism. A, arterial non-esterified FFAs (NEFAs) in the basal 5-h fasted state and during the insulin clamp. B, expression of several genes implicated in the regulation of fatty acid metabolism. mRNA was extracted from 5-h fasted livers, and qPCR was performed to determine gene expression (n = 5–6/group). C and D, high resolution respirometry was performed on mechanically permeabilized liver pieces from 5-h fasted mice. Data are presented as means ± S.E. *, p < 0.05 compared with chow-fed itga1+/+ mice; §, p < 0.05 compared with HF-fed itga1+/+ mice. CSA, citrate synthase activity.
To determine the contribution of mitochondrial fatty acid-supported respiration to TG and DAG accumulation in both chow- and HF-fed itga1+/+ and itga1−/− mice, we assessed mitochondrial oxygen consumption in mechanically permeabilized liver pieces. Mechanically permeabilized liver pieces were used in lieu of isolated mitochondria to ensure that the cytoskeletal attachments were conserved and that the mitochondria were not stressed during the functional measurements. ADP-stimulated (state 3) respiration in the presence of palmitoylcarnitine and malate was increased in itga1−/− mice compared with itga1+/+ mice regardless of diet (Fig. 7D). There was no difference in glutamate/malate-stimulated state 3 respiration (Fig. 7C) or citrate synthase activity (data not shown) between groups. Thus, the observed decrease in TG and DAG accumulation in HF-fed itga1−/− mice is linked to increased capacity for mitochondrial fatty acid-supported respiration.
DISCUSSION
Integrin α1β1 is a collagen-binding integrin found on the hepatocyte (15). The results from this study show that integrin α1 protein expression was increased in hepatocytes upon HF feeding. The goal of this study was to determine whether increased expression of the integrin α1 subunit with HF feeding promotes hepatic insulin action and protects against metabolic impairments in HF-fed C57BL/6J mice. We found that integrin α1β1 signaling protects against diet-induced hepatic insulin resistance, as it promoted hepatic insulin action. Insulin clamp studies showed that hepatic glucose production was completely resistant to suppression by an insulin stimulus in HF-fed itga1−/− mice, and this was associated with decreased insulin signaling. Despite severe hepatic insulin resistance, HF-fed itga1−/− mice exhibited a 50% reduction in hepatic TG and DAG accumulation compared with their wild-type littermates. The reduction in hepatic lipids was associated with a combination of decreased FFA availability upon insulin stimulation, decreased gene expression of the fatty acid transporter Cd36, and increased mitochondrial fatty acid utilization.
The hepatocyte is the most abundant cell type and is a major site of hepatic insulin action. Thus, it was important to determine the effects of HF feeding on integrin α1 protein expression in isolated hepatocytes. Our study shows that integrin α1 protein expression is increased in hepatocytes isolated from wild-type mice after 16 weeks of HF feeding.
To determine the role of integrin α1 in glucose and lipid metabolism, itga1+/+ and itga1−/− mice were fed either a chow or HF diet for 16 weeks. The whole-body deletion of integrin α1 in HF-fed mice resulted in a small decrease in fat mass (∼8%). Glucose transported into the fat cell provides the three-carbon backbone essential for the esterification of FFAs (40). Previous studies from our laboratory showed that HF-fed itga1−/− mice have decreased insulin-stimulated adipose tissue glucose uptake (17). Thus, it is reasonable to propose that this reduction in glucose uptake contributes to the small decrease in fat mass in the HF-fed itga1−/− mice.
The measurement of basal 5-h fasting blood glucose and insulin levels revealed that fasting insulin levels were elevated in HF-fed itga1−/− mice, indicating impaired glucose metabolism and potentially insulin resistance. Oral glucose tolerance was assessed in mice with an indwelling gastric catheter for glucose delivery and an arterial catheter for blood sampling. There was no difference in either the glycemic or insulin responses during the oral GTT between genotypes. To determine whether the increase in integrin α1 protein expression improves hepatic insulin sensitivity in HF-fed mice, insulin clamps were used to assess insulin action. Consistent with our previous report (17) HF-fed itga1−/− mice exhibited a lower glucose infusion rate compared with HF-fed itga1+/+ mice. Hepatic glucose production was suppressed by ∼50% in HF-fed itga1+/+, whereas it was not suppressed in itga1−/− mice. This indicates a more severe hepatic insulin resistance in HF-fed itga1−/− mice. There was no difference in glucose disappearance rates during the insulin clamp. This indicates that the difference in the glucose infusion rate during the insulin clamp is due to complete loss of hepatic insulin sensitivity and not decreased insulin-stimulated glucose disposal from tissues such as skeletal muscle. The absence of skeletal muscle insulin resistance in HF-fed itga1−/− mice supports a previous observation from our laboratory showing that there is no difference in skeletal muscle insulin sensitivity between HF-fed itga1+/+ and itga1−/− mice (17).
Glucose tolerance is determined by several factors in addition to insulin resistance. These include glucose effectiveness (i.e. the ability of glucose to suppress endogenous glucose production and stimulate glucose uptake (41)) and insulin secretion and clearance. Because insulin concentrations were equal between genotypes during the oral GTT, it is probable that impaired insulin action is offset by differences in glucose effectiveness. This study highlights the value of considering both glucose tolerance and insulin action as distinct readouts when determining the metabolic phenotype of novel mouse models.
The complete lack of suppression of hepatic glucose production during the insulin clamp in HF-fed itga1−/− mice was associated with decreased insulin-stimulated IRS1 and Akt activation, followed by no insulin-mediated suppression of G6pc gene expression and increased insulin-mediated Pepck gene expression. This occurred in the absence of differences in insulin-mediated Foxo1 phosphorylation at Ser253 in whole liver homogenate. There was also no difference in insulin receptor phosphorylation. This suggests that there may be some aspect of integrin α1β1 signaling that promotes hepatic insulin action downstream of the insulin receptor in HF-fed mice (11). To further investigate how the deletion of integrin α1 in HF-fed mice results in decreased IRS1 and Akt activation, we assessed activation of several signaling molecules in the MAPK signaling pathway, including ERK1/2 and JNK1/2. Previous studies imply a role for JNK activation in the development of hepatic insulin resistance by decreasing insulin-induced tyrosine phosphorylation of IRS1 (42). In contrast, we found that activation of this pathway was down-regulated in HF-fed itga1−/− mice compared with HF-fed itga1+/+ mice, indicating that the absence of integrin α1 dampens MAPK signaling despite hepatic insulin resistance, and JNK activation is not responsible for the observed decrease in IRS1 Tyr phosphorylation in HF-fed itga1−/− mice.
There is considerable cross-talk between the insulin and integrin signaling cascades (9, 11–13). A potential mediator of this cross-talk is the integrin-specific signaling molecule FAK. FAK has been implicated in the regulation of hepatic insulin signaling (14, 37, 43). Decreased FAK signaling is associated with decreased hepatic IRS1 and Akt phosphorylation (37). Furthermore, fa/fa rats treated with a TNF-α-neutralizing agent exhibited increased hepatic FAK phosphorylation associated with decreased hepatic glucose output (43). In contrast to these studies, our findings show that down-regulation of FAK phosphorylation is not essential for the severe insulin resistance in insulin-clamped HF-fed itga1−/− mice compared with their wild-type littermates. There was a tendency for increased FAK activation in the basal 5-h fasted itga1−/− mice; however, this trend was equivalent regardless of diet. It is notable, however, that we did see a striking decrease in FAK phosphorylation in livers from 5-h fasted HF-fed mice compared with chow-fed mice regardless of genotype. This suggests that HF feeding facilitates an integrin α1β1-independent inhibition of FAK activation. Although the hepatic insulin resistance seen in the HF-fed itga1−/− mice cannot be attributed to decreased FAK phosphorylation, this may be an independent mechanism whereby HF feeding decreases hepatic insulin action in wild-type mice.
To further elucidate the mechanisms of the severe hepatic insulin resistance in the HF-fed itga1−/− mice, we measured hepatic lipid accumulation in our mouse models. Hepatic TG accumulation (or its lipogenic metabolites) can decrease hepatic insulin action (5, 44). Surprisingly, we found that HF-fed itga1−/− mice exhibited a 50% reduction in hepatic TG and DAG accumulation compared with their wild-type littermates. These data are important for several reasons. First, they suggest that integrin α1β1 promotes TG and DAG accumulation in the liver. Second, hepatic steatosis is dissociated from insulin resistance in HF-fed itga1−/− mice. This makes it a unique model of hepatic insulin resistance. It also supports other studies (45–48) that show that insulin resistance can be dissociated from lipid accumulation and can exist in the absence of increased TG and DAG.
To identify a potential mechanism whereby HF-fed itga1−/− mice exhibit decreased TG and DAG accumulation, we examined indices of TG synthesis and breakdown in both the HF-fed itga1+/+ and itga1−/− mice. These include lipogenesis, VLDL secretion, circulating FFAs, and mitochondrial fatty acid respiration (39, 44, 45). The results from this study show that the expression of the lipogenic genes Srebp-1c, Fas, Acc1, Scd1, and Elovl6 was increased. This indicates that the decrease in hepatic lipid accumulation is independent of decreased expression of these key lipogenic genes. We propose this is the result of a feedback mechanism that exists to up-regulate lipogenic pathways when hepatic lipid stores are low. This supports the notion that the liver promotes the incorporation of lipid metabolites into neutral lipid droplets to protect against the deleterious effects of a HF diet.
TG synthesis is regulated by the step-limiting enzyme DGAT (diglyceride acyltransferase). Contrary to the increase in genes involved in lipogenesis, we found that the gene expression of two predominant isoforms of DGAT (Dgat1 and Dgat2) was decreased. This finding may explain, at least in part, why hepatic DAG and TG accumulation is decreased in HF-fed itga1−/− mice. This conclusion is supported by previously published work by Chen et al. (49). In this study, the global deletion of DGAT1 resulted in a decrease in hepatic DAG levels. To determine whether a critical substrate for the DGAT reaction is altered, we measured hepatic glycerol 3-phosphate levels. Our results indicate that glycerol 3-phosphate levels are increased in livers of HF-fed itga1−/− mice (35.6 ± 4.7 and 48.4 ± 1.8 nmol/mg of liver), suggesting that the observed decrease in lipid accumulation is independent of hepatic glycerol 3-phosphate levels.
The majority of liver TGs are derived from circulating FFAs (39, 50). Here, we have shown that HF-fed itga1−/− mice have decreased circulating FFAs compared with HF-fed itga1+/+ mice during an insulin clamp. This indicates that insulin suppresses circulating FFAs to a greater extent in HF-fed itga1−/− mice, suggesting that insulin-mediated lipolysis (i.e. hydrolysis of triacylglycerol in adipocytes) is blunted, and this may account for the decreased lipid accumulation.
CD36 is key for FFA transport into the hepatocyte, and CD36 expression is correlated with liver fat content (51, 52). Our results show that liver Cd36 gene expression was in fact decreased in itga1−/− mice, consistent with the reduction in lipid accumulation. Additionally, CD36 is a known transcriptional target of PPARG (53). In further support of the decrease in Cd36 gene expression, our studies show that the gene expression of Pparg was also decreased.
FFAs are esterified into TGs or oxidized by the mitochondria in the hepatocyte (54). When the supply of FFAs exceeds the amount the mitochondria can oxidize, hepatic fat accumulation occurs. To determine the contribution of the mitochondria to fatty acid utilization in the chow- and HF-fed itga1+/+ and itga1−/− mice, palmitoylcarnitine/malate-stimulated state 3 respiration was measured in mechanically permeabilized liver pieces. The results show that palmitoylcarnitine-stimulated state 3 respiration was higher in itga1−/− mice regardless of diet. This suggests that hepatic mitochondria in itga1−/− mice have a greater capacity to utilize fatty acid substrates. Hepatic mitochondrial oxygen flux was not different in chow- and HF-fed mice. This result is supported by a study by Satapati et al. (55), who also showed no difference in hepatic mitochondrial palmitoylcarnitine/malate-supported state 3 respiration at 16 weeks of HF feeding in mice. The differences in mitochondrial respiration (i.e. fatty acid utilization by the mitochondria) occurred in the absence of differences in the expression of genes involved in the regulation of fatty acid oxidation (i.e. Ppara and Cpt-1a). Overall, the results show that the decrease in hepatic lipid accumulation in the HF-fed itga1−/− mice is due to regulation at multiple sites. Hepatic FFA availability and uptake of FFAs are decreased, whereas FFA utilization by the mitochondria is increased.
Here, we introduce for the first time that integrin α1 is up-regulated in hepatocytes as an important adaptive response to overnutrition and that this may protect against more severe insulin resistance. This novel concept is illustrated in Fig. 8. In HF-fed wild-type mice, the combination of both insulin and integrin α1β1 signaling leads to IRS1 and Akt phosphorylation, resulting in the partial suppression of hepatic glucose output. In contrast, when integrin α1 is absent, insulin-mediated IRS1 and Akt activation is decreased. This results in the complete absence of insulin-induced suppression of hepatic glucose production. Interestingly, not only is the profound insulin resistance observed in HF-fed itga1−/− mice independent of increased hepatic TG and DAG concentrations, it occurs in the presence of a striking decrease in these lipids. This is attributed to alterations in FFA metabolism. In HF-fed wild-type mice, circulating FFAs are taken up by the liver and utilized for the synthesis of DAG and TG. In HF-fed itga1−/− mice, circulating FFAs are lower, and those that are available within the hepatocyte are utilized to a greater extent by the mitochondria for mitochondrial respiration.
FIGURE 8.

Model whereby integrin α1β1 protects against severe hepatic insulin resistance while promoting TG accumulation. Integrin α1 protein expression increases when wild-type mice are fed a HF diet. This leads to increased integrin α1β1 cell signaling upon collagen binding. Upon insulin stimulation, the combination of both insulin and integrin α1β1 signaling leads to the phosphorylation and subsequent activation of IRS1 and Akt. This results in the partial suppression of hepatic glucose output. Circulating FFAs are taken up by the liver and utilized primarily for the synthesis of DAG and TG, while some may be shunted toward the mitochondria for mitochondrial (mt) respiration. In contrast, when integrin α1-null mice are fed a HF diet and insulin levels are high, the absence of integrin signaling leads to decreased IRS1 and Akt activation. This results in no suppression of hepatic glucose output. Circulating FFAs are lower, and the available FFAs are utilized primarily by the mitochondria for mitochondrial respiration. This results in decreased DAG and TG levels.
The mouse model utilized in these studies was a whole-body deletion of the integrin α1 subunit. The liver is a heterogeneous tissue composed of many different cell types, including, but not limited to, hepatocytes, sinusoidal lining cells, endothelial cells, stellate cells, and Kupffer cells. The hepatocyte is the most abundant cell type, making up ∼80% of the cells in the liver, and is a major site of hepatic insulin action and lipid metabolism (56). Thus, it seems highly likely that the observed metabolic phenotype is a result of changes within the hepatocyte. In summary, this study has shown for the first time the novel role of integrin α1β1 in the regulation of hepatic glucose and lipid metabolism under conditions of overnutrition in vivo. Furthermore, the interaction of integrin and insulin signaling is critical in determining the scope and severity of insulin resistance.
Acknowledgments
We thank the Vanderbilt Mouse Metabolic Phenotyping Center Lipid Core for performing the liver DAG analysis and the Vanderbilt Mouse Metabolic Phenotyping Center Pathology Core for Oil Red O staining.
This work was supported, in whole or in part, by National Institutes of Health Grants R37 DK050277, R01 DK054902, and U24 DK059637 (to D. H. W.); Grant R01 DK095761 (to A. P.); and Grant DK20593 (to the Vanderbilt Diabetes Research and Training Center). This work was also supported by Veterans Affairs Merit Review 1I01BX002025-01 (to A. P.) and the Vanderbilt Molecular Endocrinology Training Program.
- HF
- high fat
- TG
- triglyceride
- FAK
- focal adhesion kinase
- DAG
- diglyceride (diacylglycerol)
- FFA
- free fatty acid
- GTT
- glucose tolerance test
- qPCR
- quantitative PCR.
REFERENCES
- 1. Pehling G., Tessari P., Gerich J. E., Haymond M. W., Service F. J., Rizza R. A. (1984) Abnormal meal carbohydrate disposition in insulin-dependent diabetes. Relative contributions of endogenous glucose production and initial splanchnic uptake and effect of intensive insulin therapy. J. Clin. Invest. 74, 985–991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. DeFronzo R. A., Del Prato S. (1996) Insulin resistance and diabetes mellitus. J. Diabetes Complications 10, 243–245 [DOI] [PubMed] [Google Scholar]
- 3. Puri P., Baillie R. A., Wiest M. M., Mirshahi F., Choudhury J., Cheung O., Sargeant C., Contos M. J., Sanyal A. J. (2007) A lipidomic analysis of nonalcoholic fatty liver disease. Hepatology 46, 1081–1090 [DOI] [PubMed] [Google Scholar]
- 4. Sanyal A. J., Campbell-Sargent C., Mirshahi F., Rizzo W. B., Contos M. J., Sterling R. K., Luketic V. A., Shiffman M. L., Clore J. N. (2001) Nonalcoholic steatohepatitis: association of insulin resistance and mitochondrial abnormalities. Gastroenterology 120, 1183–1192 [DOI] [PubMed] [Google Scholar]
- 5. Samuel V. T., Liu Z. X., Qu X., Elder B. D., Bilz S., Befroy D., Romanelli A. J., Shulman G. I. (2004) Mechanism of hepatic insulin resistance in non-alcoholic fatty liver disease. J. Biol. Chem. 279, 32345–32353 [DOI] [PubMed] [Google Scholar]
- 6. Day C. P., James O. F. (1998) Steatohepatitis: a tale of two “hits”? Gastroenterology 114, 842–845 [DOI] [PubMed] [Google Scholar]
- 7. Wada T., Miyashita Y., Sasaki M., Aruga Y., Nakamura Y., Ishii Y., Sasahara M., Kanasaki K., Kitada M., Koya D., Shimano H., Tsuneki H., Sasaoka T. (2013) Eplerenone ameliorates the phenotypes of metabolic syndrome with NASH in liver-specific SREBP-1c Tg mice fed high-fat and high-fructose diet. Am. J. Physiol. Endocrinol. Metab. 305, E1415–E1425 [DOI] [PubMed] [Google Scholar]
- 8. Jaskiewicz K., Rzepko R., Sledzinski Z. (2008) Fibrogenesis in fatty liver associated with obesity and diabetes mellitus type 2. Dig. Dis. Sci. 53, 785–788 [DOI] [PubMed] [Google Scholar]
- 9. Hynes R. O. (2009) The extracellular matrix: not just pretty fibrils. Science 326, 1216–1219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Moser M., Legate K. R., Zent R., Fässler R. (2009) The tail of integrins, talin, and kindlins. Science 324, 895–899 [DOI] [PubMed] [Google Scholar]
- 11. Guilherme A., Czech M. P. (1998) Stimulation of IRS-1-associated phosphatidylinositol 3-kinase and Akt/protein kinase B but not glucose transport by β1-integrin signaling in rat adipocytes. J. Biol. Chem. 273, 33119–33122 [DOI] [PubMed] [Google Scholar]
- 12. Zong H., Bastie C. C., Xu J., Fassler R., Campbell K. P., Kurland I. J., Pessin J. E. (2009) Insulin resistance in striated muscle-specific integrin receptor β1-deficient mice. J. Biol. Chem. 284, 4679–4688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Guilherme A., Torres K., Czech M. P. (1998) Cross-talk between insulin receptor and integrin α5β1 signaling pathways. J. Biol. Chem. 273, 22899–22903 [DOI] [PubMed] [Google Scholar]
- 14. Lebrun P., Mothe-Satney I., Delahaye L., Van Obberghen E., Baron V. (1998) Insulin receptor substrate-1 as a signaling molecule for focal adhesion kinase pp125FAK and pp60src. J. Biol. Chem. 273, 32244–32253 [DOI] [PubMed] [Google Scholar]
- 15. Volpes R., van den Oord J. J., Desmet V. J. (1991) Distribution of the VLA family of integrins in normal and pathological human liver tissue. Gastroenterology 101, 200–206 [DOI] [PubMed] [Google Scholar]
- 16. Nejjari M., Couvelard A., Mosnier J. F., Moreau A., Feldmann G., Degott C., Marcellin P., Scoazec J. Y. (2001) Integrin up-regulation in chronic liver disease: relationship with inflammation and fibrosis in chronic hepatitis C. J. Pathol. 195, 473–481 [DOI] [PubMed] [Google Scholar]
- 17. Kang L., Ayala J. E., Lee-Young R. S., Zhang Z., James F. D., Neufer P. D., Pozzi A., Zutter M. M., Wasserman D. H. (2011) Diet-induced muscle insulin resistance is associated with extracellular matrix remodeling and interaction with integrin α2β1 in mice. Diabetes 60, 416–426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gardner H., Kreidberg J., Koteliansky V., Jaenisch R. (1996) Deletion of integrin α1 by homologous recombination permits normal murine development but gives rise to a specific deficit in cell adhesion. Dev. Biol. 175, 301–313 [DOI] [PubMed] [Google Scholar]
- 19. Ayala J. E., Bracy D. P., McGuinness O. P., Wasserman D. H. (2006) Considerations in the design of hyperinsulinemic-euglycemic clamps in the conscious mouse. Diabetes 55, 390–397 [DOI] [PubMed] [Google Scholar]
- 20. Finegood D. T., Bergman R. N., Vranic M. (1988) Modeling error and apparent isotope discrimination confound estimation of endogenous glucose production during euglycemic glucose clamps. Diabetes 37, 1025–1034 [DOI] [PubMed] [Google Scholar]
- 21. Steele R., Wall J. S., De Bodo R. C., Altszuler N. (1956) Measurement of size and turnover rate of body glucose pool by the isotope dilution method. Am. J. Physiol. 187, 15–24 [DOI] [PubMed] [Google Scholar]
- 22. Folch J., Lees M., Sloane Stanley G. H. (1957) A simple method for the isolation and purification of total lipids from animal tissues. J. Biol. Chem. 226, 497–509 [PubMed] [Google Scholar]
- 23. Morrison W. R., Smith L. M. (1964) Preparation of fatty acid methyl esters and dimethylacetals from lipids with boron fluoride-methanol. J. Lipid Res. 5, 600–608 [PubMed] [Google Scholar]
- 24. Kuznetsov A. V., Strobl D., Ruttmann E., Königsrainer A., Margreiter R., Gnaiger E. (2002) Evaluation of mitochondrial respiratory function in small biopsies of liver. Anal. Biochem. 305, 186–194 [DOI] [PubMed] [Google Scholar]
- 25. Hepple R. T., Baker D. J., Kaczor J. J., Krause D. J. (2005) Long-term caloric restriction abrogates the age-related decline in skeletal muscle aerobic function. FASEB J. 19, 1320–1322 [DOI] [PubMed] [Google Scholar]
- 26. Koliwad S. K., Streeper R. S., Monetti M., Cornelissen I., Chan L., Terayama K., Naylor S., Rao M., Hubbard B., Farese R. V., Jr. (2010) DGAT1-dependent triacylglycerol storage by macrophages protects mice from diet-induced insulin resistance and inflammation. J. Clin. Invest. 120, 756–767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Livak K. J., Schmittgen T. D. (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 25, 402–408 [DOI] [PubMed] [Google Scholar]
- 28. Dixon L. J., Flask C. A., Papouchado B. G., Feldstein A. E., Nagy L. E. (2013) Caspase-1 as a central regulator of high fat diet-induced non-alcoholic steatohepatitis. PLoS ONE 8, e56100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Chen X., Abair T. D., Ibanez M. R., Su Y., Frey M. R., Dise R. S., Polk D. B., Singh A. B., Harris R. C., Zent R., Pozzi A. (2007) Integrin α1β1 controls reactive oxygen species synthesis by negatively regulating epidermal growth factor receptor-mediated Rac activation. Mol. Cell. Biol. 27, 3313–3326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Best J. D., Kahn S. E., Ader M., Watanabe R. M., Ni T. C., Bergman R. N. (1996) Role of glucose effectiveness in the determination of glucose tolerance. Diabetes Care 19, 1018–1030 [DOI] [PubMed] [Google Scholar]
- 31. Mattila E., Pellinen T., Nevo J., Vuoriluoto K., Arjonen A., Ivaska J. (2005) Negative regulation of EGFR signalling through integrin-α1β1-mediated activation of protein tyrosine phosphatase TCPTP. Nat. Cell Biol. 7, 78–85 [DOI] [PubMed] [Google Scholar]
- 32. Vuori K., Ruoslahti E. (1994) Association of insulin receptor substrate-1 with integrins. Science 266, 1576–1578 [DOI] [PubMed] [Google Scholar]
- 33. Vulin A. I., Jacob K. K., Stanley F. M. (2005) Integrin activates receptor-like protein tyrosine phosphatase α, Src, and Rho to increase prolactin gene expression through a final phosphatidylinositol 3-kinase/cytoskeletal pathway that is additive with insulin. Endocrinology 146, 3535–3546 [DOI] [PubMed] [Google Scholar]
- 34. Fujita M., Ieguchi K., Davari P., Yamaji S., Taniguchi Y., Sekiguchi K., Takada Y. K., Takada Y. (2012) Cross-talk between integrin α6β4 and insulin-like growth factor-1 receptor (IGF1R) through direct α6β4 binding to IGF1 and subsequent α6β4-IGF1-IGF1R ternary complex formation in anchorage-independent conditions. J. Biol. Chem. 287, 12491–12500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Pozzi A., Moberg P. E., Miles L. A., Wagner S., Soloway P., Gardner H. A. (2000) Elevated matrix metalloprotease and angiostatin levels in integrin α1 knockout mice cause reduced tumor vascularization. Proc. Natl. Acad. Sci. U.S.A. 97, 2202–2207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Shi M., Pedchenko V., Greer B. H., Van Horn W. D., Santoro S. A., Sanders C. R., Hudson B. G., Eichman B. F., Zent R., Pozzi A. (2012) Enhancing integrin α1 inserted (I) domain affinity to ligand potentiates integrin α1β1-mediated down-regulation of collagen synthesis. J. Biol. Chem. 287, 35139–35152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Bisht B., Srinivasan K., Dey C. S. (2008) In vivo inhibition of focal adhesion kinase causes insulin resistance. J. Physiol. 586, 3825–3837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Löster K., Vossmeyer D., Hofmann W., Reutter W., Danker K. (2001) α1 Integrin cytoplasmic domain is involved in focal adhesion formation via association with intracellular proteins. Biochem. J. 356, 233–240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Donnelly K. L., Smith C. I., Schwarzenberg S. J., Jessurun J., Boldt M. D., Parks E. J. (2005) Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J. Clin. Invest. 115, 1343–1351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Summers S. A., Whiteman E. L., Birnbaum M. J. (2000) Insulin signaling in the adipocyte. Int. J. Obes. Relat. Metab. Disord. 24, Suppl. 4, S67–S70 [DOI] [PubMed] [Google Scholar]
- 41. Tonelli J., Kishore P., Lee D. E., Hawkins M. (2005) The regulation of glucose effectiveness: how glucose modulates its own production. Curr. Opin. Clin. Nutr. Metab. Care 8, 450–456 [DOI] [PubMed] [Google Scholar]
- 42. Hirosumi J., Tuncman G., Chang L., Görgün C. Z., Uysal K. T., Maeda K., Karin M., Hotamisligil G. S. (2002) A central role for JNK in obesity and insulin resistance. Nature 420, 333–336 [DOI] [PubMed] [Google Scholar]
- 43. Cheung A. T., Wang J., Ree D., Kolls J. K., Bryer-Ash M. (2000) Tumor necrosis factor-α induces hepatic insulin resistance in obese Zucker (fa/fa) rats via interaction of leukocyte antigen-related tyrosine phosphatase with focal adhesion kinase. Diabetes 49, 810–819 [DOI] [PubMed] [Google Scholar]
- 44. Stefan N., Häring H. U. (2011) The metabolically benign and malignant fatty liver. Diabetes 60, 2011–2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Postic C., Girard J. (2008) Contribution of de novo fatty acid synthesis to hepatic steatosis and insulin resistance: lessons from genetically engineered mice. J. Clin. Invest. 118, 829–838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Monetti M., Levin M. C., Watt M. J., Sajan M. P., Marmor S., Hubbard B. K., Stevens R. D., Bain J. R., Newgard C. B., Farese R. V., Sr., Hevener A. L., Farese R. V., Jr. (2007) Dissociation of hepatic steatosis and insulin resistance in mice overexpressing DGAT in the liver. Cell Metab. 6, 69–78 [DOI] [PubMed] [Google Scholar]
- 47. Yamaguchi K., Yang L., McCall S., Huang J., Yu X. X., Pandey S. K., Bhanot S., Monia B. P., Li Y. X., Diehl A. M. (2007) Inhibiting triglyceride synthesis improves hepatic steatosis but exacerbates liver damage and fibrosis in obese mice with nonalcoholic steatohepatitis. Hepatology 45, 1366–1374 [DOI] [PubMed] [Google Scholar]
- 48. Matsuzaka T., Shimano H., Yahagi N., Kato T., Atsumi A., Yamamoto T., Inoue N., Ishikawa M., Okada S., Ishigaki N., Iwasaki H., Iwasaki Y., Karasawa T., Kumadaki S., Matsui T., Sekiya M., Ohashi K., Hasty A. H., Nakagawa Y., Takahashi A., Suzuki H., Yatoh S., Sone H., Toyoshima H., Osuga J., Yamada N. (2007) Crucial role of a long-chain fatty acid elongase, Elovl6, in obesity-induced insulin resistance. Nat. Med. 13, 1193–1202 [DOI] [PubMed] [Google Scholar]
- 49. Chen H. C., Smith S. J., Ladha Z., Jensen D. R., Ferreira L. D., Pulawa L. K., McGuire J. G., Pitas R. E., Eckel R. H., Farese R. V., Jr. (2002) Increased insulin and leptin sensitivity in mice lacking acyl CoA:diacylglycerol acyltransferase 1. J. Clin. Invest. 109, 1049–1055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Murthy V. K., Shipp J. C. (1979) Synthesis and accumulation of triglycerides in liver of diabetic rats. Effects of insulin treatment. Diabetes 28, 472–478 [DOI] [PubMed] [Google Scholar]
- 51. Buqué X., Cano A., Miquilena-Colina M. E., García-Monzón C., Ochoa B., Aspichueta P. (2012) High insulin levels are required for FAT/CD36 plasma membrane translocation and enhanced fatty acid uptake in obese Zucker rat hepatocytes. Am. J. Physiol. Endocrinol. Metab. 303, E504–E514 [DOI] [PubMed] [Google Scholar]
- 52. He J., Lee J. H., Febbraio M., Xie W. (2011) The emerging roles of fatty acid translocase/CD36 and the aryl hydrocarbon receptor in fatty liver disease. Exp. Biol. Med. 236, 1116–1121 [DOI] [PubMed] [Google Scholar]
- 53. Chawla A., Barak Y., Nagy L., Liao D., Tontonoz P., Evans R. M. (2001) PPAR-γ-dependent and -independent effects on macrophage-gene expression in lipid metabolism and inflammation. Nat. Med. 7, 48–52 [DOI] [PubMed] [Google Scholar]
- 54. Begriche K., Massart J., Robin M. A., Bonnet F., Fromenty B. (2013) Mitochondrial adaptations and dysfunctions in nonalcoholic fatty liver disease. Hepatology 58, 1497–1507 [DOI] [PubMed] [Google Scholar]
- 55. Satapati S., Sunny N. E., Kucejova B., Fu X., He T. T., Méndez-Lucas A., Shelton J. M., Perales J. C., Browning J. D., Burgess S. C. (2012) Elevated TCA cycle function in the pathology of diet-induced hepatic insulin resistance and fatty liver. J. Lipid Res. 53, 1080–1092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Postic C., Magnuson M. A. (2000) DNA excision in liver by an albumin-Cre transgene occurs progressively with age. Genesis 26, 149–150 [DOI] [PubMed] [Google Scholar]