

Significance
Peroxiredoxin II (PrxII) is the major antioxidant enzyme in red blood cells (RBCs) and is occasionally hyperoxidized and inactivated during elimination of H2O2. The abundance of hyperoxidized Prx was recently shown to oscillate with a period of ∼24 h in human RBCs, reflecting the operation of a circadian clock. Here we show that the oscillation of hyperoxidized PrxII in mouse RBCs is driven both by hyperoxidation mediated by H2O2 produced during hemoglobin-dependent O2 transport and by degradation of the hyperoxidized enzyme by the proteasome, rather than by reduction of the hyperoxidized PrxII by sulfiredoxin. About 1% of PrxII undergoes daily oscillation of hyperoxidation and degradation, leading to a gradual loss of PrxII during RBC aging.
Abstract
The catalytic cysteine of the typical 2-Cys Prx subfamily of peroxiredoxins is occasionally hyperoxidized to cysteine sulfinic acid during the peroxidase catalytic cycle. Sulfinic Prx (Prx–SO2H) is reduced back to the active form of the enzyme by sulfiredoxin. The abundance of Prx–SO2H was recently shown to oscillate with a period of ∼24 h in human red blood cells (RBCs). We have now investigated the molecular mechanism and physiological relevance of such oscillation in mouse RBCs. Poisoning of RBCs with CO abolished Prx–SO2H formation, implicating H2O2 produced from hemoglobin autoxidation in Prx hyperoxidation. RBCs express the closely related PrxI and PrxII isoforms, and analysis of RBCs deficient in either isoform identified PrxII as the hyperoxidized Prx in these cells. Unexpectedly, RBCs from sulfiredoxin-deficient mice also exhibited circadian oscillation of Prx–SO2H. Analysis of the effects of protease inhibitors together with the observation that the purified 20S proteasome degraded PrxII–SO2H selectively over nonhyperoxidized PrxII suggested that the 20S proteasome is responsible for the decay phase of PrxII–SO2H oscillation. About 1% of total PrxII undergoes daily oscillation, resulting in a gradual loss of PrxII during the life span of RBCs. PrxII–SO2H was detected in cytosolic and ghost membrane fractions of RBCs, and the amount of membrane-bound PrxII–SO2H oscillated in a phase opposite to that of total PrxII–SO2H. Our results suggest that membrane association of PrxII–SO2H is a tightly controlled process and might play a role in the tuning of RBC function to environmental changes.
Peroxiredoxins (Prxs) are a family of ubiquitous peroxidases found across all kingdoms of life that reduce peroxides and exist as obligatory homodimers with a subunit molecular size of 20–30 kDa (1). Mammalian cells express six isoforms of Prx: four typical 2-Cys Prxs (PrxI to PrxIV), one atypical 2-Cys Prx (PrxV), and one 1-Cys Prx (PrxVI). Catalysis by all Prxs is initiated by reaction of the active cysteine (referred to as the peroxidatic Cys–SH, or CP–SH) with peroxides to form a sulfenic acid (CP–SOH) intermediate. This intermediate of typical 2-Cys Prxs reacts with another cysteine residue (the resolving Cys–SH, or CR–SH) of the paired subunit to form an intermolecular disulfide, CP–S–S–CR, which is subsequently reduced by thioredoxin and thioredoxin reductase (1). The cysteine sulfenic acid intermediate formed during the catalytic cycle occasionally undergoes hyperoxidation to cysteine sulfinic acid (Cys–SO2H), resulting in inactivation of peroxidase function (1, 2). The sulfinic form of typical 2-Cys Prxs is reduced back to the active form by sulfiredoxin (Srx) in a process that consumes ATP and cellular thiols (3–5). There is no known mechanism for reduction of the sulfinic form of atypical 2-Cys Prx or 1-Cys Prx.
To adapt to cyclical changes in environmental cues arising from daily cycles of light and darkness, most organisms have developed endogenous biological clocks with a period of ∼24 h (6). These clocks regulate many aspects of physiology including the sleep−wake cycle, body temperature, feeding, metabolism, and hormone and neurotransmitter secretion in mammals (7). Genetic studies of biological clocks in various model organisms have shown that many genes and gene products, which are not evolutionarily conserved across distinct phyla, are organized to support a transcription−translation feedback loop that oscillates every 24 h (6, 8, 9). Subsequent studies revealed that circadian oscillation can occur in the absence of nuclear events, however (10, 11). This transcription-independent circadian oscillation has been difficult to study in mammalian cells containing a nucleus, but it was demonstrated by the detection of a self-sustained oscillation of hyperoxidized Prx (Prx–SO2H) with a period of ∼24 h in anuclear human red blood cells (RBCs) (12). The circadian variation of Prx–SO2H has since been detected in a wide range of aerobic organisms including fungi, worms, and flies as well as mammals (13–17). The physiological relevance of these observations has remained mostly unknown, however.
During oxygen transport, hemoglobin (Hb) in RBCs undergoes autoxidation to produce superoxide, which then undergoes dismutation to hydrogen peroxide (18, 19). Catalase, glutathione peroxidase (GPx), and Prxs are responsible for the elimination of H2O2 in RBCs (19–21). Compared with catalase and GPx, which do not have a specific binding site for H2O2, Prxs function more effectively at low H2O2 concentrations because they do possess a high-affinity binding site for this molecule (22). Prx II is also much more abundant than the other two enzymes.
RBCs express three Prx isoforms: PrxI, PrxII, and PrxVI (20, 21). Which isoform of Prx is responsible for the oscillation of hyperoxidized Prx in RBCs and the mechanism of such oscillation have remained unknown, however. The expression of Srx, which is responsible for reduction of the sulfinic form of typical 2-Cys Prxs, is generally induced in cells by oxidative stress. However, Srx cannot be induced in anuclear RBCs. In addition, certain organisms, such as Caenorhabditis elegans and Neurospora crassa, do not express Srx but still exhibit a circadian rhythm of Prx hyperoxidation (16). We have now studied Prx hyperoxidation in mouse RBCs to identify the mechanism, as well as to gain insight into the physiological role, of circadian Prx hyperoxidation in RBCs.
Results
Circadian Oscillation of PrxII Hyperoxidation in Mouse RBCs.
Although a previous study showed that human RBCs can be entrained (synchronized to an external cycle) by exposure to temperature cycles for 48 h (12), we found that similar procedures were not effective for mouse RBCs, mostly because the mouse cells are more susceptible to hemolysis than are human RBCs. To identify optimal conditions for the study of circadian oscillation of Prx hyperoxidation in mouse RBCs, we isolated a RBC fraction from whole blood of B6 mice by centrifugation, washed the RBC pellet with PBS (PBS), and incubated the cells at various densities in three different basal media: Krebs-Henseleit buffer, DMEM (DMEM), and RPMI medium 1640. Lysates prepared from RBCs exposed to the various incubation conditions were then subjected to immunoblot analysis with antibodies that recognize a specific sequence surrounding the CP–SO2H of 2-Cys Prxs. We found that oscillation of Prx–SO2H could be conveniently monitored when RBCs were incubated in a modified DMEM (containing d-glucose at 4.5 g/L, sodium bicarbonate at 3.7 g/L, and sodium pyruvate at 110 mg/L, and with an osmolarity of 330 mOsm/L) at a density of 1.5 × 107 cells/50 μL in the dark and at 37 °C under an atmosphere of 5% CO2 and 20% O2 in air. The osmolarity of mouse plasma (327 mOsm/L) is higher than that of human plasma (280 mOsm/L) (23). Under these optimized conditions, the abundance of Prx–SO2H in mouse RBCs gradually decreased, achieving a nadir at ∼30 min after transfer of the isolated RBCs to the modified DMEM, and it then gradually increased (Fig. S1A). The gradual increase proved to be the beginning of the rise phase of circadian oscillation (Fig. 1). In the experiment shown in Fig. 1, RBCs from three B6 mice were incubated separately under the optimized conditions. After 30 min (when the abundance of Prx–SO2H had reached its lowest level), the cells were sampled every 3 h and subjected to immunoblot analysis. RBCs from all three mice exhibited clear rhythmicity of Prx hyperoxidation with a period of ∼24 h. A similar 24-h oscillation of Prx–SO2H abundance was also observed when RBCs were maintained at 32 °C (Fig. S1 B and C).
Fig. 1.
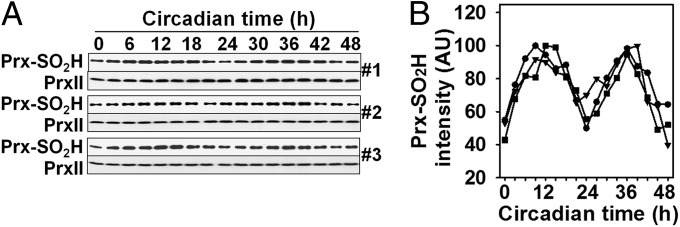
Circadian oscillation of Prx–SO2H in mouse RBCs. (A) RBCs from three mice (#1, #2, and #3) were incubated in modified DMEM at 37 °C in the dark under 5% CO2 and 20% O2 in air as described in Materials and Methods. After incubation for 30 min (time 0), the cells were sampled every 3 h for 48 h and subjected to immunoblot analysis with antibodies to the sulfinic form of 2-Cys Prxs and to PrxII. (B) The intensity of the Prx–SO2H bands in A was normalized by that of the corresponding PrxII band and then expressed in arbitrary units (AU) relative to the corresponding maximum value, with ●, ▼, and ■ representing mice #1, #2, and #3, respectively.
RBCs express two 2-Cys Prxs (PrxI and PrxII) and 1-Cys Prx (PrxVI), with PrxII being highly abundant in these cells (20, 21). Hyperoxidized 2-Cys Prxs can be detected by immunoblot analysis with antibodies generated in response to a sulfonylated peptide modeled on the conserved peroxidatic cysteine residue (CP). Given that the amino acid sequence surrounding CP is identical for PrxI and PrxII, the antibodies to the sulfinic form of 2-Cys Prxs react with both PrxI–SO2H and PrxII–SO2H, which differ by only one amino acid residue in size and cannot be separated by SDS–polyacrylamide gel electrophoresis (PAGE). To evaluate the contributions of PrxI and PrxII to the Prx–SO2H band observed in Fig. 1A, we isolated RBCs from PrxI−/− or PrxII−/− mice and incubated them under the optimized conditions. PrxI-deficient RBCs exhibited circadian oscillation of Prx–SO2H similar to that apparent for wild-type cells, whereas Prx–SO2H was not detected in PrxII-deficient RBCs (Fig. S2). These results suggested that the rhythmic hyperoxidation of Prx observed in RBCs largely reflects modification of PrxII. The Prx–SO2H detected by the antibodies to 2-Cys Prx–SO2H in RBCs is hereafter referred to as PrxII–SO2H.
The active-site cysteine residues of PrxVI and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) are also susceptible to hyperoxidation (21). PrxI/II–SO2H and PrxVI–SO2H can be readily differentiated as they are different in size, and the antibodies to their sulfinic forms are specific to each isoform. Immunoblot analysis with antibodies that recognize the sulfinic forms of PrxVI or GAPDH indicated that both PrxVI–SO2H and GAPDH–SO2H are present in RBCs but do not show circadian rhythm (Fig. S3).
Effect of Hb Autoxidation on PrxII Hyperoxidation in RBCs.
As a result of the affinity of Hb for carbon monoxide being much greater than that for oxygen, CO prevents Hb autoxidation and the subsequent production of superoxide and H2O2. We therefore tested the effect of CO on PrxII hyperoxidation in RBCs. Exposure of RBCs to CO largely abolished PrxII hyperoxidation and its circadian oscillation (Fig. 2 and Fig. S4A). Conversion of oxyhemoglobin to carboxyhemoglobin during the incubation of RBCs under an atmosphere of 100% CO was apparent both by visual inspection (development of a bright red color) and spectrophotometry (Fig. S4B).
Fig. 2.

Effect of CO on PrxII hyperoxidation in RBCs. RBCs isolated from three mice were exposed to CO (○, ▽, and □ for mice #1, #2, and #3, respectively) or to room air (●, ▼, and ■ for mice #1, #2, and #3, respectively) for 30 min and then incubated and analyzed for PrxII hyperoxidation as in Fig. 1. The immunoblots are shown in Fig. S4A.
Circadian Oscillation of PrxII–SO2H in RBCs of Srx+/+ and Srx−/− Mice.
Srx catalyzes the reduction of sulfinic typical 2-Cys Prxs. To examine the role of Srx in PrxII–SO2H rhythmicity, we incubated RBCs from age- and sex-matched Srx+/+ and Srx−/− littermates under the optimal conditions. To directly compare the PrxII–SO2H band intensities between Srx+/+ and Srx−/− mice, RBC lysates derived from those littermates were analyzed on the same gel and treated identically during all steps. Both wild-type and Srx-deficient RBCs showed a similar pattern of PrxII–SO2H oscillation (Fig. 3 and Fig. S5). This finding was unexpected, given the role of Srx in the reduction of PrxII–SO2H. We compared the amounts of PrxII and Srx in RBCs with those in mouse embryonic fibroblasts, mouse leukemic monocytes-macrophages (RAW 264.7 cells), the mouse embryonic fibroblast cell line NIH 3T3, and mouse mononuclear cells (MNCs) obtained during RBC preparation. Immunoblot analysis indicated that the abundance of Srx in RBCs is 10–50% of that in the other mouse cells, whereas the abundance of PrxII in RBCs is 10–30 times that in the other cell types (Fig. S6A). The ratio of Srx to PrxII is thus unusually low in RBCs. The amount of Srx in RBCs did not change during a 24-h time period (Fig. S5).
Fig. 3.

Circadian oscillation of PrxII–SO2H in RBCs from Srx+/+ and Srx−/− mice. RBCs isolated from three Srx+/+ (●, ▼, and ■ for mice #1, #2, and #3, respectively) or Srx−/− (○, ∇, and □ for mice #1, #2, and #3, respectively) mice were incubated and analyzed for PrxII hyperoxidation as in Fig. 1. The immunoblots are shown in Fig. S5.
To estimate the extent of PrxII hyperoxidation in RBCs during the circadian cycle, we prepared a standard RBC lysate, in which ∼10% of PrxII is hyperoxidized (Fig. S6B), by incubating RBCs in the presence of glucose oxidase and glucose, which produces H2O2 during the glucose oxidase-catalyzed conversion of glucose to glucolactone using molecular oxygen. Comparison with immunoblot intensities of the standard lysate indicated that the extent of PrxII hyperoxidation in RBCs incubated under the optimal conditions for 0 or 12 h was ∼0.7% and ∼1.6%, respectively, for wild-type cells and ∼1.7% and ∼2.8%, respectively, for Srx-deficient cells (Fig. S6C).
Selective Degradation of PrxII–SO2H by the 20S Proteasome.
Oxidatively modified proteins have been found to be selectively degraded in mammalian cells (24, 25). In many instances, this degradation is mediated in an ATP-independent manner by the 20S proteasome, which recognizes hydrophobic residues of its substrates that are exposed as a result of oxidative modification. To determine whether proteasome-dependent proteolysis contributes to the decay phase of the circadian oscillation of PrxII–SO2H abundance, we exposed RBCs to proteasome inhibitors (MG132 and lactacystin) at circadian time 9.5 h, just before the peak of PrxII hyperoxidation. The addition of MG132, lactacystin, or both agents blocked the decay phase of PrxII–SO2H abundance (Fig. 4A and Fig. S7A).
Fig. 4.

Prevention of the decay phase of circadian PrxII–SO2H oscillation in RBCs by proteasome inhibitors, and selective degradation of PrxII–SO2H by the 20S proteasome in vitro. (A) RBCs from three mice were incubated and analyzed as in Fig. 1, with the exception that the cells were incubated in the absence of both agents (●) or the presence of the proteasome inhibitors MG132 (○), lactacystin (▼), or both agents (△), each at a final concentration of 25 μM, beginning at circadian time 9.5 h. The immunoblots are shown in Fig. S7A. Data are means ± SD for cells from the three mice. (B–D) A 1:1 mixture of hyperoxidized and nonhyperoxidized PrxII was incubated for the indicated times in triplicate with a purified 20S proteasome fraction in the absence or presence of proteasome inhibitors (PI: MG132 plus lactacystin). The incubation mixtures were then subjected to immunoblot analysis with antibodies to the sulfinic form of 2-Cys Prxs and to PrxII (B). Immunoblot intensities of PrxII–SO2H (C) and PrxII (D) bands in B were normalized by the corresponding value obtained for the incubation performed in the absence of inhibitors at 0 h and are presented in arbitrary units. Data in C and D are means ± SD of triplicates from the representative experiment.
To examine further the role of the 20S proteasome in the degradation of PrxII–SO2H, we partially purified the 20S proteasome complex from lysates of mouse RBCs by gel filtration chromatography (Fig. S7B). We prepared a 1:1 mixture of hyperoxidized and nonhyperoxidized PrxII and incubated the mixture with the 20S proteasome fraction. Immunoblot analysis showed that the band intensity for PrxII–SO2H decreased with time, resulting in its almost complete disappearance after incubation for 4 h (Fig. 4 B and C). The band intensity for total PrxII also decreased with time but remained at ∼50% of the initial level after 4 h (Fig. 4 B and D). The presence of MG132 and lactacystin completely inhibited the degradation of both PrxII–SO2H and total PrxII. These results thus suggested that the 20S proteasome is able to degrade hyperoxidized PrxII selectively relative to nonhyperoxidized PrxII, and that this degradation is responsible for the decay phase of the circadian oscillation of PrxII–SO2H in RBCs. The gradual decrease of Prx II-SO2H observed during the first 30 min incubation in the modified DMEM (see Fig. S1A) is also likely due to proteolysis as the decrease was blocked by proteasome inhibitors (Fig. S7 C and D).
RBCs also express Ca2+-dependent proteases such as calpain and caspase. To test whether any of these enzymes might contribute to PrxII–SO2H degradation, we exposed RBCs to the Ca2+ chelators EGTA and 1,2-bis (o-aminophenoxy) ethane-N,N,N′,N′-tetra-acetic acid acetoxymethyl ester (BAPTA-AM) as well as to a mixture of calpain inhibitors at a circadian time of 9.5 h. None of these agents had a measurable effect on the decay phase of PrxII–SO2H oscillation (Fig. S7 E and F), excluding a role for Ca2+-dependent proteases in circadian PrxII–SO2H degradation.
Age-Dependent Loss of PrxII in RBCs.
Given that mature RBCs are not able to perform protein synthesis, the daily cycle of hyperoxidation and degradation of PrxII would be expected to result in a gradual decline in the amount of this protein during the lifetime of these cells, which is 30–40 d in the mouse. An age-dependent increase in the density of RBCs allows the cells to be fractionated according to their mean age by discontinuous Percoll gradient centrifugation. We separated RBCs from wild-type and Srx-deficient mice into four fractions (F1 to F4) by this approach (Fig. 5A). Equal amounts of protein from unfractionated RBCs and from each of the four fractions were then subjected to immunoblot analysis with various antibodies. The band intensity for PrxII–SO2H increased with increasing RBC density (age) for both wild-type and Srx-deficient cells (Fig. 5 B and C). In contrast, the band intensity for PrxII decreased with increasing RBC age for both genotypes (Fig. 5 B and C). These results suggested that circadian oscillation of PrxII–SO2H inevitably results in a decrease in the amount of total PrxII as RBCs age.
Fig. 5.

Age-dependent loss of PrxII in mouse RBCs. (A) Fresh RBCs isolated from Srx+/+ or Srx−/− mice were separated into four fractions (F1 to F4) on the basis of their density (age) on a discontinuous Percoll gradient. (B) RBCs from each of three (#1, #2, and #3) Srx+/+ or Srx−/− mice either before (T) or after (F1 to F4) fractionation as in A were subjected to immunoblot analysis with antibodies to the sulfinic form of 2-Cys Prxs, to PrxII, to PrxVI–SO2H, to PrxVI, to GAPDH–SO2H, to GAPDH, and to β-actin (loading control). (C) Immunoblot intensities of PrxII–SO2H and PrxII bands in B were normalized by that of the corresponding β-actin band and then expressed in arbitrary units relative to the corresponding maximum value. Data are means ± SD of the three independent experiments. *, P < 0.05; +, P < 0.01; #, P < 0.001; Srx+/+ F1 vs. the other fractions in PrxII intensity.
Similar to the results obtained for PrxII–SO2H, the band intensities for PrxVI–SO2H and GAPDH–SO2H were higher in Srx-deficient RBCs than in wild-type cells, and they increased with increasing RBC age (Fig. 5B). This is likely because ablation of Srx reduced the amount of PrxII by ∼20% and increased the amount of PrxII−SO2H by ∼70% (Fig. 5C), which would result in increased level of H2O2, leading to more hyperoxidation of Prx VI and GAPDH. In contrast to PrxII, however, the band intensities for PrxVI and GAPDH did not change during RBC aging. These results corroborated our observation that PrxVI–SO2H and GAPDH–SO2H do not exhibit circadian fluctuation (Fig. S3).
Circadian-Dependent Changes in the Abundance of Ghost Membrane-Associated PrxII–SO2H.
PrxII is present mostly in the cytosol but is also found associated with the ghost membrane of RBCs (26). We examined whether the amount of ghost membrane-associated PrxII–SO2H shows circadian oscillation. Sampling of RBCs at 6-h intervals for 48 h revealed that the amount of ghost membrane-associated PrxII–SO2H first decreased in a stepwise manner, reaching its lowest level at 12 h, increased in a stepwise manner to reach a maximum at 24 h, and then went down at 36 h and up again at 48 h (Fig. 6A). The amount of ghost membrane-associated PrxII–SO2H thus appeared to oscillate in a phase opposite to that of total PrxII–SO2H. We then examined the distribution of PrxII–SO2H and total PrxII in more detail after 0, 12, and 24 h of incubation. The amount of PrxII–SO2H in whole RBC lysates (as well as that in the cytosol) increased ∼2.5-fold from 0 h to 12 h before decreasing again at 24 h, whereas the amount of ghost membrane-associated PrxII–SO2H decreased by ∼60% from 0 h to 12 h before increasing again at 24 h (Fig. 6 B and C). The amount of ghost membrane-associated PrxII–SO2H was ∼10% and ∼1.5% of total PrxII–SO2H at 0 h and 12 h, respectively. The amount of PrxII associated with the ghost membrane was estimated to be ∼5% of total PrxII and did not change substantially with time (Fig. 6 A and B). Exposure of RBCs to the slow production of H2O2 in glucose oxidase-containing medium resulted in an increase in the amounts of PrxII–SO2H in the whole cell lysate and cytosolic fraction, but a marked decrease in the amount of that associated with the ghost membrane (Fig. S8). These results suggested that increased PrxII hyperoxidation in RBCs either associated with circadian oscillation or resulting from exposure to an oxidative environment is accompanied by a concurrent decline in the amount of membrane-associated PrxII–SO2H.
Fig. 6.

Circadian-dependent changes in the abundance of ghost membrane-associated PrxII–SO2H. (A) RBCs isolated from three mice (#1, #2, and #3) were incubated as in Fig. 1 and sampled every 6 h for 48 h for preparation of ghosts by hypotonic hemolysis. Ghost membranes were then isolated by centrifugation and subjected to immunoblot analysis with antibodies to the sulfinic form of 2-Cys Prxs and to PrxII. (B) RBCs sampled at 0, 12, and 24 h were lysed, and the lysates as well as the cytosolic and ghost membrane fractions derived therefrom were subjected to immunoblot analysis with antibodies to the sulfinic form of 2-Cys Prxs, to PrxII, and to β-actin (ghost marker). The volume of samples loaded onto the gel was adjusted to represent a cell ratio of 1:1:30 for the whole lysate, cytosolic fraction, and ghost membranes, respectively. (C) Immunoblot intensities of PrxII–SO2H bands in B were normalized by the corresponding value for the whole lysate at 0 h and then expressed in arbitrary units. Data are means ± SD for cells from the three mice.
Discussion
Mouse and human RBCs differ markedly in certain physiological characteristics such as cell size, antioxidant content, and life span (30–40 d versus 120–150 d, respectively) (27). In contrast to the well-established culture conditions for human RBCs, similar protocols for mouse RBCs have not been well described. By trial and error, we found that the circadian rhythm of Prx hyperoxidation could be monitored for 48 h by maintenance of mouse RBCs in a modified version of DMEM. Experimental conditions used for human RBCs by O’Neil and Reddy (12) and mouse RBCs in this study are compared in Table S1. On incubation of isolated mouse RBCs under the optimal conditions, the level of Prx–SO2H declined to a nadir after ∼30 min and then increased gradually to initiate the rising phase of circadian oscillation.
PrxII, like other Prx isoforms, exists as an obligatory homodimer arranged in a head-to-tail manner. The PrxII dimer undergoes further oligomerization to form a decamer and higher molecular weight complexes in cells. Fractionation of human RBC lysates by nonreducing SDS/PAGE revealed that almost all PrxII molecules migrated as a monomer if N-ethyl maleimide (NEM) was included in the lysis buffer to alkylate CP–SH. In the absence of NEM, a large proportion of PrxII molecules migrated as a dimer, suggesting that PrxII exists mainly in the thiol form in RBCs but forms an intermolecular disulfide (CP–S–S–CR) as the result of air oxidation after lysis (28). In a previous study of circadian rhythms in human RBCs (12), RBC lysates were analyzed by nonreducing SDS/PAGE without NEM treatment. PrxI and PrxII were thus detected as monomers, dimers, and multimers with similar band intensities, and the circadian oscillation of Prx–SO2H was most apparent for the dimeric form. The analysis of circadian oscillation therefore focused on the dimer-associated Prx–SO2H detected on nonreducing SDS gels. In our experiments, however, we analyzed RBC proteins on reducing SDS gels to detect all Prx molecules as monomers, and we observed pronounced circadian oscillation of the monomeric Prx–SO2H. Our analysis of RBCs from PrxI or PrxII knockout mice also showed that PrxII is responsible for the circadian oscillation of Prx–SO2H. PrxI–SO2H was thus not detected in RBCs from PrxII−/− mice, likely reflecting either the low abundance of PrxI or its insensitivity to hyperoxidation in these cells. Hyperoxidized forms of PrxVI and GAPDH were detected in RBCs, but they did not exhibit circadian oscillation.
Consistent with Hb autoxidation being the major source of H2O2 in RBCs, we found that CO poisoning blocked PrxII–SO2H formation in RBCs. Unexpectedly, however, we also found that the circadian oscillation of PrxII–SO2H was still apparent in RBCs from Srx−/− mice. Whereas ablation of Srx increased the basal level of PrxII–SO2H, it affected neither the amplitude nor the decay rate of the oscillation. These results suggest that the reduction of PrxII–SO2H by Srx is so slow in RBCs that it is inconsequential to the daily changes in PrxII–SO2H abundance, but that it exerts a cumulative effect during the life span of these cells. This situation thus contrasts with the Srx-dependent circadian oscillation of PrxIII–SO2H apparent in the mouse adrenal cortex, where cholesterol is converted to corticosterone in response to stimulation with adrenocorticotropic hormone (ACTH) (17). Corticosterone synthesis is accompanied by the cytochrome P450-mediated generation of H2O2 in mitochondria, and this H2O2 is eliminated by the mitochondrion-specific isoform of Prx, PrxIII, resulting in the production of PrxIII–SO2H. Corticosterone production undergoes daily oscillation, which in turn results in PrxIII–SO2H oscillation, at the peak of which, >70% of PrxIII is inactivated through hyperoxidation. PrxIII inactivation leads to the accumulation of H2O2 in mitochondria and its overflow into the cytosol, where it triggers activation of p38 mitogen-activated protein kinase and consequent down-regulation of both corticosterone synthesis and H2O2 production. In addition, ACTH stimulation greatly increases the expression of Srx, which eventually translocates into mitochondria and mediates the decay phase of PrxIII–SO2H oscillation. This Srx-dependent reversible hyperoxidation of PrxIII thus provides for a negative feedback regulatory mechanism for steroidogenesis independently of the hypothalamic-pituitary-adrenal axis (17). The reduction of hyperoxidized Prx by Srx is a slow process (kcat = 0.2–0.5 min–1 at 30 °C) (29), and it is especially slow in RBCs, likely because the Srx concentration in these cells is much lower than that in other cell types and, unlike in nucleated cells, Srx expression cannot be induced in anuclear RBCs. Furthermore, given that Srx binds to both nonhyperoxidized and hyperoxidized typical 2-Cys Prxs (30), the high concentration of PrxII in RBCs would also be expected to slow the Srx reaction.
Selective degradation of damaged proteins, which is important for maintenance of optimal cellular function, is mediated by both the 26S proteasome and the 20S proteasome, which are respectively dependent on and independent of ubiquitin and ATP. Mild oxidative stress promotes disassembly of the 26S proteasome into the 20S core proteasome and the 19S regulatory particle. The concentration of the free 20S core is >10 times that of the 26S proteasome in RBCs (31), in which the ubiquitin- and ATP-independent degradation of oxidatively damaged proteins has been observed. The 20S proteasome has been suggested to recognize hydrophobic residues exposed as a result of mild oxidation of proteins (25). Hyperoxidation of Prx also exposes hydrophobic patches that drive oligomerization (32). Our data now show that the 20S proteasome degrades hyperoxidized PrxII selectively over nonhyperoxidized PrxII, and that this degradation is responsible for the decay phase of the circadian oscillation of PrxII–SO2H.
The amplitude of PrxII–SO2H oscillation in RBCs was estimated to be ∼0.9% of total PrxII; that is, ∼0.9% of PrxII is depleted daily in RBCs under our experimental conditions. Fractionation of RBCs on the basis of cell density (age) immediately after their isolation from mouse blood revealed that the amount of PrxII in the most aged RBC fraction was ∼30% less than that in the least aged fraction, suggesting that the circadian-dependent degradation of PrxII in RBCs also occurs in vivo. Reduction of PrxII–SO2H by Srx requires ATP (4, 5), which is produced through energy-inefficient pathways in RBCs, which lack mitochondria. Given that the daily loss of such a small proportion of the abundant PrxII pool is likely inconsequential to the overall antioxidant capacity of RBCs, degradation of PrxII–SO2H, instead of its reactivation through the costly Srx reaction, might be economic for the short-lived cells.
PrxII in RBCs is present mostly in the cytosol but also associates with the ghost membrane (26). Given that Hb near the membrane first senses the demand of tissues for O2, the deoxygenation of Hb likely occurs predominantly near the plasma membrane, resulting in a concomitant local increase in H2O2 concentration followed by hyperoxidation of membrane-associated PrxII. The amount of PrxII–SO2H thus oscillates likely because the supply of O2 to tissues undergoes circadian cycling. We found that PrxII–SO2H is also distributed between the cytosol and ghost membrane, that this distribution exhibits circadian variation, and that the amount of membrane-associated PrxII–SO2H decreases when the total amount of PrxII–SO2H increases in RBCs. This inverse relation between total and membrane-associated PrxII–SO2H was also observed when total PrxII hyperoxidation was increased by exposure of RBCs to H2O2. Membrane-bound PrxII is likely also a primary target for H2O2 molecules that permeate the cell from outside. The amount of total (hyperoxidized plus nonhyperoxidized) PrxII associated with the membrane remained unchanged during the circadian cycle. Given that membrane-bound PrxII is critical for the protection of membrane lipids from oxidative damage (26), replacement of inactivated PrxII with active enzyme would be expected to be important for RBC function. The inverse relation between total and membrane-associated PrxII–SO2H suggests that such replacement is not dependent on thermodynamic equilibrium but rather is tightly controlled, likely through binding of PrxII–SO2H to as yet unidentified proteins. In what is, to our knowledge, the first example of a protein interaction dependent on the hyperoxidized state of a Prx, the protein disulfide isomerase ERp46 was recently identified as a binding partner of PrxII–SO2H in T cells and endothelial cells (33). The role of membrane-localized PrxII–SO2H is only speculative at the present time. Given that hyperoxidation of 2-Cys Prxs results in loss of their peroxidase activity but a concomitant gain of chaperone function, membrane-bound PrxII–SO2H might be required to protect neighboring proteins from oxidative denaturation.
Materials and Methods
Materials.
Rabbit polyclonal antibodies to PrxI, to PrxII, to the sulfinic-sulfonic forms of 2-Cys Prxs (SI Materials and Methods), to Prx VI, to the sulfinic-sulfonic forms of PrxVI, to Srx, to GAPDH, or to the sulfinic-sulfonic forms of GAPDH were described previously (21). Percoll and a Superose 6 column were obtained from Amersham Bioscience, glucose oxidase and Krebs−Henseleit buffer were from Sigma-Aldrich, and horseradish peroxidase-conjugated secondary antibodies and ECL detection reagents were from Pierce. MG132, lactacysitn, and a 20S proteasome assay kit were obtained from Cayman Chemical, and PBS, DMEM, and RPMI 1640 were from WelGENE. FBS and penicillin-streptomycin were from HyClone.
Animals.
Mice (8–12 wk of age; body mass of 20–25 g) were housed in a temperature-controlled room with a 12-h-light, 12-h-dark cycle and allowed free access to food and water. PrxI−/−, PrxII −/−, and Srx−/− mice were as described previously (17, 34). All animal experiments were performed according to protocols approved by the Institutional Animal Care and Use Committee of Ewha Womans University.
Optimization of Culture Conditions for Mouse RBCs.
Mice were anesthetized by i.p. injection with a combination of Zoletil (50 µg/kg) and Rompun (1 µL/kg). Whole blood (∼0.5 mL) was collected from the abdominal vein with a 1-mL syringe, transferred to a 1.5-mL tube containing 100 µL of acid citrate dextrose solution (7.3 g of anhydrous citric acid, 22.0 g of sodium citrate dihydrate, and 24.5 g of dextrose dissolved in 1,000 mL of distilled water), and centrifuged at 1,500 × g for 10 min at room temperature to remove plasma and white blood cells. The RBC pellet was washed three times with sterile PBS, and 0.3 mL of the final washed pellet was suspended in 3 mL of one of three different media (Krebs-Henseleit buffer, DMEM, or RPMI medium 1640). The resulting RBC suspension was dispensed into 1.5-mL Eppendorf tubes (50 µL per tube), which were then maintained at 37 °C in complete darkness under an atmosphere of 5% CO2 and 20% O2 in air. Tubes were removed from the incubator at various times for the addition of 80 μL of 2× SDS sample buffer containing 10 mM NEM. They were then incubated at 95 °C for 10 min before storage at –80 °C until immunoblot analysis. The basal Krebs−Henseleit buffer was supplemented or not with glucose (2 g/L), sodium bicarbonate (1.6 g/L), or sodium pyruvate (110 mg/L). DMEM was modified or not by the addition of sodium pyruvate (110 mg/L). All three media were supplemented with 0.1% BSA, penicillin (100 U/mL), and streptomycin (100 mg/mL). These experiments allowed us to determine that circadian oscillation of Prx hyperoxidation in mouse RBCs can be studied adequately with modified DMEM (containing d-glucose at 4.5 g/L, sodium bicarbonate at 3.7 g/L, and sodium pyruvate at 110 mg/L and with an osmolarity of 330 mOsm/L) at a density of 1.5 × 107 cells/50 μL in the dark at 37 °C under an atmosphere of 5% CO2 and 20% O2 in air.
Immunoblot Analysis.
RBC proteins were fractionated by reducing SDS/PAGE on a 14% gel and then transferred eletrophoretically to a nitrocellulose membrane for immunoblot analysis with various antibodies. Immune complexes were detected with horseradish peroxidase-conjugated secondary antibodies and ECL reagents. Band intensities on X-ray film were measured with the use of densitometry software (TINA 2.09).
Measurement of the Extent of PrxII Hyperoxidation in Mouse RBCs.
To prepare a standard RBC lysate for which the extent of PrxII hyperoxidation is quantified, we incubated mouse RBCs at a 50% hematocrit in 50 µL of DMEM containing 0.5 mU of glucose oxidase for 1 h at 37 °C with gentle shaking. The cells were then washed with PBS and lysed by ultrasonic treatment in four 30-s bursts. The resulting lysate was subjected to 2D PAGE followed by immunoblot analysis for quantitation of the extent of PrxII hyperoxidation as described (30). About 10% of PrxII was found to be hyperoxidized in the standard lysate. The extent of PrxII hyperoxidation in RBCs during circadian oscillation was estimated by comparison of the immunoblot intensities of PrxII–SO2H with those for the standard RBC lysate.
Exposure of Mouse RBCs to CO.
RBCs were washed three times with PBS, and 0.3 mL of the washed RBC pellet was suspended in 3 mL of modified DMEM and exposed for 30 min to 100% CO by direct injection of the gas stream through a line-connected needle into the RBC tube.
Fractionation of RBCs Based on Mean Age.
RBCs were washed with PBS and then layered on top of a discontinuous density gradient consisting of 85%, 80%, 76%, 72%, 69%, and 66% Percoll (from the bottom up), as described previously (35). Centrifugation of the tube at 4,000 × g for 15 min at 4 °C yielded six discrete bands, of which the two minor bands at the top and bottom of the gradient were discarded and the four middle bands (F1 to F4 for the least dense to most dense) were collected, washed three times with PBS, and subjected to immunoblot analysis.
Preparation of Ghost Membranes.
RBCs (0.15 mL) that had been washed with PBS were suspended in 10 mL of a 1:25 dilution of PBS and incubated for 30 min at 0 °C to induce hypotonic lysis. The lysate was then mixed with 2.5 mL of 5× PBS to restore isotonicity, incubated for 45 min at 37 °C, and centrifuged at 2,500 × g for 10 min at room temperature to obtain cytosolic and ghost membrane fractions. The ghost membranes were washed with PBS until the supernatant appeared free of Hb.
Supplementary Material
Acknowledgments
This study was supported by a grant from the Korean Science and Engineering Foundation [Bio R&D Program Grant M10642040001-07N4204-00110 (to S.G.R.)] and by the BK21 plus program through the National Research Foundation funded by the Ministry of Education (H.J.Y.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1401100111/-/DCSupplemental.
References
- 1.Rhee SG, Woo HA. Multiple functions of peroxiredoxins: Peroxidases, sensors and regulators of the intracellular messenger H2O2, and protein chaperones. Antioxid Redox Signal. 2011;15(3):781–794. doi: 10.1089/ars.2010.3393. [DOI] [PubMed] [Google Scholar]
- 2.Yang KS, et al. Inactivation of human peroxiredoxin I during catalysis as the result of the oxidation of the catalytic site cysteine to cysteine-sulfinic acid. J Biol Chem. 2002;277(41):38029–38036. doi: 10.1074/jbc.M206626200. [DOI] [PubMed] [Google Scholar]
- 3.Woo HA, et al. Reversing the inactivation of peroxiredoxins caused by cysteine sulfinic acid formation. Science. 2003;300(5619):653–656. doi: 10.1126/science.1080273. [DOI] [PubMed] [Google Scholar]
- 4.Biteau B, Labarre J, Toledano MB. ATP-dependent reduction of cysteine-sulphinic acid by S. cerevisiae sulphiredoxin. Nature. 2003;425(6961):980–984. doi: 10.1038/nature02075. [DOI] [PubMed] [Google Scholar]
- 5.Jeong W, Park SJ, Chang TS, Lee DY, Rhee SG. Molecular mechanism of the reduction of cysteine sulfinic acid of peroxiredoxin to cysteine by mammalian sulfiredoxin. J Biol Chem. 2006;281(20):14400–14407. doi: 10.1074/jbc.M511082200. [DOI] [PubMed] [Google Scholar]
- 6.Dunlap JC. Molecular bases for circadian clocks. Cell. 1999;96(2):271–290. doi: 10.1016/s0092-8674(00)80566-8. [DOI] [PubMed] [Google Scholar]
- 7.Mohawk JA, Green CB, Takahashi JS. Central and peripheral circadian clocks in mammals. Annu Rev Neurosci. 2012;35:445–462. doi: 10.1146/annurev-neuro-060909-153128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bell-Pedersen D, et al. Circadian rhythms from multiple oscillators: Lessons from diverse organisms. Nat Rev Genet. 2005;6(7):544–556. doi: 10.1038/nrg1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hastings MH, Maywood ES, O’Neill JS. Cellular circadian pacemaking and the role of cytosolic rhythms. Curr Biol. 2008;18(17):R805–R815. doi: 10.1016/j.cub.2008.07.021. [DOI] [PubMed] [Google Scholar]
- 10.Kitayama Y, Nishiwaki T, Terauchi K, Kondo T. Dual KaiC-based oscillations constitute the circadian system of cyanobacteria. Genes Dev. 2008;22(11):1513–1521. doi: 10.1101/gad.1661808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.O’Neill JS, et al. Circadian rhythms persist without transcription in a eukaryote. Nature. 2011;469(7331):554–558. doi: 10.1038/nature09654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.O’Neill JS, Reddy AB. Circadian clocks in human red blood cells. Nature. 2011;469(7331):498–503. doi: 10.1038/nature09702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Edgar RS, et al. Peroxiredoxins are conserved markers of circadian rhythms. Nature. 2012;485(7399):459–464. doi: 10.1038/nature11088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Olmedo M, et al. Circadian regulation of olfaction and an evolutionarily conserved, nontranscriptional marker in Caenorhabditis elegans. Proc Natl Acad Sci USA. 2012;109(50):20479–20484. doi: 10.1073/pnas.1211705109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bass J, Takahashi JS. Circadian rhythms: Redox redux. Nature. 2011;469(7331):476–478. doi: 10.1038/469476a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stangherlin A, Reddy AB. Regulation of circadian clocks by redox homeostasis. J Biol Chem. 2013;288(37):26505–26511. doi: 10.1074/jbc.R113.457564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kil IS, et al. Feedback control of adrenal steroidogenesis via H2O2-dependent, reversible inactivation of peroxiredoxin III in mitochondria. Mol Cell. 2012;46(5):584–594. doi: 10.1016/j.molcel.2012.05.030. [DOI] [PubMed] [Google Scholar]
- 18.Winterbourn CC. Free-radical production and oxidative reactions of hemoglobin. Environ Health Perspect. 1985;64:321–330. doi: 10.1289/ehp.8564321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Johnson RM, Goyette G, Jr, Ravindranath Y, Ho YS. Hemoglobin autoxidation and regulation of endogenous H2O2 levels in erythrocytes. Free Radic Biol Med. 2005;39(11):1407–1417. doi: 10.1016/j.freeradbiomed.2005.07.002. [DOI] [PubMed] [Google Scholar]
- 20.Low FM, Hampton MB, Winterbourn CC. Peroxiredoxin 2 and peroxide metabolism in the erythrocyte. Antioxid Redox Signal. 2008;10(9):1621–1630. doi: 10.1089/ars.2008.2081. [DOI] [PubMed] [Google Scholar]
- 21.Cho CS, et al. Irreversible inactivation of glutathione peroxidase 1 and reversible inactivation of peroxiredoxin II by H2O2 in red blood cells. Antioxid Redox Signal. 2010;12(11):1235–1246. doi: 10.1089/ars.2009.2701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hall A, Nelson K, Poole LB, Karplus PA. Structure-based insights into the catalytic power and conformational dexterity of peroxiredoxins. Antioxid Redox Signal. 2011;15(3):795–815. doi: 10.1089/ars.2010.3624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Silverstein E, Sokoloff L, Mickelsen O, Jay GE. Primary polydipsia and hydronephrosis in an inbred strain of mice. Am J Pathol. 1961;38(2):143–159. [PMC free article] [PubMed] [Google Scholar]
- 24.Davies KJ, Goldberg AL. Proteins damaged by oxygen radicals are rapidly degraded in extracts of red blood cells. J Biol Chem. 1987;262(17):8227–8234. [PubMed] [Google Scholar]
- 25.Grune T, Merker K, Sandig G, Davies KJ. Selective degradation of oxidatively modified protein substrates by the proteasome. Biochem Biophys Res Commun. 2003;305(3):709–718. doi: 10.1016/s0006-291x(03)00809-x. [DOI] [PubMed] [Google Scholar]
- 26.Cha MK, Yun CH, Kim IH. Interaction of human thiol-specific antioxidant protein 1 with erythrocyte plasma membrane. Biochemistry. 2000;39(23):6944–6950. doi: 10.1021/bi000034j. [DOI] [PubMed] [Google Scholar]
- 27.Mazzaccara C, et al. Age-related reference intervals of the main biochemical and hematological parameters in C57BL/6J, 129SV/EV and C3H/HeJ mouse strains. PLoS ONE. 2008;3(11):e3772. doi: 10.1371/journal.pone.0003772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Low FM, Hampton MB, Peskin AV, Winterbourn CC. Peroxiredoxin 2 functions as a noncatalytic scavenger of low-level hydrogen peroxide in the erythrocyte. Blood. 2007;109(6):2611–2617. doi: 10.1182/blood-2006-09-048728. [DOI] [PubMed] [Google Scholar]
- 29.Chang TS, et al. Characterization of mammalian sulfiredoxin and its reactivation of hyperoxidized peroxiredoxin through reduction of cysteine sulfinic acid in the active site to cysteine. J Biol Chem. 2004;279(49):50994–51001. doi: 10.1074/jbc.M409482200. [DOI] [PubMed] [Google Scholar]
- 30.Woo HA, et al. Reduction of cysteine sulfinic acid by sulfiredoxin is specific to 2-cys peroxiredoxins. J Biol Chem. 2005;280(5):3125–3128. doi: 10.1074/jbc.C400496200. [DOI] [PubMed] [Google Scholar]
- 31.Majetschak M, Sorell LT. Immunological methods to quantify and characterize proteasome complexes: Development and application. J Immunol Methods. 2008;334(1-2):91–103. doi: 10.1016/j.jim.2008.02.004. [DOI] [PubMed] [Google Scholar]
- 32.Jang HH, et al. Two enzymes in one; two yeast peroxiredoxins display oxidative stress-dependent switching from a peroxidase to a molecular chaperone function. Cell. 2004;117(5):625–635. doi: 10.1016/j.cell.2004.05.002. [DOI] [PubMed] [Google Scholar]
- 33.Pace PE, Peskin AV, Han MH, Hampton MB, Winterbourn CC. Hyperoxidized peroxiredoxin 2 interacts with the protein disulfide- isomerase ERp46. Biochem J. 2013;453(3):475–485. doi: 10.1042/BJ20130030. [DOI] [PubMed] [Google Scholar]
- 34.Lee TH, et al. Peroxiredoxin II is essential for sustaining life span of erythrocytes in mice. Blood. 2003;101(12):5033–5038. doi: 10.1182/blood-2002-08-2548. [DOI] [PubMed] [Google Scholar]
- 35.Boyer C, Kahn A, Cottreau D, Marie J. Mechanism of decrease in erythrocyte enzyme activities during red cell aging in the newborn and the adult. Nouv Rev Fr Hematol Blood Cells. 1977;18(1):229–231. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.