

Abstract
Eukaryotic elongation factor 2 kinase (eEF2K) is a Ca2+/calmodulin-dependent protein kinase. We recently demonstrated that eEF2K protein increases in mesenteric artery from spontaneously hypertensive rats (SHR). Pathogenesis of hypertension is regulated in part by vascular inflammation. We tested the hypothesis whether eEF2K mediates vascular inflammatory responses and development of hypertension. In vascular endothelial cells, small interfering RNA (siRNA) against eEF2K inhibited induction of VCAM-1 and endothelial-selectin as well as monocyte adhesion by TNF-α (10 ng/ml). eEF2K siRNA inhibited phosphorylation of JNK and NF-κB p65 as well as reactive oxygen species (ROS) production by TNF-α. In vascular smooth muscle cells, eEF2K siRNA also inhibited VCAM-1 induction and phosphorylation of JNK and NF-κB by TNF-α. In vivo, increased blood pressure in SHR and ROS production, induction of inflammatory molecules, and hypertrophy in SHR superior mesenteric artery were reduced by an eEF2K inhibitor NH125 (500 μg·kg−1·day−1). In SHR superior mesenteric artery, impairment of ACh-induced relaxation was normalized by NH125. The present results for the first time demonstrate that eEF2K mediates TNF-α-induced vascular inflammation via ROS-dependent mechanism, which is at least partly responsible for the development of hypertension in SHR.
Keywords: hypertension, inflammation, reactive oxygen species, signal transduction, vasoconstriction
calmodulin (CaM) is a central mediator of Ca2+-dependent signaling. CaM binds to Ca2+ in responses to increase in intracellular Ca2+, which activates numerous proteins including a family of CaM-dependent protein kinases (CaMKs). CaMKs such as CaMKI, CaMKII, CaMKIII, and CaMKIV phosphorylate multiple downstream targets. Recently, it was clarified that CaMKII plays pivotal roles in regulation of cardiovascular diseases. CaMKII is strongly expressed in vascular smooth muscle cells (SMCs) (19) and is responsible for ANG II-induced vascular hypertrophy in rats (19) by regulating proliferation (14) and migration (24) of vascular SMCs. In addition, it was demonstrated that CaMKIIδ mediated neointima formation after carotid ligation through regulation of vascular SMCs proliferation (21). Furthermore, we recently demonstrated that a CaMKII-dependent transcriptional regulator, histone deacetylase 4, mediates development of hypertension via vascular inflammation in spontaneously hypertensive rats (SHR) (38).
Eukaryotic elongation factor (eEF) 2 kinase (eEF2K), also known as CaMKIII, is a unique member of CaMK family protein (30), and its activity is normally dependent on Ca2+ and CaM. eEF2K prevents binding of eEF to the ribosome via phosphorylation of eEF and thus prevents further translation (15). The activity of eEF2K is selectively increased in proliferating cells, whereas the inhibition of the kinase remained the cells in G0/G1-S and decreased viability (7, 10).
In the previous study, we compared expression level of several CaM-related proteins with almost unknown function in vasculature. We found that protein expression of eEF2K increased in mesenteric artery from SHR compared with Wistar Kyoto rats (WKY) (37). It is well known that vascular inflammation and reactive oxygen species (ROS) production regulate the development of hypertension (17). However, it remains to be clarified how eEF2K controls hypertensive vascular diseases via inflammation and/or ROS production. We therefore tested the hypothesis whether eEF2K affects vascular endothelial and smooth muscle inflammatory states and development of hypertension. Here we, for the first time, demonstrate that eEF2K mediates ROS-dependent vascular inflammation and is at least in part responsible for the development of hypertension via propagating vascular hypertrophy and endothelial dysfunction in SHR.
MATERIALS AND METHODS
Approval of animal study.
Animal care and treatment were conducted in conformity with the institutional guidelines of The Kitasato University and the National Institutes of Health Guide for the Care and Use of Laboratory Animals. This animal study was approved by ethical committee of School of Veterinary Medicine, The Kitasato University.
Materials.
Reagent sources were as follows: TNF-α (Roche Applied Science, Mannheim, Germany); noradrenaline (NA) (Sigma-Aldrich, St. Louis, MO); ACh (Daiich-Sankyo Pharmaceutical, Tokyo); eEF2K inhibitor (NH125) 1-benzyl-3-cetyl-2-methylimidazolium iodide (Cayman, Ann Arbor, MI); and eEF2K inhibitor (A-484954) 7-amino-1-cyclopropyl-3-ethyl-2,4-dioxo-1,2,3,4-tetrahydropyrido[2,3-d] pyrimidine-6-carboxamide (Merck KGaA, Darmstadt, Germany). It was reported that NH125 inhibits eEF2K activity via inhibition of α-kinase, a catalytic domain of eEF2K (2, 31). On the other hand, A-484954 is a novel small molecule inhibitor identified from a chemical library (9). It is a cell-permeable pyrido-pyrimidinedione derivative that inhibits eEF2K in an ATP-competitive but CaM-independent manner. TNF-α, NA, and ACh were dissolved in distilled water. NH125 and A-484954 were dissolved in DMSO (final concentration for in vitro use was 0.1%).
Antibody sources were as follows: phospho-JNK (Thr183/Tyr185) (No. PR-V7932) (Promega, Madison, WI); phospho-NF-κB p65 (Ser536) (No. 3033), total JNK (No. 9252) (Cell Signaling, Beverly, MA); total NF-κB p65 (sc-8008), VCAM-1 (sc-1504) (Santa Cruz Biotech, Santa Cruz, CA); endothelial-selectin (E-selectin) (No. BRA16) (R&D System, Minneapolis, MN); total actin (No. MAB1501) (Sigma-Aldrich); α-actin (No. M0851) (Dako, Glostrup, Denmark); total eEF2K (No. GTX107879) (Gene Tex, Irvine, CA); and phospho-eEF2K (No. A0071) (Assay Biotechnology, Sunnyvale, CA).
Culture of human umbilical vein endothelial cells.
Human umbilical vein endothelial cells (ECs) (HUVECs) were obtained from Kurabo (Osaka, Japan) and cultured in Medium 200 supplemented with low serum growth supplement (Cascade Biologics, Portland, OR) as described previously (39). Cells at passage from 3 to 10 were used.
Culture of rat mesenteric arterial SMCs.
Male Wistar rats (7 to 9 wk old) were anesthetized with urethane (1.5 g/kg ip) and euthanized by exsanguination. The superior mesenteric artery was isolated. SMCs isolated from mesenteric artery by an explant method were cultured in DMEM supplemented with 10% FBS (Invitrogen, Carlsbad, CA) (40). Passages 4 to 20 SMCs at 80 to 90% confluence were growth arrested by incubating in DMEM containing 0.5% FBS for 24 h before stimulation.
Western blotting.
Western blotting was performed as described previously (37). Protein lysates were obtained by homogenizing HUVECs, SMCs, or tissue samples with Triton-based lysis buffer containing 1% Triton X-100, 20 mmol/l Tris (pH 7.4), 150 mmol/l NaCl, 1 mM EDTA, 1 mmol/l EGTA, 2.5 mmol/l sodium pyrophosphate, 1 mmol/l β-glycerol phosphate, 1 mmol/l NA3VO4, 1 μg/ml leupeptin, and 0.1% protease inhibitor cocktail (Nacalai Tesque, Kyoto, Japan). Protein concentration was determined using a bicinchoninic acid method (Pierce, Rockford, IL). Equal amount of proteins (8–10 μg) were separated by SDS-PAGE (7.5%) and transferred to a nitrocellulose membrane (Pall, Ann Arbor, MI). After being blocked with 3% BSA (for phosphorylation-specific antibodies) or 0.5% skim milk (for others), membranes were incubated with primary antibodies at 4°C overnight and then visualized using horseradish peroxidase-conjugated secondary antibodies (1:10,000 dilution, 1 h at room temperature) and the EZ-ECL system (Biological industries, Kibbutz Beit Hesmek, Israel). Equal loading of protein was confirmed by measuring total protein or actin expression. We confirmed that nonphosphorylated proteins used as equal loading control did not vary among groups. The resulting bands were analyzed using CS Analyzer 3.0 software (ATTO, Tokyo, Japan).
Small interfering RNA transfection.
One day after HUVECs or SMCs were subcultured, they (30–40% confluent) were transfected for 24 h with small interfering RNA (siRNA) against eEF2K (eEF2K siRNA for HUVECs, AAGUAAUCAAAUCCUUUGGtt, and eEF2K siRNA for SMCs, ACUAAAAUCAUGGACUGCCtt; Nippon EGT, Toyama, Japan) or nonsilencing control siRNA (Qiagen, Valencia, CA) using Lipofectamine 2000 (Invitrogen) dissolved in Optimem (Invitrogen) at a final concentration of 40 nmol/l (36). HUVECs or SMCs were then recovered for an additional 24 h before stimulation.
Cell adhesion assays.
U937 (monocytes) cells were obtained from RIKEN Cell Bank (Tsukuba, Japan) and cultured in RPMI-1640 medium supplemented with 5% FBS (36). After HUVECs transfected with eEF2K siRNA or control siRNA (40 nmol/l, 24 h) in a 6-well culture plate were stimulated with TNF-α (10 ng/ml) for 6 h, U937 cells (∼8.5 × 105 cells/well) were co-incubated for 1 h at 37°C. Nonattached cells were removed by the gentle washing, and then the cells were fixed with 4% paraformaldehyde at 37°C for 10 min. The number of attached U937 cells was randomly counted in three areas per well at ×200 filed and averaged.
Measurement of ROS production in vitro.
Intracellular ROS production in HUVECs was examined by a fluorescence staining using 2′,7′-dichlorodihydrofluorescein diacetate (H2DCFDA) (Invitrogen) (36). After treatment for 20 min with TNF-α in the presence of eEF2K siRNA, control siRNA, or NH125 (1 μmol/l, 6 h pretreatment), HUVECs were loaded with H2DCFDA (10 μmol/l) for 30 min. Fluorescence images were obtained using a fluorescence microscope (BX-51; Olympus, Tokyo, Japan) equipped with cooled charge-coupled device camera (MicroPublisher 5.0 RTV; Roper Japan, Tokyo, Japan). The ImageJ software was used for the quantitative analysis of the images.
Animal studies.
NH125 or vehicle (100% DMSO) was subcutaneously administered to male SHR (4 wk old; Hoshino Laboratory Animals, Ibaragi, Japan) and age-matched male WKY at a dose of 300–500 μg·kg−1·day−1 for 6 wk.
Blood pressure and heart rate measurement.
Systolic blood pressure (SBP) and heart rate (HR) of SHR and WKY were measured using a tail-cuff system (Softron, Tokyo, Japan) in the conscious condition as described previously (36–38). We measured blood pressure (BP) six times for each rat, and the three stable values were averaged.
Tissue preparation.
SHR (0.25–0.29 kg: 10 wk old) and age-matched WKY (0.27–0.31 kg) were anesthetized with urethane (1.5 g/kg ip) and euthanized by exsanguination. The superior mesenteric arteries were isolated as described previously (37). After the fat and connective tissues were removed, the arterial samples were used for the extraction of protein, the measurement of isometric tension, and the histological analysis.
Histology and measurement of ROS in arterial tissues.
After isolated mesenteric artery was embedded in optimal cutting temperature compound (Sakura Finetechnical, Tokyo, Japan) and quickly frozen, the thin frozen sections (5 μm) were made using a cryostat (Leica, Solms, Germany) and then stained with hematoxylin and eosin as described previously (36). The images were obtained using a light microscope (BX-51; Olympus). For measurement of ROS, the frozen sections were loaded with H2DCFDA (5 μmol/l) for 30 min. Fluorescence images were obtained using a fluorescence microscope. The ImageJ software was used for the quantitative analysis of the images.
Measurement of smooth muscle contraction.
The contractility of endothelium-intact mesenteric artery was measured as described previously (25, 36, 38). The arterial rings were placed in normal physiological salt solution, which contained (in mmol/l) 139.9 NaCl, 5.4 KCl, 1.5 CaCl2, 1.0 MgCl2, 23.8 NaHCO3, and 5.5 glucose. EDTA, 1 μmol/l, was also added to remove the contaminating heavy metal ions, which catalyze oxidation of organic chemicals. The high K+ (72.4 mmol/l) solution was prepared by replacing NaCl with equimolar KCl. These solutions were saturated with a 95% O2-5% CO2 mixture at 37°C and pH 7.4. Smooth muscle contractility was recorded isometrically with a force-displacement transducer (Nihon Kohden, Tokyo, Japan). Each muscle ring was attached to a holder under a resting tension of 0.5 g. After equilibration for 30 min in a 3-ml organ bath, each ring was repeatedly exposed to high K+ solution until the responses became stable (60–90 min). ACh (0.1–300 nmol/l) was cumulatively applied to the arteries precontracted to the similar level with submaximal concentration of NA (100 nmol/l to 1 μmol/l).
Statistical analysis.
Data were shown as means ± SE. Statistical evaluations were performed using one-way ANOVA followed by Bonferroni's test for comparisons in more than three groups and by Student's t-test between two groups. Values of P < 0.05 were considered statistically significant. All pD2 values were calculated as the −log10EC50 by sigmoid curve fitting.
RESULTS
Effects of eEF2K inhibitor NH125 or A-484954 on TNF-α-induced inflammatory responses in HUVECs.
We first examined whether eEF2K mediates inflammatory responses in HUVECs. TNF-α (10 ng/ml, 6 h)-induced expression of VCAM-1 (Fig. 1A) and E-selectin (Fig. 1B) was significantly inhibited by a specific eEF2K inhibitor NH125 (1 μmol/l). Another eEF2K inhibitor, A-484954 (10 μmol/l), also significantly inhibited the TNF-α-induced expression of VCAM-1 (Fig. 1C) and E-selection (Fig. 1D). Activity of eEF2K is dependent on Ca2+ and CaM. In the presence of Ca2+ and CaM, eEF2K undergoes autophosphorylation on serine and threonine residues (Ser78, Ser345, Ser359, Ser366, Se377, Ser396, Ser445, Ser474, Ser491, Ser499, Thr348, Thr353) (32) (16). Among these residues, insulin-like growth factor-activated p70S6 kinase induced phosphorylation of eEF2K at Ser366, resulting in dephosphorylation of eEF2 (41). In addition, it was shown that mutation of eEF2K at Ser366, Ser78, or Thr348 decreased eEF2K activity as measured by myosin heavy chain-1 peptide as a substrate (32). Considering these reports, we focused on phosphorylation of eEF2K at Ser366 and confirmed that TNF-α (10 ng/ml, 5 min)-induced phosphorylation of eEF2K at Ser366 was significantly inhibited by each of the eEF2K inhibitor NH125 (1 μmol/l) or A-484954 (10 μmol/l) in HUVECs (Fig. 2).
Fig. 1.
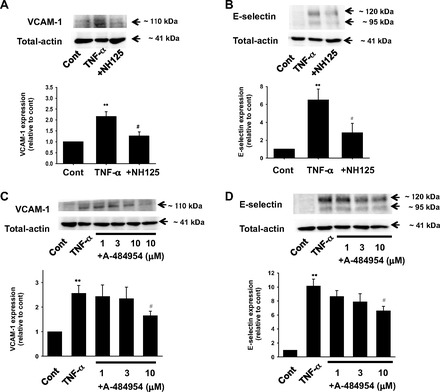
Effects of eukaryotic elongation factor (eEF) 2 kinase (eEF2K) inhibitor NH125 or A-484954 on TNF-α-induced expression of VCAM-1 (A, n = 5; C, n = 6–8) and endothelial-selectin (E-selectin; B, n = 6; D, n = 4) in cultured human umbilical vein endothelial cells (ECs) (HUVECs). After HUVECs were treated with 10 ng/ml TNF-α for 6 h in the absence or presence of NH125 (1 μmol/l, pretreatment for 30 min) or A-484954 (1–10 μmol/l, pretreatment for 30 min), expression of VCAM-1 and E-selectin was determined by Western blotting and shown as fold increase relative to control (cont; no-drug treatment). Equal protein loading was confirmed using total actin antibody. **P < 0.01 vs. cont; #P < 0.05 vs. TNF-α.
Fig. 2.
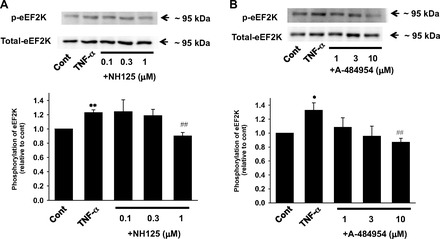
Effects of eEF2K inhibitor NH-125 (A) or A-484954 (B) on TNF-α-induced eEF2K phosphorylation in HUVECs. After HUVECs were treated with 10 ng/ml TNF-α for 5 min in the absence or presence of NH125 (0.1–1 μmol/l, pretreatment for 30 min) or A-484954 (1–10 μmol/l, pretreatment for 30 min), eEF2K phosphorylation (A, n = 4; B, n = 4) was determined by Western blotting and shown as fold increase relative to control (no drug treatment). Equal protein loading was confirmed using total eEF2K antibody. *P < 0.05, **P < 0.01 vs. cont; ##P < 0.01 vs. TNF-α.
Effects of eEF2K gene knockdown on TNF-α-induced inflammatory responses in HUVECs.
To further clarify the roles of eEF2K on inflammatory responses in HUVECs, eEF2K gene was specifically silenced by eEF2K siRNA transfection. We confirmed that eEF2K protein was significantly decreased by eEF2K siRNA (Fig. 3A). TNF (10 ng/ml, 6 h)-induced expression of VCAM-1 (Fig. 3B) and E-selectin (Fig. 3C) was significantly inhibited by eEF2K siRNA. eEF2K siRNA had no influence on basal expression of VCAM-1 and E-selectin (n = 4; data not shown). We next examined whether eEF2K gene knockdown inhibits monocyte adhesion to HUVECs. The eEF2K siRNA significantly decreased the number of monocyte adhesion to HUVECs (Fig. 3D). To explore upstream mechanisms of inhibition of adhesion molecules induction, effects of eEF2K knockdown on inflammatory signals were examined. TNF-α (10 ng/ml, 20 min)-induced phosphorylation of JNK (Fig. 4A) and NF-κB p65 (Ser536) (Fig. 4B) was significantly inhibited by eEF2K siRNA. eEF2K siRNA had no influence on basal phosphorylation of JNK and NF-κB p65 (Ser536) (n = 4; data not shown). We also confirmed that eEF2K siRNA had no effect on TNF-induced phosphorylation of p38 and ERK in HUVECs (n = 4; data not shown). To further investigate the upstream mechanisms, we examined whether eEF2K knockdown prevents TNF-α-induced ROS production in HUVECs. The eEF2K siRNA significantly inhibited the TNF-α (10 ng/ml, 20 min)-induced ROS production (Fig. 4C). We also confirmed that TNF-α (10 ng/ml, 20 min)-induced phosphorylation of JNK (Fig. 5A) and NF-κB p65 (Ser536) (Fig. 5B) was significantly inhibited by NH125 (1 μmol/l). NH125 also significantly inhibited TNF-α (10 ng/ml, 20 min)-induced ROS production (Fig. 5C).
Fig. 3.
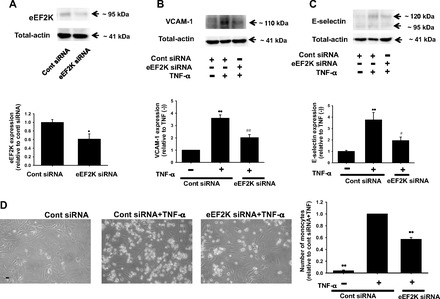
Effects of eEF2K gene knockdown on TNF-α-induced inflammatory responses in HUVECs. Knockdown of eEF2K gene by small interfering RNA (siRNA) is shown (A). After HUVECs were transfected with eEF2K-specific siRNA (eEF2K siRNA) or nonsilencing control siRNA (40 nmol/l; 24 h), total cell lysates were harvested. Expression of eEF2K protein (n = 6) was determined by Western blotting and shown as fold change relative to control siRNA. Equal protein loading was confirmed using total-actin antibody. *P < 0.05 vs. control siRNA. Effects of eEF2K knockdown on TNF-α-induced expression of VCAM-1 (B) and E-selectin (C) are also shown. After HUVECs were transfected with eEF2K siRNA or control siRNA, they were treated with 10 ng/ml TNF-α for 6 h. Expression of VCAM-1 (n = 4) and E-selectin (n = 6) was determined by Western blotting and shown as fold increase relative to control siRNA without TNF-α stimulation. Equal protein loading was confirmed using total-actin antibody. **P < 0.01 vs. control siRNA without TNF-α stimulation; #P < 0.05, ##P < 0.01 vs. cont siRNA + TNF-α. Effects of eEF2K knockdown on monocyte (U937 cells) adhesion to HUVECs (D) are also shown. After HUVECs were transfected with eEF2K siRNA or control siRNA, TNF-α (10 ng/ml, 6 h) was treated. After U937 cells were added for 1 h to HUVECs (n = 4), nonadherent cells were removed and the number of adhering U937 cells was randomly counted in 3 areas (×200 fields) and averaged. Scale bar, 50 μm. The number of U937 cells adhering to HUVECs was shown as fold increase relative to control siRNA with TNF-α stimulation. **P < 0.01 vs. cont siRNA + TNF-α.
Fig. 4.
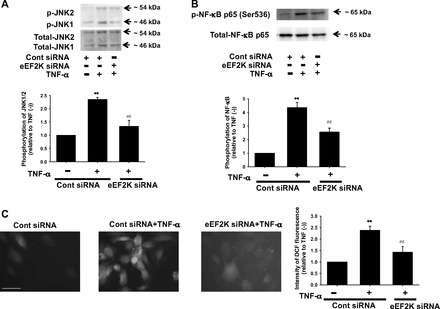
Effects of eEF2K knockdown on TNF-α-induced phosphorylation of JNK (A) and NF-κB (B), and reactive oxygen species (ROS) production (C) in HUVECs. After transfection with eEF2K siRNA or control siRNA (40 nmol/l, 24 h), HUVECs were stimulated with 10 ng/ml TNF-α for 20 min. Phosphorylation of JNK (n = 4) and NF-κB p65 (Ser536) (n = 4) was determined by Western blotting and shown as fold increase relative to control siRNA without TNF-α stimulation. Equal protein loading was confirmed by using total antibody. ROS production was determined by a fluorescence staining using 2′,7′-dichlorodihydrofluorescein diacetate (H2DCFDA). After HUVECs were treated with 10 ng/ml TNF-α for 20 min in the presence of eEF2K siRNA or control siRNA (n = 4), they were loaded with H2DCFDA (10 μmol/l) for 30 min. Images were obtained using a fluorescence microscope. Scale bar, 50 μm. Fluorescent intensity was measured using ImageJ software and shown as fold increase relative to control siRNA without TNF-α stimulation. **P < 0.01 vs. control siRNA without TNF-α; ##P < 0.01 vs. cont siRNA + TNF-α.
Fig. 5.
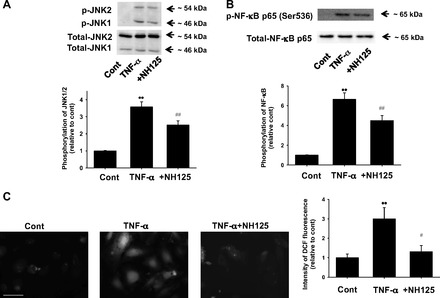
Effects of NH125 on TNF-α-induced phosphorylation of JNK (A) and NF-κB (B), and ROS production (C) in HUVECs. After HUVECs were treated with 10 ng/ml TNF-α for 20 min in the absence or presence of NH125 (1 μmol/l, pretreatment for 6 h), phosphorylation of JNK (n = 8) and NF-κB p65 (Ser536) (n = 8) was determined by Western blotting and shown as fold increase relative to control. Equal protein loading was confirmed using total actin antibody. **P < 0.01 vs. cont; ##P < 0.01 vs. TNF-α. ROS production was determined by an H2DCFDA staining. After HUVECs were treated with 10 ng/ml TNF-α for 20 min in the absence or presence of NH125 (1 μmol/l, pretreatment for 6 h), they were loaded with H2DCFDA (10 μmol/l) for 30 min (n = 4). Images were obtained using a fluorescence microscope. Scale bar, 50 μm. Fluorescent intensity was measured using ImageJ software and shown as fold increase relative to control. **P < 0.01 vs. cont; #P < 0.05 vs. TNF-α.
Effects of CaM inhibitor W-7 on TNF-α-induced inflammatory responses in HUVECs.
eEF2K is a CaM-dependent protein kinase known as CaMKIII. We thus investigated whether CaM regulates eEF2K-mediated inflammatory responses in HUVECs. TNF-α (10 ng/ml, 6 h)-induced expression of VCAM-1 (Fig. 6A) and E-selectin (Fig. 6B) was significantly inhibited by a CaM inhibitor, W-7 (10 μmol/l). W-7 also inhibited TNF-α (10 ng/ml, 20 min)-induced phosphorylation of JNK (Fig. 6C) and NF-κB p65 (Ser536) (Fig. 6D). We confirmed that W-7 (10 μmol/l) inhibited TNF-α (10 ng/ml, 5 min)-induced phosphorylation of eEF2K (Fig. 6E).
Fig. 6.
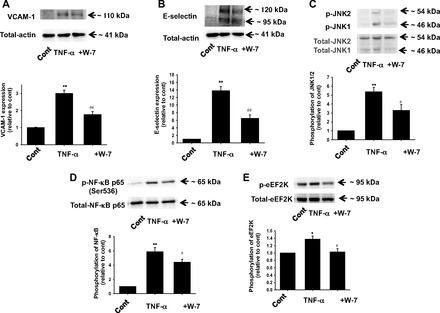
Effects of calmodulin (CaM) inhibitor W-7 on TNF-α-induced inflammatory responses in HUVECs. Effects of W-7 on TNF-α-induced expression of VCAM-1 (A) and E-selectin (B) in HUVECs are shown. After HUVECs were treated with 10 ng/ml TNF-α for 6 h in the absence or presence of W-7 (10 μmol/l, pretreatment for 30 min), expression of VCAM-1 (n = 3) and E-selectin (n = 4) was determined by Western blotting and shown as fold increase relative to control. Equal protein loading was confirmed using total-actin antibody. Effects of W-7 on TNF-α-induced phosphorylation of JNK (C), NF-κB p65 (Ser536) (D), and eEF2K (E) are also shown. After HUVECs were treated with 10 ng/ml TNF-α for 5–20 min in the absence or presence of W-7 (10 μM, pretreatment for 30 min), phosphorylation of JNK (n = 6), NF-κB (n = 6), and eEF2K (n = 4) was determined by Western blotting and shown as fold increase relative to control. Equal protein loading was confirmed using total-actin, JNK, NF-κB, and eEF2K antibody. *P < 0.05, **P < 0.01 vs. cont; #P < 0.05, ##P < 0.01 vs. TNF-α.
Effects of eEF2K knockdown on TNF-α-induced inflammatory responses in rat mesenteric arterial SMCs.
We next examined whether eEF2K also mediates inflammatory responses in SMCs by using siRNA. We confirmed that eEF2K protein was significantly decreased by eEF2K siRNA transfection (Fig. 7A). TNF-α (10 ng/ml, 24 h)-induced VCAM-1 expression (Fig. 7B) was significantly inhibited by eEF2K siRNA. The eEF2K siRNA also inhibited TNF-α (10 ng/ml, 20 min)-induced phosphorylation of JNK (Fig. 7C) and NF-κB p65 (Ser536) (Fig. 7D).
Fig. 7.
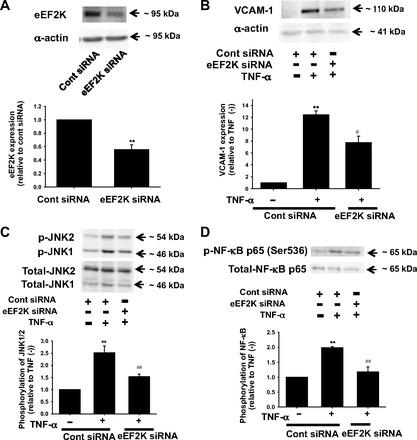
Effects of eEF2K knockdown on TNF-α-induced inflammatory responses in rat mesenteric arterial smooth muscle cells (SMCs). Knockdown of eEF2K gene by siRNA in SMCs (A) is shown. After SMCs were transfected with eEF2K siRNA or control siRNA (40 nmol/l, 24 h), total cell lysates were harvested. Expression of eEF2K protein (n = 3) was determined by Western blotting and shown as fold change relative to control siRNA. Equal protein loading was confirmed using total-α-actin antibody. **P < 0.01 vs. control siRNA. Effects of eEF2K knockdown on TNF-α-induced VCAM-1 expression (B) are also shown. After SMCs were transfected with eEF2K siRNA or control siRNA, they were treated with 10 ng/ml TNF-α for 24 h. VCAM-1 expression was determined by Western blotting and shown as fold increase relative to control siRNA without TNF-α stimulation. Equal protein loading was confirmed using α-actin antibody. Effects of eEF2K knockdown on TNF-α-induced phosphorylation of JNK (C) and NF-κB p65 (Ser536) (D) are also shown. After transfection with eEF2K siRNA or control siRNA (40 nmol/l, 24 h), SMCs were stimulated with 10 ng/ml TNF-α for 20 min. Phosphorylation of JNK (n = 6) and NF-κB (n = 4) was determined by Western blotting and shown as fold increase relative to control siRNA without TNF-α stimulation. Equal protein loading was confirmed using total antibody. **P < 0.01 vs. control siRNA without TNF-α; #P < 0.05, ##P < 0.01 vs. cont siRNA + TNF-α.
Effects of long-term NH125 treatment on BP and physical parameters of SHR.
We next examined effects of long-term NH125 treatment (for 6 wk) on BP of SHR (from 4 wk old to 10 wk old). The SBP was significantly higher in SHR than WKY (at 10 wk old; Fig. 8A). NH125 (500 μg·kg−1·day−1) significantly decreased the SBP in SHR. Treatment of WKY with NH125 (500 μg·kg−1·day−1) had no influence on the SBP. The HR tended to be higher in SHR than WKY (Fig. 8B). Treatment of SHR or WKY with NH125 had no significant influence on the HR except for one-time point (5 wk old). Body weight (BW) was not significantly different between SHR and WKY (Fig. 8C). Treatment of SHR or WKY with NH125 had no significant influence on the BW. We also examined effects of lower concentration of NH125. Treatment of SHR with NH125 (300 μg·kg−1·day−1) significantly decreased the SBP in SHR (Fig. 9A). Treatment of SHR with NH125 (300 μg·kg−1·day−1) had no influence on the BW (Fig. 9B).
Fig. 8.
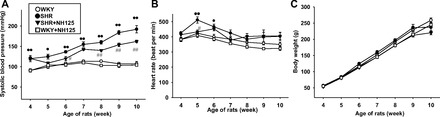
Effects of long-term NH125 treatment on blood pressure (BP; A), heart rate (HR; B), and body weight (BW; C) of spontaneously hypertensive rats (SHR). NH125 was administered to SHR (4 wk old) or age-matched Wistar Kyoto rats (WKY) subcutaneously at a dose of 500 μg·kg−1·day−1 for 6 wk (n = 6). Systolic BP (SBP) and HR (in beats/min) of SHR and WKY were measured using a tail-cuff system at weekly intervals. Results were expressed as means ± SE. *P < 0.05, **P < 0.01 vs. WKY; #P < 0.05, ##P < 0.01 vs. SHR.
Fig. 9.
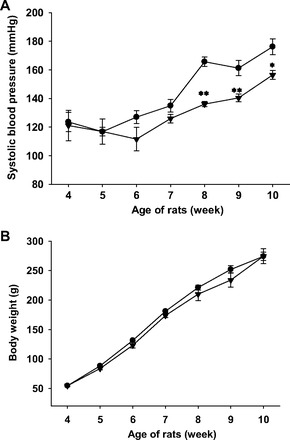
Effects of lower concentration of NH125 on BP (A) and BW (B) of SHR. NH125 was administered to SHR (4 wk old) subcutaneously at a dose of 300 μg·kg−1·day−1 for 6 wk (n = 4). SBP was measured using a tail-cuff system at weekly intervals. Results were expressed as means ± SE. *P < 0.05, **P < 0.01 vs. SHR.
Effects of long-term NH125 treatment on inflammatory responses and ROS production in mesenteric artery from SHR.
To check whether eEF2K mediates vascular inflammation in vivo, we examined effects of long-term NH125 treatment on the expression of VCAM-1 and E-selectin in isolated mesenteric artery from SHR. We confirmed that NH125 treatment significantly inhibited the increased eEF2K expression in SHR (Fig. 10A). The expression of VCAM-1 (Fig. 10B) and E-selectin (Fig. 10C) significantly increased in SHR (10 wk old) compared with that in WKY. NH125 (500 μg·kg−1·day−1, 6 wk) significantly reduced the increased expression of VCAM-1 and E-selectin. NH125 had no influence on the expression of VCAM-1 and E-selectin in WKY. We next examined whether long-term NH125 treatment attenuates ROS production. In mesenteric artery from SHR (10 wk old), fluorescence intensity of H2DCFDA significantly increased compared with WKY (Fig. 10D). NH125 significantly inhibited the increased ROS production in SHR. NH125 had no influence on the ROS level in WKY.
Fig. 10.
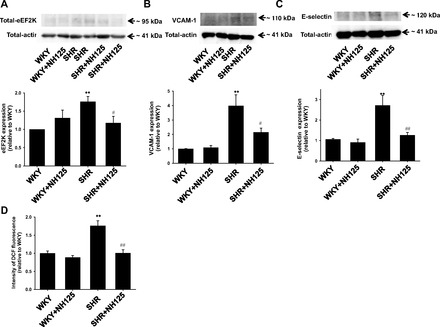
Effects of long-term NH125 treatment on expression of eEF2K (A), VCAM-1 (B), and E-selectin (C), and ROS production (D) in isolated mesenteric artery. After NH125 was administered to SHR (4 wk old) or age-matched WKY subcutaneously at a dose of 500 μg·kg−1·day−1 for 6 wk, superior mesenteric artery was harvested. Expression of eEF2K, VCAM-1, and E-selectin was determined by Western blotting and shown as fold increase relative to WKY (n = 6). Equal protein loading was confirmed using total-actin antibody. The frozen sections (5 μm) of the mesenteric arteries were made. They were loaded with H2DCFDA (5 μmol/l) for 30 min, and the fluorescence images were obtained. Fluorescent intensity (n = 6) was shown as fold increase relative to WKY. **P < 0.01 vs. WKY; #P < 0.05, ##P < 0.01 vs. SHR.
Effect of long-term NH125 treatment on ACh-induced endothelium-dependent relaxation in mesenteric artery from SHR.
We next examined effects of long-term NH125 treatment on ACh-induced endothelium-dependent relaxation. In mesenteric artery from WKY (10 wk old), ACh (0.1–300 nmol/l) induced relaxation in a concentration-dependent manner (Fig. 11A). In mesenteric artery from SHR, the ACh-induced relaxation was significantly impaired. NH125 (500 μg·kg−1·day−1, 6 wk) significantly normalized the impaired ACh-induced relaxation in SHR. NH125 had no influence on the ACh-induced relaxation in WKY. The PD2 values for the ACh-induced relaxation were as follows: WKY, 8.5, n = 6; SHR, 7.4, n = 6; SHR + NH125, 8.5, n = 4; WKY + NH125, 8.2, n = 5.
Fig. 11.
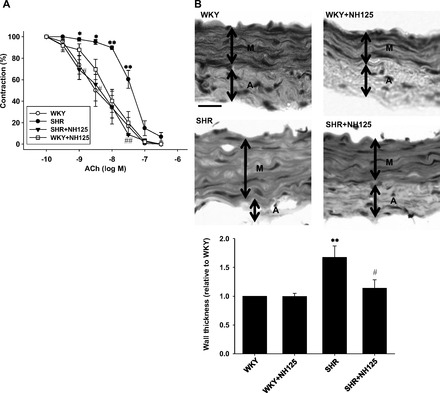
Effects of long-term NH125 treatment on ACh-induced endothelium-dependent relaxation (A) and vascular hypertrophy (B) in mesenteric artery from SHR. A: after NH125 was administered to SHR (4 wk old) or age-matched WKY subcutaneously at a dose of 500 μg·kg−1·day−1 for 6 wk, superior mesenteric artery was harvested. Concentration-contraction relationships to ACh were shown (n = 4–6). ACh (0.1–300 nmol/l) was cumulatively added after the precontraction induced by submaximal doses of noradrenaline had reached a steady state. 100%, noradrenaline-induced precontraction. Results were expressed as means ± SE. B: after NH125 was administered to SHR (4 wk old) or age-matched WKY subcutaneously at a dose of 500 μg·kg−1·day−1 for 6 wk, superior mesenteric artery was harvested. The frozen sections (5 μm) were stained with hematoxylin and eosin. Thickness of arterial media (n = 6) was shown as fold increase relative to WKY. Representative photomicrographs were shown. Arterial media (M) and adventitia (A) lesions were shown. Scale bar, 50 μm. *P < 0.05, **P < 0.01 vs. WKY; #P < 0.05, ##P < 0.01 vs. SHR.
Effects of long-term NH125 treatment on hypertrophy of mesenteric artery from SHR.
To further explore mechanisms of the reduction of BP in SHR by NH125, we examined whether long-term NH125 treatment affects hypertrophy. In mesenteric artery from SHR, wall thickness of arterial media significantly increased compared with WKY (Fig. 11B). NH125 (500 μg·kg−1·day−1, 6 wk) inhibited the increased wall thickness in SHR. NH125 had no influence on the wall thickness in WKY.
DISCUSSION
In the present study, we examined whether eEF2K mediates vascular inflammatory responses and development of hypertension. The major findings of the present study are that inhibition of eEF2K prevents TNF-α-induced expression of VCAM-1 and E-selectin and activation of JNK and NF-κB as well as ROS production in cultured vascular ECs (Figs. 1, 3, 4, and 5). It was also shown that inhibition of eEF2K prevents TNF-α-induced expression of VCAM-1 as well as activation of JNK and NF-κB in SMCs (Fig. 7). We have previously shown that antioxidant, N-acetyl l-cysteine prevented TNF-α-induced ROS, VCAM-1, and phosphorylation of JNK and NF-κB, suggesting that ROS mediates inflammatory responses in vascular SMCs and ECs (39, 40). In addition, we showed that a CaM inhibitor, W-7, inhibited TNF-α-induced activation of eEF2K, JNK, and NF-κB as well as TNF-α-induced expression of VCAM-1 and E-selectin in ECs (Fig. 6), implying that CaM may regulate eEF2K-mediated inflammatory responses in ECs. Collectively, our results indicate that TNF-α activates eEF2K in a CaM-dependent manner and that the activated eEF2K promotes ROS-dependent inflammation through induction of pro-inflammatory molecules (VCAM-1 and E-selectin) via activation of JNK and NF-κB in vascular ECs and SMCs. It was also suggested that eEF2K may be at least in part responsible for the development of hypertension in SHR likely via ROS-dependent inflammation, endothelial dysfunction, and hypertrophy (Figs. 8–11).
eEF2K is a CaM-dependent protein kinase also known as CaMKIII, the main function of which is to inhibit transition of transfer RNA from the A to P site in the ribosome via inhibition of eEF2 activity (33). Recently, it was reported that eEF2K activity was associated with promotion of autophagy, a process favoring cancer cell survival. In glioblastoma cell lines, eEF2K overexpression enhanced autophagy, whereas eEF2K siRNA decreased it (13, 42). The activity of eEF2K was increased in fresh human tumor samples and several cancer cell lines (42). Thus evidence that eEF2K has a potential target for anticancer therapy is emerging (43). eEF2K activity was also greater in proliferating cells (4, 5), especially during the S phase of the cell cycle (34). Similarly, it is important for development of hypertension to promote the proliferation of SMCs and subsequent vascular hypertrophy (6). In this study, we showed that an eEF2K inhibitor reduced the increased BP (Figs. 8 and 9) and vascular hypertrophy (Fig. 11) in SHR. These results indicate that eEF2K may at least partly play a pivotal role not only in the cancer cell survival but also in the development of hypertension.
NH125 is an inhibitor of eEF2K and was shown to inhibit proliferation of different cancer cells (9). In this study, we used NH125 at the concentrations of 300–500 μg·kg−1·day−1 for the treatment in vivo. Although there was no report so far that NH125 was used for rats, NH125 was used for the treatment of mice at the concentration of 1 mg/kg and it exerted anticancer effects (3). The present results for the first time showed that NH125 (500 μg·kg−1·day−1) can reduce the increased BP, expression of inflammatory molecules, ROS production, and hypertrophy as well as endothelial dysfunction in SHR (Figs. 8, 10, and 11). The BP-reducing effect of NH125 was confirmed by a lower concentration (300 μg·kg−1·day−1) (Fig. 9). Although we didn′t investigate the level of circulating cytokines or anti-oxidant capacity in vivo, it was reported that these parameters were associated with the development of hypertension (1, 20). Therefore, it might be possible that NH125 may affect the circulating inflammatory cytokine level as well as antioxidant property. It should be noted that the abdominal fat deposition seemed to decrease in SHR but not WKY treated with NH125 (at 10 wk old). Although the mechanism remains to be determined, exploring the effects of eEF2K on adipocyte metabolism may be an important future target.
ROS production in cardiovascular tissues is associated with the development of hypertension (28). It was reported in vascular ECs that TNF-α-induced ROS production was inhibited by NADPH oxidase (NOX) inhibitors, diphenylene iodonium and apocynin (22). In the same report, it was demonstrated that diphenylene iodonium and apocynin inhibited TNF-α-induced phosphorylation of JNK and NF-κB as well as VCAM-1 expression and monocyte adhesion. These data suggest that TNF-α induced ROS production via activation of NOX, which mediated VCAM-1 expression and monocyte adhesion via activation of JNK and NF-κB signaling. It was further reported in vascular ECs that an activator of nuclear transcription factor, peroxisome proliferator-activated receptor, rosiglitazone, activated AMP-activated protein kinase, which prevented high glucose-induced NOX activation through protein kinase C (PKC) inhibition (8). In the present study, we demonstrated that eEF2K siRNA inhibited TNF-α-induced ROS production in HUVECs (Fig. 4). Therefore, eEF2K may promote ROS production via NOX activation thorough PKC pathway.
In this study, NH125 treatment normalized the impaired ACh-induced endothelium-dependent relaxation in mesenteric artery from SHR (Fig. 11). We have recently shown that neither NA-induced contraction nor sodium nitroprusside-induced endothelium-independent relaxation significantly changed in mesenteric artery from SHR (10 wk old) compared with WKY (26). It was also shown that endothelial nitric oxide synthase expression did not change in mesenteric artery from SHR compared with WKY (26). Because our previous study also demonstrated that expression of NOX-1 and NOX-2 increased in mesenteric artery from SHR compared with WKY (26), it might be possible that eEF2K mediates ROS production through upregulation of NOX1 and/or NOX2, which may lead to decrease in endothelial nitric oxide synthase activity and subsequent impairment of ACh-induced relaxation.
It was reported that VCAM-1 induction causes the adhesion of monocytes to vascular cells. Augmented interaction between monocytes and vascular cells can stimulate the production of various inflammatory mediators including IL-6 and monocyte chemoattractant protein-1, which propagates the inflammatory responses (27). In addition, it was reported that ROS and inflammation caused endothelial dysfunction, which led to the impairment of ACh-induced relaxation in vasculature of SHR (35). These reports imply that expression of VCAM-1 may at least partly control impairment of ACh-induced endothelium-dependent relaxation in SHR via inflammatory responses. There are reports showing that serum level of soluble VCAM-1 increased in atherosclerosis and hypertension (12, 29). It was reported that soluble VCAM-1 promoted proliferation and migration of vascular SMCs in SHR (18). Because proliferation and migration of SMCs mediate the development of hypertension through medial thickening (11, 23), it is suggested that expression of VCAM-1 may at least partly control vascular hypertrophy in SHR.
Essential hypertension is a major risk factor for cardiovascular diseases. However, the initiating causative mechanisms are still unknown. In this study, we for the first time demonstrate that eEF2K may be at least in part responsible for the development of hypertension in SHR, likely via ROS-dependent inflammation, hypertrophy, and endothelial dysfunction (Fig. 12). The present study also provides novel mechanistic insights into the role of eEF2K, which controls inflammatory signals in vasculature (Fig. 12). Our results suggest eEF2K as a novel molecular target for the prevention and treatment of essential hypertension.
Fig. 12.
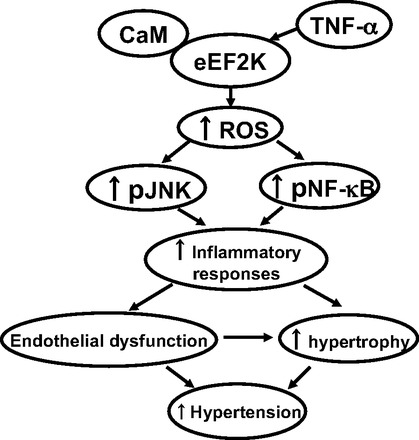
Summary of the present results. eEF2K mediates ROS-dependent inflammatory responses via activation of JNK and NF-κB in vascular ECs and SMCs in a CaM-dependent manner, which may be at least in part responsible for the development of hypertension via propagating endothelial dysfunction and hypertrophy in SHR.
GRANTS
This study was supported in part by a Grant for Scientific Research from the Japan Society for the Promotion of Science.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: T.U., M.O., and H.Y. provided conception and design of research; T.U. performed experiments; T.U. and H.Y. analyzed data; T.U., M.O., Y.H., and H.Y. interpreted results of experiments; T.U. prepared figures; T.U. drafted manuscript; T.U., M.O., Y.H., and H.Y. approved final version of manuscript; Y.H. and H.Y. edited and revised manuscript.
ACKNOWLEDGMENTS
We thank Dr. Taisuke Kogane, Dr. Tomoka Morita, Ryo Niijima, and Tomoki Sakatsume for technical assistance.
REFERENCES
- 1.Amoureux S, Lorgis L, Sicard P, Girard C, Rochette L, Vergely C. Vascular BDNF expression and oxidative stress during aging and the development of chronic hypertension. Fundam Clin Pharmacol 26: 227–234, 2012 [DOI] [PubMed] [Google Scholar]
- 2.Arora S, Yang JM, Kinzy TG, Utsumi R, Okamoto T, Kitayama T, Ortiz PA, Hait WN. Identification and characterization of an inhibitor of eukaryotic elongation factor 2 kinase against human cancer cell lines. Cancer Res 63: 6894–6899, 2003 [PubMed] [Google Scholar]
- 3.Arora S, Yang JM, Utsumi R, Okamoto T, Kitayama T, Hait WN. P-glycoprotein mediates resistance to histidine kinase inhibitors. Mol Pharmacol 66: 460–467, 2004 [DOI] [PubMed] [Google Scholar]
- 4.Bagaglio DM, Cheng EH, Gorelick FS, Mitsui K, Nairn AC, Hait WN. Phosphorylation of elongation factor 2 in normal and malignant rat glial cells. Cancer Res 53: 2260–2264, 1993 [PubMed] [Google Scholar]
- 5.Bagaglio DM, Hait WN. Role of calmodulin-dependent phosphorylation of elongation factor 2 in the proliferation of rat glial cells. Cell Growth Differ 5: 1403–1408, 1994 [PubMed] [Google Scholar]
- 6.Bucher B, Travo P, Stoclet JC. Smooth muscle cell hypertrophy and hyperplasia in the thoracic aorta of spontaneously hypertensive rats. Cell Biol Int Rep 8: 567–577, 1984 [DOI] [PubMed] [Google Scholar]
- 7.Calberg U, Nilsson A, Skog S, Palmquist K, Nygard O. Increased activity of the eEF-2 specific, Ca2+ and calmodulin dependent protein kinase III during the S-phase in Ehrlich ascites cells. Biochem Biophys Res Commun 180: 1372–1376, 1991 [DOI] [PubMed] [Google Scholar]
- 8.Ceolotto G, Gallo A, Papparella I, Franco L, Murphy E, Iori E, Pagnin E, Fadini GP, Albiero M, Semplicini A, Avogaro A. Rosiglitazone reduces glucose-induced oxidative stress mediated by NAD(P)H oxidase via AMPK-dependent mechanism. Arterioscler Thromb Vasc Biol 27: 2627–2633, 2007 [DOI] [PubMed] [Google Scholar]
- 9.Chen Z, Gopalakrishnan SM, Bui MH, Soni NB, Warrior U, Johnson EF, Donnelly JB, Glaser KB. 1-Benzyl-3-cetyl-2-methylimidazolium iodide (NH125) induces phosphorylation of eukaryotic elongation factor-2 (eEF2): a cautionary note on the anticancer mechanism of an eEF2 kinase inhibitor. J Biol Chem 286: 43951–43958, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cheng EH, Gorelick FS, Czernik AJ, Bagaglio DM, Hait WN. Calmodulin-dependent protein kinases in rat glioblastoma. Cell Growth Differ 6: 615–621, 1995 [PubMed] [Google Scholar]
- 11.Daniel EE, Kwan CY, Lee RM, Smeda J. Early structural changes in precapillary vessels in hypertension and their relationship to functional changes. J Cardiovasc Pharmacol 6, Suppl 4: S671–S682, 1984 [DOI] [PubMed] [Google Scholar]
- 12.Glowinska B, Urban M, Peczynska J, Florys B. Soluble adhesion molecules (sICAM-1, sVCAM-1) and selectins (sE selectin, sP selectin, sL selectin) levels in children and adolescents with obesity, hypertension, and diabetes. Metabolism 54: 1020–1026, 2005 [DOI] [PubMed] [Google Scholar]
- 13.Hait WN, Wu H, Jin S, Yang JM. Elongation factor-2 kinase: its role in protein synthesis and autophagy. Autophagy 2: 294–296, 2006 [DOI] [PubMed] [Google Scholar]
- 14.House SJ, Ginnan RG, Armstrong SE, Singer HA. Calcium/calmodulin-dependent protein kinase II-δ isoform regulation of vascular smooth muscle cell proliferation. Am J Physiol Cell Physiol 292: C2276–C2287, 2007 [DOI] [PubMed] [Google Scholar]
- 15.Jozwiak J, Jozwiak S, Grzela T, Lazarczyk M. Positive and negative regulation of TSC2 activity and its effects on downstream effectors of the mTOR pathway. Neuromolecular Med 7: 287–296, 2005 [DOI] [PubMed] [Google Scholar]
- 16.Knebel A, Haydon CE, Morrice N, Cohen P. Stress-induced regulation of eukaryotic elongation factor 2 kinase by SB 203580-sensitive and -insensitive pathways. Biochem J 367: 525–532, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Koeners MP, Braam B, Joles JA. Perinatal inhibition of NF-kappaB has long-term antihypertensive effects in spontaneously hypertensive rats. J Hypertens 29: 1160–1166, 2011 [DOI] [PubMed] [Google Scholar]
- 18.Lee HM, Kim HJ, Won KJ, Choi WS, Park SH, Song H, Park PJ, Park TK, Lee CK, Kim B. Soluble form of vascular cell adhesion molecule 1 induces migration and proliferation of vascular smooth muscle cells. J Vasc Res 45: 259–268, 2008 [DOI] [PubMed] [Google Scholar]
- 19.Li H, Li W, Gupta AK, Mohler PJ, Anderson ME, Grumbach IM. Calmodulin kinase II is required for angiotensin II-mediated vascular smooth muscle hypertrophy. Am J Physiol Heart Circ Physiol 298: H688–H698, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li L, Yi-Ming W, Li ZZ, Zhao L, Yu YS, Li DJ, Xia CY, Liu JG, Su DF. Local RAS and inflammatory factors are involved in cardiovascular hypertrophy in spontaneously hypertensive rats. Pharmacol Res 58: 196–201, 2008 [DOI] [PubMed] [Google Scholar]
- 21.Li W, Li H, Sanders PN, Mohler PJ, Backs J, Olson EN, Anderson ME, Grumbach IM. The multifunctional Ca2+/calmodulin-dependent kinase II delta (CaMKIIdelta) controls neointima formation after carotid ligation and vascular smooth muscle cell proliferation through cell cycle regulation by p21. J Biol Chem 286: 7990–7999, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liang CJ, Wang SH, Chen YH, Chang SS, Hwang TL, Leu YL, Tseng YC, Li CY, Chen YL. Viscolin reduces VCAM-1 expression in TNF-alpha-treated endothelial cells via the JNK/NF-kappaB and ROS pathway. Free Radic Biol Med 51: 1337–1346, 2011 [DOI] [PubMed] [Google Scholar]
- 23.Luscher TF. [Hypertension and vascular diseases: molecular and cellular mechanisms]. Schweiz Med Wochenschr 125: 270–282, 1995 [PubMed] [Google Scholar]
- 24.Mercure MZ, Ginnan R, Singer HA. CaM kinase II δ2-dependent regulation of vascular smooth muscle cell polarization and migration. Am J Physiol Cell Physiol 294: C1465–C1475, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Morita T, Okada M, Hara Y, Yamawaki H. Mechanisms underlying impairment of endothlium-dependent relaxation by fetal bovine serum in organ-cultured rat mesenteric artery. Eur J Pharmacol 668: 401–406, 2011 [DOI] [PubMed] [Google Scholar]
- 26.Mukohda M, Okada M, Hara Y, Yamawaki H. Methylglyoxal accumulation in arterial walls causes vascular contractile dysfunction in spontaneously hypertensive rats. J Pharmacol Sci 120: 26–35, 2012 [DOI] [PubMed] [Google Scholar]
- 27.Orr AW, Hastings NE, Blackman BR, Wamhoff BR. Complex regulation and function of the inflammatory smooth muscle cell phenotype in atherosclerosis. J Vasc Res 47: 168–180, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Paravicini TM, Touyz RM. Redox signaling in hypertension. Cardiovasc Res 71: 247–258, 2006 [DOI] [PubMed] [Google Scholar]
- 29.Parissis JT, Venetsanou KF, Mentzikof DG, Kalantzi MV, Georgopoulou MV, Chrisopoulos N, Karas SM. Plasma levels of soluble cellular adhesion molecules in patients with arterial hypertension. Correlations with plasma endothelin-1. Eur J Intern Med 12: 350–356, 2001 [DOI] [PubMed] [Google Scholar]
- 30.Parmer TG, Ward MD, Yurkow EJ, Vyas VH, Kearney TJ, Hait WN. Activity and regulation by growth factors of calmodulin-dependent protein kinase III (elongation factor 2-kinase) in human breast cancer. Br J Cancer 79: 59–64, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pavur KS, Petrov AN, Ryazanov AG. Mapping the functional domains of elongation factor-2 kinase. Biochemistry 39: 12216–12224, 2000 [DOI] [PubMed] [Google Scholar]
- 32.Pyr Dit Ruys S, Wang X, Smith EM, Herinckx G, Hussain N, Rider MH, Vertommen D, Proud CG. Identification of autophosphorylation sites in eukaryotic elongation factor-2 kinase. Biochem J 442: 681–692, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ryazanov AG, Shestakova EA, Natapov PG. Phosphorylation of elongation factor 2 by EF-2 kinase affects rate of translation. Nature 334: 170–173, 1988 [DOI] [PubMed] [Google Scholar]
- 34.Smith EM, Proud CG. cdc2-Cyclin B regulates eEF2 kinase activity in a cell cycle- and amino acid-dependent manner. EMBO J 27: 1005–1016, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Toth P, Csiszar A, Sosnowska D, Tucsek Z, Cseplo P, Springo Z, Tarantini S, Sonntag WE, Ungvari Z, Koller A. Treatment with the cytochrome P450 omega-hydroxylase inhibitor HET0016 attenuates cerebrovascular inflammation, oxidative stress and improves vasomotor function in spontaneously hypertensive rats. Br J Pharmacol 168: 1878–1888, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Usui T, Okada M, Hara Y, Yamawaki H. Death-associated protein kinase 3 mediates vascular inflammation and development of hypertension in spontaneously hypertensive rats. Hypertension 60: 1031–1039, 2012 [DOI] [PubMed] [Google Scholar]
- 37.Usui T, Okada M, Hara Y, Yamawaki H. Exploring calmodulin-related proteins, which mediate development of hypertension, in vascular tissues of spontaneous hypertensive rats. Biochem Biophys Res Commun 405: 47–51, 2011 [DOI] [PubMed] [Google Scholar]
- 38.Usui T, Okada M, Mizuno W, Oda M, Ide N, Morita T, Hara Y, Yamawaki H. HDAC4 mediates development of hypertension via vascular inflammation in spontaneous hypertensive rats. Am J Physiol Heart Circ Physiol 302: H1894–H1904, 2012 [DOI] [PubMed] [Google Scholar]
- 39.Usui T, Yamawaki H, Kamibayashi M, Okada M, Hara Y. CV-159, a unique dihydropyridine derivative, prevents TNF-induced inflammatory responses in human umbilical vein endothelial cells. J Pharmacol Sci 113: 182–191, 2010 [DOI] [PubMed] [Google Scholar]
- 40.Usui T, Yamawaki H, Kamibayashi M, Okada M, Hara Y. Mechanisms underlying the anti-inflammatory effects of the Ca2+/calmodulin antagonist CV-159 in cultured vascular smooth muscle cells. J Pharmacol Sci 113: 214–223, 2010 [DOI] [PubMed] [Google Scholar]
- 41.Wang X, Li W, Williams M, Terada N, Alessi DR, Proud CG. Regulation of elongation factor 2 kinase by p90(RSK1) and p70 S6 kinase. EMBO J 20: 4370–4379, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wu H, Yang JM, Jin S, Zhang H, Hait WN. Elongation factor-2 kinase regulates autophagy in human glioblastoma cells. Cancer Res 66: 3015–3023, 2006 [DOI] [PubMed] [Google Scholar]
- 43.Wu H, Zhu H, Liu DX, Niu TK, Ren X, Patel R, Hait WN, Yang JM. Silencing of elongation factor-2 kinase potentiates the effect of 2-deoxy-D-glucose against human glioma cells through blunting of autophagy. Cancer Res 69: 2453–2460, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]