

Abstract
Androgen antagonists or androgen deprivation is a primary therapeutic modality for the treatment of prostate cancer. Invariably, however, the disease becomes progressive and unresponsive to androgen ablation therapy (hormone refractory). The molecular mechanisms by which the androgen antagonists inhibit prostate cancer proliferation are not fully defined. In this report, we demonstrate that SIRT1, a nicotinamide adenosine dinucleotide-dependent histone deacetylase linked to the regulation of longevity, is required for androgen antagonist-mediated transcriptional repression and growth suppression. Androgen antagonist-bound androgen receptor (AR) recruits SIRT1 and NCoR to AR-responsive promoters and deacetylates histone H3 locally at the PSA promoter. Furthermore, SIRT1 down-regulation by siRNA or by pharmacological means increased the sensitivity of androgen-responsive genes to androgen stimulation, enhanced the sensitivity of prostate cancer cell proliferative responses to androgens, and decreased the sensitivity of prostate cancer cells to androgen antagonists. In this study, we demonstrate the ligand-dependent recruitment of a class III HDAC into a co-repressor transcriptional complex, and a necessary functional role for a class III HDAC as a transcriptional co-repressor in AR antagonist-induced transcriptional repression. Collectively, these findings identify SIRT1 as a co-repressor of AR and elucidate a new molecular pathway relevant to prostate cancer growth and approaches to therapy.
Keywords: Androgen Antagonists, Androgen Receptor, SIRT1, Histone Deacetylase, Prostate cancer
INTRODUCTION
Prostate cancer is the second leading cause of cancer death in men [1–3]. The androgen receptor (AR), a hormone-dependent transcription factor, plays a major role in promoting the development and progression of prostate cancer [4–7]. Androgen ablation and blockade of androgen actions through the androgen receptor remain the mainstay of treatment for advanced prostate cancer [8, 9]. While initial responses to androgen deprivation are the norm, most tumors eventually recur in what is termed an androgen-independent (refractory) state [10–12].
The transcriptional activity of the AR is modulated by nuclear co-regulatory proteins, known as co-activators and co-repressors [13–15]. Upon activation by ligands, such as dihydrotestosterone (DHT), the AR translocates to the nucleus, whereupon it binds to androgen-response elements (ARE) on target genes and regulates their transcription [16–18]. The balance of co-repressors and co-activators in the AR complex determines AR transcriptional activity [19, 20]. Binding of androgens induces recruitment of co-activators such as SRC-1, TIF2/GRIP1–1 and ACTR/AIB1/RAC3/pCIP, as well as p300, CBP and pCAF, which contain intrinsic histone acetylase (HAT) activity [21–30]. p160 appears to mediate the binding of the AR to the HAT complex [16, 17]. In contrast, binding of AR antagonists induces AR to form a complex with co-repressors, such as NCoR, SMRT and HDAC-1, -2 and -3 [24, 25, 31–34]. While many co-activators of the AR have been identified and well-studied, the currently known AR co-repressors are fewer and less well-characterized, and in some cases their necessity in transcriptional repression has not yet been established [13, 15]. Identification of new co-repressors and developing an understanding of the precise mechanisms underlying the regulation of AR function is of critical importance for the design and development of novel therapies and pharmaceutical targets for treating prostate cancer.
SIRT1 is a mammalian NAD-dependent deacetylase belonging to the class III histone deacetylase (HDAC) family [35]. Recent studies have demonstrated that SIRT1 plays a role in a wide variety of processes including stress responses [36], metabolism [37], apoptosis [38], embryogenesis [39], calorie restriction and aging [40, 41]. SIRT1 binds to, and regulates the activity of, several transcription factors, including p53 [42–44], FOXO1, FOXO3a, and FOXO4 [45–47], PPARr [48], HES-1 and HEY-2 [49], MyoD [50], CTIP2 [51], NF-κB [52], and PGC1a [53].
In the present study, we establish SIRT1 as a specific co-repressor of the androgen receptor. We find that androgen antagonists induce recruitment of SIRT1 to AR-responsive promoters and that AR-dependent transcriptional suppression by androgen antagonists requires SIRT1. We demonstrate that SIRT1 suppresses AR-dependent gene transcription through its deacetylase activity, and alters local histone H3 acetylation. Furthermore, we find that SIRT1 is required for androgen antagonist-mediated growth suppression and demonstrate that down-regulation or suppression of SIRT1 activity increases the sensitivity of prostate cancer cells to the transcriptional and proliferative activities of androgens.
RESULTS
SIRT1 suppresses AR-dependent gene transcription
To determine if the SIRT1 could regulate androgen-dependent and -independent transcription, we assessed the effect of altering SIRT1 activity on the regulation of AR-mediated transcription of two androgen-responsive promoter-reporter vectors, a human prostate-specific antigen (PSA) promoter-driven reporter (PSA-LUC) and a mouse mammary tumor virus LTR-driven-reporter (MMTV-LUC), in a human prostate cancer (LNCaP) cell line. Firstly, a Cell-based HDAC activity assays were performed to confirm that the SIRT1 agonist and SIRT1 inhibitor were affecting HDAC enzymatic activity in vivo, at the concentrations utilized. These studies demonstrated that exposure to resveratrol increased NAD-dependent HDAC activity greater than 2.5-fold (p<0.05), whereas nicotinamide exposure diminished enzymatic activity by 3.3-fold (p<0.05) (Fig. 1A). Secondly, we analyzed the effect of these SIRT1 modulator on AR dependent and independent transcription. Dihydrotestosterone (DHT)-induced luciferase activity of PSA-LUC was suppressed 16-fold (p<0.01) by treatment of the transfected cells with resveratrol, an agonist of SIRT1 (Fig. 1B). Conversely, PSA-LUC activity was induced 3.5-fold (p<0.01) by exposure to the SIRT1 inhibitor nicotinamide (NAM) and 3-fold by Sirtinol, a structurally-unrelated SIRT1 inhibitor (data not shown). Similar effects were seen using pMMTV-LUC as the reporter (data not shown). In the absence of DHT, NAM or resveratrol treatment did not result in significant change in the PSA-LUC activity. Immunoblot assays demonstrated that neither resveratrol nor nicotinamide treatment changed AR protein levels (Fig. 1B). Collectively, these results demonstrated that pharmacological agents capable of regulating SIRT1 deacetylase activity modulated AR-mediated PSA- and pMMTV-promoter driven gene activity.
Figure 1. SIRT1 represses AR-dependent gene transcription.

A) Cell-based NAD-dependent HDAC activity assay of cells treated with 50 μM resveratrol or 10 mM NAM for 24 hr. B) Upper panel: The effects of resveratrol and nicotinamide on PSA-LUC transcription. LNCaP cells cultured in media containing charcoal-stripped serum were transfected with the PSA-LUC reporter vector plasmid. Transfected cells were then exposed to resveratrol (RES) at 50 μM or nicotinamide (NAM) at 10 mM for 24 hr, and treated with 10 nM DHT or vehicle-treated (Veh) for 24 hr before assay of luciferase activity, expressed here in arbitrary units. Insert shows the results from vehicle treatment, with an expanded y-axis. Lower panel: Immunoblot analysis of AR protein levels in cells treated with nicotinamide or resveratrol. C) Upper panel: Effects of SIRT1 over-expression or SIRT1 knockdown on PSA-LUC transcription. LNCaP cells were co-transfected with PSA-LUC and an empty control vector (pcDNA3.1), or a wt-SIRT1 expression vector (pcDNA3.1-SIRT1), or a vector expressing SIRT1 siRNA (pSUPER-SIRT1), or the empty siRNA expression vector (pSUPER), then treated with 10 nM DHT or vehicle-treated for 24 hr, and cells were harvested for luciferase assay. Insert shows the results from vehicle treatment, with an expanded y-axis. Lower panel: Immunoblot analysis of SIRT1 protein levels in cells transfected with wt-SIRT (pCDNA3.1-SIRT1), or DN-SIRT1 (pCDNA3.1-H363Y), or empty vector (pCDNA3.1), or empty siRNA vector (pSUPER), or SIRT1 siRNA expression vector (pSUPER-SIRT1). Cell extracts were normalized for protein content, separated by PAGE, transferred, probed with an anti-SIRT1 antibody or anti-AR antibody or an anti-β-actin antibody, and developed with a chemoluminescence kit. D) The effect of resveratrol or nicotinamide on endogenous AR-dependent and –independent PSA gene transcription. LNCaP cells cultured in media containing charcoal-stripped serum were exposed to 10 mM nicotinamide (NAM), 50 uM resveratrol (RES) or vehicle (control) for 2 hr, and treated with 10 nM DHT or vehicle-treated for 48 hr. Transcript levels of PSA was measured by quantitative RT-PCR analysis of RNA extracted from the cells. Transcript levels are expressed relative to β-actin transcripts. E) The effect of resveratrol or nicotinamide on endogenous AR-dependent and –independent KLK2 gene transcription. LNCaP cells were cultured in media containing charcoal-stripped serum were exposed to 10 mM nicotinamide (NAM), 50 uM resveratrol (RES) or vehicle (control) for 2 hr, and treated with 10 nM DHT or vehicle-treated for 48 hr. Transcript levels of KLK2 were measured by quantitative RT-PCR analysis of RNA extracted from the cells. Transcript levels are expressed relative to β-actin transcripts. F) Upper panel: SIRT1 knockdown increases endogenous AR-dependent PSA genes transcription. Transcript levels of PSA were measured by quantitative RT-PCR analysis of RNA extracted from empty vector-transfected LNCaP cells (Ctrl) and LNCaP cell lines in which SIRT1 expression levels had been knocked down by stable expression of siRNA (RNAi). The cells were cultured under androgen-deprivation conditions for 3 days, followed by treatment with DHT or vehicle for 48 hr. Lower panel: Immunoblot analysis of SIRT1 and AR protein levels in an empty vector-transfected LNCaP cell line (Ctrl) and LNCaP cell lines in which SIRT1 expression levels had been knocked down by stable expression of siRNA (RNAi). G) SIRT1 knockdown increases endogenous AR-dependent KLK2 gene transcription. Transcript levels of KLK2 were measured by quantitative RT-PCR analysis of RNA extracted from empty vector-transfected LNCaP cells (Ctrl) and a LNCaP cell line in which SIRT1 expression levels had been knocked down by stable expression of siRNA (RNAi). The cells were cultured under androgen-deprivation conditions for 3 days, followed by treatment with DHT or vehicle for 48 hr. H) SIRT1 deacetylase activity is required for SIRT1 effects on AR-dependent gene transcription. LNCaP cells cultured in media containing charcoal-stripped serum were co-transfected with the PSA-LUC vector plus an empty vector (Vector), or a SIRT1 expression vector (SIRT1), or a dominant-negative SIRT1 vector (H363Y), or SIRT1 expression vector plus DN-SIRT1 vectors (SIRT1+H363Y), then treated with 10 nM DHT or vehicle, and harvested for assay of luciferase activity. Insert shows the results from vehicle treatment, with an expanded y-axis. In all relevant figures, relative luciferase activities were normalized to β-gal activity to control for transfection efficiency. The error bars represent the SEM. Asterisks indicate significant differences between two groups (** p<0.01, *p<0.05).
We next tested the role of SIRT1 in AR-dependent and -independent gene transcription, using ectopic over-expression of SIRT1, or SIRT1 knockdown by RNAi, in LNCaP cells. Co-transfection of PSA-LUC with a SIRT1 expression vector reduced DHT-stimulated PSA-LUC transcription by 3-fold (p<0.01) (Fig. 1C). Conversely, SIRT1 knockdown by expression of SIRT1 RNAi resulted in a 2.5-fold (p<0.01) increase in DHT-stimulated PSA-LUC transcription. In parallel studies, ectopic expression of SIRT1 reduced pMMTV-LUC transcription by 7.5-fold, while SIRT1 knockdown by RNAi induced a 3-fold (p<0.01) increase in pMMTV-LUC transcription (data not shown). The levels of SIRT1 protein in cells transfected with the SIRT1 expression vector, or with SIRT1 RNAi, were measured and confirmed increased or decreased levels of SIRT1 protein, respectively. SIRT1 over-expression or down-regulation did not affect AR protein expression levels (Fig. 1C). To assess the specificity of SIRT1 in affecting promoter activity, the effect of altering SIRT1 activity on expression of an SV40 promoter-driven luciferase reporter was studied. The activity of this vector after transient transfection was not affected by over-expression of SIRT1 (data not shown).
The effect of SIRT1 activity on endogenous AR-responsive genes was next examined. The PSA and KLK2 genes were chosen for study because they are well-recognized targets of AR-dependent transcriptional regulation in vivo [54]. Inhibition of SIRT1 activity by NAM treatment induced PSA transcript levels by 3-fold (Fig. 1D) and KLK2 transcripts by 4-fold (Fig. 1E) in the presence of DHT (p<0.05). Conversely, activation of SIRT1 by exposure to resveratrol suppressed both PSA and KLK2 transcripts below basal levels by 31-fold and 5-fold (p<0.05), respectively (Figs. 1D & E). The ability of SIRT1 to suppress transcription of endogenous AR-responsive genes was confirmed independently by analysis of cell lines in which SIRT1 had been stably knocked-down by transfection with a vector expressing a SIRT1 hairpin (see Fig. 1F for SIRT1 and AR protein levels in these cells). In comparison to control-transfected cells, knockdown of SIRT1 resulted in induction of PSA transcripts by 3.6-fold (Fig. 1F) and KLK2 transcripts by 4.7-fold (p<0.05) (Fig. 1G), in the presence of DHT. In the absence of DHT, a minimal but non-significant induction of the very low basal levels of transcription was detected after treatment with NAM and in the SIRT1 knockdown cell lines, although no further suppression was detected after exposure to resveratrol (Figs. 1D, E, F & G). Collectively, these experiments demonstrate that SIRT1 specifically suppresses AR-dependent gene transcription. SIRT1 may also exert a modest repressive effect on AR target genes in the absence of DHT, but the levels of basal expression are so low in the absence of DHT that the significance of any such effect is difficult to discern.
The deacetylase activity of SIRT1 is required for suppression of AR-dependent transcription
SIRT1 is a NAD-dependent histone deacetylase, the enzymatic activity of which is induced by resveratrol [55] and reduced by nicotinamide [56]. Our finding that AR-dependent transcription is regulated by these reciprocal modulators of SIRT1 deacetylase activity is consistent with the enzymatic activity of SIRT1 being responsible for this effect. To independently demonstrate that SIRT1 regulates AR-dependent transcription through its deacetylase activity, we employed a dominant-negative mutant that abolishes the deacetylase activity of SIRT1 (SIRT1H363Y - mutation of His363 to Tyr). Co-expression of SIRT1 H363Y protein in LNCaP cells failed to suppress PSA-LUC transcription (Fig. 1H), or pMMTV-LUC transcription (data not shown), whereas co-transfection of wt-SIRT1 in parallel inhibited transcription, as previously demonstrated. When both wt-SIRT1 and SIRT1H363Y expression vectors were co-transfected, the repressive effect of wt-SIRT1 expression was blocked by the dominant-negative mutant. These results establish that the suppression of AR-mediated transcription by SIRT1 requires its deacetylase activity.
SIRT1 associates with the PSA promoter and acts as a co-repressor of AR
To investigate the mechanism underlying SIRT1 regulation of AR-dependent gene transcription, we first determined if SIRT1 associates with known AR-binding sites in the promoter regions of the PSA gene. Three sets of PCR primer pairs were generated to amplify genomic fragments (~150 bp in size) encompassing the promoter, the enhancer, and a control region distal to the PSA gene (7 kb upstream of the start site). LNCaP cells were cultured under androgen deprivation for 3 days, followed by treatment with DHT or bicalutamide (CDX), an AR antagonist. Chromatin immunoprecipitation (ChIP) assays were performed using antibodies against SIRT1 and AR. SIRT1 and AR proteins were both detected at the promoter and enhancer regions of the PSA gene (Figs. 2A, B, C & D). Exposure to the androgen-antagonist bicalutamide increased the recruitment of SIRT1 both to promoter and enhancer regions (Figs. 2A & B). In contrast, treatment with DHT did not induce SIRT1 occupancy (Figs. 2A & B). AR recruitment to both regions was induced by exposure to either DHT or bicalutamide (Figs. 2C & D). Binding of SIRT1 or AR to a control DNA region 7 kb upstream of the PSA gene was not observed (data not shown). These results indicate that SIRT1 and AR bind to the same general promoter/enhancer regions of the PSA gene and that SIRT1 recruitment to the promoter and enhancer is stimulated by androgen-antagonists.
Figure 2. ChIP analysis of the endogenous PSA gene promoter region.

LNCaP cells were cultured in charcoal-stripped serum (S) for three days and certain cultures were treated for 4 hr with DHT (10 nM) (D) or bicalutamide (15 μM) (CDX), or vehicle control (S). ChIP assays were performed using primers sets which amplified the PSA promoter region, the enhancer region, or a distal region upstream of known PSA regulatory elements. Immunoprecipitations were carried out using antibodies directed against SIRT1, androgen receptor (AR), NCoR, polymerase II (Pol II) and an irrelevant protein (Rag1). The bound and input DNA were analyzed by ABI 7500 Real-Time PCR system (Applied Biosystems, Foster City, California) by the ΔΔ Ct method. The results are presented as the relative level of the protein associated with the PSA promoter or enhancer, normalized to irrelevant control antibody and input DNA. A) SIRT1 on the PSA promoter. B) SIRT1 on the PSA enhancer. C) AR on the PSA promoter. D) AR on the PSA enhancer. E) Pol II on the PSA promoter. F) Pol II on the PSA enhancer. G) NCoR on the PSA promoter. H) NCoR on the PSA enhancer. I) Immunoblot analysis of AR protein levels in DU145 cells transfected with wt-AR or empty vector. J) Bicalutamide-induced recruitment of SIRT1 to the endogenous PSA promoter requires the AR. DU145 cells were cultured in DMEM+10% charcoal-treated FBS. The cells were transfected with AR or mock-transfected, and treated with bicalutamide (CDX) or DHT (D). ChIP assays were performed using antibodies directed against SIRT1.
Parallel ChIP assays examining Pol II recruitment to the promoter and enhancer as a marker for active transcription demonstrated increases in Pol II occupancy after exposure to DHT, but not when bicalutamide was present (Figs. 2E & F). Pol II recruitment was inversely related to occupancy by SIRT1, and bicalutamide uncoupled AR occupancy at the promoter from Pol II recruitment.
Nuclear receptor co-repressor (NCoR) is an established repressor of AR-driven transcription, and is known to physically associate with the AR [32, 57, 58]. SIRT1 has been shown to interact with NCoR and suppress the transcriptional activity of certain genes, such as the PPAR-receptor [48]. To determine whether NCoR occupancy parallels SIRT1 occupancy at the PSA enhancer and promoter, ChIP assays were conducted with an anti-NCoR antibody. We observed increased occupancy by NCoR at both the endogenous PSA promoter and enhancer sites in the presence of bicalutamide (Figs. 2G & H).
Finally, we determined whether the recruitment of SIRT1 to the PSA promoter by androgen antagonists required the androgen receptor. ChIP assays were performed using anti-SIRT1 antibody in DU145 cells, an androgen receptor-negative prostate cancer cell line, with or without transfection of an androgen receptor expression vector, in the presence of DHT or CDX (see Fig. 2I for AR protein expression levels in DU145 cells after transfection). Exposure to DHT did not induce SIRT1 occupancy at the PSA promoter, whether or not the AR was expressed (Fig. 2J) Bicalutamide treatment did not induce SIRT1 occupancy at the PSA promoter in control (empty-vector transfected) AR-negative DU145 cells (Fig. 2J). When AR was introduced into the cells by transfection, however, exposure to bicalutamide stimulated recruitment of SIRT1 to the PSA promoter approximately 4-fold more efficiently than in the absence of AR. These results suggest that antagonist-bound AR mediates SIRT1 occupancy at the PSA promoter.
These results are consistent with a model in which SIRT1 and NCoR complex with the AR at the PSA promoter/enhancer under situations of transcriptional silencing by androgen antagonists, thus facilitating co-repression, in which NCoR acts as a co-repressor adaptor for the AR and may bridge SIRT1 with the AR at the PSA promoter/enhancer.
Histone H3 in the PSA promoter region is a potential target of SIRT1
Several lines of evidence support the importance of local histone acetylation in transcriptional activation by nuclear receptors [16, 17, 59]. The acetylation level of histone H3 at the PSA promoter has been reported to increase after exposure to DHT [58]. We examined the acetylation state of histone H3 at the PSA promoter and enhancer using an anti-acetylated-histone H3 antibody in a ChIP assay. Exposure to DHT produced a marked increase in local histone H3 acetylation. Enhancement of SIRT1 activity by resveratrol treatment reversed the H3 acetylation induced by DHT (Figs. 3A & B). To determine whether SIRT1 itself was required for these ligand-dependent changes in local histone acetylation, we examined local H3 acetylation in cell lines in which SIRT1 levels had been knocked down by SIRT1 hairpin RNA expression (Fig. 3C). SIRT1 knockdown elevated the levels of acetylated histone H3 at the enhancer and promoter compared to levels in control vector-transfected LNCaP cells, resulting in approximately 4-fold more acetylated H3 in the SIRT1 knockdown cells at the PSA enhancer, and 2-fold more at the PSA promoter (p<0.05), in response to DHT (Fig. 3C). These results suggest that histone H3 in the PSA promoter/enhancer regions may be one direct target of SIRT1 activity (or alternatively, and less likely, that SIRT1 is influencing the activity of other co-regulatory histone acetylases or deacetylases at AR-responsive promoters).
Figure 3. Androgen and antagonists alter histone H3 acetylation at the PSA promoter.
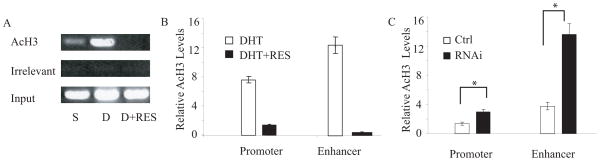
A) Histone H3 acetylation at the PSA promoter. Cross-linked chromatin was extracted from cells cultured under androgen-deprivation conditions and then treated with 10 nM DHT (D), or DHT plus 25 μM resveratrol (D+RES), or vehicle (S) for 4 hr. Anti-Histone H3 antibody (AcH3) or an irrelevant antibody (anti-RAG) were used for immunoprecipitation. The ethidium-stained PCR products of the ChIP assay are shown. B) Quantitative PCR results from the same ChIP assays, analyzing both the PSA promoter and enhancer. C) Quantitative PCR analysis of ChIP assays for acetylated histone H3 at the PSA promoter or enhancer in the presence of DHT in control-transfected LNCaP cells (Ctrl) and in LNCaP cell lines in which SIRT1 expression levels had been knocked down by stable expression of siRNA (RNAi). Asterisks (*) indicates significant differences between two groups (p<0.05).
SIRT1 is required for bicalutamide-mediated transcriptional suppression of the PSA promoter and growth suppression by bicalutamide
The finding that bicalutamide exposure induces recruitment of SIRT1 to the PSA promoter, coincident with changes in local promoter histone acetylation and transcriptional repression, raised the possibility of a functional role for SIRT1 in the transcriptional repression induced by androgen antagonists. To test for a necessary role for SIRT1 in bicalutamide-mediated transcription repression, the activity of the androgen-responsive PSA-LUC vector transfected into SIRT1-knockdown LNCaP cells or control (empty-vector)-transfected LNCaP cells was assessed. Treatment with bicalutamide suppressed DHT-induced PSA-LUC transcriptional activity in cells expressing SIRT1 by 80%, but only suppressed transcription by 28% in SIRT1 knockdown cells (p<0.05) (Fig. 4A) (see Fig. 4B for SIRT1 and AR protein levels in these cells). To verify that the observed defect of bicalutamide-mediated transcription suppression in the SIRT1-knockdown cells was indeed due to the suppression of SIRT1, we preformed a rescue experiment by reintroducing wt-SIRT1 vector back into the stable SIRT1-knockdown cell lines and tested for restoration of the activity of bicalutamide. Whereas treatment with bicalutamide suppressed DHT-induced PSA-LUC transcriptional activity by a non-significant 25% in SIRT1-deficient cells co-transfected with an empty-vector and PSA-LUC, bicalutamide suppressed DHT-induced PSA-LUC transcriptional activity by 55% in the cells co-transfected with wtSIRT1 and PSA-LUC (p<0.05) (Fig. 4C). However, when co-transfected with a dominant-negative mutant that abolishes the deacetylase activity of SIRT1 (SIRT1H363Y) and PSA-LUC, bicalutamide did not significantly suppress DHT-induced PSA-LUC transcriptional activity (see Fig. 4D for levels of wtSIRT1, SIRT1H363Y, and AR protein expression in these cells). These studies demonstrate that the defect in bicalutamide-mediated transcriptional suppression in the knockdown cells is due to deficiency of SIRT1 deacetylate activity rather than to an off-target effect of siRNA, consistent with the results independently obtained using chemical inhibitors of SIRT1.
Figure 4. Reversal of bicalutamide-mediated transcriptional and growth suppression of cells by SIRT1 depletion.

A) SIRT1 knockdown impairs bicalutamide-mediated PSA transcription repression. A SIRT1 knockdown cell line (RNAi) and control-transfected LNCaP cells (Ctrl) were cultured in charcoal-stripped serum, transfected with the PSA-LUC reporter vector, and then treated with DHT (1 nM) (D), or DHT plus bicalutamide (10 μM) (D+CDX), or vehicle (S). Cells were harvested after 24 hr and lysates were assayed for luciferase activity. The data are presented as a percent of the activity obtained in DHT alone (assigned the value of 100). B) Immunoblot analysis of SIRT1, AR and β-actin levels in control-transfected LNCaP cells (Ctrl) and a LNCaP line in which SIRT1 had been knocked down by stable expression of siRNA (RNAi). C) SIRT1 over-expression can partially rescue the defect of bicalutamide-mediated PSA transcription suppression induced by SIRT1 depletion. SIRT1 knockdown cells were cultured in charcoal-stripped serum, co-transfected with PSA-LUC and empty-vector (RNAi) or with PSA-LUC and SIRT1 expression vector (RNAi+SIRT1wt), or with PSA-LUC and SIRT1 catalytic inactive mutant (RNAi+SIRT1H363Y) and then treated with DHT (1 nM) (D), or DHT plus bicalutamide (10 μM) (D+CDX), or vehicle (S). Cells were harvested after 24 hr and lysates were assayed for luciferase activity. The data are presented as a percent of the activity obtained with exposure to DHT alone (assigned the value of 100). D) Immunoblot analysis of SIRT1 and AR protein levels in SIRT1 knockdown cell lines transfected with empty vector (RNAi), or wt-SIRT (SIRT1wt), or catalytic inactive mutant (SIRT1H363Y). E) SIRT1 is required for bicalutamide-mediated cell growth suppression. Parental empty-vector transfected LNCaP cells (Ctrl) or LNCaP cells in which SIRT1 had been stably knocked down by siRNA expression (RNAi) were cultured in charcoal-stripped serum three days, and exposed to DHT (1 nM) for another three days, then treated with addition of bicalutamide (2.5 μM) (D+CDX) or vehicle (D) for 48 hr. Viable cells were quantitated by MTS assay, and the results expressed relative to the values obtained from wells cultured without added bicalutamide (assigned as value of 100). Asterisks (*) indicates significant differences between two groups (p<0.05).
Taken together with the data on Pol II recruitment shown in Fig. 2E & 2F, these results suggest that SIRT1 is required for bicalutamide-mediated AR-dependent transcriptional suppression. Parallel studies using treatment with nicotinamide to suppress SIRT1 activity confirmed the requirement for SIRT in the action of androgen antagonists (data not shown).
Bicalutamide suppresses both AR-dependent transcription and prostate cancer cell proliferation. We next assessed whether SIRT1 plays a role in bicalutamide-mediated prostate cancer cell growth suppression, using the SIRT1 partial-knockdown LNCaP cells and control (empty-vector-transfected) cells. Exposure to bicalutamide suppressed AR-dependent growth by 60% in the control (empty-vector) transfected cells, but induced only 30% growth suppression in the stable SIRT1 partial knockdown cells (p<0.05) (Fig. 4E). Together with the studies presented above, these findings suggest that SIRT1 is required both for bicalutamide-mediated transcriptional suppression of AR-responsive genes, and for bicalutamide-mediated prostate cancer cell growth suppression.
SIRT1 down-regulation enhances LNCaP cell sensitivity to DHT with respect to AR-dependent gene transcription and cell growth
It has been proposed that the inevitable androgen “independence” and antagonist insensitivity that occurs during the progression of prostate cancer may in some cases be the result of an acquired hyper-sensitivity of the AR to androgen, resulting in the ability of the tumor cells to respond to very low levels of androgen [3, 12, 60]. The studies described above demonstrate that SIRT1 can regulate AR-responsive genes in a ligand-dependent fashion. We asked, therefore, whether alterations in SIRT1 levels or activity could increase the sensitivity of hormone-responsive cells to androgen. We determined the relative androgen-sensitivity of AR-responsive genes, using the androgen-responsive PSA promoter-LUC vector in control-transfected LNCaP cells and in stable SIRT1-knockdown LNCaP cell lines. Cells were androgen-deprived for 72 hr, transfected with the PSA-LUC reporter, and treated with various concentrations of DHT in the absence or presence of the SIRT1-inhibitor nicotinamide. At very low concentrations of DHT, ranging from 0.01 to 0.1 nM, there was minimal induction of PSA-LUC activity above basal levels in the control (empty-vector)-transfected LNCaP cells (Fig. 5A). In contrast, both the SIRT1 knockdown cells and NAM-treated control-transfected LNCaP cells demonstrated significant increases in PSA-LUC transcription at these low doses of DHT. For example, at 0.1 nM DHT, PSA-LUC transcription in SIRT1-knockdown or nicotinamide-treated cells was induced by over 10-fold (p<0.05), compared to parental LNCaP cells. Thus, inhibition of SIRT1 activity by two independent methods enhanced the transcriptional sensitivity of the androgen-responsive PSA promoter to DHT.
Figure 5. SIRT1 depletion increases the sensitivity of AR-dependent gene transcription and cell proliferation to DHT.

A) SIRT1 knockdown increases the sensitivity of AR-dependent gene transcription to DHT. Parental empty-vector transfected LNCaP cells (Ctrl) or LNCaP cells in which SIRT1 had been stably knocked down by siRNA expression (RNAi) were cultured in charcoal-stripped serum, transfected with the PSA-LUC reporter vector, and then treated with nicotinamide (NAM) or vehicle (Control), and exposed to the indicated concentrations of DHT (0–1 nM). Cells were harvested after 48 hr and assayed for luciferase activity. B) Immunoblot analysis of SIRT1, AR and β-actin levels in control-transfected LNCaP cells (Ctrl) and three LNCaP lines in which SIRT1 had been knocked down by stable expression of siRNA (RNAi-1, RNAi-2 and RNAi-3). C) SIRT1 knockdown increases the sensitivity of the proliferative response of LNCaP cells to DHT. Control, empty-vector transfected LNCaP cells (Ctrl) or LNCaP cell lines in which SIRT1 had been stably knocked-down by siRNA expression (lines RNAi-1, RNAi-2 and RNAi-3) were made quiescent by culture in charcoal-stripped serum, and exposed to the indicated concentrations of DHT (0–10 nM). Viable cells were quantitated at 72 hr by MTT assay, and the results expressed relative to values obtained from plates cultured without added DHT (assigned an arbitrary value of 1). In all relevant figures, relative luciferase activities were normalized to β-gal activity to control for transfection efficiency. The error bars represent the SEM. Asterisks (*) indicates significant differences between two groups (p<0.05).
To determine if SIRT1 knockdown increased the sensitivity of hormone-responsive cells to the mitogenic effects of androgen, the androgen-responsiveness of control (empty-vector)-transfected LNCaP cells and three SIRT1-knockdown LNCaP cell lines (Fig. 5B) was compared. After androgen-deprivation, cell lines were treated with varying concentrations of DHT for 6 days and their proliferation was assessed. Each of the three SIRT1-knockdown cell lines exhibited significantly enhanced sensitivity to the mitogenic effects of DHT at concentrations less than 1 nM (Fig. 5C). For example, at concentrations of 0.05 nM DHT, the SIRT1 knockdown cell line RNAi-2 proliferated 300%, compared to only approximately 50% for control cells (p<0.05). Maximal proliferation was observed at 0.5 nM DHT in the RNAi-2 cell line, compared to 1 nM DHT in the control cells. Similar degrees of sensitization to DHT were observed in the RNAi-1 line and other SIRT1-knockdown cell lines (Fig. 5C). These data demonstrate that suppression of SIRT1 increases the sensitivity of hormone-responsive prostate cancer cells to the mitogenic effects of DHT.
DISCUSSION
In this study, we demonstrate a role for SIRT1 in the regulation of androgen receptor-dependent gene transcription and androgen-dependent prostate cancer cell growth. We identify SIRT1 as a novel co-repressor of AR suppressing AR-dependent gene transcription. In addition, we demonstrate that SIRT1 is recruited to androgen-response elements (AREs) by AR antagonists and is required for bicalutamide-mediated transcriptional repression and prostate cancer cell growth suppression. We further demonstrate that down-regulation of SIRT1 increases the sensitivity of prostate cancer cells to the proliferative and transcriptional actions of androgens. Finally, we provide evidence to suggest that the mechanism of SIRT1-mediated transcriptional inhibition on AR-responsive genes may be due to local deacetylation of histone H3 in AR-dependent gene promoters.
AR co-repressor complexes play a critical role in regulating AR activity with precision and efficiency [15]. To date, several co-repressors of AR have been characterized, including NCoR, SMRT, and HDACs 1–3 [24, 25, 31–34]. HDACs 1–3 are Class I deacetylases, and form a holo-co-repressor complex with AR and NCoR at the PSA promoter, suppressing AR-dependent transcription [24, 25]. SIRT1 is a member of the Class III HDAC family, the deacetylase activity of which is NAD-dependent. We report herein that SIRT1 is required for antagonist (bicalutamide)-mediated transcription suppression of AR-dependent genes. To our knowledge, this is the first functional demonstration of ligand-induced recruitment of a class III HDAC to transcriptionally repress any promoter, and the first demonstration of a necessary role for a class III HDAC as a transcriptional co-repressor in steroid hormone-responsive gene regulation. Taken together with the report of Zhu, et al., our data indicate that both class I HDACs (1, 2 and 3) and class III HDACs (SIRT1) are required for androgen antagonist-mediated transcriptional repression [24]. We also found that SIRT1 inhibits AR-dependent transcription tonically during transcriptional activation in response to androgens, in that modulation of SIRT1 activity influences the amplitude of the transcriptional response. Several biochemical and molecular genetic studies have shown that a chimeric AR/co-activator/co-repressor complex exists at the promoter at the onset of the androgenic response. Co-repressors (HDAC1, SMRT) and co-activators (TIP60) have simultaneously been identified in this complex [61]. The co-repressor elements may attenuate agonist-induced transactivation, acting transiently as part of a cycle of cofactors recruited to target promoters by ligand-bound receptors. We suggest that SIRT1 may be another element in the incremental and constant regulation of AR activity, playing a role to overcome or reduce co-activator-mediated effects upon the receptor, thereby preventing excessive gene expression and finely tuning the transcriptional response. The dramatic recruitment of SIRT1 to ARE-containing promoter and enhancer elements following exposure to androgen antagonists, and the finding that SIRT1 is required for transcriptional repression of exogenous and endogenous AR-responsive promoters, clearly demonstrates its functional necessity for the action of androgen antagonists.
Resveratrol is recognized as an agonist of SIRT1 [62], and is a natural chemopreventive agent in prostate cancer models [63, 64]. Both SIRT1 over-expression and exposure to resveratrol suppress AR-dependent transcription, although their efficiency in suppressing AR-dependent transcription differs. Resveratrol suppresses transcription by 16-fold, whereas SIRT1 over-expression represses it by approximately 2.5-fold. There are several possible mechanisms accounting for this difference. Firstly, although resveratrol is a agonist of SIRT1, it also targets a number of other proteins and signaling pathways [65]. It is therefore possible that Resveratrol may be acting on additional proteins contributing to transcriptional repression. In addition, as prostate cancer cell lines take up DNA with low efficiency, the level of SIRT1 expression may partially limit the effects of SIRT1 over-expression on the PSA gene transcription. Another possible explanation could be that resveratrol might be acting not directly on SIRT1, but rather is blocking androgen binding to the AR, particularly in prostate cancer cell lines in which the AR is mutated and exhibits relaxed ligand specificity, such as LNCaP cells. Blocking androgen binding to the AR would then impair AR translocation, prevent recruitment of AR to androgen-response elements (AREs), and suppress AR-mediated transcriptional activity. As shown in Fig. S1, we have ruled out this possible mechanisms by demonstrating that exposure to resveratrol does not affect AR binding to the PSA promoter in response to androgens. To exclude the possibility that resveratrol acts differentially on the mutated AR in LNCaP cells compared to wt-AR, we also carried out a PSA-reporter assay using a 293 cell line, transfected and expressing wt–AR, in the presence or absence of resveratrol. Exposure to resveratrol produced similar levels of suppression on the transcriptional activity of this wt-AR as it did on the activity of the mutant AR in LNCaP cells (Fig. S2).
Unlike the thyroid hormone and retinoid receptors, the androgen receptor does not bind to and repress target gene transcription in the absence of ligand [66]. Instead, the switch to transcriptional repression is induced by binding of an androgen antagonist. Antagonist-bound AR is then rendered unable to bind co-activator proteins, and is also newly able to interact with, and recruit, transcriptional repressors, including an NCoR holo-repressor complex containing TBL/TBLR1, HDACs 1–3, Brg1, and Sin3 [24]. NCoR and HDACs 1–3 appear to be required for transcriptional repression by androgen antagonists [24]. The state of gene repression is correlated with histone deacetylation by co-repressors and their associated histone deacetylases [24, 34, 66, 67]. We observed significant recruitment of SIRT1 to the PSA promoter in the presence of bicalutamide and found that SIRT1 is required for bicalutamide-mediated transcription suppression and growth suppression. This recruitment was dependent on the presence of the AR. We found that local deacetylation of histone H3 and transcriptional suppression of AR-responsive genes was dependent upon SIRT1 recruitment to the promoter. There is no evidence that SIRT1 can bind to DNA directly, and targeted deacetylation of histones at promoters is thought to occur through interaction and recruitment of HDACs by specific DNA-binding proteins. Our results suggest that NCoR, a known adaptor protein for SIRT1, work as a co-repressor adaptor for AR, may bridge SIRT1 with AR at the PSA promoter, and thus making SIRT1 available to deacetylate H3 histones at the promoter and suppress AR-dependent transcription. In unpublished studies, we have found that the AR associates with SIRT1 in a ligand-dependent fashion in cell lysates, and that NCoR can bind to SIRT1, also in an antagonist-dependent fashion, raising the possibility that the AR directly recruits the NCoR/SIRT1 complex when bound to androgen antagonists. Our observation that new recruitment of SIRT1 to the PSA promoter requires the AR is consistent with such a model. A rigorous testing of this model is in progress. It is also possible that SIRT1 may act on additional protein targets to achieve transcriptional repression. Because p300, a AR co-activator, is a deacetylation target of SIRT1 [68], it is tempting to speculate that SIRT1 may act in part through deacetylation of p300 to control AR-dependent transcription. It is also possible that SIRT1 inhibits AR-dependent transcription in part through deacetylation of the AR itself. In unpublished studies, we find that SIRT1 can associate with AR and inhibition of SIRT1 increases the level of acetylation of the AR. Interestingly, an independent report published [69] during the submission of this paper indicates that SIRT1 can partially regulate AR-dependent transcription through deacetylation of the AR.
The formation of an active co-activator complex on the PSA promoter in response to androgens involves the recruitment of AR to two discrete regions of the gene, an enhancer (containing ARE III) and a promoter region (containing AREs I and II) [25, 70]. AR recruitment to the PSA promoter and enhancer regions in response to androgen antagonists, however, remains controversial. One prior study has suggested that formation of an AR repressor complex involves only the promoter region [25], as no recruitment of NCoR, SMRT, HDAC1 or HDAC2 was observed in the enhancer region after exposure to an antagonist, while another study found NCoR to be recruited to both the promoter and the enhancer [58]. We also observed recruitment of SIRT1 and NCoR to both the enhancer and the promoter in response to antagonist stimulation in our studies. The reason for these disparate findings is not clear, although each study used different primer sets in the ChIP, and neither of the prior studies attempted to determine if binding of a co-repressor complex to the enhancer region was functionally important.
Androgen depletion, or blockade of androgen signaling, represents the major therapeutic strategy for the treatment of advanced prostate cancer. Invariably, however, the disease becomes progressive and unresponsive to androgen ablation therapy (hormone-refractory) within an average of 18 months [71]. This evolution occurs through one of several discrete and incompletely-understood molecular mechanisms, some of which permit AR signaling in the absence of, or at very low concentrations of, androgens [6, 72, 73]. We report herein that down-regulation of SIRT1 increases the sensitivity of LNCaP cells to DHT, as assessed by at least two functional outcomes. Down-regulation of SIRT1 activity by pharmacological or genetic means increased the sensitivity of cells to transcriptional activation of AR-responsive target genes by androgens. In parallel, the proliferation of androgen-responsive cells (which is not necessarily directly linked to the transcriptional activation of AR-regulated genes like PSA and KLK), was also significantly enhanced at low concentrations of DHT by SIRT1 inhibition. Conversely, we also show that SIRT1 is required for bicalutamide-mediated growth suppression, suggesting that loss of SIRT1 might contribute to the development of antagonist-resistance in prostate cancer Thus, loss of SIRT1 increases the sensitivity of prostate cancer cells to growth in response to androgens, while simultaneously decreasing their sensitivity to the anti-proliferative effects of androgen antagonists.
These findings suggest the possibility of a role for SIRT1 loss in the evolution to hormone-refractory prostate cancer. It is perhaps noteworthy that global histone modification in prostate tumor tissues, including acetylation of H3, predict risk of prostate cancer recurrence [74], and we found that SIRT1 down-regulation increases the acetylation level of H3 at AR-dependent gene promoters. We also note that the SIRT1 expression level is higher in the AR-dependent cell line LNCaP than in the AR-independent cell lines DU145 and PC3 (data not shown). In other studies not reported here, we found that higher concentrations of bicalutamide can decrease SIRT1 protein levels in LNCaP cells. Chronic bicalutamide treatment in patients may thus result in depletion of SIRT1 protein levels in prostate cancer cells, thereby increasing their sensitivity to circulating androgens and their progression to androgen-independence.
SIRT1 has been shown to play roles in aging [40], and in diseases and pathways related to aging, such as diabetes [75], fat mobilization [48], and insulin signaling [76, 77]. Prostate cancer is considered a disease of aging, as its incidence increases with age more rapidly than do other types of cancer [78]. A link between cellular aging and SIRT1 protein expression in humans has been proposed, as the expression of endogenous SIRT1 protein progressively decreases during replication/aging of normal human fibroblasts in culture [79]. Similarly, fetal tissues show higher expression of SIRT1 than adult tissues [79]. It is therefore possible that the age-related decline in SIRT1 expression in humans may result in abnormal AR activity or function, promoting prostate cancer development during aging, and this hypothesis is currently being investigated.
MATERIALS AND METHODS
Cells, plasmids, and antibodies
LNC aP and DU145 cells were obtained from the American Type Culture Collection (Manassas, Virginia). LNCaP cells were maintained in RPMI 1640 medium with 10% fetal bovine serum (FBS) or in 10% charcoal-treated FBS (HyClone, CO). DU145 cells were maintained in DMEM medium with 10% FBS. The human SIRT1 expression vector pcDNA3.1-SIRT1 was generated by subcloning SIRT1 cDNA into a pcDNA3.1 (+) based V5His vector, generating a C-terminal V5His-tagged fusion protein, using standard PCR-based strategies. The deacetylase-defective SIRT1(H363Y) mutant expression plasmid was generously provided by Dr. Melanie Ott (UCSF, CA). The SIRT1 RNAi vectors (pSUPER. retro. puro-SIRT1 and pSUPER. retro. neo-SIRT1) were generously provided by Dr. F. Picard (Laval University, Canada) and Dr. Melanie Ott respectively. The pSUPER. retro. puro-SIRT1 vector contains SIRT1 sequence (5′-GATGAAGTTGACCTCCTCA-3′) and the puromycin-resistance gene, and the pSUPER. retro. neo-SIRT1 vector contains (5′-CTTGTACGACGAAGACGA-3′) and the neomycin-resistance gene. The AR expression vector was provided by Dr. Marco Marcelli (Baylor College of Medicine, Houston, TX). The reporter plasmid PSA-LUC, containing the luciferase gene under the control of a fragment of the human PSA gene promoter, was provided by Dr. A.O Brinkmann (Erasmus University, MC, Netherlands). The pMMTV-LUC reporter plasmid, containing the luciferase gene driven by the mouse mammary tumor virus long terminal repeat, was provided by Dr. R. Spanjaard (Boston University, Boston, MA).
Antibodies to SIRT1 (#05-707), androgen receptor (PG-21, #06-680), and acetyl-histone H3 (#06-599) were purchased from Upstate (Lake Placid, NY). The rabbit polyclonal SIRT1 antibody was generously provided by Dr. Roy A Frye (VA Medical Center, Pittsburgh, PA). Antibodies to NCoR (H303, sc-8994), Pol II (A-10, sc-17798), AR (441, sc-7305) and RAG-1(sc-363) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Antibody to β-actin (A-2066) was purchased from Sigma-Aldrich (St. Louis, MO).
DHT (A-8380), Nicotinamide (N3376), Resveratrol (R5010), and NAD (N1636) were purchased from Sigma-Aldrich, and bicalutamide (Casodex, s210183) was provided by AstraZeneca, UK.
Retroviral infection and establishment of stable SIRT1-knockdown cell lines
The Phoenix packaging cell line was transfected with either the pSUPER. retro. puro-SIRT1, pSUPER. retro. neo-SIRT1 or the pSUPER. retro vectors separately, using Lipofectamine plus (Invitrogen, Carlsbad, CA). After 48 h, the medium containing retrovirus was collected, filtered, treated with polybrene and transferred to LNCaP cell cultures. Infected cells were selected with G418 or puromycin plus G418 for isolation of stably-infected colonies. SIRT1-knockdown stable cell line RNAi-2 was isolated from pSUPER. retro. neo-SIRT1 infected cells and is G418-resistant. SIRT1-knockdown stable cell lines RNAi-1 and RNAi-3 were selected from cells co-infected with pSUPER. retro. neo-SIRT1 and pSUPER. retro. puro-SIRT1, and are puromycin and G418 double-resistant.
Luciferase Assay
LNCaP cells were cultured in 6-well plates in RPMI1640 with 10% charcoal-treated FBS (HyClone Laboratories, Inc., Logan, UT) (androgen-deprivation conditions) for 3 days, then co-transfected with 1 μg of PSA-LUC or pMMTV-LUC reporter vectors together with SIRT1 expression vectors, using Lipofectamine plus (Invitrogen, Carlsbad, CA). Fresh medium was added after overnight transfection. Transfected cells were exposed to resveratrol or NAM or vehicle for 24 hr, then treated with DHT or vehicle control for 24 hr before assay for luciferase activity (Promega Luciferase Assay System, #E1500). The relative luciferase activities were normalized to the activity of a co-transfected β-gal expression vector, as an internal control for transfection efficiency. Each experiment was performed in triplicate and repeated a minimum of three times. These values were used to determine standard deviations (SDs), with error bars indicating +/−SEM.
Cell-based HDAC assay
Cell-based HDAC assays were performed as described [55]. LNCaP cells were cultured under androgen deprivation conditions for 3 days, then treated with resveratrol or nicotinamide. Cells were washed with PBS, and lysed with 1x HDAC lysis buffer. Equal amounts of lysates were analyzed for enzyme activity using the HDAC Fluos de Lys™ Fluorescent Assay System (BIOMOL, #AK-500).
Real-Time RT-PCR
Total cellular RNA was isolated using Trizol reagent (Invitrogen Life Technologies, Carlsbad, CA) according to the manufacturer’s instructions. A two-step RT-PCR method was employed to synthesize single-stranded cDNA (SuperScript TM III First Strand kit, Invitrogen, 18080-051). Target genes were analyzed by real-time PCR using Applied Biosystems 7500 Fast Real-Time PCR System with SYBR Green I dye (Applied Biosystems, 4309155). The primers used were: PSA forward, TGCCCACTGCATCAGGAACA; PSA reverse: GTCCAGCGTCCAGCACACAG; KLK2 forward: CCTGGCAGGTGGCTGTGTAC; KLK2 reverse: TGTGCCGACCCAGCCA, β-actin forward: GAGAAAATCTGGCACCACACC; β-actin reverse: ATACCCCTCGTAGATGGGCAC. PCR reactions were performed in triplicate. β-actin mRNA abundance was analyzed in each sample. The PSA and KLK2 mRNA levels were normalized to the β-actin mRNA level. The specificities of the RT-PCR products were monitored by melting curve analysis and also verified by agarose gel electrophoresis.
Chromatin Immunoprecipitation
LNCaP cells (107 cells) were grown in RPMI-medium 1640 (GIBCO-BRL, Gaithersburg, MD) supplemented with 10% charcoal-dextran-stripped fetal bovine serum. After 3 days of cultivation, cells were treated with DHT or bicalutamide for 4 hr, then trypsinized and washed twice with PBS. DU145 (2×107 cells) were cultured in DMEM+10%FCS, transfected with 20 μg of AR expression vector or empty vector using Lipofectamine plus (Invitrogen, Carlsbad, CA). Fresh medium was added after overnight transfection. Transfected cells were cultured for another 48 hr then treated with bicalutamide or vehicle for 4 hr, trypsinized and washed twice with PBS. The harvested cells (LNCaP or DU145) were resuspended in 10 ml of culture medium, cross-linked with 1% formaldehyde at RT for 10 min, and washed three times with ice-cold PBS. The pellets were then resuspended in 0.4 ml of lysis buffer (1% SDS, 10 mM EDTA, 50 mM Tris-HCl, pH 8.0, 1 mM PMSF and protease inhibitor cocktail (Roche Molecular Biochemicals), incubated 20 min at RT, sonicated 4 times for 10 sec each followed by a 1 min pulse, at 20% power (Fisher Sonic Dismembrator, Model 550). The samples were centrifuged for 10 min, and the supernatants were collected and diluted 5-fold in dilution buffer (1% Triton X-100, 2 mM EDTA, 150 mM NaCl, 20 mM Tris-HCl, pH 8.1 and protein inhibitor cocktail) followed by immuno-clearing with protein A/G-Sepharose (50 μl of 50% slurry) for 1 hr at 4°C. One hundred μl of the supernatant was reserved as input. Immunoprecipitation was performed for 1 hr at RT with specific antibodies (anti-SIRT1; anti-AR; anti-AcH3; anti-N-CoR; anti-Pol II; or irrelevant control antibody anti-RAG-1). Seventy μl of protein A/G-Sepharose and 1 μg of salmon sperm DNA were added, and the incubation was continued overnight at 4°C. Sepharose beads were washed sequentially for 5 min each in Wash I (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris-HCl, pH 8.0, 150 mM NaCl, and 1 mM PMSF), Wash II (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris-HCl, pH 8.1, 500 mM NaCl and 1 mM PMSF), Wash III (1% NP-40, 0.25 M LiCl, 1 mM EDTA, 10 mM Tris-HCl, pH 8.0 and 1 mM PMSF), and TE buffer. Beads were then extracted twice with 1% SDS, 0.1 M NaHCO3, for 15 min at RT, with rotation. Eluates were pooled and heated to 65°C in 0.2M NaCl for 6 hr to reverse the formaldehyde cross-linking. The samples were treated with Protein K for 1 hr at 37°C and the DNA fragments were purified by phenol/chloroform extraction, followed by ethanol precipitation at −20°C for 10 min and the precipitates were washed with 70% ethanol. For PCR, a 2 μl aliquot of the total 50 μl of extracted DNA was amplified in 21–25 PCR cycles for gel analysis, or in real-time PCR, two μl aliquot of the total 50 μl of IP and input DNA were analyzed using Applied Biosystems 7500 Fast Real-Time PCR System with SYBR Green I dye (Applied Biosystems, 4309155). Triplicate PCR reactions for each sample were preformed and each ChIP assay was performed on at least three independent experiments. The primer sequences were as follows: PSA promoter forward: ACAATCTCCTGAGTGCTGGTGT; PSA promoter reverse: GCAGAGGAGACATGCCCA G; PSA enhancer forward: GAGAATTGCCTCC CAACACTG; PSA enhancer reverse: TGCCAGA CACAGTCGATCG; Distal control primer forward, TTCACCGTGTTGGCCAGG; Distal control primer reverse: ATGGTGGCTCACGCC TG.
Immunoblots
Cells (5×106) treated with different reagents were lysed in 200 μl 1% NP-40 immunoblotting lysis buffer. The samples were separated on 8% PAGE gels and further analyzed after transfer by immunoblot analysis with anti-AR (441), -SIRT1 (# 05-707) or –β-actin antibodies.
Cell viability assay
MTT (M2128, Sigma) or MTS (Promega, G3580) assays were used to quantitate cell viability. Cells were plated at a density of 4 ×104 cells/well on 24-well plates (for MTT assay) or 104 cells/well on 96-well plates (for MTS assay) and cultured under androgen-deprivation conditions for 3 days, followed by treatment with different concentrations of DHT for another 3 days. MTT (50 mg/ml) was added to the medium for 3 h, then the supernatant was removed and the formazone crystals were dissolved using DMSO. The absorbance was read at 690 nm on an ELISA plate reader. For the MTS assay, 20 μl of CellTiter96Aqueous one solution reagent was added to the medium for 1 hr, and the absorbance was read at 490 nm.
Acknowledgments
This work was supported by grants from the American Cancer Society (IRG-72-001-27-IRG) (Y.D.), BUSM Department of Medicine Pilot Project Grant Award (Y.D.), the National Cancer Institute (CA101992-03) and the Karin Grunebaum Cancer Research Foundation (D.V.F.).
We sincerely thank Dr Margie Oettinger (Genetic Department of Harvard Medical School) for her continuous support. We thank Dr. A Brinkmann (Brasmus, MC, Netherlands), Dr. M. Marcelli (Baylor College of Medicine), Dr. M. Ott (UCSF, CA), Dr. F. Picard (Laval University, Canada), Dr. S. Qin (Brigham and Women’s Hospital, Harvard Medical School), Dr. R.A. Frye (VA Medical Center, Pittsburgh, PA), Dr. D. Sinclair (Department of Pathology, Harvard Medical School), and Drs. R. Spanjaard and X. Zhao (Boston University School of Medicine) for reagents and helpful discussions.
Footnotes
The Endocrine Society NIH Statement: “This is an un-copyedited author manuscript copyrighted by The Endocrine Society. This may not be duplicated or reproduced, other than for personal use or within the rule of “Fair Use of Copyrighted Materials” (section 107, Title 17, U.S. Code) without permission of the copyright owner, The Endocrine Society. From the time of acceptance following peer review, the full text of this manuscript is made freely available by The Endocrine Society at http://www.endojournals.org/. The final copy edited article can be found at http://www.endojournals.org/. The Endocrine Society disclaims any responsibility or liability for errors or omissions in this version of the manuscript or in any version derived from it by the National Institutes of Health or other parties. The citation of this article must include the following information: author(s), article title, journal title, year of publication and DOI.”
Disclosure statement: The authors have nothing to disclose
References
- 1.Chen CD, Welsbie DS, Tran C, Baek SH, Chen R, Vessella R, Rosenfeld MG, Sawyers CL. Molecular determinants of resistance to antiandrogen therapy. Nat Med. 2004;10(1):33–9. doi: 10.1038/nm972. [DOI] [PubMed] [Google Scholar]
- 2.Boring CC, Squires TS, Tong T, Montgomery S. Cancer statistics, 1994. CA Cancer J Clin. 1994;44(1):7–26. doi: 10.3322/canjclin.44.1.7. [DOI] [PubMed] [Google Scholar]
- 3.Deutsch E, Maggiorella L, Eschwege P, Bourhis J, Soria JC, Abdulkarim B. Environmental, genetic, and molecular features of prostate cancer. Lancet Oncol. 2004;5(5):303–13. doi: 10.1016/S1470-2045(04)01468-8. [DOI] [PubMed] [Google Scholar]
- 4.Chang CS, Kokontis J, Liao ST. Molecular cloning of human and rat complementary DNA encoding androgen receptors. Science. 1988;240(4850):324–6. doi: 10.1126/science.3353726. [DOI] [PubMed] [Google Scholar]
- 5.Hirawat S, Budman DR, Kreis W. The androgen receptor: structure, mutations, and antiandrogens. Cancer Invest. 2003;21(3):400–17. doi: 10.1081/cnv-120018232. [DOI] [PubMed] [Google Scholar]
- 6.Heinlein CA, Chang C. Androgen receptor in prostate cancer. Endocr Rev. 2004;25(2):276–308. doi: 10.1210/er.2002-0032. [DOI] [PubMed] [Google Scholar]
- 7.Brinkmann AO, Blok LJ, de Ruiter PE, Doesburg P, Steketee K, Berrevoets CA, Trapman J. Mechanisms of androgen receptor activation and function. J Steroid Biochem Mol Biol. 1999;69(1–6):307–13. doi: 10.1016/s0960-0760(99)00049-7. [DOI] [PubMed] [Google Scholar]
- 8.Sharifi N, Farrar WL. Androgen receptor as a therapeutic target for androgen independent prostate cancer. Am J Ther. 2006;13(2):166–70. doi: 10.1097/00045391-200603000-00013. [DOI] [PubMed] [Google Scholar]
- 9.Roscigno M, Sangalli M, Mazzoccoli B, Scattoni V, Da Pozzo L, Rigatti P. Medical therapy of prostate cancer. Minerva Urol Nefrol. 2005;57(2):71–84. [PubMed] [Google Scholar]
- 10.Nishiyama T, Suzuki K, Yamana K, Tonegawa E, Wako K, Takahashi K. Stepping-stones to the further advancement of androgen-deprivation therapy for prostate cancer. Expert Rev Anticancer Ther. 2006;6(2):259–68. doi: 10.1586/14737140.6.2.259. [DOI] [PubMed] [Google Scholar]
- 11.Oh WK. Secondary hormonal therapies in the treatment of prostate cancer. Urology. 2002;60(3 Suppl 1):87–92. doi: 10.1016/s0090-4295(02)01581-9. discussion 93. [DOI] [PubMed] [Google Scholar]
- 12.Hobisch A, Fritzer A, Comuzzi B, Fiechtl M, Malinowska K, Steiner H, Bartsch G, Culig Z. The androgen receptor pathway is by-passed in prostate cancer cells generated after prolonged treatment with bicalutamide. Prostate. 2006;66(4):413–20. doi: 10.1002/pros.20365. [DOI] [PubMed] [Google Scholar]
- 13.Heinlein CA, Chang C. Androgen receptor (AR) coregulators: an overview. Endocr Rev. 2002;23(2):175–200. doi: 10.1210/edrv.23.2.0460. [DOI] [PubMed] [Google Scholar]
- 14.Culig Z, Comuzzi B, Steiner H, Bartsch G, Hobisch A. Expression and function of androgen receptor coactivators in prostate cancer. J Steroid Biochem Mol Biol. 2004;92(4):265–71. doi: 10.1016/j.jsbmb.2004.10.003. [DOI] [PubMed] [Google Scholar]
- 15.Wang L, Hsu CL, Chang C. Androgen receptor corepressors: an overview. Prostate. 2005;63(2):117–30. doi: 10.1002/pros.20170. [DOI] [PubMed] [Google Scholar]
- 16.Glass CK, Rosenfeld MG. The coregulator exchange in transcriptional functions of nuclear receptors. Genes Dev. 2000;14(2):121–41. [PubMed] [Google Scholar]
- 17.McKenna NJ, O’Malley BW. Combinatorial control of gene expression by nuclear receptors and coregulators. Cell. 2002;108(4):465–74. doi: 10.1016/s0092-8674(02)00641-4. [DOI] [PubMed] [Google Scholar]
- 18.Hermanson O, Glass CK, Rosenfeld MG. Nuclear receptor coregulators: multiple modes of modification. Trends Endocrinol Metab. 2002;13(2):55–60. doi: 10.1016/s1043-2760(01)00527-6. [DOI] [PubMed] [Google Scholar]
- 19.Gregory CW, He B, Johnson RT, Ford OH, Mohler JL, French FS, Wilson EM. A mechanism for androgen receptor-mediated prostate cancer recurrence after androgen deprivation therapy. Cancer Res. 2001;61(11):4315–9. [PubMed] [Google Scholar]
- 20.Fernandes I, White JH. Agonist-bound nuclear receptors: not just targets of coactivators. J Mol Endocrinol. 2003;31(1):1–7. doi: 10.1677/jme.0.0310001. [DOI] [PubMed] [Google Scholar]
- 21.Blanco JC, Minucci S, Lu J, Yang XJ, Walker KK, Chen H, Evans RM, Nakatani Y, Ozato K. The histone acetylase PCAF is a nuclear receptor coactivator. Genes Dev. 1998;12(11):1638–51. doi: 10.1101/gad.12.11.1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hong H, Kohli K, Trivedi A, Johnson DL, Stallcup MR. GRIP1, a novel mouse protein that serves as a transcriptional coactivator in yeast for the hormone binding domains of steroid receptors. Proc Natl Acad Sci U S A. 1996;93(10):4948–52. doi: 10.1073/pnas.93.10.4948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Spencer TE, Jenster G, Burcin MM, Allis CD, Zhou J, Mizzen CA, McKenna NJ, Onate SA, Tsai SY, Tsai MJ, O’Malley BW. Steroid receptor coactivator-1 is a histone acetyltransferase. Nature. 1997;389(6647):194–8. doi: 10.1038/38304. [DOI] [PubMed] [Google Scholar]
- 24.Zhu P, Baek SH, Bourk EM, Ohgi KA, Garcia-Bassets I, Sanjo H, Akira S, Kotol PF, Glass CK, Rosenfeld MG, Rose DW. Macrophage/cancer cell interactions mediate hormone resistance by a nuclear receptor derepression pathway. Cell. 2006;124(3):615–29. doi: 10.1016/j.cell.2005.12.032. [DOI] [PubMed] [Google Scholar]
- 25.Shang Y, Myers M, Brown M. Formation of the androgen receptor transcription complex. Mol Cell. 2002;9(3):601–10. doi: 10.1016/s1097-2765(02)00471-9. [DOI] [PubMed] [Google Scholar]
- 26.Chen H, Lin RJ, Schiltz RL, Chakravarti D, Nash A, Nagy L, Privalsky ML, Nakatani Y, Evans RM. Nuclear receptor coactivator ACTR is a novel histone acetyltransferase and forms a multimeric activation complex with P/CAF and CBP/p300. Cell. 1997;90(3):569–80. doi: 10.1016/s0092-8674(00)80516-4. [DOI] [PubMed] [Google Scholar]
- 27.Li J, O’Malley BW, Wong J. p300 requires its histone acetyltransferase activity and SRC-1 interaction domain to facilitate thyroid hormone receptor activation in chromatin. Mol Cell Biol. 2000;20(6):2031–42. doi: 10.1128/mcb.20.6.2031-2042.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Onate SA, Tsai SY, Tsai MJ, O’Malley BW. Sequence and characterization of a coactivator for the steroid hormone receptor superfamily. Science. 1995;270(5240):1354–7. doi: 10.1126/science.270.5240.1354. [DOI] [PubMed] [Google Scholar]
- 29.Shen HC, Buchanan G, Butler LM, Prescott J, Henderson M, Tilley WD, Coetzee GA. GRIP1 mediates the interaction between the amino- and carboxyl-termini of the androgen receptor. Biol Chem. 2005;386(1):69–74. doi: 10.1515/BC.2005.009. [DOI] [PubMed] [Google Scholar]
- 30.Chakravarti D, V, LaMorte J, Nelson MC, Nakajima T, Schulman IG, Juguilon H, Montminy M, Evans RM. Role of CBP/P300 in nuclear receptor signalling. Nature. 1996;383(6595):99–103. doi: 10.1038/383099a0. [DOI] [PubMed] [Google Scholar]
- 31.Chen JD, Evans RM. A transcriptional co-repressor that interacts with nuclear hormone receptors. Nature. 1995;377(6548):454–7. doi: 10.1038/377454a0. [DOI] [PubMed] [Google Scholar]
- 32.Hodgson MC, Astapova I, Cheng S, Lee LJ, Verhoeven MC, Choi E, Balk SP, Hollenberg AN. The androgen receptor recruits nuclear receptor CoRepressor (N-CoR) in the presence of mifepristone via its N and C termini revealing a novel molecular mechanism for androgen receptor antagonists. J Biol Chem. 2005;280(8):6511–9. doi: 10.1074/jbc.M408972200. [DOI] [PubMed] [Google Scholar]
- 33.Liao G, Chen LY, Zhang A, Godavarthy A, Xia F, Ghosh JC, Li H, Chen JD. Regulation of androgen receptor activity by the nuclear receptor corepressor SMRT. J Biol Chem. 2003;278(7):5052–61. doi: 10.1074/jbc.M206374200. [DOI] [PubMed] [Google Scholar]
- 34.Nagy L, Kao HY, Chakravarti D, Lin RJ, Hassig CA, Ayer DE, Schreiber SL, Evans RM. Nuclear receptor repression mediated by a complex containing SMRT, mSin3A, and histone deacetylase. Cell. 1997;89(3):373–80. doi: 10.1016/s0092-8674(00)80218-4. [DOI] [PubMed] [Google Scholar]
- 35.Gasser SM, Cockell MM. The molecular biology of the SIR proteins. Gene. 2001;279(1):1–16. doi: 10.1016/s0378-1119(01)00741-7. [DOI] [PubMed] [Google Scholar]
- 36.Smith J. Human Sir2 and the ‘silencing’ of p53 activity. Trends Cell Biol. 2002;12(9):404–6. doi: 10.1016/s0962-8924(02)02342-5. [DOI] [PubMed] [Google Scholar]
- 37.Bordone L, Guarente L. Calorie restriction, SIRT1 and metabolism: understanding longevity. Nat Rev Mol Cell Biol. 2005;6(4):298–305. doi: 10.1038/nrm1616. [DOI] [PubMed] [Google Scholar]
- 38.Giannakou ME, Partridge L. The interaction between FOXO and SIRT1: tipping the balance towards survival. Trends Cell Biol. 2004;14(8):408–12. doi: 10.1016/j.tcb.2004.07.006. [DOI] [PubMed] [Google Scholar]
- 39.McBurney MW, Yang X, Jardine K, Hixon M, Boekelheide K, Webb JR, Lansdorp PM, Lemieux M. The mammalian SIR2alpha protein has a role in embryogenesis and gametogenesis. Mol Cell Biol. 2003;23(1):38–54. doi: 10.1128/MCB.23.1.38-54.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Guarente L, Picard F. Calorie restriction--the SIR2 connection. Cell. 2005;120(4):473–82. doi: 10.1016/j.cell.2005.01.029. [DOI] [PubMed] [Google Scholar]
- 41.Guarente L. Calorie restriction and SIR2 genes--towards a mechanism. Mech Ageing Dev. 2005;126(9):923–8. doi: 10.1016/j.mad.2005.03.013. [DOI] [PubMed] [Google Scholar]
- 42.Langley E, Pearson M, Faretta M, Bauer UM, Frye RA, Minucci S, Pelicci PG, Kouzarides T. Human SIR2 deacetylates p53 and antagonizes PML/p53-induced cellular senescence. Embo J. 2002;21(10):2383–96. doi: 10.1093/emboj/21.10.2383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vaziri H, Dessain SK, Ng Eaton E, Imai SI, Frye RA, Pandita TK, Guarente L, Weinberg RA. hSIR2(SIRT1) functions as an NAD-dependent p53 deacetylase. Cell. 2001;107(2):149–59. doi: 10.1016/s0092-8674(01)00527-x. [DOI] [PubMed] [Google Scholar]
- 44.Cheng HL, Mostoslavsky R, Saito S, Manis JP, Gu Y, Patel P, Bronson R, Appella E, Alt FW, Chua KF. Developmental defects and p53 hyperacetylation in Sir2 homolog (SIRT1)-deficient mice. Proc Natl Acad Sci U S A. 2003;100(19):10794–9. doi: 10.1073/pnas.1934713100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.van der Horst A, Tertoolen LG, de Vries-Smits LM, Frye RA, Medema RH, Burgering BM. FOXO4 is acetylated upon peroxide stress and deacetylated by the longevity protein hSir2(SIRT1) J Biol Chem. 2004;279(28):28873–9. doi: 10.1074/jbc.M401138200. [DOI] [PubMed] [Google Scholar]
- 46.Nemoto S, Fergusson MM, Finkel T. Nutrient availability regulates SIRT1 through a forkhead-dependent pathway. Science. 2004;306(5704):2105–8. doi: 10.1126/science.1101731. [DOI] [PubMed] [Google Scholar]
- 47.Brunet A, Sweeney LB, Sturgill JF, Chua KF, Greer PL, Lin Y, Tran H, Ross SE, Mostoslavsky R, Cohen HY, Hu LS, Cheng HL, Jedrychowski MP, Gygi SP, Sinclair DA, Alt FW, Greenberg ME. Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science. 2004;303(5666):2011–5. doi: 10.1126/science.1094637. [DOI] [PubMed] [Google Scholar]
- 48.Picard F, Kurtev M, Chung N, Topark-Ngarm A, Senawong T, Machado De Oliveira R, Leid M, McBurney MW, Guarente L. Sirt1 promotes fat mobilization in white adipocytes by repressing PPAR-gamma. Nature. 2004;429(6993):771–6. doi: 10.1038/nature02583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Takata T, Ishikawa F. Human Sir2-related protein SIRT1 associates with the bHLH repressors HES1 and HEY2 and is involved in HES1- and HEY2-mediated transcriptional repression. Biochem Biophys Res Commun. 2003;301(1):250–7. doi: 10.1016/s0006-291x(02)03020-6. [DOI] [PubMed] [Google Scholar]
- 50.Fulco M, Schiltz RL, Iezzi S, King MT, Zhao P, Kashiwaya Y, Hoffman E, Veech RL, Sartorelli V. Sir2 regulates skeletal muscle differentiation as a potential sensor of the redox state. Mol Cell. 2003;12(1):51–62. doi: 10.1016/s1097-2765(03)00226-0. [DOI] [PubMed] [Google Scholar]
- 51.Senawong T, V, Peterson J, Avram D, Shepherd DM, Frye RA, Minucci S, Leid M. Involvement of the histone deacetylase SIRT1 in chicken ovalbumin upstream promoter transcription factor (COUP-TF)-interacting protein 2-mediated transcriptional repression. J Biol Chem. 2003;278(44):43041–50. doi: 10.1074/jbc.M307477200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yeung F, Hoberg JE, Ramsey CS, Keller MD, Jones DR, Frye RA, Mayo MW. Modulation of NF-kappaB-dependent transcription and cell survival by the SIRT1 deacetylase. Embo J. 2004;23(12):2369–80. doi: 10.1038/sj.emboj.7600244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rodgers JT, Lerin C, Haas W, Gygi SP, Spiegelman BM, Puigserver P. Nutrient control of glucose homeostasis through a complex of PGC-1alpha and SIRT1. Nature. 2005;434(7029):113–8. doi: 10.1038/nature03354. [DOI] [PubMed] [Google Scholar]
- 54.Lilja H. Biology of prostate-specific antigen. Urology. 2003;62(5 Suppl 1):27–33. doi: 10.1016/s0090-4295(03)00775-1. [DOI] [PubMed] [Google Scholar]
- 55.Howitz KT, Bitterman KJ, Cohen HY, Lamming DW, Lavu S, Wood JG, Zipkin RE, Chung P, Kisielewski A, Zhang LL, Scherer B, Sinclair DA. Small molecule activators of sirtuins extend Saccharomyces cerevisiae lifespan. Nature. 2003;425(6954):191–6. doi: 10.1038/nature01960. [DOI] [PubMed] [Google Scholar]
- 56.Revollo JR, Grimm AA, Imai S. The NAD biosynthesis pathway mediated by nicotinamide phosphoribosyltransferase regulates Sir2 activity in mammalian cells. J Biol Chem. 2004;279(49):50754–63. doi: 10.1074/jbc.M408388200. [DOI] [PubMed] [Google Scholar]
- 57.Cheng S, Brzostek S, Lee SR, Hollenberg AN, Balk SP. Inhibition of the dihydrotestosterone-activated androgen receptor by nuclear receptor corepressor. Mol Endocrinol. 2002;16(7):1492–501. doi: 10.1210/mend.16.7.0870. [DOI] [PubMed] [Google Scholar]
- 58.Kang Z, Janne OA, Palvimo JJ. Coregulator recruitment and histone modifications in transcriptional regulation by the androgen receptor. Mol Endocrinol. 2004;18(11):2633–48. doi: 10.1210/me.2004-0245. [DOI] [PubMed] [Google Scholar]
- 59.Chen H, Lin RJ, Xie W, Wilpitz D, Evans RM. Regulation of hormone-induced histone hyperacetylation and gene activation via acetylation of an acetylase. Cell. 1999;98(5):675–86. doi: 10.1016/s0092-8674(00)80054-9. [DOI] [PubMed] [Google Scholar]
- 60.Harada S, Keller ET, Fujimoto N, Koshida K, Namiki M, Matsumoto T, Mizokami A. Long-term exposure of tumor necrosis factor alpha causes hypersensitivity to androgen and anti-androgen withdrawal phenomenon in LNCaP prostate cancer cells. Prostate. 2001;46(4):319–26. doi: 10.1002/1097-0045(20010301)46:4<319::aid-pros1039>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- 61.Gaughan L, I, Logan R, Cook S, Neal DE, Robson CN. Tip60 and histone deacetylase 1 regulate androgen receptor activity through changes to the acetylation status of the receptor. J Biol Chem. 2002;277(29):25904–13. doi: 10.1074/jbc.M203423200. [DOI] [PubMed] [Google Scholar]
- 62.Howitz KT, Bitterman KJ, Cohen HY, Lamming DW, Lavu S, Wood JG, Zipkin RE, Chung P, Kisielewski A, Zhang LL, Scherer B, Sinclair DA. Small molecule activators of sirtuins extend Saccharomyces cerevisiae lifespan. Nature. 2003;425(6954):191–6. doi: 10.1038/nature01960. [DOI] [PubMed] [Google Scholar]
- 63.Baur JA, Sinclair DA. Therapeutic potential of resveratrol: the in vivo evidence. Nat Rev Drug Discov. 2006;5(6):493–506. doi: 10.1038/nrd2060. [DOI] [PubMed] [Google Scholar]
- 64.Delmas D, Lancon A, Colin D, Jannin B, Latruffe N. Resveratrol as a chemopreventive agent: a promising molecule for fighting cancer. Curr Drug Targets. 2006;7(4):423–42. doi: 10.2174/138945006776359331. [DOI] [PubMed] [Google Scholar]
- 65.Aziz MH, Nihal M, Fu VX, Jarrard DF, Ahmad N. Resveratrol-caused apoptosis of human prostate carcinoma LNCaP cells is mediated via modulation of phosphatidylinositol 3′-kinase/Akt pathway and Bcl-2 family proteins. Mol Cancer Ther. 2006;5(5):1335–41. doi: 10.1158/1535-7163.MCT-05-0526. [DOI] [PubMed] [Google Scholar]
- 66.Hu X, Lazar MA. Transcriptional repression by nuclear hormone receptors. Trends Endocrinol Metab. 2000;11(1):6–10. doi: 10.1016/s1043-2760(99)00215-5. [DOI] [PubMed] [Google Scholar]
- 67.Zhang Y, Akinmade D, Hamburger AW. The ErbB3 binding protein Ebp1 interacts with Sin3A to repress E2F1 and AR-mediated transcription. Nucleic Acids Res. 2005;33(18):6024–33. doi: 10.1093/nar/gki903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bouras T, Fu M, Sauve AA, Wang F, Quong AA, Perkins ND, Hay RT, Gu W, Pestell RG. SIRT1 deacetylation and repression of p300 involves lysine residues 1020/1024 within the cell cycle regulatory domain 1. J Biol Chem. 2005;280(11):10264–76. doi: 10.1074/jbc.M408748200. [DOI] [PubMed] [Google Scholar]
- 69.Fu M, Liu M, Sauve AA, Jiao X, Zhang X, Wu X, Powell MJ, Yang T, Gu W, Avantaggiati ML, Pattabiraman N, Pestell TG, Wang F, Quong AA, Wang C, Pestell RG. Hormonal Control of Androgen Receptor Function through SIRT1. Mol Cell Biol. 2006;26(21):8122–35. doi: 10.1128/MCB.00289-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Louie MC, Yang HQ, Ma AH, Xu W, Zou JX, Kung HJ, Chen HW. Androgen-induced recruitment of RNA polymerase II to a nuclear receptor-p160 coactivator complex. Proc Natl Acad Sci U S A. 2003;100(5):2226–30. doi: 10.1073/pnas.0437824100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Cronauer MV, Schulz WA, Burchardt T, Anastasiadis AG, de la Taille A, Ackermann R, Burchardt M. The androgen receptor in hormone-refractory prostate cancer: relevance of different mechanisms of androgen receptor signaling (Review) Int J Oncol. 2003;23(4):1095–102. doi: 10.3892/ijo.23.4.1095. [DOI] [PubMed] [Google Scholar]
- 72.Harada S, Keller ET, Fujimoto N, Koshida K, Namiki M, Matsumoto T, Mizokami A. Long-term exposure of tumor necrosis factor alpha causes hypersensitivity to androgen and anti-androgen withdrawal phenomenon in LNCaP prostate cancer cells. Prostate. 2001;46(4):319–26. doi: 10.1002/1097-0045(20010301)46:4<319::aid-pros1039>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- 73.Culig Z, Hoffmann J, Erdel M, Eder IE, Hobisch A, Hittmair A, Bartsch G, Utermann G, Schneider MR, Parczyk K, Klocker H. Switch from antagonist to agonist of the androgen receptor bicalutamide is associated with prostate tumour progression in a new model system. Br J Cancer. 1999;81(2):242–51. doi: 10.1038/sj.bjc.6690684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Seligson DB, Horvath S, Shi T, Yu H, Tze S, Grunstein M, Kurdistani SK. Global histone modification patterns predict risk of prostate cancer recurrence. Nature. 2005;435(7046):1262–6. doi: 10.1038/nature03672. [DOI] [PubMed] [Google Scholar]
- 75.Moynihan KA, Grimm AA, Plueger MM, Bernal-Mizrachi E, Ford E, Cras-Meneur C, Permutt MA, Imai S. Increased dosage of mammalian Sir2 in pancreatic beta cells enhances glucose-stimulated insulin secretion in mice. Cell Metab. 2005;2(2):105–17. doi: 10.1016/j.cmet.2005.07.001. [DOI] [PubMed] [Google Scholar]
- 76.Leibiger IB, Berggren PO. Cell Metab. 2005;2(2):80–2. doi: 10.1016/j.cmet.2005.07.006. [DOI] [PubMed] [Google Scholar]
- 77.Lemieux ME, Yang X, Jardine K, He X, Jacobsen KX, Staines WA, Harper ME, McBurney MW. The Sirt1 deacetylase modulates the insulin-like growth factor signaling pathway in mammals. Mech Ageing Dev. 2005;126(10):1097–105. doi: 10.1016/j.mad.2005.04.006. [DOI] [PubMed] [Google Scholar]
- 78.Kennedy BJ. Aging and cancer. Oncology (Williston Park) 2000;14(12):1731–3. discussion 1734, 1739–40. [PubMed] [Google Scholar]
- 79.Michishita E, Park JY, Burneskis JM, Barrett JC, Horikawa I. Evolutionarily conserved and nonconserved cellular localizations and functions of human SIRT proteins. Mol Biol Cell. 2005;16(10):4623–35. doi: 10.1091/mbc.E05-01-0033. [DOI] [PMC free article] [PubMed] [Google Scholar]