

Abstract
Fus1 is a tumor suppressor protein with recently described immunoregulatory functions. Although its role in sterile inflammation is being elucidated, its role in regulating immune responses to infectious agents has not been examined. We used here a murine model of Acinetobacter baumannii pneumonia to identify the role of Fus1 in antibacterial host defenses. We found that the loss of Fus1 in mice results in significantly increased resistance to A. baumannii pneumonia. We observed earlier and more robust recruitment of neutrophils and macrophages to the lungs of infected Fus1−/− mice, with a concomitant increase in phagocytosis of invading bacteria and more rapid clearance. Such a prompt and enhanced immune response to bacterial infection in Fus1−/− mice stems from early activation of proinflammatory pathways (NF-κB and phosphatidylinositol 3-kinase/Akt/mammalian target of rapamycin [mTOR]), most likely due to significantly increased mitochondrial membrane potential and mitochondrial reactive oxygen species production. Significant early upregulation of interleukin-17 (IL-17) in Fus1−/− immune cells was also observed, together with significant downregulation of IL-10. Depletion of neutrophils eliminates the enhanced antibacterial defenses of the Fus1−/− mice, suggesting that ultimately it is the enhanced immune cell recruitment that mediates the increased resistance of Fus1−/− mice to A. baumannii pneumonia. Taken together, our data define the novel role for Fus1 in the immune response to A. baumannii pneumonia and highlight new avenues for immune modulating therapeutic targets for this treatment-resistant nosocomial pathogen.
INTRODUCTION
The mitochondrial protein Fus1 was first identified as a tumor suppressor involved in the pathobiology of tumors associated with chronic inflammation, such as lung cancer and mesothelioma (1–3). However, biological and molecular functions of Fus1 in healthy tissues were not recognized until recently. Studying a Fus1 mouse knockout model, we identified an immunoregulatory function of Fus1 as these mice showed aberrations in immune cell subsets, developed an SLE-like autoimmunity, and showed spontaneous tumors with age (4). Moreover, Fus1−/− mice demonstrate an altered acute inflammatory response to asbestos. Fus1−/− immune cells exhibit altered dynamics of pro- and anti-inflammatory cytokines (gamma interferon [IFN-γ], tumor necrosis factor alpha [TNF-α], interleukin-1α [IL-1α], IL-1β, and IL-10) and profound basal and asbestos-induced changes in major mitochondrial parameters (reactive oxygen species [ROS] production, membrane mitochondrial potential, and UCP2 expression) (5).
The studies described above delineated roles for Fus1 in autoimmunity, tumorigenesis, and other sterile inflammatory reactions. However, the contribution of Fus1 to the inflammatory response to infectious agents is not yet defined. Recent research has highlighted mitochondria as a centrally positioned hub for regulation of innate and adaptive immune responses (6). Mounting evidence suggests that mitochondria facilitate antibacterial immunity by generating ROS and contribute to innate immune activation following cellular damage and stress (7). Thus, we hypothesized that Fus1, as a mitochondrial resident and immunoregulatory protein, modulates antibacterial host defense.
Acinetobacter baumannii is an opportunistic pathogen of growing importance in the hospital setting. This organism is particularly problematic due to its propensity to acquire antibiotic resistance determinants, resulting in high rates of multidrug resistance. In fact, current multidrug resistance rates range from 48 to 85% of isolates, with the highest rates in Asia and Eastern Europe (8–12). The growing burden of A. baumannii-associated disease has prompted investigation of this organism's pathogenic mechanisms, as well as the components of the host immune response necessary for protection against A. baumannii infection. Neutrophils play a critical role in defense against A. baumannii infection (13). Depletion of neutrophils, loss of neutrophil function, or disruption of signaling pathways that promote neutrophil recruitment to the lungs renders hosts more susceptible to A. baumannii infection (13–16). Conversely, increased recruitment of neutrophils to the lungs promotes A. baumannii clearance and host resistance to infection (17). Like neutrophils, alveolar macrophages are important in mediating direct clearance of A. baumannii through phagocytosis of bacteria in the lungs but also play an important role in initiating signaling pathways that recruit neutrophils to the lung (18). Given that Fus1 modulates inflammatory parameters that are typically induced upon A. baumannii infection and the availability of a reproducible and clinically relevant A. baumannii pneumonia model, we sought to define the role for Fus1 in host defenses against A. baumannii lung infections.
MATERIALS AND METHODS
Animals and housing conditions.
Fus1−/− mice were generated by A.I. in the laboratory of M. Lerman (NCI-Frederick). The mice were extensively backcrossed to obtain a homogenous 129sv genetic background in the laboratory of S. Andersen (NCI-Frederick), and mating pairs were obtained from his laboratory. All animal experiments were performed according to a protocol approved by the Institutional Animal Care and Use Committee at Vanderbilt University. The mice were fed a standard diet. The animals were housed four per cage in standard cages and under a 12-h-light/12-h-dark light cycle. The animals had ad libitum access to drinking water and the normal diet throughout the experiment.
Experimental infections and tissue processing.
Seven- to nine-week-old male mice were used for all experiments unless stated otherwise. Mice were infected using an A. baumannii pneumonia protocol established in our laboratory (19, 20). Briefly, mice were anesthetized with an intraperitoneal injection of Avertin. Log-phase bacteria were suspended in phosphate-buffered saline (PBS) to a final density of 107 CFU/μl, and 30 μl of this suspension was inoculated intranasally into anesthetized mice. Where indicated, neutrophils were depleted by intraperitoneal injection of 250 μg of rat IgG2b anti-Gr-1 monoclonal antibody RB6-8C5 in 100 μl of PBS 24 h prior to infection with a repeated injection at the time of infection. For these experiments, the bacteria were resuspended at 106 CFU/μl to avoid the increased mortality observed with higher inoculation doses. For all experiments, mice were euthanized at the indicated time points postinfection, and lungs and spleens were aseptically harvested. For quantification of bacterial burdens, the lungs were homogenized, and the resulting homogenate was serially diluted and plated onto nonselective agar for determination of the CFU. Bacterial burdens are expressed as CFU/g of lung tissue. For the isolation of BALF cells, mice were euthanized, and the lungs were cannulized and washed three times with 0.5 to 1.0 ml of sterile PBS. When quantitative bacteriology was not required, lungs were either processed for microscopy, or tissues were flash frozen and later used for RNA and protein isolation. For microscopic analyses, the lungs were gently inflated and fixed in 10% formalin, followed by paraffin embedding and sectioning. Sections were stained with hematoxylin and eosin (H&E) for histological examination.
Cytospin preparation.
BALF cells were routinely pretreated with ammonium-chloride-potassium (ACK) buffer to remove erythrocytes. A total of 105 leukocytes were washed in cold 2% fetal calf serum-PBS twice and diluted in 100 μl of cold 1% bovine serum albumin-PBS. The cells were loaded into filter/slide cassettes and spun at 900 to 1,000 rpm for 5 min. Filters were carefully removed from their slides, and the cells were immediately fixed in 95% alcohol for 10 min and then air dried. Cells were stained with a Diff Quick kit, dried, and covered with cover glasses.
Real-time PCR.
Total RNA was extracted from BALF cells by using a miniRNA isolation kit (Qiagen, Inc.). First strand DNA was built on RNA template using First Strand Kit (Invitrogen, Inc.), followed by real-time PCRs using SYBR green PCR kit (Bio-Rad, Inc.). All primer pairs for real-time PCR were obtained from Origene, Inc. The expression values were normalized to the GusB housekeeping gene.
Cytokine analysis.
Approximately 100 mg of lung tissue was homogenized in radioimmunoprecipitation assay (RIPA) lysis buffer supplemented with a protease inhibitor cocktail. Total protein concentrations in lysates were determined by using a BCA quantification kit according to the manufacturer's recommendations (Pierce). Samples were normalized according to total protein concentration. Cytokine concentrations in lung lysates were determined by using a Becton Dickinson (BD) cytokine bead array mouse inflammation kit (IL-6, IL-10, TNF-α, IL-12p70, IFN-γ, and MCP-1) and a FACSCalibur flow cytometer according to the manufacturer's protocols. Cytokine concentrations in lung lysates were calculated from a standard curve produced using a set of purified IL-6, IL-10, TNF-α, IL-12p70, IFN-γ, and MCP-1 standards.
Complete blood analysis.
Automated blood cell counts, including white blood cells and red blood cells, hemoglobin, hematocrit, indicies, and platelet counting were performed in the Histopathology Core Laboratory, Vanderbilt University School of Medicine (VUMC).
Western blotting.
For Western blotting, lung lysates were prepared by using RIPA lysis buffer supplemented with protease inhibitor cocktail (Sigma, Inc.). A total of 20 to 40 μg of protein was loaded per lane. After electrophoresis using SDS-PAGE 4 to 20% gels followed by semidry transfer, nitrocellulose membranes were blocked with PBS containing 0.1% Tween 20 and 5% skimmed milk (TPBS–5% milk) and then probed overnight with primary antibodies (1:1,000 in TPBS–5% milk). The pNFkB, pERK1/2, total ERK1/2, PTEN, and phospho-S6 ribosomal protein anti-rabbit antibodies and NF-κB anti-mouse antibodies were from Cell Signaling, Inc. The S6 ribosomal protein anti-mouse antibodies were from Santa Cruz Biotechnology, Inc. The membranes were then washed with TPBS and incubated for 1 h in TPBS–5% milk containing the corresponding peroxidase-conjugated secondary antibody (1:10,000). After a washing step in TPBS, enhanced chemiluminescence (Pierce) was performed to visualize the peroxidase-coated bands.
Cell preparation and flow cytometric analysis of ΔΨ and ROS.
Single-cell suspensions were prepared from BALF cells, and spleens were depleted of red blood cells by using ACK buffer. Stained cells were analyzed with a FACSCalibur (BD). CellQuest (BD) and FlowJo (Tree Star, Inc., Ashland, OR) software were used for the acquisition and analysis of data, respectively. The production of ROS was assessed using oxidation-sensitive 2′-7′-dichlorodihydrofluorescein diacetate (CM-H2DCFDA). The ΔΨ was estimated by using 5,5′,6,6′-tetrachloro-1,1′,3,3′ tetraethylbenzimidazolocarbocyanine iodide (JC-1; Invitrogen), a potential-dependent J-aggregate-forming lipophilic cation. JC-1 is selectively accumulated in mitochondria, where it forms monomers (green fluorescence, 527 nm) or aggregates, at high transmembrane potentials (red fluorescence, 590 nm). The cells were incubated with 0.5 μM JC-1 for 15 to 30 min at 37°C and then washed with PBS before flow cytometry. Treatment of cells with a protonophore CCCP, 5 μM carbonyl cyanide m-chlorophenylhydrazone (Sigma-Aldrich, St. Louis, MO), for 15 to 30 min at 37°C resulted in decreased JC-1 fluorescence and was used as a positive control for the disruption of ΔΨ.
RESULTS
Fus1−/− mice are more resistant to A. baumannii infection than Fus1+/+ mice.
In order to determine the role for Fus1 in the immune response against A. baumannii, we first infected Fus1+/+ and Fus1−/− mice with A. baumannii using a pneumonia model previously established in our laboratory. Male Fus1−/− mice were found to be more resistant to A. baumannii infection than Fus1+/+ males (Fig. 1a). Interestingly, the same effect was not observed in female mice (Fig. 1b). Notably, female mice exhibited higher bacterial burdens on average compared to males, suggesting a reduced ability to clear A. baumannii infection that may mask any effect of Fus1 on bacterial killing in the lung. The results of these infections demonstrate that the loss of Fus1 leads to increased resistance to A. baumannii infection and supports a possible immunomodulatory role for this protein in antibacterial host defenses.
Fig 1.
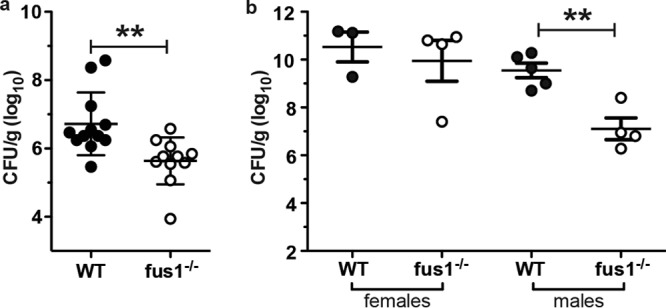
Loss of Fus1 function leads to reduced susceptibility to A. baumannii infection. (a) Bacterial burden in lungs of male Fus1+/+ (WT) and Fus1−/− (KO) mice infected with A. baumannii and harvested at 48 hpi. **, P < 0.01 as determined by two-tailed Student t test. (b) Bacterial burden in the lungs of male and female Fus1+/+ and Fus1−/− mice infected and harvested at 48 hpi with A. baumannii.
Histopathological analysis shows early neutrophil recruitment and earlier clearance of A. baumannii in Fus1−/− mice.
Histopathological analysis of lungs and complete blood analysis performed on mice prior to A. baumannii challenge did not show any significant difference between WT and KO mice (data not shown). However, at 4 h postinfection (hpi) Fus1−/− and Fus1+/+ mice developed different inflammatory responses to A. baumannii infection in the lungs. Fus1+/+ lungs were characterized by prominent margination of leukocytes in pulmonary vessels and by the presence of significant areas of perivascular edema associated with a large number of organisms in the perivenous space (Fig. 2a). In Fus1−/− lungs at this time point, neutrophils were already present in the interstitium with minimal leukocyte margination observed in blood vessels, suggesting earlier neutrophil recruitment to Fus1−/− lungs. Inflammatory infiltrates in the parenchyma of Fus1+/+ lungs at 8 hpi consisted primarily of neutrophils with few macrophages. Notably, there were numerous free bacteria within alveolar spaces observed, whereas bacterial phagocytosis was seen rarely (Fig. 2b). In the Fus1−/− lungs at 8 hpi, the inflammatory infiltrate in the alveoli was composed of both neutrophils and macrophages. Free bacteria were not observed within the alveolar space of Fus1−/− lungs, whereas phagocytosis of bacteria by macrophages was common, indicating earlier clearance of bacteria by macrophages. Finally, at 48 hpi the Fus1+/+ lungs still showed extensive inflammation in the interstitium that consisted of neutrophils and macrophages, and their cytoplasm was filled with phagocytosed bacteria (Fig. 2c). In Fus1−/− lungs at this time point, only small patches of inflammation remained, and they consisted of mostly neutrophils and fewer macrophages. Remarkably, lungs of Fus1−/− mice were completely free of visible bacteria by 48 hpi. These histopathological observations clearly indicate that in Fus1−/− mice, leukocytes are recruited into infected tissues earlier; they actively phagocytose bacteria and clear the lungs at earlier time points than observed in Fus1+/+ mice.
Fig 2.

Histopathological analysis of A. baumannii-induced immune response in Fus1+/+ (WT) and Fus1−/− (KO) lungs at different time points after infection. (a) 4 hpi, H&E staining. The upper row shows different magnifications of Fus1−/− lungs distinguished by the presence of neutrophils in the interstitium with minimal leukocyte margination observed in blood vessels. In contrast, the bottom row illustrates the late response to bacterial challenge in Fus1+/+ lungs characterized by margination of leukocytes (black arrows) and perivascular edema (red arrows) in pulmonary vessels. (b) 8 hpi, H&E staining. In the left panel, Fus1−/− lungs are presented with alveolar inflammatory infiltrate composed of neutrophils and macrophages with phagocytosed or bound bacteria (arrows) and no trace of free-floating bacteria within the alveolar spaces. In the right panel, Fus1+/+ lungs are characterized by inflammatory infiltrates consisting primarily of neutrophils, numerous free bacteria within alveolar spaces (arrows), and no macrophage-associated bacteria. (c) 48 hpi, H&E staining. In the upper row, Fus1−/− lungs are presented with areas of inflammation that are small and consist of neutrophils and fewer macrophages; bacteria are not observed. In the bottom row, Fus1+/+ lungs are presented with extensive inflammation in the interstitium that consists of neutrophils and macrophages filled with bound or phagocytosed bacteria.
Total cell and macrophage counts were increased in BALF from Fus1−/− mice compared to Fus1+/+ mice after infection.
An antibacterial immune response is characterized by an early cell infiltration into infected organs. Thus, we counted immune cells in the lungs of Fus1+/+ and Fus1−/− mice at 8 and 36 hpi. The total cell count was increased in BALF from infected Fus1−/− mice at both 8 and 36 hpi (Fig. 3a and b). Although neutrophils accounted for the majority of the cells in BALF at 8 hpi, there was a significant increase in the relative ratio of macrophages in BALF from Fus1−/− mice at this early time point (Fig. 3c). Large, foamy macrophages loaded with ingested bacteria were identified in cytospin preparations from the BALF of Fus1−/− mice, suggesting that early macrophage activation and phagocytosis is impeded by functional Fus1 (Fig. 3d).
Fig 3.

Early immune response to A. baumannii infection in Fus1−/− mice is characterized by abundant immune cell infiltrate and a higher number of macrophages. (a and b) Total numbers of immune infiltrate cells per lung at 8 and 36 hpi, respectively, infected with A. baumannii. (c and d) Differential count of macrophages in BALF cells (c) and cytospin prep of BALF cells (d) at 8 hpi of Fus1+/+ (WT) and Fus1−/− (KO) mice. Note the larger size of Fus1−/− macrophages (red arrows) and Fus1−/− macrophage-associated bacteria (insets). The data represent means ± the standard errors (*, P ≤ 0.05; **, P ≤ 0.005) as determined by using the Student t test (n = 3 to 5 mice per group).
BALF cells of infected Fus1+/+ and Fus1−/− mice differ in production of pro- and anti-inflammatory cytokines.
Fus1−/− mice exhibit an increased ability to clear A. baumannii from the lung likely secondary to increased recruitment of neutrophils and macrophages to the site of infection. In models of sterile inflammation, loss of Fus1 leads to alterations in pro- and anti-inflammatory cytokines. We examined the abundance of IL-6, IL-10, IL-12p70, TNF-α, IFN-γ, and MIP-1 in the lungs of A. baumannii-infected mice at 12 and 48 hpi. We also examined the expression of IL-17 and IL-10 through real-time PCR on RNA isolated from BALF cells at 36 hpi. Interestingly, we found no statistically significant differences in the abundance of proinflammatory cytokines at either 12 or 48 hpi, although there was a trend toward increased IFN-γ at 12 hpi and of IL-12p70 at 48 hpi (Fig. 4a). However, RNA analyses demonstrated a significant increase in IL-17 transcript at 36 hpi (Fig. 4b). Interestingly, IL-10, which has both pro- and anti-inflammatory properties, was significantly increased in Fus1−/− lungs at 12 hpi, but decreased compared to Fus1+/+ by 48hpi (Fig. 4a). The latter finding is consistent with transcriptional analyses, which also demonstrated decreased significantly IL-10 expression at 36 hpi (Fig. 4c).
Fig 4.

Cytokine analysis in lung lysates and BALF cells from Fus1+/+ (WT) and Fus1−/− (KO) mice at different time points postinfection. (a) Cytokine quantification in whole-lung lysates from A. baumannii-infected Fus1−/− and Fus1+/+ mice at 12 and 48 hpi. *, P < 0.05 (one-way analysis of variance). (b and c) Real-time PCR analysis of IL-17 and IL-10 transcript levels in BALF cells collected from Fus1−/− and Fus1+/+ lungs at 36 hpi with A. baumannii. Total RNA was isolated from BALF cells, and expression of the mRNA was analyzed by real-time PCR and normalized to the GusB gene. The bar graph shows relative levels of cytokine expression. The data represent means ± the standard errors (*, P ≤ 0.05) as determined by using the Student t test comparing Fus1+/+ to Fus1−/− (n = 3 to 5 mice per group).
BALF cells from A. baumannii-infected Fus1−/− mice have higher levels of reactive oxygen species (ROS), mitochondrial membrane potential (ΔΨ) and UCP2 expression.
In our earlier work, we showed that Fus1 activity in mitochondria is crucial for maintenance of basal and stress-induced (asbestos exposure) mitochondrial parameters, including ROS production, ΔΨ, and the expression of uncoupling protein 2 (Ucp2) (5). Since ROS produced in monocytes protect mice from bacterial infections (21), we measured the number of high ROS-producing cells in BALF cells at 12 hpi, which corresponds to the time point of maximal monocyte infiltration. Immune cells infiltrating lungs of Fus1−/− mice encompassed twice as many cells producing high ROS levels as those from Fus1+/+ mice (not shown). Interestingly, at 48 hpi we did not find such a difference in ROS-producing cells between the wild type (WT) and knockout mice (KO), suggesting that an early increase in ROS production is crucial for early bacterial clearance. We also measured ΔΨ in splenocytes and BALF cells of Fus1+/+ and Fus1−/− mice at 36 hpi with A. baumannii. We found that whereas the ΔΨ value of splenocytes from infected Fus1−/− mice did not show a statistically significant difference from Fus1+/+ mice, the ΔΨ value of BALF cells demonstrated a pronounced difference compared to Fus1+/+ mice (Fig. 5a).
Fig 5.

Fus1−/− BALF cells show alterations of mitochondrial membrane potential (ΔΨ) and UCP2 expression after A. baumannii infection. (a) Evaluation of infection-induced changes of mitochondrial membrane potential (ΔΨ) in BALF cells and splenocytes from Fus1+/+ (WT) and Fus1−/− (KO) mice at 36 hpi. ΔΨ was measured via fluorescence-activated cell sorting analysis of cells stained with potential-dependent lipophilic cationic dye, JC-1, that is selectively accumulated in mitochondria, where it forms monomers at low ΔΨ or forms aggregates at high ΔΨ, which results in different fluorescence colors. Curves show right-shift (hyperpolarization) in FL-2 fluorescence observed in Fus1−/− cells (gray line) compared to wild-type cells (black line). (b) Expression of Ucp2 in BALF cells of infected mice at 36 hpi was assessed by real-time RT-PCR and normalized to GusB. All data represent means ± the standard errors. *, P < 0.05; **, P < 0.01 (Fus1+/+ versus Fus1−/−; n = 3 to 5 mice per group).
Mitochondrial UCP2 controls immune cell activation via regulation of ΔΨ that, in turn, is positively correlated with ROS production (22, 23). Given the contribution of mROS to bacterial killing and the observed increase in ΔΨ in Fus1−/− cells harvested from A. baumannii-infected mice, we analyzed Ucp2 expression in BALF cells from Fus1+/+ and Fus1−/− mice at 36 hpi via real-time reverse transcription-PCR (RT-PCR). We found that the Ucp2 expression level was significantly higher in Fus1−/− cells, suggesting that these cells are responding to increased mitochondrial membrane potential (Fig. 5b). Taken together, these data demonstrate that loss of Fus1 function leads to increased ΔΨ and ROS levels during the immune response to bacteria.
Early activation of antibacterial defense pathways in Fus1−/− lungs.
Since our histopathological findings clearly demonstrate earlier infiltration of neutrophils and macrophages into Fus1−/− lungs upon A. baumannii infection, we analyzed activation of molecular pathways involved in early lymphocyte recruitment in the infected lungs. Protein lysates from A. baumannii-infected lungs of Fus1+/+ and Fus1−/− mice were analyzed at 4 hpi for activation of the phosphatidylinositol 3-kinase (PI3K)/Akt/mammalian target of rapamycin (mTOR) pathway, the NF-κB pathway, and the PI3K antagonist, phosphatase, and tensin homolog (PTEN) pathway.
Fus1−/− lungs responded to infection with an early decrease in the PI3K antagonist PTEN level, while no PTEN changes were observed in Fus1+/+ mice at this time point (Fig. 6a). Consistent with this finding, activation or phosphorylation of the PI3K/Akt/mTOR pathway based on the S6/pS6 ratio was significantly higher in Fus1−/− lungs compared to Fus1+/+ lungs (Fig. 6a). Finally, NF-κB activation (p65/pp65) in Fus1−/− lungs was increased, while no significant activation of the NF-κB was observed in Fus1+/+ mice at this early time point after infection (Fig. 6a).
Fig 6.
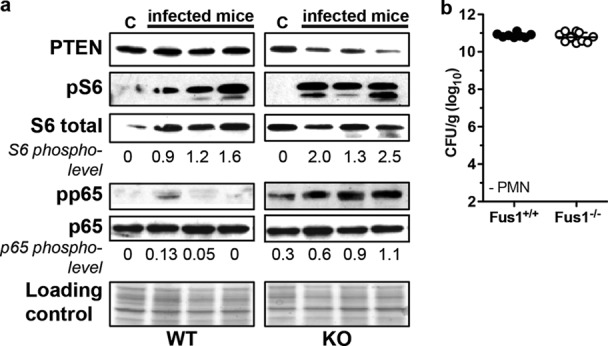
Early activation of antibacterial pathways in Fus1−/− lungs. (a) Activation of markers of immune response to bacterial infection was assessed by Western blotting on total lung lysates obtained from infected Fus1+/+ (WT) and Fus1−/− (KO) mice at 4 hpi and compared to control (lanes C, uninfected) mice. Ponceau S staining of the membrane after protein transfer is shown as a loading control. PTEN/PI3K, NF-κB, and mTOR pathways were evaluated. Activation levels of NF-κB and mTOR pathways were assessed by Western blot analysis of p65 and S6 protein activation. Phosphorylation of p65 (pp65) and ribosomal protein S6 (pS6) was determined and normalized to total proteins (the activation index is shown under each lane). Three mice (1 to 3) were examined in each infected group. (b) Depletion of neutrophils restores the susceptibility of Fus1−/− mice to A. baumannii infection. The bacterial burdens in Fus1+/+ and Fus1−/− mice after antibody-mediated neutrophil depletion and subsequent infection with A. baumannii were determined. The bacterial burdens are expressed as CFU/g of lung tissue.
Neutrophil depletion eliminates differences in resistance to A. baumannii infection between Fus1+/+ and Fus1−/− mice.
Our molecular analysis showed that loss of Fus1 clearly predisposes lung tissues to early antibacterial responses due to early activation of molecular machinery involved in recruitment of neutrophils. Neutrophils are critical for host resistance to A. baumannii infection (13, 15, 24). To further examine the role for early recruitment of neutrophils in resistance to infection in the Fus1−/− mice, we depleted neutrophils prior to infection with A. baumannii via intraperitoneal injection of anti-neutrophil antibodies. Neutrophil depletion eliminated the differential clearance of A. baumannii between Fus1+/+ and Fus1−/− mice (Fig. 6b). Taken together, our data demonstrate that loss of Fus1 promotes both neutrophil- and macrophage-mediated antibacterial defenses in the lung.
DISCUSSION
A. baumannii is a nosocomial pathogen capable of causing life-threatening community and health care-associated infections. Despite significant health problems associated with this pathogen, the components of the host immune response necessary for protection against A. baumannii infection are incompletely understood. We identify here that the loss of the mitochondrial protein Fus1 augments the host response to A. baumannii, resulting in earlier bacterial clearance and the resolution of infection.
Upon infection of the murine lung, A. baumannii induces a robust inflammatory response including recruitment of neutrophils, activation of local macrophages, and the production proinflammatory mediators (13, 18). A delay in the early pulmonary recruitment of neutrophils in A/J mice is associated with high susceptibility to A. baumannii infection (24). Based on this finding, it would seem plausible that the reverse of this situation would also be true. Here we have demonstrated that recruitment of lymphocytes into infected lungs of Fus1−/− mice occurs earlier and more robustly than in WT mice and that this confers augmented resistance of Fus1−/− mice to A. baumannii infection. Recognition of bacterial lipopolysaccharides by Toll-like receptors (TLRs) induces airway epithelial NF-κB activation, which promotes the production of chemokines involved in neutrophil recruitment and activation (25, 26). Through this mechanism, engagement of TLRs regulates host defense following infections caused by Acinetobacter baumannii, Pseudomonas aeruginosa, and Mycoplasma pneumoniae (27–29). Our analysis of NF-κB activation in lung tissues of infected animals showed that at 4 hpi with A. baumannii, NF-κB was significantly phosphorylated in Fus1−/− mice, whereas no phosphorylation was observed in WT mice. Moreover, lung tissues of control Fus1−/− mice also showed some level of NF-κB activation (Fig. 6a), suggesting that the earlier recruitment of lymphocytes in response to infection may be due to a basal preactivated NF-κB state. In agreement with these data, we recently showed that in Fus1−/− CD4+ T cells, activation of the NF-κB pathway is prominent at a basal level that we associated with a perturbed mitochondrial Ca2+ homeostasis and other coupled mitochondrial parameters in these cells (30).
NF-κB activation depends on several factors associated with mitochondrial activities, one of which is ROS production (31). Earlier, we showed that Fus1 KO immune cells produce higher ROS in response to asbestos than WT immune cells (5). Here, we found that at 12 hpi of Fus1−/− mice contained twice as many cells producing higher levels of ROS than WT mice, suggesting that further activation of NF-κB early after infection in Fus1−/− mice may be due to increased ROS production. Likewise, earlier activation/phosphorylation of the PI3K/Akt/mTOR pathways (Fig. 6a) that is involved in chemotaxis of neutrophils may also be due to higher ROS production in Fus1−/− mice (32–34). These data introduce the possibility that increased ROS production in Fus1−/− mice contributes to increased NF-κB and mTOR pathway activity, which in turn increases neutrophil recruitment (26, 27, 35–37).
It is established that activation of NF-κB results in downregulation of PTEN expression (38–40). The downregulation of PTEN leads to increased recruitment of macrophages and elevated levels of proinflammatory cytokines and chemokines in inflamed lungs, which, in turn, is thought to induce increased neutrophil recruitment to the lungs. Conversely, depleting PTEN significantly delays apoptosis and enhances the bactericidal capability of recruited neutrophils (41). Consistent with these known roles for PTEN in regulating inflammatory responses in the lungs, we observed decreased PTEN expression in Fus1−/− lungs following A. baumannii infection compared to Fus1+/+ lungs. In response to pathogens and other inflammatory stimuli, neutrophils form neutrophil extracellular traps (NETs), which capture and kill extracellular microbes (42). Deficient NET formation predisposes humans to severe infection (43, 44). The mammalian target of rapamycin (mTOR) pathway regulates NET formation and thus is crucial for neutrophil antibacterial defense (45). Although a direct role for NET formation in defense against A. baumannii infection has not been shown, NETs contain significant levels of the Zn- and Mn-binding protein complex, calprotectin (46, 47). Calprotectin plays an important role in defense against A. baumannii infection through chelation of nutrient Mn and Zn (19). We also showed recently that expression of S100A8 and S100A9, the components of calprotectin complex, is 3-fold higher in Fus1−/− T cells than in Fus1+/+ cells (30). Interestingly, while other groups have shown that in WT C57BL/6 mice IL-17 does not play a significant role in defense against A. baumannii infection, we have shown significant upregulation of IL-17 in Fus1−/− mice after A. baumannii intranasal challenge. Given that calprotectin is positively regulated by IL-17, it is possible that in Fus1-deficient mice, significant upregulation of IL-17 and mTOR coordinately increase calprotectin-mediated inhibition of A. baumannii proliferation in the lung (48). It is possible that these conflicting data regarding the role for IL-17 in defense against A. baumannii infection may be due to differences in the genetic background of the mice. However, it is also possible that under normal conditions, the level of IL-17 induced by A. baumannii is insufficient to contribute significantly to A. baumannii defenses. It is only when greater levels of IL-17 are produced that this cytokine exerts a beneficial effect on pathogen clearance.
Analyzing Fus1-dependent mitochondrial parameters (5), we found that the membrane mitochondrial potential (ΔΨ) and UCP2 expression in Fus1−/− immune cells infiltrated into infected lung are higher than in Fus1+/+ cells. UCP2 is a mitochondrial membrane protein, which regulates ΔΨ (49). UCP2 is upregulated in phagocytes engulfing apoptotic cells. Consistent with this observation, loss of UCP2 reduces phagocytic capacity, whereas UCP2 overexpression enhances phagocytosis (50). Macrophages from Ucp2-deficient mice are impaired in phagocytosis in vitro, and Ucp2-deficient mice have profound in vivo defects in clearing dying cells in the thymus and testes (51, 52). Here, we show that at 36 hpi Fus1−/− BALF cells, consisting mostly of phagocytes, have increased ΔΨ and high compensatory expression of UCP2 compared to Fus1+/+ cells. Since UCP2 expression in phagocytes is positively linked to enhanced clearance of apoptotic bodies, it is tempting to speculate that upregulation of UCP2 is one of the mechanisms mediating enhanced clearance of A. baumannii from Fus1−/− lungs.
Fus1 has primarily been recognized for roles in tumorigenesis, autoimmunity and sterile inflammatory processes. Here we demonstrate that Fus1 also plays a role in host defenses. Loss of Fus1 leads to enhanced antibacterial host defenses through enhanced immune cell recruitment, increased IL-17 production and increased mROS and ΔΨ generation. Our data suggest that Fus1 may act to reduce inflammatory responses through regulation of mitochondrial inflammatory pathway, perhaps as a means of preventing excessive tissue damage. Notably, Fus1−/− mice have improved antibacterial host defenses without a significant reciprocal increase in collateral tissue damage. Taken together, these data raise the intriguing possibility that Fus1-dependent pathways may be targets for immune modulating therapeutics for the treatment of A. baumannii infections.
ACKNOWLEDGMENTS
This study was supported by National Institute of Health grants 1R21ES017496 (A.I.), R01 AI101171 (E.P.S.), and T32 GM07347 (Vanderbilt University Medical Scientist Training Program) and by the Howard Hughes Medical Institute (International Student Research Fellowship to M.I.H.).
Footnotes
Published ahead of print 16 September 2013
REFERENCES
- 1.Ivanova AV, Ivanov SV, Prudkin L, Nonaka D, Liu Z, Tsao A, Wistuba I, Roth J, Pass HI. 2009. Mechanisms of FUS1/TUSC2 deficiency in mesothelioma and its tumorigenic transcriptional effects. Mol. Cancer 8:91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Prudkin L, Behrens C, Liu DD, Zhou X, Ozburn NC, Bekele BN, Minna JD, Moran C, Roth JA, Ji L, Wistuba II. 2008. Loss and reduction of FUS1 protein expression is a frequent phenomenon in the pathogenesis of lung cancer. Clin. Cancer Res. 14:41–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wistuba II, Behrens C, Virmani AK, Mele G, Milchgrub S, Girard L, Fondon JW, III, Garner HR, McKay B, Latif F, Lerman MI, Lam S, Gazdar AF, Minna JD. 2000. High resolution chromosome 3p allelotyping of human lung cancer and preneoplastic/preinvasive bronchial epithelium reveals multiple, discontinuous sites of 3p allele loss and three regions of frequent breakpoints. Cancer Res. 60:1949–1960 [PubMed] [Google Scholar]
- 4.Ivanova AV, Ivanov SV, Pascal V, Lumsden JM, Ward JM, Morris N, Tessarolo L, Anderson SK, Lerman MI. 2007. Autoimmunity, spontaneous tumourigenesis, and IL-15 insufficiency in mice with a targeted disruption of the tumour suppressor gene Fus1. J. Pathol. 211:591–601 [DOI] [PubMed] [Google Scholar]
- 5.Uzhachenko R, Issaeva N, Boyd K, Ivanov SV, Carbone DP, Ivanova AV. 2012. Tumour suppressor Fus1 provides a molecular link between inflammatory response and mitochondrial homeostasis. J. Pathol. 227:456–469 [DOI] [PubMed] [Google Scholar]
- 6.West AP, Shadel GS, Ghosh S. 2011. Mitochondria in innate immune responses. Nat. Rev. Immunol. 11:389–402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.West AP, Brodsky IE, Rahner C, Woo DK, Erdjument-Bromage H, Tempst P, Walsh MC, Choi Y, Shadel GS, Ghosh S. 2011. TLR signaling augments macrophage bactericidal activity through mitochondrial ROS. Nature 472:476–480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Erdem I, Ozgultekin A, Sengoz Inan A, Dincer E, Turan G, Ceran N, Ozturk Engin D, Senbayrak Akcay S, Akgun N, Goktas P. 2008. Incidence, etiology, and antibiotic resistance patterns of gram-negative microorganisms isolated from patients with ventilator-associated pneumonia in a medical-surgical intensive care unit of a teaching hospital in Istanbul, Turkey (2004–2006). Jpn. J. Infect. Dis. 61:339–342 [PubMed] [Google Scholar]
- 9.Garza-González E, Llaca-Díaz JM, Bosques-Padilla FJ, González GM. 2010. Prevalence of multidrug-resistant bacteria at a tertiary-care teaching hospital in Mexico: special focus on Acinetobacter baumannii. Chemotherapy 56:275–279 [DOI] [PubMed] [Google Scholar]
- 10.Gaynes R, Edwards JR, System NNIS. 2005. Overview of nosocomial infections caused by gram-negative bacilli. Clin. Infect. Dis. 41:848–854 [DOI] [PubMed] [Google Scholar]
- 11.Jean S-S, Hsueh P-R. 2011. High burden of antimicrobial resistance in Asia. Int. J. Antimicrob. Agents 37:291–295 [DOI] [PubMed] [Google Scholar]
- 12.Jean S-S, Hsueh P-R, Lee W-S, Chang H-T, Chou M-Y, Chen I-S, Wang J-H, Lin C-F, Shyr J-M, Ko W-C, Wu J-J, Liu Y-C, Huang W-K, Teng L-J, Liu C-Y. 2009. Nationwide surveillance of antimicrobial resistance among non-fermentative Gram-negative bacteria in intensive care units in Taiwan: SMART programme data 2005. Int. J. Antimicrob. Agents 33:266–271 [DOI] [PubMed] [Google Scholar]
- 13.van Faassen H, KuoLee R, Harris G, Zhao X, Conlan JW, Chen W. 2007. Neutrophils play an important role in host resistance to respiratory infection with Acinetobacter baumannii in mice. Infect. Immun. 75:5597–5608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Breslow JM, Meissler JJ, Jr, Hartzell RR, Spence PB, Truant A, Gaughan J, Eisenstein TK. 2011. Innate immune responses to systemic Acinetobacter baumannii infection in mice: neutrophils, but not interleukin-17, mediate host resistance. Infect. Immun. 79:3317–3327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Qiu H, KuoLee R, Harris G, Chen W. 2009. Role of NADPH phagocyte oxidase in host defense against acute respiratory Acinetobacter baumannii infection in mice. Infect. Immun. 77:1015–1021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tsuchiya T, Nakao N, Yamamoto S, Hirai Y, Miyamoto K, Tsujibo H. 2012. NK1.1+ cells regulate neutrophil migration in mice with Acinetobacter baumannii pneumonia. Microbiol. Immunol. 56:107–116 [DOI] [PubMed] [Google Scholar]
- 17.Zhao L, KuoLee R, Harris G, Tram K, Yan H, Chen W. 2011. c-di-GMP protects against intranasal Acinetobacter baumannii infection in mice by chemokine induction and enhanced neutrophil recruitment. Int. Immunopharmacol. 11:1378–1383 [DOI] [PubMed] [Google Scholar]
- 18.Qiu H, KuoLee R, Harris G, Van Rooijen N, Patel GB, Chen W. 2012. Role of macrophages in early host resistance to respiratory Acinetobacter baumannii infection. PLoS One 7:e40019. 10.1371/journal.pone.0040019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hood MI, Mortensen BL, Moore JL, Zhang Y, Kehl-Fie TE, Sugitani N, Chazin WJ, Caprioli RM, Skaar EP. 2012. Identification of an Acinetobacter baumannii zinc acquisition system that facilitates resistance to calprotectin-mediated zinc sequestration. PLoS Pathog. 8:e1003068. 10.1371/journal.ppat.1003068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jacobs AC, Hood I, Boyd KL, Olson PD, Morrison JM, Carson S, Sayood K, Iwen PC, Skaar EP, Dunman PM. 2010. Inactivation of phospholipase D diminishes Acinetobacter baumannii pathogenesis. Infect. Immun. 78:1952–1962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pizzolla A, Hultqvist M, Nilson B, Grimm MJ, Eneljung T, Jonsson IM, Verdrengh M, Kelkka T, Gjertsson I, Segal BH, Holmdahl R. 2012. Reactive oxygen species produced by the NADPH oxidase 2 complex in monocytes protect mice from bacterial infections. J. Immunol. 188:5003–5011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Emre Y., Nubel T. Uncoupling protein UCP2: when mitochondrial activity meets immunity. FEBS Lett. 584:1437–1442 [DOI] [PubMed] [Google Scholar]
- 23.Rousset S, Emre Y, Join-Lambert O, Hurtaud C, Ricquier D, Cassard-Doulcier AM. 2006. The uncoupling protein 2 modulates the cytokine balance in innate immunity. Cytokine 35:135–142 [DOI] [PubMed] [Google Scholar]
- 24.Qiu H, KuoLee R, Harris G, Chen W. 2009. High susceptibility to respiratory Acinetobacter baumannii infection in A/J. mice is associated with a delay in early pulmonary recruitment of neutrophils. Microbes Infect. Inst. Pasteur 11:946–955 [DOI] [PubMed] [Google Scholar]
- 25.Pantano C, Ather JL, Alcorn JF, Poynter ME, Brown AL, Guala AS, Beuschel SL, Allen GB, Whittaker LA, Bevelander M, Irvin CG, Janssen-Heininger YM. 2008. Nuclear factor-κB activation in airway epithelium induces inflammation and hyperresponsiveness. Am. J. Respir. Crit. Care Med. 177:959–969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Poynter ME, Irvin CG, Janssen-Heininger YM. 2003. A prominent role for airway epithelial NF-κB activation in lipopolysaccharide-induced airway inflammation. J. Immunol. 170:6257–6265 [DOI] [PubMed] [Google Scholar]
- 27.Chen SM, Cheng DS, Williams BJ, Sherrill TP, Han W, Chont M, Saint-Jean L, Christman JW, Sadikot RT, Yull FE, Blackwell TS. 2008. The nuclear factor kappa-B pathway in airway epithelium regulates neutrophil recruitment and host defence following Pseudomonas aeruginosa infection. Clin. Exp. Immunol. 153:420–428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jiang D, Nelson ML, Gally F, Smith S, Wu Q, Minor M, Case S, Thaikoottathil J, Chu HW. 2012. Airway epithelial NF-κB activation promotes Mycoplasma pneumoniae clearance in mice. PLoS One 7:e52969. 10.1371/journal.pone.0052969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.March C, Regueiro V, Llobet E, Moranta D, Morey P, Garmendia J, Bengoechea JA. 2010. Dissection of host cell signal transduction during Acinetobacter baumannii-triggered inflammatory response. PLoS One 5:e10033. 10.1371/journal.pone.0010033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Uzhachenko R, Ivanov SV, Yarbrough WG, Medzhitov R, Ivanova AV. Fus1 is a novel regulator of mitochondrial calcium handling, Ca2+-coupled mitochondrial parameters and Ca2+-dependent NFAT and NF-κB pathways in CD4+ T cells. Antiox. Redox Signal., in press [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gloire G, Legrand-Poels S, Piette J. 2006. NF-κB activation by reactive oxygen species: fifteen years later. Biochem. Pharmacol. 72:1493–1505 [DOI] [PubMed] [Google Scholar]
- 32.Khajah M, Millen B, Cara DC, Waterhouse C, McCafferty DM. 2011. Granulocyte-macrophage colony-stimulating factor (GM-CSF): a chemo-attractive agent for murine leukocytes in vivo. J. Leukoc. Biol. 89:945–953 [DOI] [PubMed] [Google Scholar]
- 33.Liu L, Das S, Losert W, Parent CA. 2010. mTORC2 regulates neutrophil chemotaxis in a cAMP- and RhoA-dependent fashion. Developmental Cell 19:845–857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pani G. 2010. P66SHC and ageing: ROS and TOR? Aging 2:514–518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Alcamo E, Mizgerd JP, Horwitz BH, Bronson R, Beg AA, Scott M, Doerschuk CM, Hynes RO, Baltimore D. 2001. Targeted mutation of TNF receptor I rescues the RelA-deficient mouse and reveals a critical role for NF-κB in leukocyte recruitment. J. Immunol. 167:1592–1600 [DOI] [PubMed] [Google Scholar]
- 36.Sadikot RT, Han W, Everhart MB, Zoia O, Peebles RS, Jansen ED, Yull FE, Christman JW, Blackwell TS. 2003. Selective I κB kinase expression in airway epithelium generates neutrophilic lung inflammation. J. Immunol. 170:1091–1098 [DOI] [PubMed] [Google Scholar]
- 37.Skerrett SJ, Liggitt HD, Hajjar AM, Ernst RK, Miller SI, Wilson CB. 2004. Respiratory epithelial cells regulate lung inflammation in response to inhaled endotoxin. Am. J. Physiol. Lung Cell. Mol. Physiol. 287:L143–L152 [DOI] [PubMed] [Google Scholar]
- 38.Chow JY, Ban M, Wu HL, Nguyen F, Huang M, Chung H, Dong H, Carethers JM. 2010. TGF-β downregulates PTEN via activation of NF-κB in pancreatic cancer cells. Am. J. Physiol. Gastrointest. Liver Physiol. 298:G275–G282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vasudevan KM, Gurumurthy S, Rangnekar VM. 2004. Suppression of PTEN expression by NF-κB prevents apoptosis. Mol. Cell. Biol. 24:1007–1021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Xia D, Srinivas H, Ahn YH, Sethi G, Sheng X, Yung WK, Xia Q, Chiao PJ, Kim H, Brown PH, Wistuba II, Aggarwal BB, Kurie JM. 2007. Mitogen-activated protein kinase kinase-4 promotes cell survival by decreasing PTEN expression through an NF κB-dependent pathway. J. Biol. Chem. 282:3507–3519 [DOI] [PubMed] [Google Scholar]
- 41.Li Y, Jia Y, Pichavant M, Loison F, Sarraj B, Kasorn A, You J, Robson BE, Umetsu DT, Mizgerd JP, Ye K, Luo HR. 2009. Targeted deletion of tumor suppressor PTEN augments neutrophil function and enhances host defense in neutropenia-associated pneumonia. Blood 113:4930–4941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, Weinrauch Y, Zychlinsky A. 2004. Neutrophil extracellular traps kill bacteria. Science 303:1532–1535 [DOI] [PubMed] [Google Scholar]
- 43.Bianchi M, Hakkim A, Brinkmann V, Siler U, Seger RA, Zychlinsky A, Reichenbach J. 2009. Restoration of NET formation by gene therapy in CGD controls aspergillosis. Blood 114:2619–2622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yost CC, Cody MJ, Harris ES, Thornton NL, McInturff AM, Martinez ML, Chandler NB, Rodesch CK, Albertine KH, Petti CA, Weyrich AS, Zimmerman GA. 2009. Impaired neutrophil extracellular trap (NET) formation: a novel innate immune deficiency of human neonates. Blood 113:6419–6427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.McInturff AM, Cody MJ, Elliott EA, Glenn JW, Rowley JW, Rondina MT, Yost CC. 2012. Mammalian target of rapamycin regulates neutrophil extracellular trap formation via induction of hypoxia-inducible factor 1α. Blood 120:3118–3125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bianchi M, Niemiec MJ, Siler U, Urban CF, Reichenbach J. 2011. Restoration of anti-Aspergillus defense by neutrophil extracellular traps in human chronic granulomatous disease after gene therapy is calprotectin-dependent. J. Allergy Clin. Immunol. 127:1243–1252.e1247 [DOI] [PubMed] [Google Scholar]
- 47.Urban CF, Ermert D, Schmid M, Abu-Abed U, Goosmann C, Nacken W, Brinkmann V, Jungblut PR, Zychlinsky A. 2009. Neutrophil extracellular traps contain calprotectin, a cytosolic protein complex involved in host defense against Candida albicans. PLoS Pathog. 5:e1000639. 10.1371/journal.ppat.1000639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liang SC, Tan X-Y, Luxenberg DP, Karim R, Dunussi-Joannopoulos K, Collins M, Fouser LA. 2006. Interleukin (IL)-22 and IL-17 are coexpressed by Th17 cells and cooperatively enhance expression of antimicrobial peptides. J. Exp. Med. 203:2271–2279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhang J, Khvorostov I, Hong JS, Oktay Y, Vergnes L, Nuebel E, Wahjudi PN, Setoguchi K, Wang G, Do A, Jung HJ, McCaffery JM, Kurland IJ, Reue K, Lee WN, Koehler CM, Teitell MA. 2011. UCP2 regulates energy metabolism and differentiation potential of human pluripotent stem cells. EMBO J. 30:4860–4873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Park D, Han CZ, Elliott MR, Kinchen JM, Trampont PC, Das S, Collins S, Lysiak JJ, Hoehn KL, Ravichandran KS. 2011. Continued clearance of apoptotic cells critically depends on the phagocyte Ucp2 protein. Nature 477:220–224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Arsenijevic D, Onuma H, Pecqueur C, Raimbault S, Manning BS, Miroux B, Couplan E, Alves-Guerra MC, Goubern M, Surwit R, Bouillaud F, Richard D, Collins S, Ricquier D. 2000. Disruption of the uncoupling protein-2 gene in mice reveals a role in immunity and reactive oxygen species production. Nat. Genet. 26:435–439 [DOI] [PubMed] [Google Scholar]
- 52.Blanc J, Alves-Guerra MC, Esposito B, Rousset S, Gourdy P, Ricquier D, Tedgui A, Miroux B, Mallat Z. 2003. Protective role of uncoupling protein 2 in atherosclerosis. Circulation 107:388–390 [DOI] [PubMed] [Google Scholar]