

Abstract
BACE1 and presenilin (PS)/γ-secretase play a major role in Alzheimer's disease pathogenesis by regulating amyloid-β peptide generation. We recently showed that these secretases also regulate the processing of voltage-gated sodium channel auxiliary β-subunits and thereby modulate membrane excitability. Here, we report that KCNE1 and KCNE2, auxiliary subunits of voltage-gated potassium channels, undergo sequential cleavage mediated by either α-secretase and PS/γ-secretase or BACE1 and PS/γ-secretase in cells. Elevated α-secretase or BACE1 activities increased C-terminal fragment (CTF) levels of KCNE1 and 2 in human embryonic kidney (HEK293T) and rat neuroblastoma (B104) cells. KCNE-CTFs were then further processed by PS/γ-secretase to KCNE intracellular domains. These KCNE cleavages were specifically blocked by chemical inhibitors of the secretases in the same cell models. We also verified our results in mouse cardiomyocytes and cultured primary neurons. Endogenous KCNE1- and KCNE2-CTF levels increased by 2- to 4-fold on PS/γ-secretase inhibition or BACE1 overexpression in these cells. Furthermore, the elevated BACE1 activity increased KCNE1 processing and shifted KCNE1/KCNQ1 channel activation curve to more positive potentials in HEK cells. KCNE1/KCNQ1 channel is a cardiac potassium channel complex, and the positive shift would lead to a decrease in membrane repolarization during cardiac action potential. Together, these results clearly showed that KCNE1 and KCNE2 cleavages are regulated by BACE1 and PS/γ-secretase activities under physiological conditions. Our results also suggest a functional role of KCNE cleavage in regulating voltage-gated potassium channels.—Sachse, C. C., Kim, Y. H., Agsten, M., Huth, T., Alzheimer, C., Kovacs, D. M., and Kim, D. Y. BACE1 and presenilin/γ-secretase regulate proteolytic processing of KCNE1 and 2, auxiliary subunits of voltage-gated potassium channels.
Keywords: MinK, MiRP, Alzheimer's disease, KCNQ1
Alzheimer's disease (AD) is characterized by the formation of senile plaques and neurofibrillary tangles in specific brain regions. In addition to cognitive deficits, patients with AD also frequently exhibit severe personality changes and various psychiatric symptoms (1, 2). Epileptic and myoclonic seizures are commonly observed in patients with AD, especially with presenilin (PS) familial Alzheimer's disease (FAD) mutations (3). These functional disturbances may reflect altered neuronal excitability and neural network dysfunction (4, 5). However, the underlying mechanism is not fully known.
β-Site of amyloid precursor protein (APP)-cleaving enzyme 1 (BACE1; β-secretase, memapsin-2) and PS/γ-secretase generate amyloid β (Aβ) peptides found in senile plaques (6, 7). These two secretases sequentially cleave APP to generate a soluble APP extracellular domain (sAPPβ), APP intracellular domain (AICD), and the short toxic Aβ peptides. BACE1 and PS/γ-secretase are intensively investigated as prime drug targets for treating patients with AD (8, 9). Therefore, it is very important to understand the physiological roles of these secretases for developing safe therapeutic inhibitors without serious side effects.
Previously, we and other groups found that the voltage-gated sodium (Nav) channel β2-subunit (Navβ2) undergoes proteolytic processing mediated by BACE1, a disintegrin and metalloproteinase domain-containing protein 10 (ADAM10), and PS/γ-secretase (10–12). Later, we found that BACE1 and PS/γ-secretase activities promote release of the intracellular domain of Navβ2 (β2-ICD), increase mRNA and protein levels of the channel-forming Nav1.1 α subunit, and therefore modulate neuronal membrane excitability in neuroblastoma cells and mouse hippocampal neurons (13, 14). We and others have also shown that BACE1 plays an essential role in regulating Nav channel metabolism and neuronal membrane excitability in mouse brains under physiological conditions (14–16).
Similar to Nav channels, voltage-gated potassium (Kv) channels regulate membrane excitability in neuronal and nonneuronal cells (17). Kv channels consist of 4 pore-forming α subunits and various regulatory accessory subunits (18). KCNE proteins [MinK and MinK-related peptides (MiRPs)] are a group of Kv channel auxiliary subunits that belong to type I membrane proteins (18, 19). Similar to Navβ2, KCNEs interact with α subunits of Kv channels and regulate the channel function (18, 19). Five KCNE family proteins are currently known (KCNE1–5) and are widely expressed in various tissues and cell types, including the heart and brain tissues. The importance of KCNEs is underscored by the facts that genetic mutations of KCNEs are linked to human diseases, such as long QT syndrome, deafness, and periodic paralysis (19).
Here, we report that KCNE1 and KCNE2 undergo sequential cleavage mediated by either α-secretase and PS/γ-secretase or BACE1 and PS/γ-secretase. We also explored whether BACE1-mediated cleavage of KCNE can modulate Kv channel function using an in vitro model system.
MATERIALS AND METHODS
Antibodies and reagents
DAPT, TAPI-1, and BACE inhibitor IV were purchased from Calbiochem (EMD Millipore, Billerica, MA, USA). PMA was obtained from Sigma-Aldrich (St. Louis, MO, USA), GM6001 was obtained from Millipore, and epoxomicin from Enzo Life Sciences (Farmingdale, NY, USA). BACE1 inhibitor DR9 was a kind gift from Dr. Jordan Tang (University of Oklahoma Health Science Center, Oklahoma City, OK, USA). The following antibodies were purchased for this study: anti-GAPDH (EMD Millipore), BACE1 poly antibody (Abcam, Cambridge, MA, USA), anti-KCNQ1 antibody [University of California–Davis (UC Davis)/U.S. National Institutes of Health (NIH) NeuroMab Facility, Davis, CA, USA], anti-KCNE1 (Alomone Laboratories, Jerusalem, Israel), anti-KCNE2 (Alomone Laboratories), and anti-V5 (Invitrogen, Grand Island, NY, USA).
Constructs
Full-length human KCNE1 and KCNE2 were obtained from the Dana-Farber/Harvard Cancer Center DNA Resource Core (Boston, MA, USA). KCNE1 was cloned into pcDNA6.1 (Invitrogen) with a C-terminal V5 tag by using the Gateway cloning system (Invitrogen). KCNE2 was PCR-amplified and cloned to pcDNA3.2 (Invitrogen) with a C-terminal V5 tag. The primers used for PCR amplification were 5′-CACCTGAAGCGCTGAAATGAGCACTTTATCCAATTTCACACAGACGC-3′ and 5´-GGGGGACATTTTGAACCCAGCCGCACC-3´. An expression plasmid vector harboring full-length human BACE1 (GenBank: AF190725) containing a C-terminal myc tag has been described previously (10). To express BACE1 in neuronal cells, we also constructed a lentiviral CSCW-hBACE1-IRES-GFP vector. The parental CSCW-IGs vector was provided by the Massachusetts General Hospital viral core (http://www2.massgeneral.org/ncs/neuro_core_VectorDevelopmentandProduction.htm). For electrophysiology, human KCNQ1 and KCNE1 in pFrog constructs were kindly provided by Dr. Michael Schwake (Institute of Biochemistry, University of Kiel, Kiel, Germany), while human BACE1 constructs were kindly provided by Dr. Michael Willem (Department of Biochemistry, Ludwig Maximilians University München, Munich, Germany).
Cell culture, transfection, and drug treatments
B104 rat neuroblastoma and human embryonic kidney (HEK)293T cells were maintained in Dulbecco's modified Eagle medium (DMEM; Invitrogen), supplemented with 10% FBS and 1% penicillin/streptomycin solution (Invitrogen) in 5% CO2 at 37°C. For selection and maintenance of stable cell lines, 0.2 mg/ml G418 (Mediatech, Manassas, VA, USA) or 0.01 mg/ml blasticidin (Invitrogen) was added to complete DMEM medium. Effectene (Qiagen, Valencia, CA, USA) was routinely used for transfecting cell lines. For drug treatments, cells were maintained at 37°C in a humidified 5% CO2 atmosphere. PMA was used at a 1 μM concentration for 1 or 3 h, epoxomicin was used at a 1 μM concentration for 3 h, and DAPT was used at a 0.5 μM concentration for 3 h.
Primary mouse neuronal and neonatal cardiomyocyte cultures
Mouse primary hippocampal/cortical neuronal cultures were prepared and maintained in Neurobasal medium (Invitrogen) supplemented with B27 and 0.5 mM l-glutamine, as described previously (10). The neuronal cultures were maintained for 14 d in the supplemented Neurobasal medium. Mouse cardiomyocytes were isolated from neonatal mice by using a cardiomyocyte isolation kit (Cellutron Life Technologies, Baltimore, MD, USA). The dissociated neonatal cardiomyocytes were plated on 6-well plates precoated with SureCoat (Cellutron Life Technologies) and maintained in a CO2 incubator for 3–5 d. The beating cardiomyocytes were detected from 2 d after plating. To block PS/γ-secretase activity, the cells were treated with 1–5 μM DAPT for 3 d. For BACE1 overexpression, the cells were infected with CSCW-BACE1-IRES-GFP lentiviral vector (MOI=1) for 3 d. The expression levels were monitored by GFP signals using a fluorescence microscope. To block endogenous BACE1 or α-secretase activity, 1 μM BACE1 inhibitor IV or GM6001 (Millipore) were treated to the cells for 3 d. All animal procedures were approved by the subcommittee on research animal care (SRAC) at Massachusetts General Hospital.
Western blot analysis
For immunoblotting, cell extracts were prepared by directly extracting cells in a buffer containing 10 mM Tris (pH 7.6), 2 mM EDTA, 150 mM NaCl, 0.25% P-40, 1% Triton X-100, and a protease inhibitor mixture (Roche Molecular Biochemicals, Minneapolis, MN, USA), followed by a spin at 16,000 g. Proteins (20–100 μg) were resolved on 4–12% or 12% Bis/Tris gels (Invitrogen), depending on the individual experiment as described. Primary antibodies were used at the following dilutions: GAPDH, 1:1000 (Chemicon, Temecula, CA, USA), BACE1 poly antibody, 1:1000 (Abcam), anti-KCNQ1 antibody, 1:200 (UC Davis/NIH NeuroMab Facility), anti-KCNE1, 1:250 with 5% BSA (Alomone Labs), anti-KCNE2, 1:250 with 1% BSA (Alomone Labs), and anti-V5, 1:5000 with 5% BSA (Invitrogen). The blots were visualized by enhanced chemiluminescence (ECL). The images were captured by using BioMax film (Kodak, Rochester, NY, USA) or the VersaDoc imaging system (Bio-Rad, Hercules, CA, USA) and quantitated by using QuantityOne software (Bio-Rad).
Electrophysiology
One day prior to transfection, cells were plated in 35-mm dishes (Falcon; BD Biosciences, Bedford, MA, USA). For transient transfection, Nanofectin (PAA, Pasching, Austria) was applied according to the manufacturer's protocol, using 1 μg of cDNA of each construct and 0.5 μg of enhanced green fluorescent protein (c-EGFP; Clontech, Mountain View, CA, USA). Transfected cells were identified using an inverted fluorescent microscope (Axiovert 40; Zeiss, Jena, Germany) with a fiber optic-coupled light source (UVICO; Rapp OptoElectronic, Hamburg, Germany). Current signals were recorded in whole-cell patch-clamp mode at room temperature 2–3 d post-transfection. Recordings were started 3 min after whole-cell access was obtained. Data were sampled at 20 kHz and filtered at 5 kHz, using an Axopatch 200B amplifier in combination with a Digidata 1322A interface and pClamp10.2 software (all from Molecular Devices/MDS Analytical Technologies, Sunnyvale, CA, USA). Electrodes were made from borosilicate glass (Harvard Apparatus, Edenbridge, UK), using a DMZ-Universal Puller (Zeitz, Munich, Germany). External solution contained (in mM): 145 NaCl, 4 KCl, 2 CaCl2, 2 MgCl2, 10 d-glucose, and 10 HEPES, adjusted to pH 7.4 with NaOH. Pipette resistance in bath solution was 1.8–2.6 MΩ, while access resistance was typically <5 MΩ before series resistance compensation (75%). The internal solution was composed of (in mM) 5 NaCl, 120 KCl, 2 MgCl2, 1 CaCl2, 5 EGTA, and 10 HEPES, adjusted to pH 7.2 with KOH. Chemicals were purchased from Sigma-Aldrich (Deisenhofen, Germany). To determine the voltage dependence of current activation, we first performed a leak correction. The potassium conductance at each command potential was calculated with the use of the equation G = I/(V − Erev), where I is the peak amplitude of the potassium current (at t = 10 s after activation) at membrane potential V, and where Erev is the equilibrium potential for potassium ions under our experimental conditions (−86.5 mV). G was fitted by a standard sigmoidal (Boltzmann) relation of the form G/Gmax=1/[1 + exp((Vmid − V)/k)], where Vmid is the midpoint of activation and k is the slope factor. Data analysis was performed using pClamp 10.2 and Origin Pro8.1G software (OriginLab Corp., Northhampton, MA, USA).
Statistical analyses
All statistical analyses were performed using a 2-tailed Student's t test or 1-way ANOVA followed by a post hoc Tukey's test. Error bars represented in graphs denote the se.
RESULTS
KCNE1 and KCNE2 are sequentially cleaved by α-secretase and PS/γ-secretase activities
Previously, our laboratory identified a group of novel PS/γ-secretase substrate proteins via an unbiased protein sequence database search (10, 20–22). This unbiased screen was based on a putative amino acid sequence homology of the known PS/γ-secretase substrate proteins. In this screen, we found that select auxiliary subunits of Nav and Kv channels, Navβ2, and KCNE1/2 harbor putative PS/γ-secretase and BACE1 cleavage sites close to their transmembrane domains (Fig. 1). In the follow-up studies, we found that α-secretase, BACE1, and PS/γ-secretase indeed cleave Navβ2 in neuronal cells and thereby regulate neuronal membrane excitability (10, 13, 14). Next, we explored whether these secretases also cleave KCNE1 and KCNE2 and possibly modulate electrical properties of Kv channels.
Figure 1.
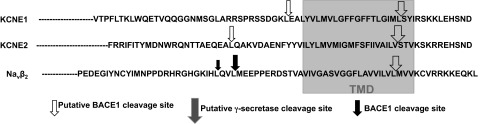
Putative BACE1 and PS/γ-secretase cleavage sites of human KCNE1 and KCNE2.
As summarized in Fig. 2A, common characteristics of PS/γ-secretase substrate proteins include extracellular domain cleavage by α-secretase alone or both α-secretase and BACE1 activities, which generate soluble extracellular domains and C-terminal fragments (CTFs); accumulation of membrane-tethered CTFs in response to PS/γ-secretase inhibition; and generation/release of ICDs that is regulated by PS/γ-secretase activity (13, 20, 23). We have previously shown that modulators of these pathways can be used to identify novel PS/γ-secretase substrate proteins (10, 20–22).
Figure 2.
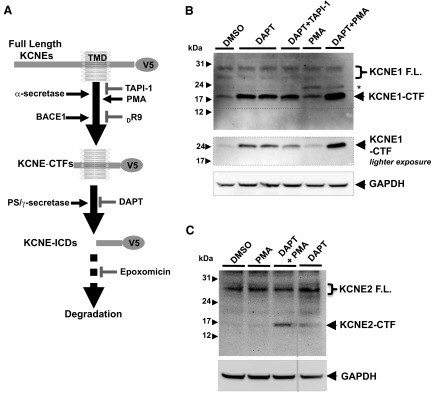
KCNE1 and KCNE2 undergo sequential cleavage mediated by α- and PS/γ-secretases. A) Schematic diagram showing sequential cleavage of KCNE1 and KCNE2 by BACE1, α-, and PS/γ-secretases. α-Secretase inhibitor (TAPI-1) or BACE1 inhibitor (DR9) decreases generation of KCNE C-terminal fragments (KCNE-CTFs), while PMA, an α-secretase activator, increases KCNE-CTF levels. KCNE-CTFs are then cleaved by PS/γ-secretases to generate KCNE intracellular domains (KCNE-ICDs). B) Western blot analysis of human KCNE1 full-length (F.L.) and its CTF expressed in B104 cells. KCNE1-CTF levels were increased by treatment with DAPT and further elevated by cotreatment with PMA, while partially decreased by TAPI-1. C) Western blot analysis of human KCNE2 F.L. and its CTF expressed in B104 cells. Similar to KCNE1, KCNE2-CTF levels are also increased by DAPT treatment and further elevated by cotreatment with PMA.
To test whether KCNEs are cleaved by these secretases, we made plasmid constructs designed to express human KCNE1 and KCNE2 with C-terminal V5 epitope tag. We then generated rat B104 neuroblastoma cells stably expressing either KCNE1 or KCNE2. Using these cells as model systems, we first investigated whether the KCNE1 yield KCNE1 CTF products (KCNE1-CTFs) that can be further cleaved by PS/γ-secretase activity, as shown in Fig. 2A. Employing an antibody against the C-terminal V5 tag, Western blot analysis showed that multiple full-length KCNE1 bands migrated around 29-kDa and shorter 17-kDa KCNE1-CTF bands (Fig. 2B). The size of this fragment was consistent with the predicted molecular weight of the KCNE1-CTF/V5 if ectodomain shedding occurred near the plasma membrane. DAPT, a specific γ-secretase inhibitor, further elevated KCNE1-CTF levels, while cotreatment of TAPI-1, an α-secretase inhibitor, partially blocked the increase of KCNE1-CTF levels by DAPT (Fig. 2B). KCNE1-expressing cells were also treated with PMA to enhance α-secretase-mediated cleavage. PMA treatment alone slightly increased KCNE1-CTF levels, but in conjunction with DAPT, PMA dramatically elevated KCNE1-CTF levels (Fig. 2B). These data suggest that increased KCNE1-CTFs by PMA treatment were rapidly processed by PS/γ-secretase activity in cells. We also detected an additional KCNE1-CTF band with higher molecular mass as compared to the normal KCNE1-CTF band, only it was also increased after PMA treatments (Fig. 2B, asterisk). However, levels of the high-sized KCNE1-CTF bands were not changed by DAPT cotreatment, which suggests that this high-sized KCNE1-CTF band is not further cleaved by PS/γ-secretase. Similarly, PMA treatment together with DAPT dramatically increased KCNE2-CTF levels in B104 cells (Fig. 2C). Together, these data strongly indicate that KCNE1 and KCNE2 undergo sequential cleavage mediated by α- and PS/γ-secretase activities.
PS/γ-secretase activity generates intracellular domains of KCNE1 and KCNE2
PS/γ-secretase-mediated cleavage of membrane-tethered KCNE-CTFs would generate and release ICDs of KCNEs (KCNE-ICDs), as shown in the summary diagram (Fig. 2A). Previous studies of other PS/γ-secretase substrates suggest that the released ICDs are rapidly degraded by proteasomal activities and therefore are very difficult to detect in normal cell extraction conditions (23). To detect KCNE1- and KCNE2-ICDs, we cotreated the KCNE-expressing cells with epoxomicin, a proteasome inhibitor, to block KCNE-ICD degradation in cells. To enhance α-secretase-mediated cleavage of KCNEs, we cotreated PMA together with expoxomicin. In these conditions, we were able to detect ∼14-kDa KCNE1- and KCNE2-ICD bands that completely disappear on DAPT treatment (Fig. 3). The sizes of these ICD bands match with the predicted molecular mass of KCNE-ICDs. These findings demonstrate that KCNE1- and KCNE2-CTFs undergo PS/γ-secretase-mediated cleavage to generate KCNE1- and KCNE2-ICDs, while the generated KCNE-ICDs are rapidly degraded by proteasome activities in cells.
Figure 3.
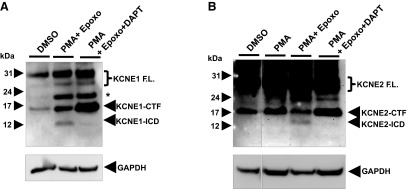
PS/γ-secretase activity regulates generation of KCNE1- and KCNE2-ICDs. A) Western blot analysis showed that KCNE1-ICD is specifically regulated by PS/γ-secretase activity in B104 cells stably expressing full-length KCNE1. Epoxomicin, a proteasome inhibitor, prevented KCNE1-ICD degradation. B) KCNE2-ICD is detected in the similar conditions.
BACE1 cleaves KCNE1 and KCNE2
Next, we tested whether KCNE1 and KCNE2 are also cleaved by BACE1. Because BACE1 activity is relatively low in cultured cell lines, we overexpressed human BACE1 in either KCNE1- or KCNE2-expressing cells described in Fig. 2 and measured KCNE1- and KCNE2-CTF levels by Western blot analysis. We found that transient BACE1 overexpression dramatically increased KCNE1- and KCNE2-CTF levels in B104 cells (Fig. 4). DAPT treatments further elevated KCNE1- and KCNE2-CTF levels that are already increased by BACE1 overexpression (Fig. 4). We further characterized BACE1-mediated KCNE2 processing in B104 cells stably coexpressing KCNE2 and BACE1. Consistent with the transient BACE1 overexpression, stable expression of BACE1 largely elevated KCNE2-CTF levels in all the stable cell lines that we tested, as compared to the control cells expressing KCNE2 alone (Supplemental Fig. S1). The increased KCNE2-CTF levels by BACE1 overexpression were not significantly affected by cotreatment with TAPI-1, an α-secretase inhibitor, confirming that BACE1 activity is directly responsible for the increase of KCNE2-CTF levels (Supplemental Fig. S1). We also found that 72 h of treatment with DR9, a cell-permeable BACE1 inhibitor, significantly decreased KCNE2-CTF levels in control B104 cells stably expressing KCNE2 alone (Supplemental Fig. S2). Together, these data demonstrate that KCNE1 and KCNE2 cleavage are regulated by BACE1 activities in cells.
Figure 4.
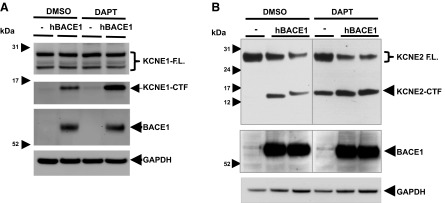
Elevated BACE1 activity increases KCNE1- and KCNE2-CTF levels in B104 rat neuroblastoma cells. A) Overexpression of human BACE1 increased KCNE1-CTF levels in B104 cells. DAPT treatment further enhanced KCNE1-CTF levels in control and BACE1-overexpressing cells. B) KCNE2-CTF levels were also elevated by BACE1 overexpression in B104 cells.
BACE1 and PS/γ-secretase activities regulate endogenous KCNE1 and KCNE2 cleavage
In Figs. 2–4, we showed that BACE1 and PS/γ-secretase activities regulate the processing of human KCNE1 and KCNE2 that are overexpressed in cultured cells. We then investigated whether BACE1 and PS/γ-secretase activities also regulate endogenous KCNE1 and KCNE2 cleavage under physiological conditions. To test endogenous KCNE1 cleavage, we used cultured mouse cardiomyocytes as a model system, since KCNE1 is highly expressed in cardiac tissues (18). The dissociated neonatal cardiomyocytes were maintained for 3–5 d and tested for KCNE1 expression by Western blot analysis. Using an antibody recognizing the KCNE1 C-terminal region, we were able to detect endogenous KCNE1 full-length (∼30-kDa) and KCNE1-CTF (∼15-kDa) bands in these cells (Fig. 5). Endogenous KCNE1-CTF size is slightly smaller than KCNE1-CTF overexpressed in B104 cells (Figs. 2–4), possibly due to lack of V5 tag. As predicted, either DAPT treatment or BACE1 overexpression specifically increased KCNE1-CTF levels as compared to the controls (Fig. 5). These results support the hypothesis that endogenous KCNE1 undergoes cleavage mediated by PS/γ-secretase activity under physiological conditions and can be cleaved by the increased BACE1 activity in cultured cardiomyocytes.
Figure 5.

BACE1 and PS/γ-secretase activities regulate endogenous KCNE1-CTF levels. A) Detection of endogenous KCNE1 full-length (F.L.) and CTF bands in cultured mouse cardiomyocytes. DAPT (5 μM) treatment increased endogenous KCNE1-CTF levels. B) Endogenous KCNE1-CTF levels were also increased by BACE1 overexpression in mouse cardiomyocytes.
Similarly, we found that endogenous KCNE2-CTF levels are also increased by either DAPT treatment or human BACE1 overexpression in cultured mouse primary neuronal cells (Fig. 6A–D). Quantitative analyses showed that both BACE1 overexpression and DAPT treatment significantly increased endogenous KCNE2-CTF levels by 2- to 4-fold (Fig. 6B, D). To explore whether the endogenous BACE1 activity regulates KCNE2 processing in neuronal cells, we treated the cultured mouse primary neurons (DIV7) with 1 μM BACE1 inhibitor IV (Calbiochem) for 3 d and analyzed the KCNE2-CTF levels by Western blot analysis (Fig. 6E, F). We found that BACE inhibitor treatments significantly decreased KCNE2-CTF levels by 55% (Fig. 6F). To test whether endogenous KCNE2 also undergoes an α-secretase-mediated cleavage as shown in Fig. 2, the mouse primary neurons were treated with GM6001 (Millipore), a pan α-secretase inhibitor. As predicted, we found a decrease of KCNE2-CTF levels in the cells treated with GM6001, as compared to DMSO control (Fig. 6G). Together, these results strongly support the hypothesis that endogenous KCNE2 undergoes sequential cleavage regulated by α-secretase, BACE1, and PS/γ-secretase activities in neuronal cells.
Figure 6.
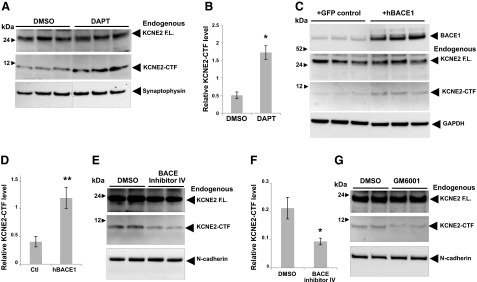
α-Secretase, BACE1, and PS/γ-secretase activities regulate KCNE2-CTF levels in cultured mouse primary neurons. A) Detection of endogenous KCNE2 full-length (F.L.) and KCNE2-CTF bands in cultured mouse primary cortical/hippocampal neurons (DIV14). DAPT treatment specifically increased 10-kDa KCNE2-CTF levels. B) Relative levels of KCNE2-CTF in panel A were quantitated (n=3/condition). *P < 0.05. C) Endogenous KCNE2-CTF levels were also elevated by BACE1 overexpression, as compared to the primary neuronal cells transfected with control GFP vectors. D) Quantitative analysis of KCNE2-CTF levels shown in panel B (n=3/condition). **P < 0.01. E–G) Mouse primary neurons were also treated with 1 μM BACE inhibitor IV (E) or 1 μM GM6001 (a pan-α-secretase inhibitor; G) for 3 d, and KCNE2-CTF levels were analyzed by Western blotting. F) Quantitative analysis of KCNE2-CTF levels in neurons treated with BACE1 inhibitor IV or DMSO control. Relative KCNE2-CTF levels were normalized by full-length KCNE2 to avoid blot-to-blot variations (n=3/condition). *P < 0.05.
BACE1 overexpression modulates KCNQ1/KCNE1 currents in HEK293T cells
Previous studies have shown that KCNE1 associates with KCNQ1 channels to form slow delayed rectifying K+ currents (IKs), which are important for the repolarization of cardiac action potentials (24, 25). Although BACE1 is mostly expressed in brain tissues, low levels of BACE1 are also detected in heart and several other tissues (26). Therefore, we tested whether BACE1-mediated cleavage of KCNE1 can influence IKs by modulating KCNQ1/KCNE1 channels.
To address this question, we transiently coexpressed human KCNQ1 and KCNE1 (without V5 tag) in HEK239T cells and performed whole-cell patch-clamp experiments with or without overexpression of human BACE1. KCNQ1, KCNE1, and BACE1 expressions were confirmed by Western blot analysis (Fig. 7A). Transient expression of KCNE1 in HEK293 cells led to multiple bands, including ∼30-kDa full-length KCNE1- and KCNE1-CTF bands (Fig. 7A). Consistent with previous findings, BACE1 overexpression significantly increased KCNE1-CTF levels, which are further enhanced by DAPT treatment (Fig. 7A). KCNQ1/KCNE1 current activation was examined using 10-s voltage steps to command potentials between −40 and +80 mV (Fig. 7B). The activation curves were plotted as described in Materials and Methods (Fig. 7C). As previously reported, coexpression of KCNE1 and KCNQ1 produces Kv currents that resemble the cardiac IKs (24, 25). Interestingly, we found that overexpression of BACE1 shifts the current activation to significantly more positive potentials, associated with a significantly flatter slope factor as compared to the controls, including the inactive BACE1 (D289N) overexpression (Fig. 7C). Half-maximal activation (Vmid) for KCNQ1/KCNE1 current was 21.7±2.0 mV (slope factor k=16.9±0.9 mV, n=19) (Fig. 7C). Coexpression of wild-type BACE1 dramatically shifted Vmid to 37.4±2.9 mV (k=17.3.3±1.1 mV, n=26) while inactive BACE1 (D289N) slightly changed to 27.3±2.8 mV (k=20.3±0.6 mV, n=13). The slope factor significance was P < 0.01. The positive shift of the activation curve indicates that less open Kv channels are available at a given potential, and this would lead to a decrease of membrane repolarization during cardiac action potential. A slight, but significant, shift of Vmid by an inactive BACE1(D289N) may suggest an additional role of inactive BACE1 protein in regulating KCNQ1/KCNE1 function, which needs further studies to clarify. Together, these data clearly suggest that BACE1 and, possibly, PS/γ-secretase activity can directly regulate KCNQ1/KCNE1 channel currents, possibly though KCNE1 processing.
Figure 7.
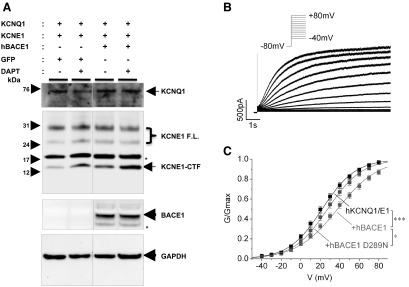
BACE1 activity modulates KCNQ1/KCNE1 current activation in HEK293T cells. Whole-cell recordings of KCNQ1 and KCNE1 transiently transfected in HEK293T cells with or without overexpressing wild-type BACE1. A) Western blot analysis detects human KCNQ1 and KCNE1 coexpression in HEK293T cells. As shown in Figs. 2 and 3, either BACE1 expression and/or DAPT treatment increases KCNE1-CTF levels. Asterisks indicate nonspecific bands that are detected when transiently expressing KCNE1, not in stable B104 cells. B) Activation of KCNQ1/E1 currents by depolarizing voltage steps from −40 to +80 mV (inset). Prepulse duration at −80 mV was 5 s. C) Normalized conductance was plotted against voltage from experiments as in panel B. Curves were fitted with a Boltzmann equation. Conductance of the cells expressing KCNQ1/KCNE1 (KCNQ1/E1, black) was compared with the cells coexpressing wild-type BACE1 (red) and inactive mutant BACE1 (D289N; green). Significance was tested for the half-maximal activation (Vmid). *P < 0.05, ***P < 0.001.
DISCUSSION
Here, we show that KCNE1 and KCNE 2, auxiliary subunits of potassium channels undergo dual-processing pathways mediated by either α/γ-secretase or BACE1/γ-secretase activities (Figs. 2–6). Using KCNE1/KCNQ1 channels as a model system, we also demonstrated that BACE1- and possibly γ-secretase-mediated processing of KCNE1 can specifically regulate the delayed-rectifier K+ current, which is essential for the repolarization of the cardiac action potential (Fig. 7). In addition to KCNQ1, KCNEs regulate multiple Kv channel α-subunits and other select voltage-gated ion channels in vitro and in vivo (18, 27). Therefore, our results suggest the interesting possibility that Alzheimer's disease-related secretases modulate the membrane excitability by cleaving auxiliary subunits of multiple voltage-gated ion channels.
Similar to Navβ subunits, KCNEs modulate Kv channel trafficking, gating, and Kv currents in vitro and in vivo (27). Previously, we and others have shown that BACE1 and PS/γ-secretase regulate Navβ subunit processing and Nav channel metabolism in vitro and in vivo (9, 12–15, 28, 29). Similarly, KCNE processing may also modulate Kv currents. In Fig. 7, we showed that the elevated BACE1 activity shifts the activation curve of KCNE1/KCNQ1 channels to more depolarized potentials. The altered channel gating properties may result from a direct change of channel structure induced by the KCNE cleavage or from an indirect signaling cascade activated on KCNE processing. In the previous Navβ2 studies, we found that the intracellular domain of Navβ2 (Navβ2-ICD) plays an important signaling role in regulating total and surface levels of Nav1.1 channel in neuronal cells (13, 14). Similarly, KCNE-ICDs shown in Fig. 3 may also play a signaling role to regulate Kv channel levels and surface expression. Therefore, it will be interesting to test whether KCNE-ICDs can mediate Kv channel metabolism similar to Navβ2-ICD on Nav channels. Further studies will be required to investigate physiological consequences of the secretase-mediated ion channel regulation.
KCNQ1/KCNE1 channels regulate IKs, the slow component of the delayed rectifying K+ current, and therefore the cardiac action potential in vivo (30–32). Mutations in KCNE1 or KCNQ1 are associated with an inherited cardiac arrhythmia (LQT syndrome) due to delayed repolarization of the cardiac action potential (33, 34). Although BACE1 expression levels are low in the heart under normal conditions, BACE1 levels might be elevated under stress conditions (26, 35, 36). Our data predict that elevated BACE1 activity may alter the voltage trajectory of cardiac action potentials by shifting IKs, as predicted by our model system (Fig. 7).
Although the cardiac function of KCNE1 has been mostly explored in previous studies, studies demonstrated that KCNE1 is also expressed in neuronal cells and regulates a neuronal Kv channel function (18, 37). Because BACE1 is expressed at high levels in neurons as compared to other cell types (38), BACE1 may have more profound roles in regulating neuronal KCNE1- and KCNE1-regulated Kv channels in neuronal cells under physiological conditions. We tried but failed to detect KCNE1 cleavage products in primary neuronal cultures, possibly due to the low sensitivity of endogenous KCNE1 antibodies used in this study (data not shown). Further studies will be required to explore a functional role of BACE1 and PS/γ-secretase in regulating neuronal KCNE1 function.
KCNEs are expressed in a wide range of tissues (18). In particular, KCNE2 is highly expressed in the brain and interacts with a number of neuronal Kv channel α subunits (27). KCNE2 regulates inactivation kinetics of KCNQ2 or KCNQ2/3 channels, which then modulate M-type K+ currents in vitro (39). Because both KCNQ2 and KCNQ3 mutations are linked to familiar neonatal convulsions and epilepsy, the alteration of KCNE2 metabolism might affect neural network activity (18). In addition, KCNE2 also modulates neuronal Kv2.1, Kv4.2, Kv4.3, and hyperpolarization-activated cyclic nucleotide-gated channels (HCNs; refs. 28, 40, 41). In Fig. 6, we showed the altered processing of endogenous KCNE2 by changing BACE1, α-secretase, or PS/γ-secretase activities. These may then modulate KCNE2-regulated neuronal ion channels in a fashion similar to that shown here for KCNE1/KCNQ1 channels (Fig. 7). In addition to KCNE2, studies showed regulatory roles of other KCNEs on neuronal Kv channels, including KCNE1 and KCNE3 (18, 37, 42, 43). Therefore, it will be very interesting to explore whether BACE1 and/or PS/γ-secretase cleave these KCNEs and regulate neuronal Kv channels, as well as KCNE2. Indeed, we have found similar putative BACE1 and PS/γ-secretase cleavage sites in KCNE3–5, as well as KCNE1 and KCNE2, shown in Fig. 1. We are currently exploring the cleavage of these KCNEs and their potential contribution to neuronal Kv channels, using the similar methods described in this study.
BACE1 levels and activity are significantly increased in brains of patients with AD (44–47). Previously, we reported increased levels of Navβ2 processing and the altered Nav1.1 levels in brains of patients with AD, particularly with high BACE1 levels (14). Similarly, the elevated BACE1 activity may also affect KCNE processing and KCNE-regulated ion channels in brains of patients with AD. Indeed, up-regulation of Kv channel α-subunits has been already observed in brain samples from patients with AD (48). Pannacione et al. (43) have reported that Aβ increases the transcription of both KCNE3 and Kv3.4 channels, which may contribute to Aβ toxicity in neuronal cells. Therefore, KCNEs and Kv may contribute to AD pathogenesis through multiple pathways.
BACE1 and PS/γ-secretase are prime drug targets for developing AD treatments. However, the presence of multiple BACE1 and/or PS/γ-secretase substrate proteins casts concerns on full inhibition of these secretases. Our study adds KCNEs to the growing list of secretase substrate proteins. Therefore, it will be very important to find a proper therapeutic window for blocking Aβ generation without affecting other substrate processing when treating patients with AD with secretase inhibitors.
Supplementary Material
Acknowledgments
The authors thank Stephanie Hartmann (University of Erlangen-Nürnberg) for her contribution in revising this manuscript and Dr. Jordan Tang (University of Oklahoma Health Sciences Center, Oklahoma City, OK, USA) for BACE1 inhibitor DR9.
This work is supported by grants from the Cure Alzheimer's fund to D.Y.K, the U.S. National Institutes of Health/National Institute on Aging to D.M.K and D.Y.K., the Dr. Ernst und Anita Bauer-Stiftung to S.H., and the Johannes und Frieda Marohn-Stiftung to T.H. and C.A.
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
- Aβ
- amyloid β
- AD
- Alzheimer's disease
- ADAM10
- a disintegrin and metalloproteinase domain-containing protein 10
- APP
- amyloid precursor protein
- BACE1
- β-site of APP-cleaving enzyme 1
- CTF
- C-terminal fragment
- DMEM
- Dulbecco's modified Eagle medium
- HEK
- human embryonic kidney
- ICD
- intracellular domain
- IKs
- slow delayed rectifying K+ current
- Kv
- voltage-gated potassium
- MiRP
- MinK-related peptide
- Nav
- voltage-gated sodium
- Navβ2
- voltage-gated sodium channel β2 subunit
- PS
- presenilin
REFERENCES
- 1. Assal F., Cummings J. L. (2002) Neuropsychiatric symptoms in the dementias. Curr. Opin. Neurol. 15, 445–450 [DOI] [PubMed] [Google Scholar]
- 2. Shepherd C., McCann H., Halliday G. M. (2009) Variations in the neuropathology of familial Alzheimer's disease. Acta Neuropathol. 118, 37–52 [DOI] [PubMed] [Google Scholar]
- 3. Larner A. J. (2010) Epileptic seizures in AD patients. Neuromolecular Med. 12, 71–77 [DOI] [PubMed] [Google Scholar]
- 4. Palop J., Chin J., Mucke L. (2006) A network dysfunction perspective on neurodegenerative diseases. Nature 443, 768–773 [DOI] [PubMed] [Google Scholar]
- 5. Verret L., Mann E. O., Hang G. B., Barth A. M. I., Cobos I., Ho K., Devidze N., Masliah E., Kreitzer A. C., Mody I., Mucke L., Palop J. J. (2012) Inhibitory interneuron deficit links altered network activity and cognitive dysfunction in Alzheimer model. Cell 149, 708–721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. De Leon M. J., Convit A., DeSanti S., Golomb J., Tarshish C., Rusinek H., Bobinski M., Ince C., Miller D. C., Wisniewski H. M. (1995) The hippocampus in aging and Alzheimer's disease. Neuroimaging Clin. N. Am. 5, 1–17 [PubMed] [Google Scholar]
- 7. Hardy J., Selkoe D. (2002) The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science 297, 353–356 [DOI] [PubMed] [Google Scholar]
- 8. Wolfe M. S. (2008) Gamma-secretase inhibition and modulation for Alzheimer's disease. Curr. Alzheimer Res. 5, 158–164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Vassar R., Kovacs D. M., Yan R., Wong P. C. (2009) The beta-secretase enzyme BACE in health and Alzheimer's disease: regulation, cell biology, function, and therapeutic potential. J. Neurosci. 29, 12787–12794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kim D., Ingano L., Carey B., Pettingell W., Kovacs D. (2005) Presenilin/γ-secretase-mediated cleavage of the voltage-gated sodium channel β2-subunit regulates cell adhesion and migration. J. Biol. Chem. 280, 23251–23261 [DOI] [PubMed] [Google Scholar]
- 11. Wong H., Sakurai T., Oyama F., Kaneko K., Wada K., Miyazaki H., Kurosawa M., De Strooper B., Saftig P., Nukina N. (2005) β Subunits of voltage-gated sodium channels are novel substrates of β-site amyloid precursor protein-cleaving enzyme (BACE1) and γ-secretase. J. Biol. Chem. 280, 23009–23017 [DOI] [PubMed] [Google Scholar]
- 12. Huth T., Schmidt-Neuenfeldt K., Rittger A., Saftig P., Reiss K., Alzheimer C. (2009) Non-proteolytic effect of beta-site APP-cleaving enzyme 1 (BACE1) on sodium channel function. Neurobiol. Dis. 33, 282–289 [DOI] [PubMed] [Google Scholar]
- 13. Kim D., Carey B., Wang H., Ingano L., Binshtok A., Wertz M., Pettingell W., He P., Lee V., Woolf C., Kovacs D. (2007) BACE1 regulates voltage-gated sodium channels and neuronal activity. Nat. Cell Biol. 9, 755–764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kim D. Y., Gersbacher M. T., Inquimbert P., Kovacs D. M. (2011) Reduced sodium channel Na(v)1.1 levels in BACE1-null mice. J. Biol. Chem. 286, 8106–8116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hu X., Zhou X., He W., Yang J., Xiong W., Wong P., Wilson C. G., Yan R. (2010) BACE1 deficiency causes altered neuronal activity and neurodegeneration. J. Neurosci. 30, 8819–8829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Huth T., Rittger A., Saftig P., Alzheimer C. (2011) β-Site APP-cleaving enzyme 1 (BACE1) cleaves cerebellar Na+ channel β4-subunit and promotes Purkinje cell firing by slowing the decay of resurgent Na+ current. Pflügers Arch. 461, 355–371 [DOI] [PubMed] [Google Scholar]
- 17. Birnbaum S. G., Varga A. W., Yuan L. L., Anderson A. E., Sweatt J. D., Schrader L. A. (2004) Structure and function of Kv4-family transient potassium channels. Physiol. Rev. 84, 803–833 [DOI] [PubMed] [Google Scholar]
- 18. McCrossan Z. A., Abbott G. W. (2004) The MinK-related peptides. Neuropharmacology 47, 787–821 [DOI] [PubMed] [Google Scholar]
- 19. Li Y., Um S., McDonald T. (2006) Voltage-gated potassium channels: regulation by accessory subunits. Neuroscientist 12, 199–210 [DOI] [PubMed] [Google Scholar]
- 20. Kim D., Ingano L., Kovacs D. (2002) Nectin-1α, an immunoglobulin-like receptor involved in the formation of synapses, is a substrate for presenilin/gamma-secretase-like cleavage. J. Biol. Chem. 277, 49976–49981 [DOI] [PubMed] [Google Scholar]
- 21. Carey B., Kim D., Kovacs D. (2007) Presenilin/γ-secretase and alpha-secretase-like peptidases cleave human MHC class I proteins. Biochem. J. 401, 121–127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Haapasalo A., Kim D., Carey B., Turunen M., Pettingell W., Kovacs D. (2007) Presenilin/γ-secretase-mediated cleavage regulates association of leukocyte-common antigen-related (LAR) receptor tyrosine phosphatase with β-catenin. J. Biol. Chem. 282, 9063–9072 [DOI] [PubMed] [Google Scholar]
- 23. Haapasalo A., Kovacs D. M. (2011) The many substrates of presenilin/γ-secretase. J. Alzheimers Dis. 25, 3–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Barhanin J., Lesage F., Guillemare E., Fink M., Lazdunski M., Romey G. (1996) K(V)LQT1 and lsK (minK) proteins associate to form the I(Ks) cardiac potassium current. Nature 384, 78–80 [DOI] [PubMed] [Google Scholar]
- 25. Sanguinetti M. C., Curran M. E., Zou A., Shen J., Spector P. S., Atkinson D. L., Keating M. T. (1996) Coassembly of K(V)LQT1 and minK (IsK) proteins to form cardiac I(Ks) potassium channel. Nature 384, 80–83 [DOI] [PubMed] [Google Scholar]
- 26. Rossner S., Apelt J., Schliebs R., Perez-Polo J. R., Bigl V. (2001) Neuronal and glial beta-secretase (BACE) protein expression in transgenic Tg2576 mice with amyloid plaque pathology. J. Neurosci. Res. 64, 437–446 [DOI] [PubMed] [Google Scholar]
- 27. Pongs O., Schwarz J. R. (2010) Ancillary subunits associated with voltage-dependent K+ channels. Physiol. Rev. 90, 755–796 [DOI] [PubMed] [Google Scholar]
- 28. Kovacs D. M., Gersbacher M. T., Kim D. Y. (2010) Alzheimer's secretases regulate voltage-gated sodium channels. Neurosci. Lett. 486, 68–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Dominguez D., Tournoy J., Hartmann D., Huth T., Cryns K., Deforce S., Serneels L., Camacho I. E., Marjaux E., Craessaerts K., Roebroek A. J., Schwake M., D'Hooge R., Bach P., Kalinke U., Moechars D., Alzheimer C., Reiss K., Saftig P., De Strooper B. (2005) Phenotypic and biochemical analyses of BACE1- and BACE2-deficient mice. J. Biol. Chem. 280, 30797–30806 [DOI] [PubMed] [Google Scholar]
- 30. Thomas G., Killeen M. J., Gurung I. S., Hakim P., Balasubramaniam R., Goddard C. A., Grace A. A., Huang C. L.-H. (2007) Mechanisms of ventricular arrhythmogenesis in mice following targeted disruption of KCNE1 modelling long QT syndrome 5. J. Physiol. (Lond.) 578, 99–114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Temple J., Frias P., Rottman J., Yang T., Wu Y., Verheijck E. E., Zhang W., Siprachanh C., Kanki H., Atkinson J. B., King P., Anderson M. E., Kupershmidt S., Roden D. M. (2005) Atrial fibrillation in KCNE1-null mice. Circ. Res. 97, 62–69 [DOI] [PubMed] [Google Scholar]
- 32. Balasubramaniam R., Grace A. A., Saumarez R. C., Vandenberg J. I., Huang C. L.-H. (2003) Electrogram prolongation and nifedipine-suppressible ventricular arrhythmias in mice following targeted disruption of KCNE1. J. Physiol. (Lond.) 552, 535–546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bianchi L., Shen Z., Dennis A. T., Priori S. G., Napolitano C., Ronchetti E., Bryskin R., Schwartz P. J., Brown A. M. (1999) Cellular dysfunction of LQT5-minK mutants: abnormalities of IKs, IKr and trafficking in long QT syndrome. Hum. Mol. Genet. 8, 1499–1507 [DOI] [PubMed] [Google Scholar]
- 34. Sesti F., Goldstein S. A. (1998) Single-channel characteristics of wild-type IKs channels and channels formed with two minK mutants that cause long QT syndrome. J. Gen. Physiol. 112, 651–663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Velliquette R. A., O'Connor T., Vassar R. (2005) Energy inhibition elevates beta-secretase levels and activity and is potentially amyloidogenic in APP transgenic mice: possible early events in Alzheimer's disease pathogenesis. J. Neurosci. 25, 10874–10883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. O'Connor T., Sadleir K. R., Maus E., Velliquette R. A., Zhao J., Cole S. L., Eimer W. A., Hitt B., Bembinster L. A., Lammich S., Lichtenthaler S. F., Hébert S. S., De Strooper B., Haass C., Bennett D. A., Vassar R. (2008) Phosphorylation of the translation initiation factor eIF2α increases BACE1 levels and promotes amyloidogenesis. Neuron 60, 988–1009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Clancy S. M., Chen B., Bertaso F., Mamet J., Jegla T. (2009) KCNE1 and KCNE3 β-subunits regulate membrane surface expression of Kv12.2 K+ channels in vitro and form a tripartite complex in vivo. PLoS One 4, e6330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Cole S., Vassar R. (2007) The Alzheimer's disease β-secretase enzyme, BACE1. Mol. Neurodegener. 2, 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Tinel N., Diochot S., Lauritzen I., Barhanin J., Lazdunski M., Borsotto M. (2000) M-type KCNQ2-KCNQ3 potassium channels are modulated by the KCNE2 subunit. FEBS Lett. 480, 137–141 [DOI] [PubMed] [Google Scholar]
- 40. Zhang M., Jiang M., Tseng G. N. (2001) minK-related peptide 1 associates with Kv4.2 and modulates its gating function: potential role as β-subunit of cardiac transient outward channel? Circ. Res. 88, 1012–1019 [DOI] [PubMed] [Google Scholar]
- 41. McCrossan Z. A., Roepke T. K., Lewis A., Panaghie G., Abbott G. W. (2009) Regulation of the Kv2.1 potassium channel by MinK and MiRP1. J. Membr. Biol. 228, 1–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Pannaccione A., Boscia F., Scorziello A., Adornetto A., Castaldo P., Sirabella R., Taglialatela M., Di Renzo G., Annunziato L. (2007) Up-regulation and increased activity of KV3.4 channels and their accessory subunit MinK-related peptide 2 induced by amyloid peptide are involved in apoptotic neuronal death. Mol. Pharmacol. 72, 665–673 [DOI] [PubMed] [Google Scholar]
- 43. McCrossan Z. A., Lewis A., Panaghie G., Jordan P. N., Christini D. J., Lerner D. J., Abbott G. W. (2003) MinK-related peptide 2 modulates Kv2.1 and Kv3.1 potassium channels in mammalian brain. J. Neurosci. 23, 8077–8091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Fukumoto H., Cheung B. S., Hyman B. T., Irizarry M. C. (2002) β-Secretase protein and activity are increased in the neocortex in Alzheimer disease. Arch. Neurol. 59, 1381–1389 [DOI] [PubMed] [Google Scholar]
- 45. Tyler S. J., Dawbarn D., Wilcock G. K., Allen S. J. (2002) α- and β-secretase: profound changes in Alzheimer's disease. Biochem. Biophys. Res. Commun. 299, 373–376 [DOI] [PubMed] [Google Scholar]
- 46. Yang L. B., Lindholm K., Yan R., Citron M., Xia W., Yang X. L., Beach T., Sue L., Wong P., Price D., Li R., Shen Y. (2003) Elevated β-secretase expression and enzymatic activity detected in sporadic Alzheimer disease. Nat. Med. 9, 3–4 [DOI] [PubMed] [Google Scholar]
- 47. Li R., Lindholm K., Yang L. B., Yue X., Citron M., Yan R., Beach T., Sue L., Sabbagh M., Cai H., Wong P., Price D., Shen Y. (2004) Amyloid beta peptide load is correlated with increased beta-secretase activity in sporadic Alzheimer's disease patients. Proc. Natl. Acad. Sci. U. S. A. 101, 3632–3637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Angulo E., Noe V., Casado V., Mallol J., Gomez-Isla T., Lluis C., Ferrer I., Ciudad C. J., Franco R. (2004) Up-regulation of the Kv3.4 potassium channel subunit in early stages of Alzheimer's disease. J. Neurochem. 91, 547–557 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.