

Abstract
Slow-channel syndrome (SCS) is an autosomal-dominant disease resulting from mutations in muscle acetylcholine (ACh) receptor subunits. The associated fatigue and muscle degeneration are proposed to result from prolonged synaptic responses that overload intracellular calcium. Single-channel studies on reconstituted receptors bearing human mutations indicate that the prolonged responses result from an increase in receptor open duration and, in some cases, increased sensitivity to ACh. We show that both of these aberrant receptor properties are recapitulated in heterozygotic zebrafish bearing an L258P mutation in the α subunit, thus affording the unique opportunity to compare the single-channel properties of mutant receptors to the synaptic currents in vivo. Whole-cell recordings revealed synaptic currents that decayed along a multiexponential time course, reflecting receptors containing mixtures of wild-type and mutant α subunits. Treatment with quinidine, an open-channel blocker used to treat the human disorder, restored fast synaptic current kinetics and the ability to swim. Quinidine block also revealed that mutant receptors generate a large steady-state current in the absence of ACh. The spontaneous openings reflected a destabilization of the closed state, leading to an apparent increase in the sensitivity of these receptors to ACh. The effective block by quinidine on synaptic currents as well as nonliganded openings points to dual sources for the calcium-dependent myopathy in certain forms of SCS.
Introduction
Much understanding of slow-channel syndrome (SCS) has emerged since the underlying mutations in muscle acetylcholine (ACh) receptor subunits were first identified (Engel et al., 1993; Ohno et al., 1995). To date, mutations in α, β, δ, and ε subunits have been reported, the majority of which are located near or within the pore-forming regions (Ohno et al., 1995; Sine et al., 1995; Engel et al., 1996a; Gomez et al., 1996; Milone et al., 1997). Mutations in the γ subunit likely escaped detection because the switch from the γ to the ε subunit enabled afflicted individuals to recover during early development (Hoffman et al., 2006). Most mutations are manifest as a destabilization of the closed state in favor of the open state, leading to prolonged channel openings (Ohno et al., 1995; Sine et al., 1995; Engel et al., 1996b; Gomez et al., 1996; Milone et al., 1997). This is causal to sustained depolarizations, excessive calcium entry, and postsynaptic degeneration (Engel et al., 1982; Gomez et al., 1997, 2002). Single-channel studies on muscle from patients (Engel et al., 1996a; Milone et al., 1997) and heterologously expressed channels bearing the relevant mutations (Sine et al., 1995; Engel et al., 1996a) point to two principal mechanisms causal to the stabilization of the open state: (1) altered intrinsic channel gating that results in a slowed channel-closing rate and (2) an increased apparent affinity to ACh causal to reopenings by channels (Sine et al., 1995; Engel et al., 1996a; Milone et al., 1997).
Recordings of spontaneous synaptic currents from patients (Engel et al., 1982; Sine et al., 1995) and transgenic mice expressing a mutant ε subunit (Gomez et al., 1996) point to multiple kinetic components of synaptic decay as causal to prolongation. In the case of α subunit mutations, assignment of kinetic components to individual receptor isoforms is complicated by the presence of two copies in the receptor. Additionally, the γ and ε subunits compete for association with α, offering even greater potential for kinetic heterogeneity. Both of these sources apply to our zebrafish model for SCS, twister, which contains a point mutation in the acetylcholine receptor α subunit. We now capitalize on the advantages of this zebrafish model, wherein recordings of single-channel currents and synaptic currents can be performed in vivo to identify the individual kinetic components. In particular, our ability to heterologously express defined wild-type and mutant receptor isoforms enables assignment of the muscle counterparts.
Finally, for the first time, it was possible to test the functional effects of rescue by quinidine on in vivo synaptic currents. On the presumption that quinidine accelerates synaptic current decay, this long-lived open-channel blocker is widely used to treat humans with SCS (Harper and Engel, 1998; Fukudome et al., 1998). We find that quinidine accelerated synaptic current decay but also showed an unexpected block of spontaneously open, nonliganded ACh receptors. Block of this standing source of depolarization and calcium entry likely represents an added restorative action by quinidine in patients afflicted with SCS.
Materials and Methods
Zebrafish (Danio rerio) were maintained in accordance with the standards set forth in the International Animal Care and Use Committee. The motility mutant twister line carries the formal nomenclature of nic1twister dbn12 (Lefebvre et al., 2004), but for the purposes of the present study will be referred to as either twi−/− for homozygous carriers or twi+/− for heterozygous carriers. Animals of either sex were used for all experiments.
The methods used to record spontaneous synaptic currents and evoked synaptic currents were identical to those previously published (Mongeon et al., 2011). Synaptic currents were recorded using a HEKA Instruments EPC-10/2 amplifier (Instrutech) and 1 μm tetrodotoxin was added for spontaneous synaptic current recordings. Synaptic currents were sampled at 10 μs and filtered at 5 kHz using Patchmaster software (Instrutech). Synaptic current decays of twi+/− were fit offline to the sum of three exponential functions by Igor Pro (WaveMetrics) using the Levenberg-Marquardt algorithm to search for the minimum χ2 value. The fit required the entire peak-to-end range for proper estimation of all three exponential components. For both single-channel and synaptic recordings, the overall mean ± SD was computed on the basis of the mean value for individual recordings. The n values refer to the number of recordings. The range of events used to compute the means are presented in the figure legends. The methods for paired motorneuron-muscle recording were identical to those previously published (Wen and Brehm, 2005).
Single-channel recordings from fast skeletal muscle were made in the on-cell configuration using an Axopatch 200B amplifier (Molecular Devices). The electrode and bath solutions were identical to those published previously (Mongeon et al., 2011). The muscle resting potential was estimated by adjusting the applied potential to eliminate all ACh-activated single-channel currents. Because the reversal potential of the ACh receptor is near 0 mV, this applied potential is taken as the muscle resting potential.
Single-channel currents from zebrafish receptors heterologously expressed in Xenopus oocytes used the outside-out patch configuration as previously published (Mongeon et al., 2011). In these recordings, the oocyte membrane potential was directly established and no corrections were required. Single-channel records from both oocytes and muscle were acquired at 100 kHz and digitally filtered at 5 kHz. Individual channel events were detected based on half-amplitude tracking with TAC software (Bruxton) and events with burst durations <200 μs were excluded. The single-channel burst duration and amplitude histograms were fit using TACfit software (Bruxton).
Macroscopic dose–response curves for applied agonists were performed using intact oocytes expressing specific receptor isoforms as previously described (Mongeon et al., 2011). The ACh was applied at a flow rate that resulted in complete exchange within 2 s. The dose–response curves for each isoform were fit by the Hill equation using IGOR Pro 5.05 (Paradiso and Brehm, 1998). Quinidine sulfate, choline chloride, and d-tubocurare were all obtained from Sigma Chemical and made fresh in recording solution before each experiment. α-Bungarotoxin was obtained from Invitrogen.
Fish movements were tracked at 1000 frames per second using a Fastcam 512-PCI camera (Photron Instruments) and were quantitated using Flote zebrafish behavioral analysis software (Burgess and Granato, 2007).
Results
Synaptic currents
The behavioral consequences of the L258P mutation are manifest as an inability of either of twi−/− or twi+/− fish to mount an effective swimming response during the first days of development (Lefebvre et al., 2004). Heterozygotic fish eventually recover the ability to swim but homozygotic fish exhibit a complete deterioration of muscle and death within the first few days of development. Consequently, twi−/− muscle cells are leaky and resistant to whole-cell patch clamp. By contrast, twi+/− fish were healthy enough at all stages for patch-clamp recordings. At 48 h postfertilization (hpf), corresponding to the height of behavioral defect, the decay time course of miniature endplate currents (mEPCs) was greatly prolonged compared with those recorded from wild-type muscle (Fig. 1A). Fit of the current decay from twi+/− muscle generally required the sum of three exponential components termed the fast (τf), intermediate (τi), and slow (τs) time constants. The average time constants corresponded to 1.1 ± 0.3 ms, 8.6 ± 2.5 ms, and 70.2 ± 22.0 ms respectively (Fig. 1A,B). This compares to 0.6 ± 0.3 ms for 48 hpf wild-type fish, determined on the basis of a single exponential fit to the synaptic current decay. The average fractional contribution to overall peak synaptic current in twi+/− corresponded to 0.45 ± 0.09 for τf, 0.40 ± 0.09 for τi and 0.15 ± 0.08 for τs (Fig. 1C).
Figure 1.

Kinetics of spontaneous endplate currents in wild-type and twi+/− fast skeletal muscle. A, Representative mEPCs recorded from 48 hpf wild-type and twi+/− fast muscle. Synaptic current decays (black) were fit peak-end (red overlay) by either a single exponential function for wild type or by the sum of three exponential functions for twi+/−. The insets show the individual components of each fit. B, The mean decay time constants computed for each recording are indicated by filled circles and the overall mean ± SD by an open circle (wild type, n = 9; twi+/−, n = 14). Between 7 and 115 events were measured for each recording. C, Fractional contribution to overall amplitude by each exponential component in twi+/−.
The influence of the complex synaptic current kinetics on action potential generation was next determined using two approaches. First, simulated command waveforms based on twi+/− and wild-type synaptic current waveforms were injected directly into 48 hpf wild-type muscle (Fig. 2A,B). The current amplitudes were based on evoked synaptic currents obtained using motorneuron-skeletal muscle-paired recording. The decay time course was based on the mean time constants and fractional contribution by each exponential component. Under current clamp, both wild-type and twi+/− synaptic waveforms consistently generated a single action potential (Fig. 2A,B). Wild-type repolarization was rapid compared with twi+/−, which exhibited a prolonged after-depolarization. Next, muscle responses were recorded in paired motorneuron-muscle current-clamp recordings (Fig. 2C,D). The motorneuron action potentials were indistinguishable between twi+/− and wild-type pairs and, once again, only a single muscle action potential was observed, despite the greatly prolonged depolarization. Thus, both approaches point to a maintained contraction in twi+/− resulting from prolonged depolarization and not from repetitive firing of muscle action potentials.
Figure 2.

Effect of prolonged synaptic currents on postsynaptic potential. A, B, Representative action potential waveforms recorded from 48 hpf wild-type fast muscle were elicited using voltage command waveforms based on the average synaptic current decays for wild type (A) and twi+/− (B) shown in Figure 1. The command waveforms are shown in gray and the associated muscle responses are shown in black (n = 5 cells; between 12 and 20 action potentials were averaged for each cell). C, D, Paired current-clamp recordings of motorneuron (gray) and muscle action potentials (black) for 48 hpf wild-type (C; n = 4 pairs; 7–28 action potentials per cell) and twi+/− (D; n = 6 pairs; 8–26 action potentials per cell) muscle.
Identifying the effects of the L258P mutation on gating of receptor isoforms
Recordings were made from zebrafish ACh receptors expressed in Xenopus oocytes to determine the effects of the α subunit mutation on single-channel function. The oocytes were injected with RNA coding for the following combinations of zebrafish subunits: αβδγ, αβδε, αtwiβδγ, or αtwiβδε. Before single-channel recordings, dose–response relations to applied ACh were generated for whole oocytes expressing the different receptor isoforms. The relationships were then fit using the Hill equation and half maximum values were determined for each isoform (Fig. 3). The average half-saturating concentrations for wild-type αβδγ and αβδε receptors corresponded to 2.4 μm and 1.7 μm respectively. By contrast, the relationships for both receptor isoforms containing two αtwi subunits were shifted to lower concentrations. The mean half maximum corresponded to 12 nm for αtwiβδγ and 39 nm for αtwiβδε. Overall, this reflects a 200-fold increase in apparent sensitivity to ACh for αtwiβδγ and a 44-fold increase for αtwiβδε.
Figure 3.

Dose–response curves obtained for receptor subunit combinations expressed in Xenopus oocytes. Oocytes expressing either αβδγ (red), αβδε (black), αtwiβδγ (green), or αtwiβδε (blue) were voltage-clamped at −50 mV and current responses to fast flow changes in ACh concentration were determined. Each point represents the mean ± SD of the percentage of peak current determined for 6–7 oocytes at each ACh concentration. The dose–response relationships between peak ACh-activated current and ACh concentration were fit to the Hill equation I = Imax/(1 + [K1/2/(A)]n) where I is the current at a given concentration of agonist A, Imax is the peak ACh-activated current, K1/2 is the half-maximal concentration, and n is the Hill coefficient.
Next, single-channel currents from identified isoforms were recorded using outside-out oocyte patches from Xenopus oocytes. Visual inspection of sample traces for each of the four isoforms tested points to large differences in burst duration between wild-type and mutant receptors (Fig. 4A–D). Openings by both αtwiβδγ (Fig. 4D) and αtwiβδε (Fig. 4B) receptors were considerably longer than wild-type αβδγ (Fig. 4C) and αβδε (Fig. 4A). Current–voltage relations obtained for both αtwiβδγ and αtwiβδε single-channel currents were linear and yielded slope conductances of 48 and 60 pS respectively (Fig. 4E). Both values were slightly lower than the αβδε and αβδγ counterparts (Fig. 4F). The burst duration histograms for αβδε and αtwiβδε were each fit with a single exponential (Fig. 4A,B) with overall mean time constants corresponding to 0.32 ± 0.05 ms and 9.27 ± 2.74 ms respectively. In the specific case of αβδε, the fast kinetics limited full resolution of the primary component of burst duration, so the mean time constant could potentially be even briefer than our estimate. The burst duration distribution for the αβδγ isoform required two exponentials for fit (Fig. 4C). The predominant component for αβδγ corresponded to 0.67 ± 0.30 ms and contributed 75 ± 11% of total events whereas the minor slower component was 2.71 ± 1.10 ms and contributed 25 ± 11% of total events. The overall mean values for both wild-type αβδε and αβδγ receptors were extracted from a large published database generated for reconstituted zebrafish receptors (Mongeon et al., 2011). The burst duration for the αtwiβδγ isoform was well fit by a single exponential with a mean value of 37.28 ± 12.66 ms, reflecting a 56-fold and 14-fold increase over the αβδγ fast and slow components respectively (Fig. 4G).
Figure 4.
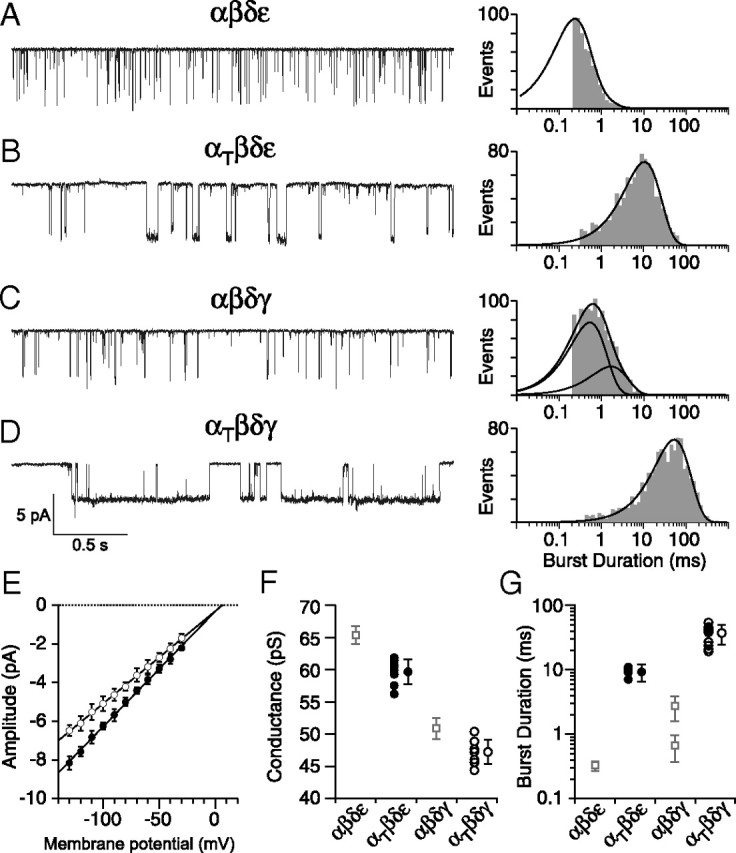
ACh-activated single-channel currents associated with different receptor isoforms. A–D, Representative single-channel recordings of ACh-activated currents from outside-out patches of Xenopus oocytes expressing either (A) αβδε, (B) αtwiβδε, (C) αβδγ, or (D) αtwiβδγ receptors. Example semilog burst duration histograms for each receptor isoform fit with either a single exponential (A, B, D) or biexponential (C) function. E, Cumulative current–voltage relations showing mean ± SD for αtwiβδε (filled circles, n = 8 recordings) and αtwiβδγ (open circles, n = 8 recordings) with linear fits. F, Scatterplot of mean slope conductances obtained for individual recordings from αtwiβδε (filled circles) and αtwiβδγ (open circles) shown alongside the overall mean ± SD for each distribution. The mean ± SD for wild-type counterparts are shown as open squares (taken from Mongeon et al., 2011). Between 31 and 111 events were used to generate each point in E and F. G, Scatterplot of mean burst duration from αtwiβδε (filled circles, n = 7 recordings) and αtwiβδγ (open circles, n = 10 recordings) receptors along with the overall mean ± SD. Between 659 and 1147 events for each recording were used to generate the individual mean values. The wild-type mean ± SD counterpart values are shown as open squares (taken from Mongeon et al., 2011).
Single-channel counterparts in muscle
Single-channel recordings of ACh-activated currents from twi+/− fast muscle were used to render assignment of the muscle receptors to specific receptor isoforms. Unfortunately, it was not possible to record ACh-activated currents from outside-out patches derived from muscle, so we turned to on-cell patch recordings. Recordings were first performed on twi+/− fish at 48 hpf, the time corresponding to the height of the motility defect and the analysis of synaptic current time course (Fig. 5A). The electrode contained 300 nm ACh to activate all of the isoforms present at this time. In three patches tested, two amplitude classes could be observed with slope conductances of 44.1 and 58.5 pS corresponding to γ-containing and ε-containing receptors respectively (Fig. A,B). The cumulative burst duration histograms for the example in Figure 5C were complex and required three exponentials for fit with mean time constants corresponding to 0.5, 3.4, and 44.8 ms. The individual burst components were next assigned to either ε-containing or γ-containing receptors by separating the event classes on the basis of amplitudes (Fig. 5D,F). The burst duration for the large amplitude class was fit by two exponentials with time constants of 0.5 and 4.8 ms (Fig. 5E). These time constants correspond to the αβδε and αtwiβδε receptors heterologously expressed in oocytes. The burst duration of the small-amplitude class (Fig. 5F) was also fit by two exponentials with time constants of 1.6 and 47.1 ms (Fig. 5G), corresponding to the αβδγ-receptor and αtwiβδγ-receptor isoforms.
Figure 5.
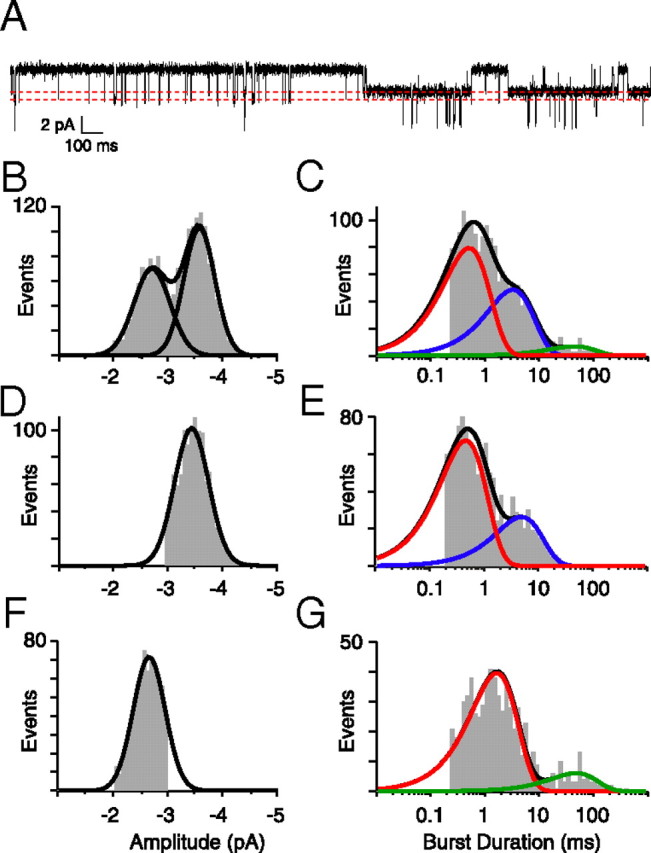
ACh-activated single-channel currents from 48 hpf twi+/− fish. A, An ∼3 s sample trace of channel openings to two amplitude levels in the presence of 300 nm ACh. The dotted lines indicate the two open levels. B, Amplitude histogram for all openings required the sum of two Gaussian distributions with means of −3.5 and −2.7 pA (n = 1796 events). C, The burst duration histogram for all openings, fit to three exponential components with time constants corresponding to 0.5, 3.4, and 44.8 ms. D, The distribution of large amplitude openings used to generate the burst duration in E (n = 1065 events). E, The burst duration for large amplitude events was fit with two exponential components with time constants corresponding to 0.5 and 4.8 ms. F, The distribution of small amplitude openings used to generate the burst duration in G (n = 731 events). G, The burst duration for small amplitude events was fit with two exponential components with time constants corresponding to 1.6 and 47.1 ms.
Further confirmation of assignment was made by recording from 24 hpf muscle, a time when ε expression is expected to be low (Fig. 6). The first recordings of ACh-activated single-channel currents came from twi−/− (Fig. 6A–C). In these fish, the αtwiβδγ isoform would be expected to be the predominant isoform. To maximize the openings at this early stage and minimize desensitization, the recordings were performed using 30 nm ACh. In seven recordings, we observed one single-amplitude class (Fig. 6A,B) that corresponded to 44 pS (Fig. 6J,K), which was similar to the 48 pS value measured for αtwiβδγ receptors. The open burst duration distribution for this class was well described by a single exponential function with time constant corresponding to 23.18 ± 3.92 ms (Fig. 6C,L). Thus, on the basis of slope conductance and burst duration, this class corresponds to αtwiβδγ receptors. Recordings from twi+/− fish showed a class with similar amplitude (Fig. 6D,E) and conductance (Fig. 6J,K). The burst duration histogram was also fit by a single exponential (Fig. 6F) with time constants similar to those of the homozygous fish (Fig. 6L).
Figure 6.

Cell-attached ACh-activated single-channel currents from 24 and 120 hpf twister muscle. A, D, G, Sample traces of ACh-activated channels in 24 hpf twi−/− (A), 24 hpf twi+/− (D), and 120 hpf twi+/− fish (G). B, E, Amplitude distributions for 24 hpf twi−/− and 24 hpf twi+/− fish fit to single Gaussian distributions. C, F, Example semilog burst duration histograms fit to a single exponential function with time constants corresponding to 24.8 ms (n = 904 events) and 23.8 ms (n = 970 events) for 24 hpf twi−/− and 24 hpf twi+/− fish, respectively. H, Amplitude distribution for 120 hpf twi+/− fish fit with a single Gaussian function. I, Example burst duration histogram for 120 hpf twi+/− fish fit by the sum of two exponential components with time constants corresponding to 0.4 and 8.3 ms (n = 1170 events). J, Cumulative current–voltage relations comparing 24 hpf twi−/− (gray open circles; n = 7), 24 hpf twi+/− (black open circles; n = 9) and 120 hpf twi+/− (filled circles; n = 10) fish. A range of 17 to 140 events was used to calculate each point on the current–voltage relationship. K, Mean slope conductance ± SD calculated for 24 hpf twi−/− (43.7 ± 4.0 pS), 24 hpf twi+/− (44.7 ± 3.1 pS), and 120 hpf (62.3 ± 3.6 pS) shown alongside individual slope conductance values. L, The mean burst durations ± SD for 24 hpf twi−/− (25.6 ± 3.4 ms; n = 5), 24 hpf twi+/− (23.2 ± 3.9 ms; n = 5), and 120 hpf twi+/− (7.4 ± 1.9 ms and 0.41 ± 0.12 ms; n = 10) are shown along with the individual cell averages. A range of 376 to 1170 events was used to calculate the burst duration. All data were obtained using 30 nm ACh.
Confirmation of the high conductance class assignments was made using 120 hpf twi+/− fish. In these fish, most receptors would be expected to contain the ε subunit. Cell recordings revealed a single amplitude class of openings (Fig. 6G,H), with a slope conductance that corresponded to 62 pS (Fig. 6J,K). The burst duration of this class required two exponentials for fit with mean time constants corresponding to 0.41 ± 0.12 ms and 7.38 ± 1.86 ms (Fig. 6I,L). These likely reflect mixed openings by αtwiβδε and αβδε receptors on the basis of conductance and burst duration.
Agonist and antagonist gating of mutant receptors
Alterations in the gating equilibrium were tested for possible involvement in heightened sensitivity to ACh for receptors harboring the twister mutation. First, we examined mutant receptors in the absence of ACh for evidence of nonliganded openings. Nonliganded openings were determined on the bases of both single-channel and macroscopic current recordings from oocytes. In both cases the recording chamber was washed thoroughly to avoid lingering low concentrations of ACh. At the single-channel level, nonliganded openings corresponding to either αβδε or αβδγ receptors were too rare to be annotated. By contrast, both αtwiβδγ and αtwiβδε receptors consistently showed single-channel openings in outside-out patches in the absence of applied ACh. The frequency of the openings for αtwiβδγ, however, was orders of magnitude greater (Fig. 7A). The burst duration was brief in the absence of ACh compared with that seen in the presence of ACh, likely reflecting further stabilization of the open state in the presence of ACh (Fig. 7A). The openings in the absence of ACh point toward altered gating rather than altered binding as responsible for increased sensitivity to ACh (Jackson, 1984; Zhou et al., 1999).
Figure 7.
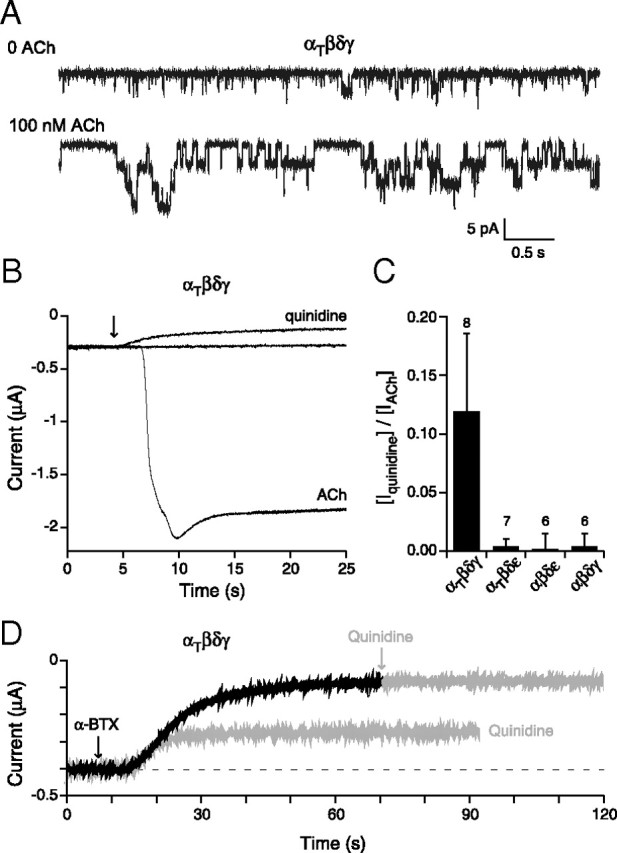
Openings by nonliganded αtwiβδγ receptors expressed in Xenopus oocytes. A, Outside-out patch recordings of αtwiβδγ receptors before (top trace) and after (bottom trace) addition of ACh (n = 6 patches). B, Recordings of macroscopic current in oocytes expressing αtwiβδγ and held at −50 mV. Shown are the baseline current in the absence of ACh, the decrease in holding current upon addition of 100 μm quinidine (arrow) and, following washout of the quinidine, the large inward current that occurred following addition of 300 nm ACh (arrow) to the oocyte. C, Overall comparisons of the effect of quinidine to reduce the holding current in the absence of ACh activation of the different receptor isoforms. The fractional peak current was determined on the basis of reduction in holding current in the presence of 100 μm quinidine relative to total ACh-activated current following washout of quinidine. The mean ± SD are shown and the number of oocytes tested is indicated. D, Traces from an oocyte expressing αtwiβδγ receptors showed decreases in holding current in response to individual application of either 1 μm α-btx (black trace) or 100 μm quinidine. The response to the first application of quinidine was allowed to recover before the oocyte was treated with α-btx. Following treatment with α-btx, the oocyte showed no response to further application of quinidine (gray arrow).
Next, we searched for evidence of a macroscopic equivalent of nonliganded ACh receptor channel openings in oocytes expressing αtwiβδγ receptors. As a measure of nonliganded openings we tested two blockers of ACh receptors, the open channel blocker quinidine and the irreversible blocker α-bungarotoxin (α-btx). α-btx was tested at 1 μm, while quinidine was tested at 100 μm corresponding to 20-fold higher than used previously in single-channel studies (Fukudome et al., 1998). This concentration was chosen to provide maximal block of spontaneous receptor openings by use of a slow channel blocker. In oocytes expressing either αβδγ or αβδε receptors, neither agent had an effect on holding current (Fig. 7C). By contrast, both agents led to large decreases in holding current in oocytes expressing αtwiβδγ receptors (Fig. 7B,D). Pretreatment with α-btx eliminated the effects of quinidine, showing that the two agents were working through the same population of ACh receptors (Fig. 7D). To estimate the fraction of total available ACh-activated current contributed by spontaneous openings, we first determined the amplitude of quinidine-sensitive holding current, then washed off the quinidine, and then treated with 300 nm ACh to activate all available receptors (Fig. 7B). The ratio of quinidine-sensitive holding current to peak ACh-activated current αtwiβδγ averaged 0.12 ± 0.07 (n = 8; Fig. 7C). This is remarkably high in light of the fact that the burst duration for spontaneous events is much briefer than in the presence of ACh (Fig. 7A,B). Only small decreases in holding current were observed following quinidine treatment for αtwiβδε receptors.
Next, choline, a very weak agonist of muscle nicotinic receptors, was tested for activation of mutant receptors (Fig. 8A). Application of 1 mm choline to oocytes expressing wild-type receptors resulted in weak fractional activations corresponding to 0.04 ± 0.01 for αβδγ and 0.03 ± 0.02 for αβδε receptors (Fig. 8A,B). By contrast, application of choline to either αtwiβδγ-receptor or αtwiβδε-receptor isoforms resulted in strong activation of inward current (Fig. 8A,B). The fractional activation for αtwiβδγ and αtwiβδε receptors corresponded to 0.92 ± 0.05 and 0.95 ± 0.04, both comparable to ACh activation (Fig. 8B). The antagonist d-tubocurare was also tested for partial agonist action (Fig. 8A). Application of 10 μm curare failed to significantly activate either αβδγ or αβδε receptors (Fig. 8B). However, significant responses were observed for both αtwiβδγ-receptor and αtwiβδε-receptor isoforms (Fig. 8A,B). The mean fractional activation corresponded to 0.17 ± 0.08 for αtwiβδγ and 0.35 ± 0.09 for αtwiβδε receptors (Fig. 8B).
Figure 8.
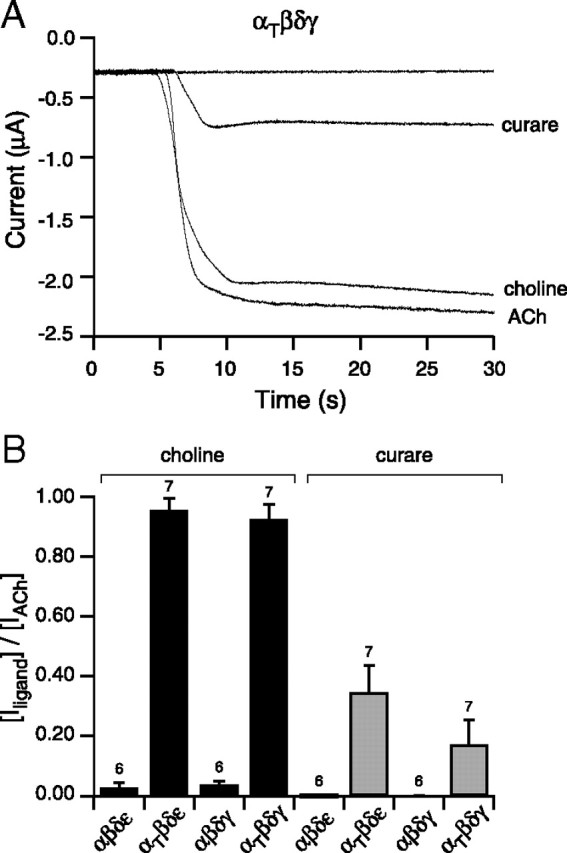
Potency of activation of αtwiβδγ receptors by ACh, choline, and curare. A, Recording of macroscopic current in oocytes expressing αtwiβδγ and held at −50 mV. Addition of either 10 μm curare, 1 mm choline, or 300 nm ACh effectively evoked an inward current. B, Overall comparisons of activation by choline (black) and curare (gray) on each of the different receptor isoforms. The bar graph reflects the mean fractional activation ± SD of peak ACh-activated current associated with either curare or choline treatment and the number of oocytes tested is indicated.
Quinidine effects on synaptic currents and nonliganded receptor openings
As quinidine is used for clinical treatment of patients afflicted with slow-channel syndrome, we were prompted to directly test the effects on twi+/− fish. First, the effects on swimming were tested before and after treatment with quinidine (Fig. 9A). As early as 48 hpf, wild-type fish could generate alternating contractions as quantitated with motion-tracking software (Fig. 9A, top). At this age, mutant fish were only able to generate a single maintained bend following mechanical stimulation (Fig. 9A, bottom). To test for improvement, a high quinidine concentration of 50 μm was added to the bath solution for 10 min, after which the fish were placed in a quinidine-free bath solution for testing. The high concentration was used to facilitate entry into the live animal from the bath solution. In all, fish showed marked improvement in 7 of 10 testing groups in the ability to perform alternating contractions like those seen in wild-type fish (Fig. 9A). Next, we tested the effects of quinidine on synaptic currents. For this purpose, mEPCs recorded from twi+/− fast muscle were compared before and after addition of 10 μm quinidine (Fig. 9B). The mean amplitude of spontaneous synaptic currents was reduced on average by 45.9% in response to quinidine. However, the most pronounced effect of the treatment was a greatly accelerated decay phase of the synaptic currents (Fig. 9B). The triple exponential decay of synaptic currents in the twi+/− fish was converted to a decay that followed a single exponential time course with a mean time constant corresponding to 0.85 ± 0.40 ms (Fig. 9B,C). There was no evidence of a slow or intermediate component of synaptic current decay at this concentration.
Figure 9.
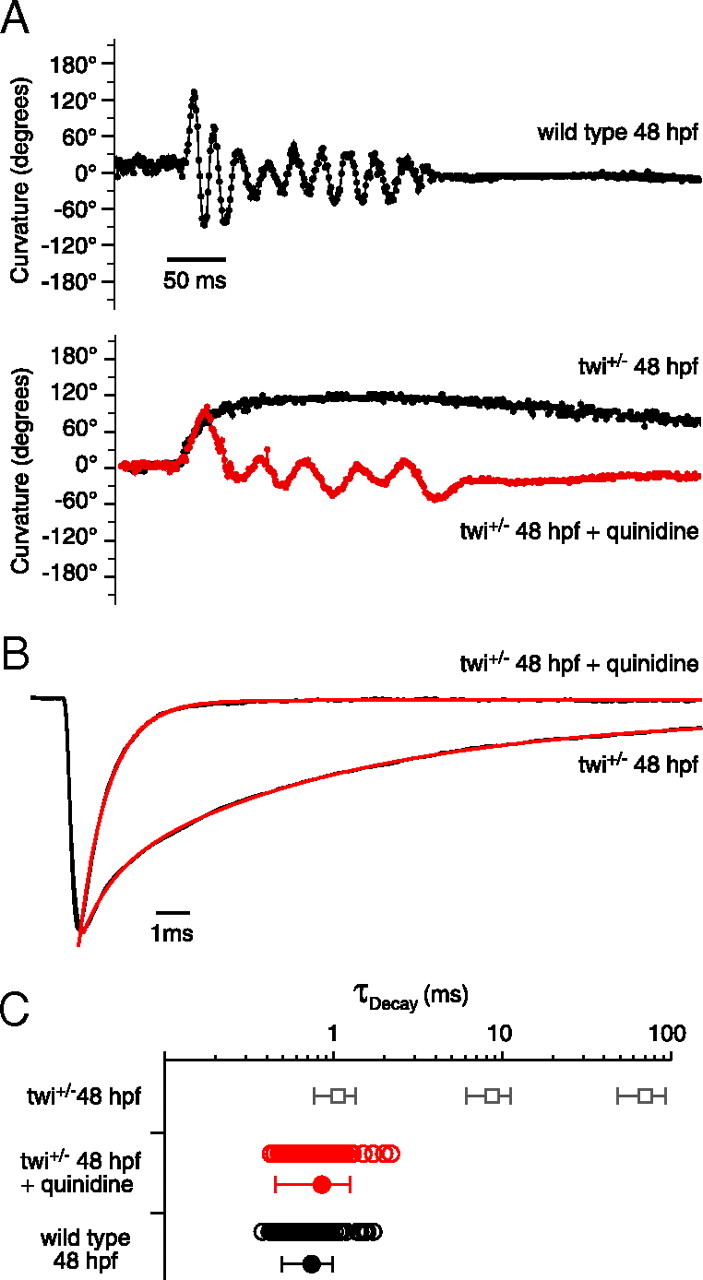
Effect of quinidine on swimming and synaptic current decay. A, Swimming behavior in 48 hpf wild type (top), twi+/− (bottom; black) and twi+/− treated with quinidine (bottom; red). Curvature is measured in 1 ms increments where each point is the sum of the head and tail bend angle relative to the central body axis. B, Normalized mEPC recorded from twi+/− 48 hpf fast muscle cell before and after addition of 10 μm quinidine. Before application, the decay was fit by a triple exponential function (red overlay) and after treatment was fit with a single exponential function (red overlay). C, Scatter plot of individual decay time constants for wild type (black open circles; n = 6 cells; 87 events), and quinidine-treated twi+/− (red open circles; n = 7 cells; 80 events) shown alongside mean ± SD (filled circles). The mean ± SD for the three twi+/− time constants (Fig. 1B) is shown as open squares for comparison.
Discussion
Slow-channel syndrome is a form of human congenital myasthenia associated with point mutations in the muscle nicotinic receptor subunits. Mutations in the α, β, δ, and ε subunits have been identified and most of these mutations are located in transmembrane domains (Ohno et al., 1995; Sine et al., 1995; Engel et al., 1996a; Gomez et al., 1996; Milone et al., 1997). Recordings from human biopsy muscle (Sine et al., 1995; Engel et al., 1996a) and findings from transgenic mice overexpressing mutant subunits (Gomez et al., 1997, 2002) have indicated prolonged synaptic potentials. However, the bulk of information regarding the functional consequences of individual mutations in humans has been extracted from single-channel studies on mutated receptors. Overall, these measurements have pointed to a panel of functional alterations that are, to a large extent, shared by the different mutations. Topping the list are altered rate constants for channel gating that lead to prolonged channel openings, the presumed underpinnings of synaptic current prolongation (Sine et al., 1995; Engel et al., 1996a; Milone et al., 1997). Additionally, some of the mutations increase the sensitivity to ACh (Sine et al., 1995; Engel et al., 1996a; Milone et al., 1997) as well as alter the desensitization kinetics (Engel et al., 1996a; Milone et al., 1997). These functional alterations lead to enhanced receptor openings, causal to prolonged contractions and calcium overload.
The zebrafish mutant twister was identified through a dominant motility screen and mapped to an L258P mutation in the pore-lining M2 domain of the α subunit (Lefebvre et al., 2004). Recordings from twi+/− revealed greatly prolonged synaptic currents, which result in a persistent depolarization. A single action potential accompanied the depolarization and subsequent firing was prevented until repolarization occurred. Because wild-type synaptic currents result in spikes that repolarize quickly, they are able to follow frequencies in excess of 200 Hz (Wen and Brehm, 2005). These findings suggest that persistent depolarization, rather than persistent firing of action potentials, is most directly involved in causing the maintained contraction.
Mutations in the α subunit differ from those in the β, δ, ε, and γ subunits by virtue of two subunit copies in the same receptor. Consequently, in slow-channel syndrome, we would expect multiple functional receptor isoforms, attributable to mixtures of wild-type and mutant α subunits. An additional layer of functional complexity is potentially introduced by the developmental switch between the γ-receptor and ε-receptor isoforms (Mongeon et al., 2011). Due to the unique opportunity to perform in vivo recordings from twi+/− muscle, it was possible to resolve three kinetic components of the synaptic decay in twi+/−. To assign synaptic components to individual receptor isoforms, we reconstituted both wild-type and ACh receptors bearing the twister mutation in Xenopus oocytes to examine single receptor gating properties.
Single-channel studies at 48 hpf formed the initial bases for assignment of the isoforms underlying individual components. At this stage, both γ-receptor and ε-receptor isoforms were present, as reflected in the two conductance classes. The overall burst duration histogram required three exponential components for fit. The fast component fell within the mixed time constants for the αβδγ and αβδε receptors (Mongeon et al., 2011). The intermediate time constant was assigned to the high conductance isoform and corresponded to the αtwiβδε, whereas the slow time constant was assigned to the low-conductance isoform and corresponded to αtwiβδγ. These assignments were further strengthened by recordings at 24 hpf, where receptor isoforms were principally γ-containing isoforms, and recordings at 120 hpf, where receptor isoforms were principally ε-containing isoforms. While the slow component of synaptic current decay matches best the mean burst duration for the αtwiβδγ receptor, overall comparisons indicate a twofold longer average for the decay. This difference is further reflected in single-channel measurements from muscle at both 24 and 48 hpf. This difference may reflect the reopening of receptors during the synaptic event. Reopening has been predicted to occur on the basis of the estimated rate constants for channel opening and ligand dissociation from single-channel studies (Colquhoun and Hawkes, 1982; Sine and Steinbach, 1986). This may be exacerbated by both the left-shifted sensitivity of mutant receptors to ACh and/or by incomplete hydrolysis of ACh, a feature associated with early synapse development allowing rebinding by ACh and persistent reopening of αtwiβδγ receptors (Kullberg et al., 1980; Nguyen et al., 1999; Drapeau et al., 2001).
In human SCS mutations, shifts in apparent affinity (Ohno et al., 1995; Engel et al., 1996a; Milone et al., 1997), the efficacy of weak agonists and competitive antagonists (Milone et al., 1997), and spontaneous openings (Ohno et al., 1995; Engel et al., 1996a; Grosman and Auerbach, 2000) have all been reported. Such changes have been proposed to result from stabilization of the open state through changes in the diliganded and nonliganded gating equilibrium constants (Jackson, 1984, 1986; Grosman and Auerbach, 2000). In the absence of agonist, wild-type receptors have a low probability of opening because the closed conformation is energetically favored (Purohit and Auerbach, 2009; Auerbach, 2010). Reducing the free energy difference between the open and closed states increases the frequency of nonliganded spontaneous openings and shifts the midpoint of the dose–response curve to lower concentrations. This allows a weak agonist, like choline, to effectively gate the receptor (Jadey et al., 2011).
Increased burst duration, spontaneous openings, enhanced sensitivity to ACh and gating by the weak agonist choline were all properties of receptors containing the mutant α subunit. Both αtwiβδγ and αtwiβδε showed left-shifted dose–response curves and effective gating by choline. However, both burst duration and the frequency of spontaneous openings were much higher for the αtwiβδγ isoform, pointing to differential interdependence of mutant α on ε versus γ subunit. For example, 30 nm ACh activates both short (αβδε-like) and long (αtwiβδε-like) burst duration components at 120 hpf in twi+/− fish. This low concentration would not be expected to activate wild-type receptors. By contrast, in 24 hpf twi+/−, there was no evidence of short (αβδγ-like) bursts at 30 nm ACh. Instead, evidence indicated only the very long αtwiβδγ bursts. This points to the possible existence of a distinction between effects of mutant α on ACh sensitivity and gating when formed with the ε instead of the γ subunit. Our ability to draw firm conclusions about the receptor isoform and functional consequences is greatly compromised by the uncertainty surrounding receptors with a single mutant α subunit. Because receptors with two mutant α subunits can function, it is reasonable to surmise that receptors with single mutant subunits can function. However, we were unable to determine whether any or all of these functional properties are shared by receptors containing two mutant α subunits versus a single mutant α. The consequences may not be shared by receptors formed through association with the γ versus ε subunit. A second source of potential complication may relate to the position of a single mutant α subunit relative to the δ subunit. In other words, the interaction of mutant α may differ when partnered with δ than when partnered with either γ or ε.
Openings by the embryonic αtwiβδγ isoform are effectively blocked by quinidine. Quinidine is a long-lived open-channel blocker used to treat humans afflicted with slow-channel syndrome (Fukudome et al., 1998; Harper and Engel, 1998). Due to the technical limitations involving human subjects, the effects on synaptic current have never been tested. The prediction that quinidine would serve as a channel blocker to accelerate synaptic current decay was based on the single-channel studies. Our recordings provide the first measure of effectiveness and substantiate the predicted effect. In response to 10 μm quinidine, the synaptic current decay time course shortened greatly and to the same extent seen for native adult wild-type receptors. The decay of synaptic current is complete and shows no sign of incomplete block, rendering it a powerful prophylactic treatment. Quinidine likely also carries a previously unappreciated source of treatment in the block of nonliganded spontaneous mutant receptor openings. For αtwiβδγ receptors, the nonliganded openings account for nearly 15% of the total available receptors. Such a large steady-state current is predicted to outweigh the total integrated current associated with low-frequency synaptic responses.
Finally, much evidence exists in support of long-term postsynaptic deterioration in humans afflicted with slow-channel syndrome (Leonard and Salpeter, 1979, 1982; Engel et al., 1982, 1996a; Gomez et al., 1996, 1997, 2002; Milone et al., 1997) and has been directly linked to the calcium entering during the prolonged depolarization associated with synaptic currents (Gomez et al., 2002). Additional contributions by spontaneous receptor openings to postsynaptic degeneration were proposed for humans with slow-channel syndrome (Engel et al., 1996a). So, as with human slow-channel syndrome, there are two potential sources for calcium overload in twi−/− fish: prolonged synaptic current and spontaneous receptor openings. The fact that quinidine so effectively blocks both sources points to a dual role for this drug in preventing muscle degeneration resulting from calcium overload.
Footnotes
This work was supported by National Institutes of Health Grant NS18205 to P.B. We thank Dr. David Dawson and Chris Alexander for generously providing Xenopus oocytes. Dr. Geng-Lin Li generated the multiexponential fitting routine for synaptic current decay. Dr. Claudio Grosman helped greatly with our interpretation of spontaneous openings and altered agonist sensitivity.
References
- Auerbach A. The gating isomerization of neuromuscular acetylcholine receptors. J Physiol. 2010;588:573–586. doi: 10.1113/jphysiol.2009.182774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgess HA, Granato M. Modulation of locomotor activity in larval zebrafish during light adaptation. J Exp Biol. 2007;210:2526–2539. doi: 10.1242/jeb.003939. [DOI] [PubMed] [Google Scholar]
- Colquhoun D, Hawkes AG. On the stochastic properties of bursts of single ion channel openings and of clusters of bursts. Philos Trans R Soc Lond B Biol Sci. 1982;300:1–59. doi: 10.1098/rstb.1982.0156. [DOI] [PubMed] [Google Scholar]
- Drapeau P, Buss RR, Ali DW, Legendre P, Rotundo RL. Limits to the development of fast neuromuscular transmission in zebrafish. J Neurophysiol. 2001;86:2951–2956. doi: 10.1152/jn.2001.86.6.2951. [DOI] [PubMed] [Google Scholar]
- Engel AG, Lambert EH, Mulder DM, Torres CF, Sahashi K, Bertorini TE, Whitaker JN. A newly recognized congenital myasthenic syndrome attributed to a prolonged open time of the acetylcholine-induced ion channel. Ann Neurol. 1982;11:553–569. doi: 10.1002/ana.410110603. [DOI] [PubMed] [Google Scholar]
- Engel AG, Hutchinson DO, Nakano S, Murphy L, Griggs RC, Gu Y, Hall ZW, Lindstrom J. Myasthenic syndromes attributed to mutations affecting the epsilon subunit of the acetylcholine receptor. Ann N Y Acad Sci. 1993;681:496–508. doi: 10.1111/j.1749-6632.1993.tb22933.x. [DOI] [PubMed] [Google Scholar]
- Engel AG, Ohno K, Milone M, Wang HL, Nakano S, Bouzat C, Pruitt JN, 2nd, Hutchinson DO, Brengman JM, Bren N, Sieb JP, Sine SM. New mutations in acetylcholine receptor subunit genes reveal heterogeneity in the slow-channel congenital myasthenic syndrome. Hum Mol Genet. 1996a;5:1217–1227. doi: 10.1093/hmg/5.9.1217. [DOI] [PubMed] [Google Scholar]
- Engel AG, Ohno K, Bouzat C, Sine SM, Griggs RC. End-plate acetylcholine receptor deficiency due to nonsense mutations in the epsilon subunit. Ann Neurol. 1996b;40:810–817. doi: 10.1002/ana.410400521. [DOI] [PubMed] [Google Scholar]
- Fukudome T, Ohno K, Brengman JM, Engel AG. AChR channel blockade by quinidine sulfate reduces channel open duration in the slow-channel congenital myasthenic syndrome. Ann N Y Acad Sci. 1998;841:199–202. doi: 10.1111/j.1749-6632.1998.tb10928.x. [DOI] [PubMed] [Google Scholar]
- Gomez CM, Maselli R, Gammack J, Lasalde J, Tamamizu S, Cornblath DR, Lehar M, McNamee M, Kuncl RW. A beta-subunit mutation in the acetylcholine receptor channel gate causes severe slow-channel syndrome. Ann Neurol. 1996;39:712–723. doi: 10.1002/ana.410390607. [DOI] [PubMed] [Google Scholar]
- Gomez CM, Maselli R, Gundeck JE, Chao M, Day JW, Tamamizu S, Lasalde JA, McNamee M, Wollmann RL. Slow-channel transgenic mice: a model of postsynaptic organellar degeneration at the neuromuscular junction. J Neurosci. 1997;17:4170–4179. doi: 10.1523/JNEUROSCI.17-11-04170.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez CM, Maselli RA, Groshong J, Zayas R, Wollmann RL, Cens T, Charnet P. Active calcium accumulation underlies severe weakness in a panel of mice with slow-channel syndrome. J Neurosci. 2002;22:6447–6457. doi: 10.1523/JNEUROSCI.22-15-06447.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grosman C, Auerbach A. Kinetic, mechanistic, and structural aspects of unliganded gating of acetylcholine receptor channels: a single-channel study of second transmembrane segment 12′ mutants. J Gen Physiol. 2000;115:621–635. doi: 10.1085/jgp.115.5.621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harper CM, Engel AG. Quinidine sulfate therapy for the slow-channel congenital myasthenic syndrome. Ann Neurol. 1998;43:480–484. doi: 10.1002/ana.410430411. [DOI] [PubMed] [Google Scholar]
- Hoffmann K, Muller JS, Stricker S, Megarbane A, Rajab A, Lindner TH, Cohen M, Chouery E, Adaimy L, Ghanem I, Delague V, Boltshauser E, Talim B, Horvath R, Robinson PN, Lochmüller H, Hübner C, Mundlos S. Escobar syndrome is a prenatal myasthenia caused by disruption of the acetylcholine receptor fetal gamma subunit. Am J Hum Genet. 2006;79:303–312. doi: 10.1086/506257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson MB. Spontaneous openings of the acetylcholine receptor channel. Proc Natl Acad Sci U S A. 1984;81:3901–3904. doi: 10.1073/pnas.81.12.3901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson MB. Kinetics of unliganded acetylcholine receptor channel gating. Biophys J. 1986;49:663–672. doi: 10.1016/S0006-3495(86)83693-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jadey SV, Purohit P, Bruhova I, Gregg TM, Auerbach A. Design and control of acetylcholine receptor conformational change. Proc Natl Acad Sci U S A. 2011;108:4328–4333. doi: 10.1073/pnas.1016617108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kullberg RW, Mikelberg FS, Cohen MW. Contribution of cholinesterase to developmental decreases in the time course of synaptic potentials at an amphibian neuromuscular junction. Dev Biol. 1980;75:255–267. doi: 10.1016/0012-1606(80)90161-x. [DOI] [PubMed] [Google Scholar]
- Lefebvre JL, Ono F, Puglielli C, Seidner G, Franzini-Armstrong C, Brehm P, Granato M. Increased neuromuscular activity causes axonal defects and muscular degeneration. Development. 2004;131:2605–2618. doi: 10.1242/dev.01123. [DOI] [PubMed] [Google Scholar]
- Leonard JP, Salpeter MM. Agonist-induced myopathy at the neuromuscular junction is mediated by calcium. J Cell Biol. 1979;82:811–819. doi: 10.1083/jcb.82.3.811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonard JP, Salpeter MM. Calcium-mediated myopathy at neuromuscular junctions of normal and dystrophic muscle. Exp Neurol. 1982;76:121–138. doi: 10.1016/0014-4886(82)90106-6. [DOI] [PubMed] [Google Scholar]
- Milone M, Wang HL, Ohno K, Fukudome T, Pruitt JN, Bren N, Sine SM, Engel AG. Slow-channel myasthenic syndrome caused by enhanced activation, desensitization, and agonist binding affinity attributable to mutation in the M2 domain of the acetylcholine receptor α subunit. J Neurosci. 1997;17:5651–5665. doi: 10.1523/JNEUROSCI.17-15-05651.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mongeon R, Walogorsky M, Urban J, Mandel G, Ono F, Brehm P. An acetylcholine receptor lacking both gamma and epsilon subunits mediates transmission in zebrafish slow muscle synapses. J Gen Physiol. 2011;138:353–366. doi: 10.1085/jgp.201110649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen PV, Aniksztejn L, Catarsi S, Drapeau P. Maturation of neuromuscular transmission during early development in zebrafish. J Neurophysiol. 1999;81:2852–2861. doi: 10.1152/jn.1999.81.6.2852. [DOI] [PubMed] [Google Scholar]
- Ohno K, Hutchinson DO, Milone M, Brengman JM, Bouzat C, Sine SM, Engel AG. Congenital myasthenic syndrome caused by prolonged acetylcholine receptor channel openings due to a mutation in the M2 domain of the epsilon subunit. Proc Natl Acad Sci U S A. 1995;92:758–762. doi: 10.1073/pnas.92.3.758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paradiso K, Brehm P. Long-term desensitization of nicotinic acetylcholine receptors is regulated via protein kinase A-mediated phosphorylation. J Neurosci. 1998;18:9227–9237. doi: 10.1523/JNEUROSCI.18-22-09227.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purohit P, Auerbach A. Unliganded gating of acetylcholine receptor channels. Proc Natl Acad Sci U S A. 2009;106:115–120. doi: 10.1073/pnas.0809272106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sine SM, Steinbach JH. Activation of acetylcholine receptors on clonal mammalian BC3H-1 cells by low concentration of agonist. J Physiol. 1986;373:129–162. doi: 10.1113/jphysiol.1986.sp016039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sine SM, Ohno K, Bouzat C, Auerbach A, Milone M, Pruitt JN, Engel AG. Mutation of the acetylcholine receptor alpha subunit causes a slow-channel myasthenic syndrome by enhancing agonist binding affinity. Neuron. 1995;15:229–239. doi: 10.1016/0896-6273(95)90080-2. [DOI] [PubMed] [Google Scholar]
- Wen H, Brehm P. Paired motor neuron-muscle recordings in zebrafish test the receptor blockade model for shaping synaptic current. J Neurosci. 2005;25:8104–8111. doi: 10.1523/JNEUROSCI.2611-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou M, Engel AG, Auerbach A. Serum choline activates mutant acetylcholine receptors that cause slow channel congenital myasthenic syndromes. Proc Natl Acad Sci U S A. 1999;96:10466–10471. doi: 10.1073/pnas.96.18.10466. [DOI] [PMC free article] [PubMed] [Google Scholar]