

Abstract
Drosophila melanogaster embryos are a source for homogeneous and stable 26S proteasomes suitable for structural studies. For biochemical characterization, purified 26S proteasomes were resolved by two-dimensional (2D) gel electrophoresis and subunits composing the regulatory complex (RC) were identified by amino acid sequencing and immunoblotting, before corresponding cDNAs were sequenced. 17 subunits from Drosophila RCs were found to have homologues in the yeast and human RCs. An additional subunit, p37A, not yet described in RCs of other organisms, is a member of the ubiquitin COOH-terminal hydrolase family (UCH). Analysis of EM images of 26S proteasomes-UCH-inhibitor complexes allowed for the first time to localize one of the RC's specific functions, deubiquitylating activity.
The masses of 26S proteasomes with either one or two attached RCs were determined by scanning transmission EM (STEM), yielding a mass of 894 kD for a single RC. This value is in good agreement with the summed masses of the 18 identified RC subunits (932 kD), indicating that the number of subunits is complete.
Keywords: protein degradation, ubiquitin, ubiquitin hydrolase, ATP-dependent proteolysis , electron microscopy
Introduction
In eukaryotic cells, the vast majority of cytosolic and nuclear proteins are degraded via the ubiquitin–proteasome pathway (Rock et al. 1994). Through a sequence of activating and ligating events, ubiquitin is covalently attached to proteins destined for degradation (for recent reviews see Varshavsky 1997; Hershko and Ciechanover 1998; Scheffner et al. 1998). Proteins carrying multiubiquitin tags are selected by the 26S proteasome and degraded in an ATP-dependent process (Coux et al. 1996; Rechsteiner 1998). The 26S proteasome is a large molecular machine built from ∼30 different subunits that has an estimated molecular mass of 2,000–3,000 kD. Two major components jointly form the 26S (or more accurately, the 30.3S) complex: the barrel-shaped proteolytic core complex (the 20S proteasome) and the regulatory complexes (RCs), which associate with either one or both ends of the core complex (Peters et al. 1993; Yoshimura et al. 1993; see also Fig. 1).
Figure 1.
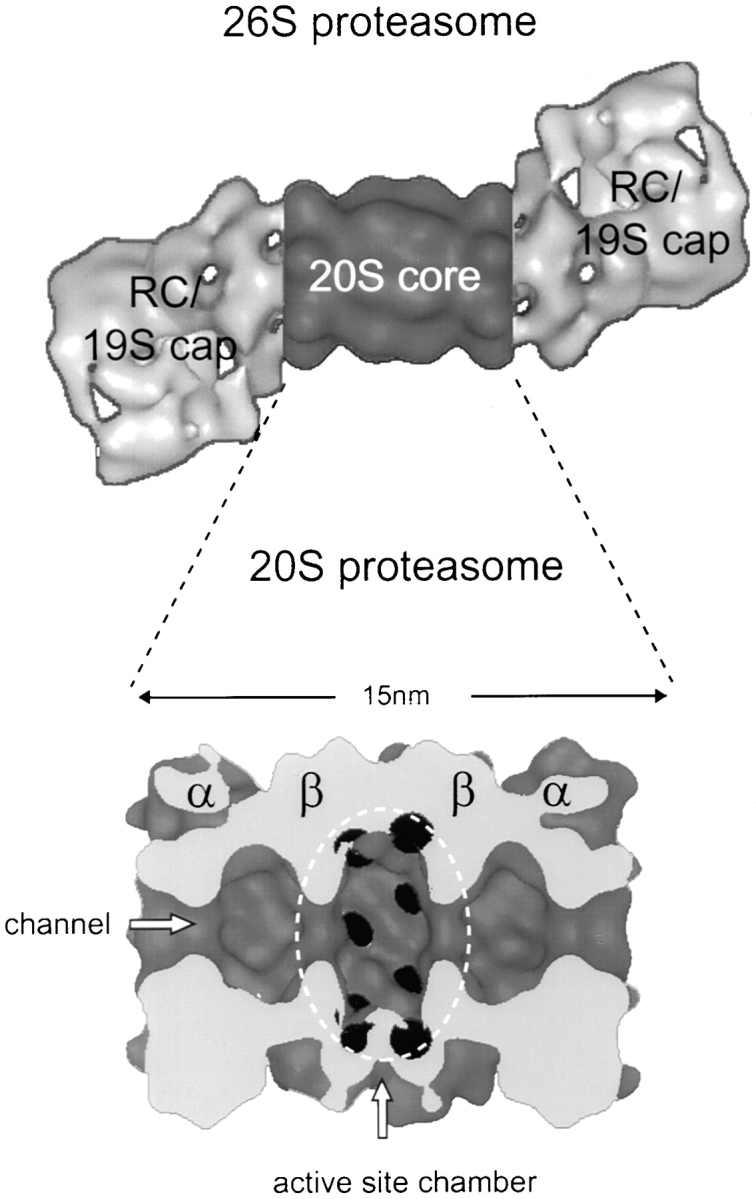
The 26S proteasome. The 3D model of the Drosophila 26S proteasome is based on EM and combined with the low-pass filtered crystal structure of the Thermoplasma 20S proteasome (Walz et al. 1998). The cut open side view of the 20S proteasome allows the view into the central cavity where the active sites (black) are located.
Whereas the structure and enzymatic mechanism of the 20S proteasome have been studied in great detail (for recent reviews see Baumeister et al. 1998; Bochtler et al. 1999; Voges et al. 1999), current understanding of the structure and function of the RC is lagging behind. The RCs serve to recognize proteins carrying multiubiquitin tags and to prepare them for degradation in the 20S proteolytic complex. The preparatory steps involve the binding of the ubiquitylated substrates, their deubiquitylation, the unfolding of the substrates, and finally, their translocation into the 20S complex (Lupas et al. 1993; Rubin and Finley 1995). Substrate unfolding is required because admission to the active site chamber inside the 20S complex is restricted to unfolded polypeptide chains (Wenzel and Baumeister 1995). At the heart of the RCs is an array of ATPases, members of the AAA family (Confalonieri and Duguet 1995; Beyer 1997), which act as reverse chaperones (Braun et al. 1999; Strickland et al. 2000). RCs of prokaryotic 20S proteasomes appear to have only a single type of AAA-ATPase (Wolf et al. 1998; Zwickl et al. 1999), which form homohexameric rings, whereas in eukaryotic RCs, six paralogs are found that are believed to assemble into heterohexameric rings. The hallmark of all proteasomal ATPases is an NH2-terminal coiled-coil domain (Lupas et al. 1993; Rechsteiner 1998). In both proteasomes and other self-compartmentalizing protein degradation machines, the proteases and the ATPases form colinear assemblies (Lupas et al. 1997; Zwickl et al. 2000). Thus, the ATPases are well placed to unfold substrates and control the gates that give access to the proteolytic compartments (Larsen and Finley 1997). Beyond the ATPases, little is known about the roles of the other ∼12 subunits of the RCs (for recent reviews see Tanaka and Tsurumi 1997; Voges et al. 1999).
Structural studies with 26S proteasomes are hampered by the low stability of the complexes, which tend to dissociate into various subcomplexes. It has been shown previously that Drosophila melanogaster embryos provide a rich source of 26S proteasomes (Udvardy 1993) and yield preparations that are sufficiently homogenous for structural studies (Walz et al. 1998). To prepare the grounds for an in-depth structural analysis of the Drosophila RC, we sought to establish a catalog of all its subunits; to assess the completeness of this catalog, we have performed quantitative mass analysis using scanning transmission EM (STEM). In the course of these studies, we identified a novel subunit that turned out to be a deubiquitylating enzyme, p37A. Taking advantage of a nonhydrolyzable substrate analogue, ubiquitin COOH-terminal aldehyde (Ub-Al), we have been able to map its location within the complex providing new insights into the sequence of events en route to substrate degradation.
Materials and Methods
Materials
Chemicals and chromatography resins for protein purification were purchased from Sigma-Aldrich, Amersham Pharmacia Biotech, Merck, BioRad, and Quiagen. Enzymes for DNA restriction and modification were obtained from New England Biolabs, Inc. and Stratagene. Oligonucleotides for PCR reactions were synthesized on an Applied Biosystems 380A DNA synthesizer.
Isolation of 26S Proteasomes from Drosophila melanogaster Embryos
26S proteasomes were purified as described previously (Udvardy 1993; Walz et al. 1998). In brief, 0–16-h Drosophila embryos (Yellow white strain) were collected at 25°C from feeding plates. After dechorionation and homogenization, the extract was clarified by centrifugation and nucleic acids were removed by precipitation with 10% streptomycin sulfate. The supernatant was fractionated with hydroxyapatite in a batch procedure, followed by anion-exchange chromatography (diethylaminoethyl cellulose, DE52, Whatman) and sucrose density gradient centrifugation (15–40% sucrose). At all stages, fractions were tested for their ability to hydrolyze Succinyl-Leu-Leu-Val-Tyr-7-amido-4-methylcoumarin (Suc-LLVY-AMC; Bachem), and only fractions containing the peaks of activity were used for further purification. Protein concentrations were determined using the BioRad protein assay with BSA as standard.
Protein Gel Electrophoresis and Immunoblotting
SDS-PAGE was performed using 12.5% separating and 3.5% stacking gels as described by Laemmli 1970.
Protein samples were resolved by nondenaturing PAGE using a modification of the method described by Hough et al. 1987. The resolving gels were 4.6% acrylamide (37.5:1), 2.3% sucrose, 90 mM Tris, 80 mM borate, 0.08 mM EDTA, pH 8.3, polymerized with 0.04% ammonium persulphate and N,N,N′,N′-tetramethyl-ethylene-diamine. The nondenaturing stacking gels contained the same buffer, but only 3.1% acrylamide (4:1). The proteins were subjected to electrophoresis in 90 mM Tris, 80 mM borate, 0.08 mM EDTA, pH 8.3, for a total of 800 V-h (50 V for 16 h) at 4°C. After electrophoresis, peptidase activity was detected by overlaying the gels with 100 μM Suc-LLVY-AMC in 5 mM MgCl2, 10 mM KCl, 0.5 mM EDTA, 30 mM Tris, pH 7.8, for 1 h at 37°C. Proteasome bands were visualized by exposure to UV light (360 nm) and photographs were taken before staining with Coomassie blue. Alternatively, proteins were transferred to nitrocellulose membranes by semidry blotting. Blots were treated with antibodies and antigen-antibody complexes were visualized by using alkaline phosphatase-conjugated anti-mouse IgG antibodies, following standard procedures (Sambrook et al. 1989).
For two-dimensional (2D) gels, 50 μg of purified 20S or 26S proteasomes were concentrated by the use of Nanosep™ microconcentrators (Pall Filtron) and resuspended in 100 μl of loading buffer (9 M urea, 4% CHAPS, 40 mM Tris, and 0.025% Bromphenol blue). Separation in the first dimension was performed with 13-cm Immobiline® Dry Strip Gels (Amersham Pharmacia Biotech) using a linear pH gradient from 3–10. After focusing for 66,500 V-h, the strips were positioned over a vertical SDS-polyacrylamide slab gel made of a 5% stacking and 12.5% separating gel, and subjected to electrophoresis using standard conditions. In addition the benzyldimethyl-n-hexadecylammonium chloride (16-BAC)/SDS-PAGE, a 2D gel electrophoresis system was used as described by Hartinger et al. 1996. Proteins were either stained in the gel with Coomassie blue or electrotransferred to polyvinylidene difluoride membranes for NH2-terminal sequencing.
Protein Sequencing
The protein-containing polyvinylidene difluoride membrane pieces were excised, cut into small pieces (3 × 3 mm), and incubated with 500 μl 0.2% polyvinylpyrolidone (PVP 30) in water for 30 min at room temperature (Patterson 1994). The supernatant was discarded and the membrane was washed six times with water and incubated with 0.1 M Tris-HCl, pH 8.0, 2 mM CaCl2, 10% acetonitrile, 1% nonylphenoxy polyethoxy ethanol (Tergitol NP-40), and 0.5 μg endoproteinase LysC (Boehringer) for 8 h at 37°C. When the NH2 terminus was blocked, protein digestion was performed in the 2D gel. Therefore, gel pieces were excised, washed twice with cleavage-buffer (12.5 mM Tris, pH 8.5, 0.5 mM EDTA, i.e., half the usual concentration), and dried. Pieces of the gel were then incubated with endoproteinase LysC for 16 h at 37°C. The resulting cleavage fragments were eluted twice with 0.1% trifluoroacetyl and once with 10% formic acid, 20% isopropanol, and 20% acetonitrile. The supernatants were dried and the peptide mixture separated on a reversed phase column Purosphere RP-18 endcapped (Merck; 1 × 150 mm). Solvent A, 0.1% trifluoroacetyl; solvent B, 0.085% trifluoroacetyl in acetonitrile. The gradient was 0–70% B over a period of 120 min at a flow rate of 60 μl/min; the detection wavelength was 206 nm. The peptides were sequenced (Edman and Begg 1967) on a pulsed liquid phase sequencer, Procise 493 (Applied Biosystems) according to the manufacturer's instructions.
DNA Sequencing
The amino acid sequences obtained from peptide analysis were used to search the databases of the Berkeley Drosophila Genome Project (http://www.fruitfly.org) for matching DNA sequences. Five of the peptides led to genes described previously. All other peptides were homologous to distinct EST cDNA clones (Rubin et al. 2000, Table I). The longest cDNA clone of each EST clot was ordered from Genome Systems, Inc. or Research Genetics, and sequenced on both strands with a 373 DNA Sequencer using the BigDye Terminator Cycle Sequencing Ready Reaction Kit (Applied Biosystems). All sequences have been submitted to Genbank/EMBL/DDBJ.
Construction and Purification of Recombinant p37A
The cDNA encoding the open reading frame for p37A was amplified by PCR, introducing an NH2-terminal NdeI and a COOH-terminal XhoI site. The amplified DNA fragment was subcloned into the prokaryotic expression vector, pET22b, fusing a (His)6-tag at the COOH terminus of the protein (Novagen Inc.). The construct was sequenced, confirming that no mutations had been introduced. Expression upon isopropyl-β-d-thiogalacto-pyranoside (Biomol) induction (1 mM, 5 h, 37°C) of Escherichia coli BL21(DE3) cells (Studier et al. 1990) yielded a His-tagged protein of ∼37 kD. The recombinant protein was purified on a nickel-nitrilotriacetic acid resin (Quiagen) and dialyzed against 50 mM Hepes, pH 7.8, 0.5 mM EDTA, 20% glycerol, 1 mM DTT.
Determination of Ubiquitin COOH-terminal Hydrolase Activity
Assays for p37A and 26S proteasome enzymatic activity were performed essentially as described for the ubiquitin COOH-terminal hydrolase (UCH)-L3 enzyme (Dang et al. 1998). 5 μl p37A (∼1 μM) and 20 μl 26S proteasomes (∼0.5 mg/ml) were incubated in 490 and 475 μl (respectively) of assay buffer (50 mM Hepes, pH 7.8, 0.5 mM EDTA, 1 mM DTT, and 0.1 mg/ml BSA) at 25°C for 1 h. The reaction was started by the addition of 5 μl of 50 μM ubiquitin COOH-terminal AMC (Ub-AMC; Affiniti) in DMSO. Reaction progress was monitored on a Perkin Elmer LS 50B luminescence spectrometer by the increase of fluorescence intensity at 460 nm (λex = 380 nm) that accompanies cleavage of AMC from Ub-AMC. For inhibition of the 20S proteolytic activity, 10 μl of a 1 mM lactacystin (Affiniti) solution in H2O were incubated with 20 μl 26S proteasomes in 70 μl assay buffer for 1 h at 25°C. 400 μl prewarmed assay buffer and 5 μl Ub-AMC (50 μM) were added to start the reaction. To test whether the p37A activity was inhibited by Ub-Al (BioTrend), protein samples were incubated with different concentrations of Ub-Al before the addition of Ub-AMC.
Ubiquitinaldehyde–Colloidal Gold Conjugate
Colloidal gold particles (3.5 nm), prepared by the method of Slot and Geuze 1985, were used with minor modifications. In brief, 80 ml of 0.012% NaAuCl4 in water and 20 ml of 0.25% tannic acid, 0.2% sodium citrate, and 1 mM potassium carbonate were heated to 60°C and rapidly mixed. The gold colloids were formed within seconds and no additional purification was needed.
Thiol groups were introduced into Ub-Al by modifying the side chain of lysine residues. Ub-Al (1.3 μg) and 2-iminothiolane (3 μg) in 20 μl of 50 mM triethanol amine, pH 7.3, were incubated for 6 h at room temperature. 15 μl of the thiolated Ub-Al solution was mixed with 100 μl of freshly prepared colloidal gold solution, incubated for 8–12 h at 4°C, and centrifuged for 10 min at 80,000 g (Airfuge; Beckman Coulter). The soft pellet was resuspended in 15 μl of distilled water.
Electron Microscopy
2 μl of purified 26S proteasomes (0.2 mg/ml) was incubated with 1 μl of gold-labeled Ub-Al and 2 μl of 20 mM Tris buffer, pH 7.2, for 7 min and applied to 100 × 400 mesh copper grids, which had been coated with carbon and glow-discharged in a plasma cleaner, for 45 s. After blotting and removing the sucrose with Tris buffer (20 mM), the preparation was negatively stained with 2% aqueous uranyl acetate for 45 s. Electron micrographs were recorded digitally (Photometrix slow scan CCD; 1024 × 1024 pixels) at 45,700× using a CM12 transmission electron microscope (Philips) at 120 kV accelerating voltage.
Image Processing
The images were transferred to a SGI workstation and analyzed using the EM software package (Hegerl 1996). Subframes containing single 26S, either with or without gold particles, were interactively extracted from the images. The two sets of complexes were separately aligned, translationally and rotationally (5 cycles), using iterative cross-correlation techniques (Baumeister et al. 1988; Phipps et al. 1991). For the set where gold labels were present, the lower cutoff of the gray level range of each frame was increased to the mean value minus 0.5× SD before alignment to minimize the contribution of the gold particles. The alignment parameters were then assigned to the original stack. Subsequently, the dataset was subjected to multivariate statistical analysis (MSA).
Scanning Transmission Electron Microscopy
For mass analysis by STEM, the 26S proteasome stock preparation (150 μg/ml) was diluted 2–8 times in buffer without sucrose, and either used directly or after cross-linking with 0.05% glutaraldehyde (final concentration) for 5 min on ice. Aliquots of the solutions were immediately adsorbed to glow-discharged thin carbon films, supported by thick perforated carbon layers on gold-coated copper grids. In the absence of cross-linking, grids were washed 4–5 times with 0.1 M ammonium acetate. The glutaraldehyde-treated samples were washed several times with quartz bidistilled water. All grids were freeze-dried overnight in the microscope pretreatment chamber.
A Vacuum Generators STEM HB-5 interfaced to a modular computer system (Tietz Video; Image Processing Systems) and operated at 80 kV was used for the measurements. Series of 512 × 512-pixel digital images were recorded at a nominal magnification of 200,000×, using doses of ∼300 electrons/nm2. The data were evaluated using the IMPSYS program package as described previously (Müller et al. 1992). To this end, the particles were classified into eight groups according to their dimensions, the selection box was kept as small as possible. Total scattering within the selection box was determined and the scattering from an equivalent area of the background support film was subtracted to calculate the particle's mass. The clearly identifiable top views of the strongly scattering 20S particles, present in all preparations, served as internal mass standards. The instrument's calibration (Müller et al. 1992) allowed the use of a single scale factor for each experiment.
The mass of the 20S particle was taken as 721 kD, i.e., the average of the calculated mass values for the yeast and human 20S proteasomes. For each experiment (two glutaraldehyde and two ammonium acetate-treated preparations), mass data from the top views were displayed in a histogram and a Gaussian curve was fitted. The position of this peak compared with 721 kD yielded a global scale factor that was subsequently applied to the whole data set. In a final step, corresponding data sets from all four experiments were pooled, displayed in histograms, and Gaussian curves were fitted. The number of complexes (n) contributing to each peak was estimated and, where relevant, the corresponding SE calculated from the SD of the Gauss curve (SE =SDn). After arithmetic operations, the resulting error was estimated as ΣS2 (where S is the SD or SE, respectively, of the individual results).
Results
Purification and Characterization of 26S Proteasomes from Drosophila Embryos
The last step of proteasome purification, the sucrose density gradient centrifugation, yielded two peaks with high peptidase activity, capable of hydrolyzing the fluorogenic peptide, Suc-LLVY-AMC. The active fractions giving rise to each peak were analyzed by SDS-PAGE (Fig. 2) and nondenaturing PAGE (Fig. 3). Fraction 10, with ∼22% sucrose, was the first fraction of peak 1 and contained pure 20S proteasomes. Fractions 13, 14, and 15 also contained 20S proteasomes, but these were contaminated with a high molecular weight protein that appeared similar to GroEL on electron micrographs. Fractions 16–20, from the second activity peak corresponding to ∼26–28% sucrose, showed a pattern characteristic of 26S proteasomes on SDS gels. Under native conditions, the Drosophila complex separated into four bands, similar to the pattern observed with mammalian and yeast proteasomes (Hoffman et al. 1992; Glickman et al. 1998a). Fluorogenic peptide overlays showed, besides 20S proteasomes, two slower migrating species; for yeast proteasomes these species were identified as complete 26S proteasomes (with RCs capping both ends of the 20S proteasome, i.e., 20S-RC2) and 26S proteasomes capped only at one end (i.e., 20S-RC1; Glickman et al. 1998a; Fig. 3 a). The coexistence of two distinct forms of the proteasome has been shown previously by EM (Peters et al. 1993; Fujinami et al. 1994; Walz et al. 1998). To determine which bands contained the RCs, the protein complexes in fraction 19 were electroblotted from the native gels onto nitrocellulose membranes and probed with several mAbs directed against various subunits of the RC and the 20S proteasome. Fig. 3 c, lane 1, shows immunoreactivity with μ3/50, an antibody directed against the RC subunit p39A, and lane 2 shows immunoreactivity with V.D5, an antibody directed against the 20S proteasome. Several other antibodies were used to verify the assignment (data not shown). These results lead to the identification of the four resolved complexes from top to bottom as follows: 26S (20S-RC2), single 26S (20S-RC1), RCs, and 20S core particles, as indicated in Fig. 3.
Figure 2.

SDS-PAGE of 26S proteasomes. Fractions 10 and 13–20 from the sucrose density gradient centrifugation were separated on a 12.5% SDS gel and the proteins were stained with Coomassie blue. Molecular weights of the marker proteins are shown on the left. For assignment of bands see Fig. 4. Fraction 10 (∼22% sucrose) contained pure 20S proteasomes, and fractions 13–20 (∼24–28%) displayed the typical pattern of subunit components exhibited by 26S proteasomes. Fractions 13–15 were contaminated with another protein complex (possibly a homologue of GroEL).
Figure 3.

Nondenaturing PAGE of 26S proteasomes. Fractions 10, 13, 16, and 19 (40 μl) from the sucrose gradients were electrophoresed for 800 V-h on 4.5% native polyacrylamide gels. a, Proteolytic activity of the resolved complexes was detected by fluorogenic peptide overlay with Suc-LLVY-AMC and the proteins were visualized by Coomassie blue stain (b; same gel). c, To further characterize the bands, duplicate samples of fraction 19 were transferred to nitrocellulose membranes and immunostained with μ3/50, an mAb directed against the RC subunit, p39A, and with another, V.D5, directed against the 20S proteasome. The analysis of a, b, and c allowed unambiguous identification of the bands as 20S proteasomes, RCs, 26S proteasomes with only one RC attached (20S-RC1), and 26S proteasomes with two RCs (20S-RC2), as indicated.
Subunit Composition of the RC
As reported previously and illustrated by Fig. 2, Drosophila 26S proteasomes are resolved on one-dimensional (1D) gels into 12 distinct bands that are in addition to the bands that arise from the 20S core complex (Udvardy 1993). According to their apparent mass, they are referred to as p110 to p37B (Haracska and Udvardy 1996). A typical 2D-electrophoresis pattern of 26S proteasomes is shown in Fig. 4. The protein pattern in the 20–30-kD range closely resembles that of purified 20S proteasomes (data not shown). The proteins in the higher molecular mass range, assigned to the RC, yield >20 distinct spots with pIs between 4.8 and 8.5. These spots were analyzed further by amino acid sequencing. Since no distinct spots could be detected for the three largest subunits, p110, p97, and p58 on the 2D gels, these proteins were sequenced from a 1D SDS gel and could be unambiguously identified. Whereas p97 and p58 were directly accessible to NH2-terminal sequencing, p110 was NH2-terminally blocked and had to be analyzed by in-gel digestion. Of the spots on the 2D gels, only E and N were accessible for NH2-terminal sequencing, the remaining spots were blocked due to posttranslational modifications.
Figure 4.
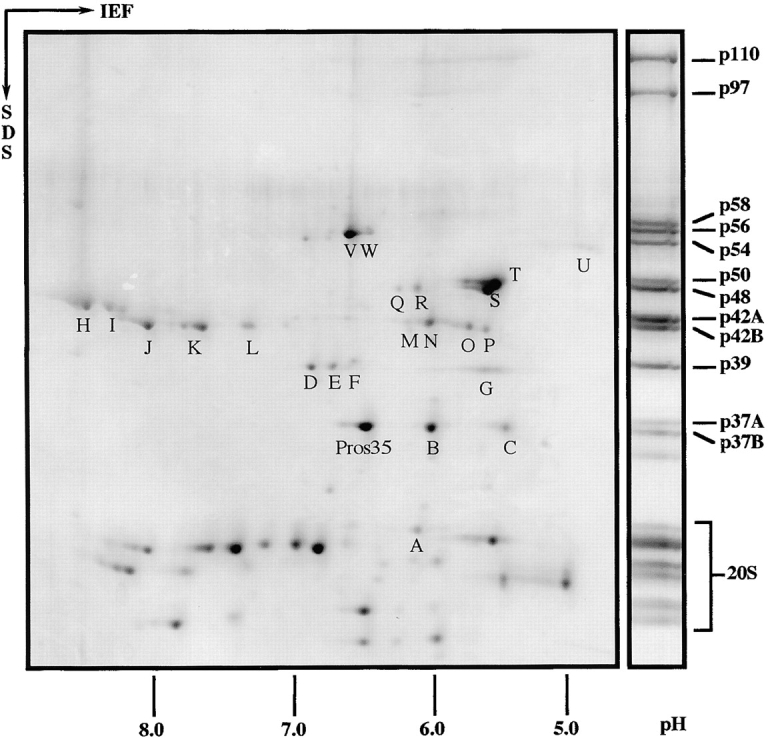
2D gel electrophoresis of Drosophila 26S proteasomes. Purified 26S proteasomes from Drosophila embryos were separated in the first dimension by isoelectric focusing (IEF) with an immobilized pH gradient from 3 to 10. Next, proteins were resolved in the second dimension using a 12.5% polyacrylamide SDS gel and stained with Coomassie blue. 26S proteasomes resolved by 1D SDS-PAGE were used as molecular weight marker and the bands were named according to the procedure of Haracska and Udvardy 1996. All spots marked with a capital were identified by peptide sequencing with the exception of spot L, which could not be sequenced, and spot U, which was identified by immunoblotting (for details see Table and Results). Low molecular weight proteins (not indicated separately) belong to the 20S core complex. With one exception, Pros35, they were not identified by peptide sequencing.
All of the peptide sequences obtained were in complete agreement with the amino acid sequences deduced from the Drosophila genome (Table ). Only one spot above the 20–30-kD range turned out not to be an RC subunit; this protein was identified as Pros35, an α-type subunit of the 20S proteasome. Spot U was not sequenced, but clearly identified as p54 by electroblotting and probing with an mAb (μ3/150).
Table 1.
Identification of the Drosophila RC Subunits
| Spot | Peptide sequence | EST cDNA clone ID | GenBank | Protein | |
|---|---|---|---|---|---|
| − | VATAVLSIAARQK | LD11427 | p110 | ||
| − | TGETKLEKKPLL | LD05942 | p97 | ||
| − | TNATDIGANDVE | M63010 | p58 (Dox-A2) | ||
| W | APQETYADIGGLD | } | U39303 | p56 (S4) | |
| V | GVILYGPPGTGK | ||||
| − | TGLTPVDSAA | LD07018 | p55 | ||
| U | S79502 | p54 (S5a) | |||
| T | SEVIRITHEIQAQNEK | LD22987 | p50 | ||
| S | EFIEVQEEYIK | GM02119 | p48A | ||
| R | FVVELADSVAP | } | LD17074 | p48B | |
| Q | QVNETGI | ||||
| O | YLIEEGG | } | GH10329 | p42A | |
| P | |||||
| N | EQGILQQGELQK | } | LD20236 | p42B | |
| M | XXVNREEQD | ||||
| H | VPDSTYEMYGGLDK | } | U97538 | p42C (DUG) | |
| I | IEELQLVVAEK | ||||
| J | SLQSVGQIVGEVL | LD04678 | p42D | ||
| K | HGEIDYEAIVK | ||||
| G | AIERISF | LD17530 | p39A | ||
| D | PSQEVSVNK | M64643 | p39B (mov34) | ||
| E | } | ||||
| F | |||||
| C | FCQCFDPYNK | LD02040 | p37A | ||
| B | MTPEQCAIK | CK01641 | p37B | ||
| A | IFQAK | LD13866 | p30 |
On 1D SDS gels, two bands are found with apparent molecular masses close to 42 kD, corresponding to four different proteins, named p42A to p42D. The proteins p42B, p42C, and p42D were also detected on 2D gels (spots H to N). However, spots O and P, in the same mass range that we assume to correspond to the fourth protein, p42A, were not always present and it was not possible to derive sequence information from them. It was also impossible to obtain sequence information for spot L, which showed very faint staining on 2D gels. Since variably migrating forms were found for several subunits (see Fig. 4 and Table ), spot L may correspond to p42D, as do spots J and K. The partially unsatisfactory resolution of the above electrophoretic system prompted the use of another gel system, 16-BAC/SDS-PAGE 2D gel electrophoresis (Hartinger et al. 1996). This gave a better separation, yielding some additional spots. Sequence comparison showed one to correspond to Rpn5/p55; no corresponding spot was found on conventional 2D gels. In the 42-kD region, no additional proteins were detected with the 16-BAC/SDS-PAGE 2D gel system.
Altogether, 18 distinct RC subunits were identified (Table ). Accordingly, the six ATPases of the AAA family (Confalonieri and Duguet 1995; Beyer 1997), which are integral components of all RCs investigated to date (DeMartino et al. 1994; Dubiel et al. 1995; Glickman et al. 1998a), are also present in the Drosophila RC. As their yeast counterparts, the six ATPases from Drosophila exhibit 40–50% sequence identity. The sequence identity between Drosophila and yeast, and between Drosophila and human ATPases varies between 64–72% and between 84–88%, respectively. This level of identity is much higher than that of the non-ATPases of the RC, which ranges from 25–44% and 42–71% between Drosophila and yeast and between Drosophila and human, respectively. Similar to the situation in yeast (Glickman et al. 1998a), the non-ATPase subunit, p37B, is an exception since it is 64% identical to yeast Rpn11 and 88% identical to human S13. Also, p110 (Rpn2/S1) is 25% identical to p97 (Rpn1/S2), and p39B (Rpn8/S12) is 30% identical to p37B (Rpn11/S13).
Table 2.
Subunit Composition of the Drosophila RC
| Drosophila RC subunits | Homologues | |||||||
|---|---|---|---|---|---|---|---|---|
| Name | Accession | AA | MW | pI | Yeast | Identity | Human | Identity |
| % | % | |||||||
| p110 | AF145303 | 1020 | 113.2 | 4.93 | Rpn2 | 38 (1–858) | S1 | 64 (3–859) |
| p97 | AF145304 | 919 | 102.3 | 5.48 | Rpn1 | 36 (65–615) | S2 | 58 (65–917) |
| p58 | M63010 | 494 | 56.0 | 9.04 | Rpn3 | 32 (12–458) | S3 | 58 (8–494) |
| p56 | U39303 | 439 | 49.3 | 6.17 | Rpt2 | 72 (24–439) | S4 | 88 (1–439) |
| p55 | AF145315 | 502 | 57.7 | 5.49 | Rpn5 | 38 (8–442) | p55 | 47 (11–455) |
| p54 | S79502 | 396 | 42.6 | 4.70 | Rpn10 | 41 (1–225) | S5a | 52 (1–381) |
| p50 | AF145305 | 428 | 47.8 | 5.20 | Rpt5 | 68 (14–428) | S6′ | 84 (9–428) |
| p48A | AF145306 | 412 | 46.9 | 5.22 | Rpt3 | 69 (34–413) | S6 | 84 (12–413) |
| p48B | AF145307 | 433 | 48.5 | 5.75 | Rpt1 | 64 (5–433) | S7 | 85 (1–433) |
| p42A | AF145308 | 389 | 45.4 | 6.06 | Rpn7 | 37 (15–389) | S10 | 70 (1–389) |
| p42B | AF145309 | 422 | 47.3 | 5.66 | Rpn6 | 42 (1–421) | S9 | 64 (2–422) |
| p42C | U97538 | 405 | 45.8 | 8.51 | Rpt6 | 71 (18–405) | S8 | 86 (6–405) |
| p42D | AF145310 | 390 | 44.2 | 8.44 | Rpt4 | 70 (8–388) | S10b | 86 (6–390) |
| P39A | AF145311 | 382 | 43.8 | 5.18 | Rpn9 | 30 (8–379) | S11 | 42 (15–381) |
| p39B | M64643 | 338 | 38.5 | 8.90 | Rpn8 | 44 (10–290) | S12 | 71 (5–290) |
| p37A | AF145312 | 324 | 37.7 | 5.12 | UCH37 | 59 (1–324) | ||
| p37B | AF145313 | 308 | 34.4 | 5.74 | Rpn11 | 64 (1–308) | S13 | 88 (1–308) |
| p30 | AF145314 | 264 | 30.2 | 5.80 | Rpn12 | 25 (25–264) | S14 | 45 (7–264) |
p37A, A Novel Subunit of the RC
One of the spots on the 2D gels yielded the protein sequence FCQCFDPYNK, and sequencing the corresponding EST cDNA clone, LD02040, revealed the existence of an open reading frame of 972 bp, which encodes a previously unreported protein of 324 amino acids, p37A. The predicted molecular mass of p37A, 37.672 kD, matches its mobility on SDS-PAGE gels. The predicted pI of 5.12 is slightly more acidic than that observed on 2D gels (Fig. 4). Drosophila p37A has no homologues among the yeast RC subunits. However, a database search revealed homologues in the human, murine, and bovine genomes, of which the human was identified as a RC subunit (Xu, W., and R.E. Cohen, personal communication). p37A is a member of the ubiquitin COOH-terminal hydrolase (UCH) family (Fig. 5; Wilkinson 1997). UCHs form a class of thiol proteases, which remove thiols, amines, peptides, and small proteins from the COOH terminus of ubiquitin (Wilkinson 1997). They are characterized by a 210–amino acid catalytic domain with four highly conserved sequence blocks, containing the four active side residues. Interestingly, p37A is closely related to human BAP1 (Jensen et al. 1998). BAP1 is a nuclear protein of 81 kD, showing significant homology to the UCHs in its 240 residue NH2-terminal domain. The COOH terminus of BAP1 is predicted to fold into a helical (possibly coiled-coil) structure, which may interact with the RING finger domain of the breast/ovarian cancer susceptibility gene product, BRCA1 (Jensen et al. 1998). Although the COOH-terminal parts of p37A (residues 252–322) and BAP1 (residues 640–716) are similar (32% identity), a coiled-coil structure for p37A is not predicted when the same algorithm (Lupas et al. 1991) is applied to the sequence.
Figure 5.

Sequence alignment of p37A with related UCHs. Ψ-Blast using default parameters (http://www.ncbi.nlm.nih.gov/blast/psiblast.cgi) was performed to search for proteins related to p37A. The identified proteins were aligned using the CLUSTAL X Multiple Sequence Alignment Program (version 1.63b; Thompson et al. 1997). UCH37 (GenBank/EMBL/DDBJ accession #AAD31528), BAP1 (GenBank/EMBL/DDBJ accession #AF045581), and p37A (GenBank/EMBL/DDBJ accession #AF145312) are more closely related to each other than to the other UCHs (AP-UCH, GenBank/EMBL/DDBJ accession #AAB52410; UCH-D, GenBank/EMBL/DDBJ accession #P35122; UCH-L1, GenBank/EMBL/DDBJ accession #P09936; UCH-L3, GenBank/EMBL/DDBJ accession #P15374; and YUH1, GenBank/EMBL/DDBJ accession #P35127). With a gap of ∼400 amino acids, the COOH-terminal region of BAP1 (residues 640–716) is again homologous to p37A and UCH37. Residues conserved among p37A, UCH37, and BAP1, as well as the other five proteins are shown in reverse type. The active site residues (denoted with arrows) are conserved in all eight proteins.
STEM Mass Mapping
It is notoriously difficult to assess the completeness of subunit catalogs of large macromolecule assemblies by 2D electrophoresis. Therefore, it is useful to determine experimentally the mass of structurally distinct subcomplexes, such as the RC, and to compare this with the summed sequence-derived mass of their subunits. Mass mapping by STEM (Engel et al. 1982) allows such measurements to be made with remarkable accuracy.
Images recorded for mass measurement from unstained 26S proteasome samples showed the preparations to be heterogeneous. Heterogeneity was not reduced by glutaraldehyde fixation. However, the bone-shaped side-view projections, typical of symmetrical 26S (20S-RC2), could be clearly distinguished from the wedge-shaped projections of asymmetrical 26S (20S-RC1) complexes. In addition, there were many ring structures with the characteristic signature of 20S proteasome particles viewed end-on, as well as less dense, almost circular projections. Since their high scattering allowed an unambiguous visual identification, the 20S proteasomes served as a convenient internal mass standard. The mass was set to 721 kD (see Materials and Methods). In this way, both slight mass differences arising from the sample preparation techniques employed, and the inherent beam-induced mass loss, could be accurately accounted for.
Pooled data sets from the four experiments were displayed in histograms and were Gaussian curve fitted (Fig. 6). The low standard deviation of ± 55 kD of the 20S proteasome particles (n = 3,853; SE = ± 0.9 kD; Fig. 6 b) illustrates the high quality of the calibration, yielding almost exactly the SD value expected (∼50 kD) from background fluctuations for the small selection box size used. The mass of the wedge-shaped species, 20S-RC1, was found to be 1,623 ± 155 kD (n = 2,003; Fig. 6 c). The mass of the 20S-RC2 complexes was 2,508 ± 196 kD (n = 286; Fig. 6 d). The population of small particles, with lower scattering power than the 20S proteasomes' almost circular projections, had a mass of 358 ± 96 kD (n = 1,394; Fig. 6 a). The above data allow three estimates to be made for the mass of the RC: considering the mass of 20S-RC2 and the mass of 20S-RC1, mass of one RC, 885 ± 250 kD (SE = ± 12 kD); considering the mass of 20S-RC1 and the calibration value for the 20S proteasome, mass of one RC 902 ± 164 kD (SE = ± 4 kD); and considering the mass of 20S-RC2 and the calibration value for the 20S proteasome, mass of one RC, 894 ± 144 kD (SE = ± 8 kD). The overall average mass of the RC is 894 ± 196 kD with a SE of ± 9 kD. Although experimental data were present in this mass range, indicating the existence of some free RCs, the absence of a well defined peak on the histogram prohibited a more direct measurement of the RC mass.
Figure 6.

STEM mass measurements. Data from the four experiments performed were individually scaled to the 20S proteasome mass, 721 kD, and pooled to yield the histograms shown. a, The peaks fall at 358 ± 96 kD (n = 1,394; for the small particles with an almost circular projection); b, 721 ± 55 kD for the 20S proteasome top-views (internal mass standard); c, 1,623 ± 155 kD (n = 2,003) for 20S-RC1; and d, 2,508 ± 196 kD (n = 286) for the bone-shaped 26S projections, 20S-RC2.
This result of 894 kD correlates well with the calculated mass of the RC (932 kD), obtained by summation of the 18 individual subunits (see Table ).
Deubiquitylating Activity of p37A and the 26S Proteasome
The cDNA encoding p37A was subcloned into pET22b, after fusing a Histidine-tag to its COOH terminus, and expressed in E. coli. The recombinant protein could be purified under native conditions and was assayed for UCH enzymatic activity using Ub-AMC as a substrate (Dang et al. 1998). Similar to the control enzyme, UCH-L3, p37A hydrolyzed the fluorogenic substrate, and was completely inhibited by Ub-Al when added at an equimolar concentration (Fig. 7 a). Purified 26S proteasomes were assayed in the same manner and also found to exhibit UCH enzymatic activity (Fig. 7 b). This activity could be blocked by the addition of Ub-Al, but was not affected by the 20S proteasome inhibitor, lactacystin (Fenteany et al. 1995), which rules out the possibility that the 20S core contributes to the release of free AMC.
Figure 7.
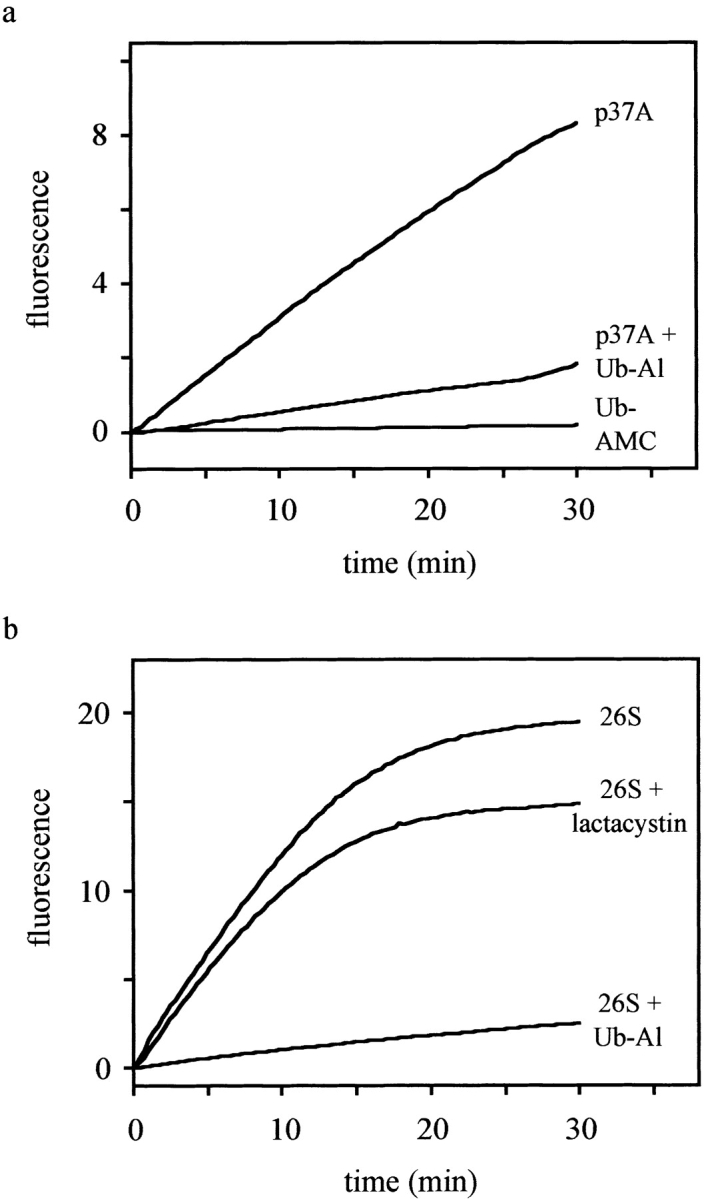
p37A and 26S proteasomes hydrolyze Ub-AMC. The fluorescence intensity (λex = 380 nm, λem = 460 nm), which is proportional to the release of AMC, was recorded and plotted as a function of time. The reactions were performed at 25°C in assay buffer (50 mM Hepes, 0.5 mM EDTA, pH 7.8, 1 mM DTT, and 0.1 mg/ml BSA) using Ub-AMC at a final concentration of 5 μM. a, The activity of p37A (∼2 nM) was measured both with and without preincubation with Ub-Al (2 nM). In a control assay, no enzyme was added to exclude self-hydrolysis of Ub-AMC. b, Similarly, 26S proteasomes (∼20 μg/ml) were preincubated with and without Ub-Al (10 nM), or with lactacystin (20 μM) before addition of Ub-AMC. The difference in the levels of fluorescence between uninhibited and lactacystin-inhibited proteasomes lies within the normally observed range of deviation.
Mapping the Position of p37A Within the RC by Electron Microscopy
In mapping of p37A, we have taken advantage of the specific binding of the UCH inhibitor, Ub-Al. The Ub-Al was conjugated to colloidal gold particles with a diameter of 3.5 nm. The gold particles are sufficiently electron dense to be clearly visible, even in negatively stained preparations. To obtain specific and stable conjugates, the side chain of lysine residues of Ub-Al was extended by reacting the terminal amino group with 2-iminothiolane to give a modified side chain with a terminal primary thiol group. Stable binding to the colloidal gold particles was then achieved through a covalent gold-sulfur bond.
An electron micrograph of negatively stained 26S proteasomes, which were labeled with Ub-Al conjugated to 3.5-nm colloidal gold particles, is shown in Fig. 8 a. About 50% of the 26S complexes have at least one bound gold particle, whereas unbound gold particles are not detected. Fig. 8 b shows a gallery of aligned 26S proteasomes carrying a gold particle on either one or both sides of the complex. 2D image analysis (averages obtained after rotational and translational alignment, followed by MSA/classification) clearly map p37A to the interface between the proximal and the distal mass of the RC (Fig. 6 c), i.e., to the interface between the two RC subcomplexes, the base and the lid (Glickman et al. 1998b).
Figure 8.
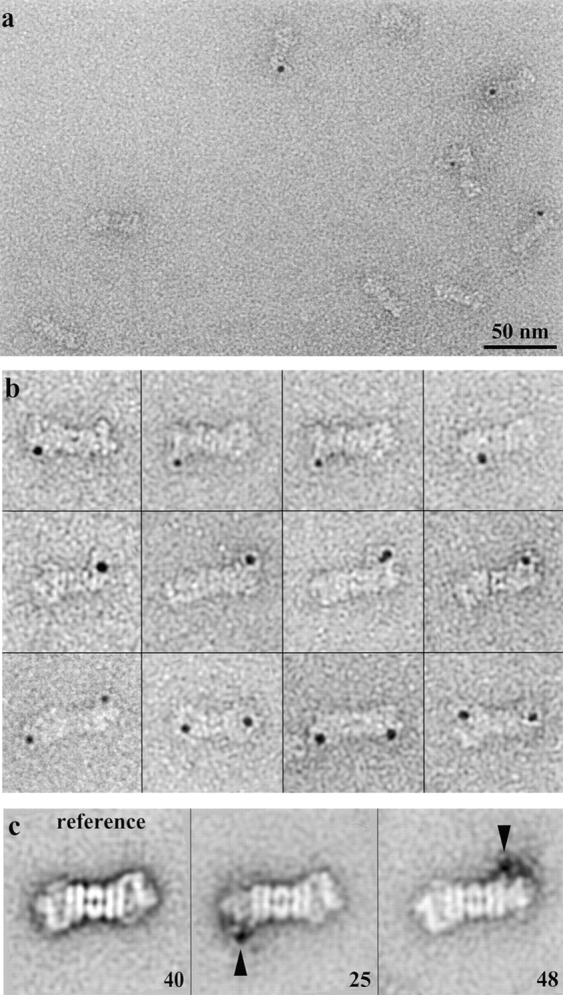
Mapping of p37A within the RC by EM. a, Electron micrograph of 26S proteasomes incubated with gold-labeled Ub-Al. Bar, 50 nm. b, Gallery of gold-labeled Ub-Al bound to the 26S proteasome after rotational and translational alignment. c, Main classes of unlabeled (reference) and gold-labeled 26S proteasomes after MSA/classification.
Discussion
Our understanding of the structure and function of the 26S proteasome advances at a relatively slow pace; even establishing the subunit composition is not a trivial task, given the labile nature of the complex. The bovine and human 26S complexes have been investigated in some detail and the primary structures of many of their ∼18 electrophoretically distinct subunits in the mass range of 20–110 kD have been reported (DeMartino et al. 1994; Dubiel et al. 1995; Tanaka and Tsurumi 1997). For yeast, a set of 18 RC subunits has been described (Glickman et al. 1998a); unfortunately, it is difficult to obtain structurally homogenous preparations of the complex from yeast. Drosophila embryos have been shown to be a particularly rich source of 26S proteasomes (Udvardy 1993), and they yield preparations that are sufficiently homogenous for structural studies (Walz et al. 1998). Initial studies with Drosophila RCs identified 12 subunits on 1D gels that were named p110 to p37 (Haracska and Udvardy 1996). However, complete primary structures have to date only been reported for p54 (Haracska and Udvardy 1995) and p42C (DUG; Mounkes and Fuller 1998).
To provide a platform for an in-depth structural analysis of the Drosophila RC, we established the subunit composition of purified 26S proteasomes by 2D gel electrophoresis and subsequent amino acid sequence analysis. In total, 18 subunits were found to constitute a single RC. 17 of them have homologues among the known yeast and mammalian RC subunits; hence, it can be assumed that none of these proteins is a contaminant and all are integral parts of the Drosophila RC. p37A, the only subunit missing in yeast, has homologues in mammalian 26S proteasomes (Xu, W., and R.E. Cohen, personal communication). The mammalian subunits, S5b (Deveraux et al. 1995) and p28 (Hori et al. 1998), as well as yeast Rpn4 (Fujimuro et al. 1998), have not been consistently found in 26S proteasomes from other organisms. In the Drosophila RC, we found no homologues of these three subunits, in agreement with their absence in Drosophila EST cDNA libraries.
It is hard to assert that the 18 proteins listed in Table represent the full complement of the Drosophila RC subunits, as subunits may be lost during purification or escape detection on 2D gels. But the close agreement between their summed masses (932 kD) and the experimentally determined mass (894 kD) suggests that the proteins derived from 2D gels indeed represent the complete set of RC subunits from our purified 26S proteasomes. It is interesting to note that in the STEM measurements, the SDs of the mass values were somewhat larger than expected from statistical background fluctuations, with the exception of the 20S particles where the SD is very close to the theoretically expected value. This indicates that the 20S core complex is stoichiometrically well defined, while the RCs display some heterogeneity. We did not find isolated and structurally well defined particles in the range of intact RCs. Instead, we found a sizeable fraction of roughly spherical particles yielding a broad peak with a maximum at 358 kD. Probably various subcomplexes of the RC (base, lid) contribute to this slightly asymmetric peak.
Most, but not all known RC subunits have been detected in all eukaryotic organisms investigated so far; hence, there seems to be a set of constitutive proteins that are essential for functional 26S proteasomes, and in addition, facultative proteins. The latter may only be expressed in certain organisms or in a tissue-dependent manner at specific developmental stages. Some of these facultative subunits may associate only transiently with the RC, and it will depend critically on the time point when a sample is taken whether they are detected or not. In fact, transient binding to the RC has been reported for Doa4 (Papa et al. 1999) and Ap-uch (Hegde et al. 1997), and also the bona fide subunit p54 is present in free and RC-bound form (Haracska and Udvardy 1995). Therefore, it appears unlikely that a universal number of subunits building the RC can be given.
Upon binding of ubiquitylated protein to the 26S proteasome, ubiquitin is usually recycled by means of deubiquitylating enzymes. Indeed, two different deubiquitylating activities have been reported to occur within the 26S proteasome. An early study described a 30-kD ubiquitin COOH-terminal hydrolase in the RC of rabbit reticulocytes that cleaves off the remnant of target proteins from the ubiquitin chains in the course of degradation (Eytan et al. 1993). More recently, an isopeptidase activity was found that shortens ubiquitin chains conjugated to target proteins by repeated removal of distal ubiquitins (Lam et al. 1997a,Lam et al. 1997b). Thus, the degradation signal of the protein is removed and poorly or erroneously ubiquitylated proteins may be rescued from proteolysis. A member of the UCH family of deubiquitylating enzymes, UCH37, is thought to be responsible for this so-called editing activity of the mammalian RC (Xu, W., and R.E. Cohen, personal communication).
We found the homologue of human UCH37 in Drosophila 26S proteasomes, which we named p37A, and expressed in E. coli for further characterization. Like other recombinant proteins of this family, it cleaves the model substrate Ub-AMC and is inhibited by Ub-Al. Thus, Drosophila p37A is probably different from 30-kD UCH found in proteasomes from rabbit reticulocytes, since the latter is insensitive to Ub-Al (Eytan et al. 1993). We do not know yet what type of conjugates Drosophila p37A prefers as substrates, but like other UCHs, it may cleave ubiquitin from peptides or small protein remnants only (Wilkinson 1997). However, it has been shown for Drosophila UCH-D (Roff et al. 1996) that UCH activity may not be restricted to small leaving groups. In addition, substrate preferences may differ between the free protein and the protein integrated into the RC. Recombinant UCH37, for instance, has typical UCH specificity, i.e., it removes an intact ubiquitin chain from a ubiquitin-protein conjugate, whereas UCH37 embedded in the 19S complex shortens a ubiquitin chain from the distal end by removing ubiquitin moieties one by one, as mentioned above (Xu, W., and R.E. Cohen, personal communication).
Since we found similar enzymatic activity and inhibition profiles with native 26S proteasomes and with recombinant p37A, we assume that p37A is at least in part responsible for the deubiquitylation of proteasome-bound conjugates. The fact that no homologue exists in the yeast genome suggests that p37A is not an essential subunit of the RC, and it cannot be excluded that there are additional subunits that exhibit deubiquitylating activity. Subunit p37B is a candidate that could confer deubiquitylating activity to 26S proteasomes, since it has some sequence similarity to ubiquitin-specific processing proteases (UBP), the second family of deubiquitylating enzymes (Wilkinson 1997). Whether p37B and its homologues, Rpn11 and S13, possess any enzymatic activity has hitherto not been shown experimentally. Since recombinant Pad1, the p37B homologue in S. pombe, shows no deubiquitylating activity (Penney et al. 1998), it remains questionable whether p37B and its homologues are indeed functional UBPs. Recently, it was shown that a sizeable fraction of Doa4, a 100-kD UBP, copurifies with yeast 26S proteasomes (Papa et al. 1999); however, a Doa4 homologue is not present in our preparations.
Besides p37A and UCH37, the only other UCH for which an association with 26S proteasomes has been reported is Ap-uch. Ap-uch is a neuron-specific protein, which is induced during long-term facilitation in Aplysia nervous tissue and which binds transiently to 26S proteasomes (Hegde et al. 1997). We did not find an Ap-uch homologue in our preparations, but could have missed it; because of its small molecular mass, it might be difficult to separate from the 14 subunits of the 20S proteasome. Whether p37A is a constitutive component of the RC, or is only expressed during embryonic stages of the Drosophila development remains to be established. UCH-D, for instance, is only present in high levels during the first four hours of embryogenesis, a rapid decline to low levels follows thereafter (Zhang et al. 1993).
Although a low-resolution three-dimensional (3D) map of the Drosophila 26S proteasome exists (Walz et al. 1998), the mapping of specific subunits to this structural framework is only in its beginnings. There is evidence from biochemical and genetic studies that the six ATPases, all members of the AAA-ATPase superfamily, are closely associated with each other; by way of analogy to other members of the AAA-ATPase family, it has been inferred that the six paralogs form a heterohexameric ring (Voges et al. 1999). Supposing that they act as reverse chaperones, unfolding substrates before their translocation into the 20S proteolytic core, they were tentatively mapped to the interface between 20S core and the remainder of the RC (Lupas et al. 1993). Recently a combined genetic, biochemical, and structural approach has provided more definitive insights into the structural organization of the RC (Glickman et al. 1998b). In yeast, the RC has been dissected into two distinct subcomplexes, the base and the lid. The base, which is proximal to the 20S core complex, indeed comprises the six ATPases and, in addition, the two largest subunits, Rpn1 and Rpn2. The eight remaining subunits were assigned to the lid. The interaction between the base and the lid is destabilized by deletion of the subunit Rpn10. Therefore, it can be assumed that Rpn10 is critically involved in providing a structural linkage. Otherwise, the role of Rpn10 is enigmatic: it binds multiubiquitin chains in vitro (Deveraux et al. 1994), but is dispensable in vivo for the degradation of ubiquitylated proteins (Van Nocker et al. 1996). This could be reconciled if one assumes that other subunits, probably in the lid, are responsible for the initial binding of ubiquitylated proteins, while Rpn10 stabilizes the interaction further downstream in the process.
Having identified p37A as a bona fide component of the Drosophila RC, we have mapped its location by EM. To this end, we have taken advantage of the specific binding of the UCH inhibitor Ub-Al to its target. By coupling Ub-Al to 3-nm colloidal gold particles, a strong signal was generated that was clearly visible, even on unprocessed electron micrographs. On averaged images, the Ub-Al gold conjugates map to the neck region of the dragon-head motif, i.e., the hinge between the base and the lid. This is the region where we also assume that Rpn10 is located (see above). Thus, it appears that both the binding of multiubiquitin chains and deubiquitylation are spatially closely related and perhaps also functionally coupled. One could envisage a scenario in which ubiquitylated proteins initially bind to the lid subcomplex. While being transferred to the base where the substrate is prepared for its feeding into the 20S core, Rpn10 prevents its escape while p37A recycles bound ubiquitin.
Acknowledgments
We thank M. Boicu for DNA sequencing, B. Wolpensinger for recording the STEM images, and M. Kania and D. Voges for proofreading of the manuscript.
This work was supported by the Deutsche Forschungsgemeinschaft (Bonn), Schwerpunktprogramm: “Struktur, Funktion und Regulation des 20S/26S Ubiquitin-Proteasomesystems,” and the Swiss National Science Foundation.
Footnotes
Abbreviations used in this paper: 1D, one-dimensional; 2D, two-dimensional; 16-BAC, benzyldimethyl-n-hexadecylammonium chloride; AMC, 7-amido-4-methylcoumarin; MSA, multivariate statistical analysis; RC, regulatory complex; STEM, scanning transmission electron microscopy; Suc-LLVY-AMC, Succinyl-Leu-Leu-Val-Tyr-AMC; Ub-Al, ubiquitin COOH-terminal aldehyde; Ub-AMC, ubiquitin COOH-terminal AMC; UBP, ubiquitin-specific processing protease; UCH, ubiquitin COOH-terminal hydrolase.
References
- Baumeister W., Dahlmann B., Hegerl R., Kopp F., Kuehn L., Pfeifer G. Electron microscopy and image analysis of the multicatalytic proteinase. FEBS Lett. 1988;241:239–245. doi: 10.1016/0014-5793(88)81069-x. [DOI] [PubMed] [Google Scholar]
- Baumeister W., Walz J., Zühl F., Seemüller E. The proteasomeparadigm of a self-compartmentalizing protease. Cell. 1998;92:3673–3680. doi: 10.1016/s0092-8674(00)80929-0. [DOI] [PubMed] [Google Scholar]
- Beyer A. Sequence analysis of the AAA protein family. Protein Sci. 1997;6:2043–2058. doi: 10.1002/pro.5560061001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bochtler M., Ditzel L., Groll M., Hartmann C., Huber R. The proteasome. Annu. Rev. Biophys. Biomol. Struct. 1999;28:295–317. doi: 10.1146/annurev.biophys.28.1.295. [DOI] [PubMed] [Google Scholar]
- Braun B.C., Glickman M., Kraft R., Dahlmann B., Kloetzel P.M., Finley D., Schmidt M. The base of the proteasome regulatory particle exhibits chaperone-like activity. Nat. Cell Biol. 1999;1:221–226. doi: 10.1038/12043. [DOI] [PubMed] [Google Scholar]
- Confalonieri F., Duguet M. A 200-amino acid ATPase module in search of a basic function. Bioessays. 1995;17:639–650. doi: 10.1002/bies.950170710. [DOI] [PubMed] [Google Scholar]
- Coux O., Tanaka K., Goldberg A.L. Structure and functions of the 20S and 26S proteasomes. Annu. Rev. Biochem. 1996;65:801–847. doi: 10.1146/annurev.bi.65.070196.004101. [DOI] [PubMed] [Google Scholar]
- Dang L.C., Melandri F.D., Stein R.L. Kinetic and mechanistic studies on the hydrolysis of ubiquitin C-terminal 7-amido-4-methylcoumarin by deubiquitinating enzymes. Biochemistry. 1998;37:1868–1879. doi: 10.1021/bi9723360. [DOI] [PubMed] [Google Scholar]
- DeMartino G.N., Moomaw C.R., Zagnitko O.P., Proske R.J., Chu-Ping M., Afendis S.J., Swaffield J.C., Slaughter C.A. PA700, an ATP-dependent activator of the 20S proteasome, is an ATPase containing multiple members of a nucleotide-binding protein family. J. Biol. Chem. 1994;269:20878–20884. [PubMed] [Google Scholar]
- Deveraux Q., Ustrell V., Pickart C., Rechsteiner M. A 26S protease subunit that binds ubiquitin conjugates. J. Biol. Chem. 1994;269:7059–7061. [PubMed] [Google Scholar]
- Deveraux Q., Jensen C., Rechsteiner M. Molecular cloning and expression of a 26S protease subunit enriched in dileucine repeats. J. Biol. Chem. 1995;270:23726–23729. doi: 10.1074/jbc.270.40.23726. [DOI] [PubMed] [Google Scholar]
- Dubiel W., Ferrell G., Rechsteiner M. Subunits of the regulatory complex of the 26S proteasome. Mol. Biol. Rep. 1995;21:27–34. doi: 10.1007/BF00990967. [DOI] [PubMed] [Google Scholar]
- Edman P., Begg G.A. A protein sequenator. Eur. J. Biochem. 1967;1:80–91. doi: 10.1007/978-3-662-25813-2_14. [DOI] [PubMed] [Google Scholar]
- Engel A., Baumeister W., Saxton W.O. Mass mapping of a protein complex with the scanning-transmission electron-microscope. Proc. Natl. Acad. Sci. USA. 1982;79:4050–4054. doi: 10.1073/pnas.79.13.4050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eytan E., Armon T., Heller H., Beck S., Hershko A. Ubiquitin C-terminal hydrolase activity associated with the 26S protease complex. J. Biol. Chem. 1993;268:4668–4674. [PubMed] [Google Scholar]
- Fenteany G., Standaert R.F., Lane W.S., Choi S., Corey E.J., Schreiber S.L. Inhibition of proteasome activities and subunit-specific amino-terminal threonine modification by lactacystin. Science. 1995;268:726–731. doi: 10.1126/science.7732382. [DOI] [PubMed] [Google Scholar]
- Finley D., Tanaka K., Mann C., Feldmann H., Hochstrasser M., Vierstra R., Johnston S., Hampton R., Haber J., Mccusker J. Unified nomenclature for subunits of the Saccharomyces cerevisiae proteasome regulatory particle. Trends Biochem. Sci. 1998;23:244–245. doi: 10.1016/s0968-0004(98)01222-5. [DOI] [PubMed] [Google Scholar]
- Fujimuro M., Tanaka K., Yokosawa H., Toh-e A. Son1p is a component of the 26S proteasome of the yeast Saccharomyces cerevisiae . FEBS Lett. 1998;423:149–154. doi: 10.1016/s0014-5793(98)00084-2. [DOI] [PubMed] [Google Scholar]
- Fujinami K., Tanahashi N., Tanaka K., Ichihara A., Cejka Z., Baumeister W., Miyawaki M., Sato T., Nakagawa H. Purification and characterization of the 26S proteasome from spinach leaves. J. Biol. Chem. 1994;269:25905–25910. [PubMed] [Google Scholar]
- Glickman M.H., Rubin D.M., Fried V.A., Finley D. The regulatory particle of the Saccharomyces cerevisiae proteasome Mol. Cell. Biol. 18 1998. 3149 3162a [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glickman M.H., Rubin D.M., Coux O., Wefes I., Pfeifer G., Cjeka Z., Baumeister W., Fried V.A., Finley D. A subcomplex of the proteasome regulatory particle required for ubiquitin-conjugate degradation and related to the COP9-signalosome and eIF3 Cell. 94 1998. 615 623b [DOI] [PubMed] [Google Scholar]
- Haracska L., Udvardy A. Cloning and sequencing a non-ATPase subunit of the regulatory complex of the Drosophila 26S protease. Eur. J. Biochem. 1995;231:720–725. doi: 10.1111/j.1432-1033.1995.tb20753.x. [DOI] [PubMed] [Google Scholar]
- Haracska L., Udvardy A. Dissection of the regulator complex of the Drosophila 26S protease by limited proteolysis. Biochem. Biophys. Res. Commun. 1996;220:166–170. doi: 10.1006/bbrc.1996.0375. [DOI] [PubMed] [Google Scholar]
- Hartinger J., Stenius K., Hogemann D., Jahn R. 16-BAC/SDS-PAGEa two-dimensional gel electrophoresis system suitable for the separation of integral membrane proteins. Anal. Biochem. 1996;240:126–133. doi: 10.1006/abio.1996.0339. [DOI] [PubMed] [Google Scholar]
- Hegde A.N., Inokuchi K., Pei W., Casadio A., Ghirardi M., Chain D.G., Martin K.C., Kandel E.R., Schwartz J.H. Ubiquitin C-terminal hydrolase is an immediate-early gene essential for long-term facilitation in Aplysia . Cell. 1997;89:115–126. doi: 10.1016/s0092-8674(00)80188-9. [DOI] [PubMed] [Google Scholar]
- Hegerl R. The EM program packagea platform for image processing in biological electron microscopy. J. Struct. Biol. 1996;116:30–34. doi: 10.1006/jsbi.1996.0006. [DOI] [PubMed] [Google Scholar]
- Hershko A., Ciechanover A. The ubiquitin system. Annu. Rev. Biochem. 1998;67:425–479. doi: 10.1146/annurev.biochem.67.1.425. [DOI] [PubMed] [Google Scholar]
- Hoffman L., Pratt G., Rechsteiner M. Multiple forms of the 20S multicatalytic and the 26S ubiquitin/ATP-dependent proteases from rabbit reticulocyte lysate. J. Biol. Chem. 1992;267:22362–22368. [PubMed] [Google Scholar]
- Hori T., Kato S., Saeki M., DeMartino G.N., Slaughter C.A., Takeuchi J., Toh-e A., Tanaka K. cdna cloning and functional analysis of p28 (Nas6p) and p40.5 (Nas7p), two novel regulatory subunits of the 26S proteasome. Gene. 1998;216:113–122. doi: 10.1016/s0378-1119(98)00309-6. [DOI] [PubMed] [Google Scholar]
- Hough R., Pratt G., Rechsteiner M. Purification of two high molecular weight proteases from rabbit reticulocyte lysate. J. Biol. Chem. 1987;262:8303–8313. [PubMed] [Google Scholar]
- Jensen D.E., Proctor M., Marquis S.T., Gardner H.P., Ha S.I., Chodosh L.A., Ishov A.M., Tommerup N., Vissing H., Sekido Y. BAP1a novel ubiquitin hydrolase which binds to the BRCA1 ring finger and enhances BRCA1-mediated cell-growth suppression. Oncogene. 1998;16:1097–1112. doi: 10.1038/sj.onc.1201861. [DOI] [PubMed] [Google Scholar]
- Laemmli U.K. Cleavage of structural proteins during assembly of the head of bacteriophage T4. Nature. 1970;227:680–683. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- Lam Y.A., DeMartino G.N., Pickart C.M., Cohen R.E. Specificity of the ubiquitin isopeptidase in the PA700 regulatory complex of 26S proteasomes J. Biol. Chem. 272 1997. 28438 28446a [DOI] [PubMed] [Google Scholar]
- Lam Y.A., Xu W., DeMartino G.N., Cohen R.E. Editing of ubiquitin conjugates by an isopeptidase in the 26S proteasome Nature. 385 1997. 737 740b [DOI] [PubMed] [Google Scholar]
- Larsen C.N., Finley D. Protein translocation channels in the proteasome and other proteases. Cell. 1997;91:431–434. doi: 10.1016/s0092-8674(00)80427-4. [DOI] [PubMed] [Google Scholar]
- Lupas A., Van Dyke M., Stock J. Predicting coiled coils from protein sequences. Science. 1991;252:1162–1164. doi: 10.1126/science.252.5009.1162. [DOI] [PubMed] [Google Scholar]
- Lupas A., Koster A.J., Baumeister W. Structural features of 26S and 20S proteasomes. Enz. Prot. 1993;47:252–273. doi: 10.1159/000468684. [DOI] [PubMed] [Google Scholar]
- Lupas A., Flanagan J.M., Tamura T., Baumeister W. Self-compartmentalizing proteases. Trends Biochem. Sci. 1997;22:399–404. doi: 10.1016/s0968-0004(97)01117-1. [DOI] [PubMed] [Google Scholar]
- Mounkes L.C., Fuller M.T. The DUG gene of Drosophila melanogaster encodes a structural and functional homolog of the Saccharomyces cerevisiae SUG1 predicted ATPase associated with the 26S proteasome. Gene. 1998;206:165–174. doi: 10.1016/s0378-1119(97)00564-7. [DOI] [PubMed] [Google Scholar]
- Müller S.A., Kenneth N.G., Bürki R., Häring R., Engel A. Factors influencing the precision of quantitative scanning transmission electron microscopy. Ultramicroscopy. 1992;46:317–334. [Google Scholar]
- Papa F.R., Amerik A.Y., Hochstrasser M. Interaction of the Doa4 deubiquitinating enzyme with the yeast 26S proteasome. Mol. Biol. Cell. 1999;10:741–756. doi: 10.1091/mbc.10.3.741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson S.D. From electrophoretically separated protein to identificationstrategies for sequence and mass analysis. Anal. Biochem. 1994;221:1–15. doi: 10.1006/abio.1994.1371. [DOI] [PubMed] [Google Scholar]
- Penney M., Wilkinson C., Wallace M., Javerzat J.P., Ferrell K., Seeger M., Dubiel W., McKay S., Allshire R., Gordon C. The pad1(+) gene encodes a subunit of the 26S proteasome in fission yeast. J. Biol. Chem. 1998;273:23938–23945. doi: 10.1074/jbc.273.37.23938. [DOI] [PubMed] [Google Scholar]
- Peters J.M., Cejka Z., Harris J.R., Kleinschmidt J.A., Baumeister W. Structural features of the 26S proteasome complex. J. Mol. Biol. 1993;234:932–937. doi: 10.1006/jmbi.1993.1646. [DOI] [PubMed] [Google Scholar]
- Phipps B.M., Hoffmann A., Stetter K.O., Baumeister W. A novel ATPase complex selectively accumulated upon heat shock is a major cellular component of thermophilic archaebacteria. EMBO (Eur. Mol. Biol. Organ.) J. 1991;10:1711–1722. doi: 10.1002/j.1460-2075.1991.tb07695.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rechsteiner M. The 26S proteasome. In: Peters J.M., Harris J.R., Finley D., editors. Ubiquitin and the Biology of the Cell. Plenum Press; New York: 1998. pp. 147–189. [Google Scholar]
- Rock K.L., Gramm C., Rothstein L., Clark K., Stein R., Dick L., Hwang D., Goldberg A.L. Inhibitors of the proteasome block the degradation of most cell proteins and the generation of peptides presented on MHC class I molecules. Cell. 1994;78:761–771. doi: 10.1016/s0092-8674(94)90462-6. [DOI] [PubMed] [Google Scholar]
- Roff M., Thompson J., Rodriguez M.S., Jacque J.M., Baleux F., Arenzana-Seisdedos F., Hay R.T. Role of IkBa ubiquitination in signal-induced activation of NFκB in vivo . J. Biol. Chem. 1996;271:7844–7850. doi: 10.1074/jbc.271.13.7844. [DOI] [PubMed] [Google Scholar]
- Rubin D.M., Finley D. The proteasomea protein-degrading organelle? Curr. Biol. 1995;5:854–858. doi: 10.1016/s0960-9822(95)00172-2. [DOI] [PubMed] [Google Scholar]
- Rubin G.M., Hong L., Brokstein P., Evans-Holm M., Frise E., Stapleton M., Harvey D.A. A Drosophila complementary DNA resource. Science. 2000;287:2222–2224. doi: 10.1126/science.287.5461.2222. [DOI] [PubMed] [Google Scholar]
- Sambrook J., Fritsch E.F., Maniatis T. Molecular Cloninga Laboratory Manual. 2nd edition. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY: 1989. [Google Scholar]
- Scheffner M., Smith S., Jentsch S. The ubiqutin-conjugation system. In: Peters J.M., Harris J.R., Finley D., editors. Ubiquitin and the Biology of the Cell. Plenum Press; NY: 1998. pp. 65–98. [Google Scholar]
- Slot J.W., Geuze H.J. A new method of preparing gold probes for multiple-labeling cytochemistry. Eur. J. Cell Biol. 1985;38:87–93. [PubMed] [Google Scholar]
- Strickland E., Hakala K., Thomas P.J., DeMartino G.N. Recognition of misfolding proteins by PA700, the regulatory subcomplex of the 26S proteasome. J. Biol. Chem. 2000;275:5565–5572. doi: 10.1074/jbc.275.8.5565. [DOI] [PubMed] [Google Scholar]
- Studier F.W., Rosenberg A.H., Dunn J.J., Dubendorf J.W. Use of T7 RNA polymerase to direct expression of cloned genes. Methods Enzymol. 1990;185:60–89. doi: 10.1016/0076-6879(90)85008-c. [DOI] [PubMed] [Google Scholar]
- Tanaka K., Tsurumi C. The 26S proteasomesubunits and functions. Mol. Biol. Rep. 1997;24:3–11. doi: 10.1023/a:1006876904158. [DOI] [PubMed] [Google Scholar]
- Thompson J.D., Gibson T.J., Plewniak F., Jeanmougin F., Higgins D.G. The ClustalX windows interfaceflexible strategies for multiple sequence alignment aided by quality analysis tools. Nucl. Acids Res. 1997;25:4876–4882. doi: 10.1093/nar/25.24.4876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Udvardy A. Purification and characterization of a multiprotein component of the Drosophila 26S (1500 kD) proteolytic complex. J. Biol. Chem. 1993;268:9055–9062. [PubMed] [Google Scholar]
- Van Nocker S., Sadis S., Rubin D.M., Glickman M., Fu H., Coux O., Wefes I., Finley D., Vierstra R.D. The multiubiquitin-chain-binding protein Mcb1 is a component of the 26S proteasome in Saccharomyces cerevisiae and plays a nonessential, substrate-specific role in protein turnover. Mol. Cell. Biol. 1996;16:6020–6028. doi: 10.1128/mcb.16.11.6020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varshavsky A. The ubiquitin system. Trends Biochem. Sci. 1997;22:383–387. doi: 10.1016/s0968-0004(97)01122-5. [DOI] [PubMed] [Google Scholar]
- Voges D., Zwickl P., Baumeister W. The 26S proteasomea molecular machine designed for controlled proteolysis. Annu. Rev. Biochem. 1999;68:1015–1068. doi: 10.1146/annurev.biochem.68.1.1015. [DOI] [PubMed] [Google Scholar]
- Walz J., Erdmann A., Kania M., Typke D., Koster A.J., Baumeister W. 26S proteasome structure revealed by 3-dimensional electron microscopy. J. Struct. Biol. 1998;121:19–29. doi: 10.1006/jsbi.1998.3958. [DOI] [PubMed] [Google Scholar]
- Wenzel T., Baumeister W. Conformational constraints in protein degradation by the 20S proteasome. J. Struct. Biol. 1995;2:199–204. doi: 10.1038/nsb0395-199. [DOI] [PubMed] [Google Scholar]
- Wilkinson K.D. Regulation of ubiquitin-dependent processes by deubiquitinating enzymes. FASEB J. 1997;11:1245–1256. doi: 10.1096/fasebj.11.14.9409543. [DOI] [PubMed] [Google Scholar]
- Wolf S., Nagy I., Lupas A., Pfeifer G., Cejka Z., Müller S.A., Engel A., De Mot R., Baumeister W. Characterization of ARC, a divergent member of the AAA ATPase family from Rhodococcus erythropolis . J. Mol. Biol. 1998;277:13–25. doi: 10.1006/jmbi.1997.1589. [DOI] [PubMed] [Google Scholar]
- Yoshimura T., Kameyama K., Takagi T., Ikai A., Tokunaga F., Koide T., Tanahashi N., Tamura T., Cejka Z., Baumeister W. Molecular characterization of the 26S proteasome complex from rat liver. J. Struct. Biol. 1993;111:200–211. doi: 10.1006/jsbi.1993.1050. [DOI] [PubMed] [Google Scholar]
- Zhang N., Wilkinson K.D., Bownes M. Cloning and analysis of expression of a ubiquitin carboxyl terminal hydrolase expressed during oogenesis in Drosophila melanogaster . Dev. Biol. 1993;157:214–223. doi: 10.1006/dbio.1993.1125. [DOI] [PubMed] [Google Scholar]
- Zwickl P., Nig D., Min Woo K., Klenk H.P., Goldberg A.L. An archaebacterial ATPase, homologous to ATPases in the eukaryotic 26S proteasome, activates protein breakdown by 20S proteasomes. J. Biol. Chem. 1999;274:26008–26014. doi: 10.1074/jbc.274.37.26008. [DOI] [PubMed] [Google Scholar]
- Zwickl P., Baumeister W., Steven A. Dis-assembly linesthe proteasome and related ATPase-assisted proteases. Curr. Opin. Struct. Biol. 2000;10:242–250. doi: 10.1016/s0959-440x(00)00075-0. [DOI] [PubMed] [Google Scholar]