

Summary
Two major decapping enzymes are involved in the decay of eukaryotic mRNA, Dcp2 and DcpS. Despite the detection of robust DcpS decapping activity in cell extract, minimal to no decapping is detected from human Dcp2 (hDcp2) in extract. We now demonstrate that one reason for the lack of detectable hDcp2 activity in extract is due to the presence of inhibitory trans factor(s). Furthermore, we demonstrate that a previously identified testis-specific protein of unknown function implicated in nonspecific X-linked mental retardation, VCX-A, can function as an inhibitor of hDcp2 decapping in vitro and in cells. VCX-A is a noncanonical cap-binding protein that binds to capped RNA but not cap structure lacking an RNA. Its cap association is enhanced by hDcp2 to further augment the ability of VCX-A to inhibit decapping. Our data demonstrate that VCX-A can regulate mRNA stability and that it is an example of a tissue-specific decapping regulator.
Introduction
Regulation of mRNA degradation is critical for the proper control of gene expression. Eukaryotic mRNAs are degraded by two major exonucleolytic decay pathways, each primarily utilizing a distinct decapping enzyme. Following the initial deadenylation of the polyadenosine (poly[A]) tail, the body of the mRNA can be degraded by one of two distinct pathways involving either 5′ end or 3′ end decay. The deadenylated mRNA can be decapped by the Dcp2 decapping enzyme exposing the 5′ end to 5′ to 3′ exonucleolytic activity or it can continually be degraded from the 3′ end to generate a cap dinucleotide that is subsequently hydrolyzed by the scavenger decapping enzyme DcpS (reviewed in Coller and Parker [2004]).
Removal of the 5′ cap from an mRNA is a highly regulated process involving both positive and negative effectors. A plethora of proteins have been identified as stimulating decapping. In yeast, the Dcp2p-interacting protein Dcp1p is required for maximal detectable decapping activity (She et al., 2004; Steiger et al., 2003). The Lsm1-7 protein complex, as well as Dhh1p and the Edc1p, Edc2p, and Edc3p proteins, has also been reported to positively impact Dcp2p decapping (reviewed in Coller and Parker [2004]). In addition, the adenosine-uracil-rich element has also been shown to promote decapping in yeast (Vasudevan and Peltz, 2001) and mammals (Fenger-Gron et al., 2005; Gao et al., 2001; Lykke-Andersen and Wagner, 2005; Stoecklin et al., 2006) as does the mammalian Edc4 (also known as Hedls and GE-1) protein (Fenger-Gron et al., 2005). hDcp2 appears to function within various multiprotein complexes that can facilitate hDcp2 decapping activity. For example, Edc4 can directly facilitate hDcp2 activity as well as promote the association of hDcp1a with hDcp2 to possibly further enhance decapping activity in cells (Fenger-Gron et al., 2005).
mRNA decapping is a regulated event. In addition to factors that activate decapping (reviewed in Simon et al. [2006]), Dcp2 decapping can also be negatively impacted. In yeast, eIF4E and the poly(A) tail both negatively affect decapping (Caponigro and Parker, 1995; Ramirez et al., 2002; Schwartz and Parker, 1999, 2000; Wilusz et al., 2001a). A network of interactions involving eIF4E, eIF4G, and Pab1p juxtaposes the two ends of the mRNA and stabilizes the cap by preventing access to decapping enzymes (Schwartz and Parker, 2000; Wells et al., 1998; Wilusz et al., 2001b). Similarly, in mammals, the eIF4E cap-binding protein inhibits decapping in vitro (Khanna and Kiledjian, 2004) and RNAs with synthetic cap structures that bind eIF4E with higher affinity are more stable in vivo (Grudzien et al., 2006). The poly(A) tail can also negatively influence decapping (Wang et al., 2002), but, in addition to the indirect association of the poly(A)-binding protein (PABP) with the cap, PABP can also directly inhibit decapping (Khanna and Kiledjian, 2004). Therefore, although a number of proteins have been shown to promote decapping, only the cap-binding protein and PABP are known factors that can inhibit decapping. However, it appeared likely that additional inhibitors are also present, since the addition of cap analog to sequester cap-binding proteins does not increase the level of decapping detected in cell extract and an unadenylated RNA is also inefficiently decapped in extract (Wang et al., 2002).
Here, we report that a protein of previously unknown function implicated in X-linked nonspecific mental retardation (MRX), variable charged X chromosome protein-A (VCX-A) (Fukami et al., 2000; Van Esch et al., 2005), is an inhibitor of decapping that can stabilize mRNA.
Results
Presence of a trans Factor Capable of Inhibiting hDcp2 Decapping
Two interesting properties of hDcp2 are the lack of detectable significant endogenous hDcp2 decapping activity in total cellular or cytoplasmic extract and the presence of only a modest level of decapping in the high-speed cytoplasmic pellet (Wang et al., 2002). This is in contrast to the robust decapping detected by the DcpS scavenger decapping activity in total extract (Liu et al., 2002; van Dijk et al., 2003; Wang and Kiledjian, 2001). Addition of cap analog competitor to sequester potential cap-binding inhibitory proteins (Khanna and Kiledjian, 2004) as well as inhibit hydrolysis of the hDcp2 decapping product m7GDP by DcpS (van Dijk et al., 2003) also failed to enhance the level of detected hDcp2 activity in extract (Wang et al., 2002). These initial observations indicated that an activity distinct from that of a canonical cap-binding protein capable of inhibiting hDcp2 was present in cytoplasmic extract and possibly fractionated away from hDcp2 in the high-speed centrifugation. The detected inhibition is distinct from the poly(A) tail-mediated repression, since unadenylated RNAs were also not decapped in total extract (Wang et al., 2002). In an effort to determine whether cell extract contains an activity that inhibits hDcp2 decapping, an increasing titration of human erythroleukemia K562 cell extract was added to a fixed amount of recombinant hDcp2 protein and decapping activity was analyzed by thin-layer chromatography (TLC) (Figure 1A). The recombinant protein was competent to decap the input RNA and produced the expected m7GDP (lane 2). However, this activity was efficiently inhibited upon the addition of cytosolic extract (lanes 3–5), but not boiled extract (lane 6), indicating that it involved a protein. These data are consistent with the presence of an inhibitor of hDcp2 decapping in cells.
Figure 1. Presence of a trans Factor Capable of Inhibiting hDcp2 Decapping in Extract.
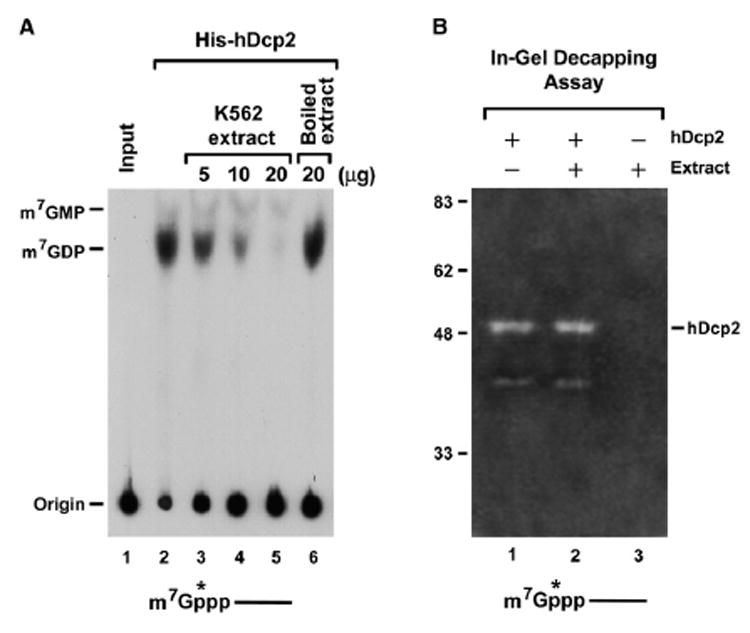
(A) Presence of a decapping inhibitor in extract. An in vitro decapping assay was carried out with 4 pmol of recombinant His-tagged hDcp2 in the absence (lane 2) or presence of K562 cell extract (lanes 3–5) and 100 μM unlabeled cold cap analog at 37°C for 30 min. The 32P-cap-labeled pcP RNA containing a 16 nucleotide G track at the 3′ end was used as substrate for the decapping reactions. Reaction products were resolved by PEI-TLC developed in 0.75 M LiCl and exposed to phosphorimager. Heat-inactivated K562 extract was used as a control (lane 6). A schematic representation of the 32P-cap-labeled pcP RNA substrate is shown on the bottom, where the line represents the RNA, the m7Gppp denotes the cap, and the asterisk indicates the position of the 32P.
(B) Twenty picomoles of recombinant His-hDcp2 protein was preincubated for 15 min at 37°C with (lane 2) or without (lane 1) 20 μg K562 cell extract and separated on an SDS-PAGE gel containing 32P-cap-labeled pcP RNA polymerized in the gel. The in-gel decapping assay was carried out as described (Wang et al., 2002), and the gel was subsequently dried and exposed to Kodak X-ray film. Decapping was monitored by formation of a zone of clearance (white band) resulting from the released 32P-labeled m7GDP product that diffuses from the gel. Recombinant His-hDcp2 generated two decapping bands that corresponded to the 48 kDa full-length His-hDcp2 and a 40 kDa truncated product as previously reported (Wang et al., 2002).
To distinguish whether the inhibition involved a covalent modification of hDcp2 or was mediated by a trans factor, an in-gel decapping assay was carried out. We reasoned that if the inhibition were a consequence of a covalent modification of hDcp2 (e.g., phosphorylation), then resolution of the protein by SDS-PAGE would not relieve the decapping inhibition within an in-gel decapping assay. Conversely, if the inhibition is a result of a trans factor that associates with and inhibits hDcp2, then separation of the proteins on SDS-PAGE gel should separate the proteins and hDcp2-mediated decapping should be detected by an in-gel assay. Recombinant hDcp2 by itself (analogous to lane 2 of Figure 1A) and hDcp2 incubated with K562 extract under conditions that inhibited its decapping activity (analogous to lane 5 of Figure 1A) were each resolved on SDS-PAGE gel containing 32P-cap-labeled RNA embedded in the gel. The gel was denatured, renatured, and subsequently incubated in decapping buffer as previously described (Wang et al., 2002). The presence of decapping activity of the renatured hDcp2 protein was assessed by the presence of a zone of radioactive signal clearing in the region of the gel that corresponded to hDcp2. Since the first phosphate following the methylguanosine is the exclusive radioactive moiety within the capped RNA, a zone of clearing arises upon the release of the radiolabeled m7GDP decapping product. Consistent with our earlier demonstration, recombinant hDcp2-mediated decapping is readily detected by this assay (Figure 1B, lane 1). Interestingly, hDcp2 incubated with cytosolic extract was also still capable of hydrolyzing the capped RNA substrate (lane 2) and this activity was comparable to untreated recombinant hDcp2 protein (compare lanes 1 and 2). The decapping was a result of the recombinant protein, since the same amount of cytosolic extract alone did not produce sufficient decapping activity to enable detection by this assay (lane 3). We therefore conclude that the observed inhibition of recombinant hDcp2 by cytoplasmic extract is a consequence of a trans factor rather than a covalent modification.
VCX-A Is an hDcp2 Decapping Inhibitor
In an initially unrelated project aimed at identifying potential RNA-binding proteins involved in human disorders, we noted that a protein implicated in MRX, termed VCX-A (Fukami et al., 2000), contained an RGG box-related motif. The RGG box was initially identified as the RNA-binding domain in the hnRNP U protein (Kiledjian and Dreyfuss, 1992) and subsequently shown to be essential for RNA binding in other proteins. Using a northwestern assay, recombinant VCX-A was confirmed to be an RNA-binding protein (Figure 2A; lanes 1 and 2). The RNA-binding property of the protein is conferred by the amino-terminal half, since the first 78 amino acids of the protein, which also contain the RGG box-like domain, were capable of binding RNA while the C-terminal 105 amino acids of VCX-A did not bind RNA (lanes 3 and 4). Interestingly, despite its general RNA-binding property, VCX-A preferentially binds to the 5′ end of a capped RNA, but not uncapped RNA containing a 32P at the same position (Figure 2B; compare lane 2 to lane 5). Binding was also not detected to cap structure lacking an RNA moiety (lane 8). In contrast, the eIF4E cap-binding protein bound to both the 5′ end of a capped RNA as well as the cap structure alone, albeit with greater affinity to the capped RNA, as expected (lanes 1 and 7). Interestingly, the cap-binding property of VCX-A is analogous to both Dcp2 and PABP, where both proteins also bind the cap only when it is linked to an RNA, but not the cap structure directly or the 5′ end of an uncapped RNA (Khanna and Kiledjian, 2004; Piccirillo et al., 2003).
Figure 2. VCX-A Is an RNA-Binding Protein and Preferentially Binds the 5′ Cap.
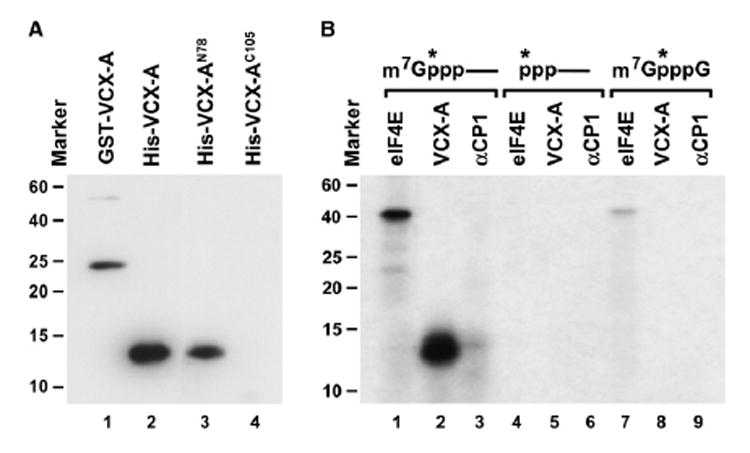
(A) Northwestern assay demonstrating the RNA-binding property of VCX-A. Ten picomoles of each recombinant protein consisting of the GST fusion or His-tagged full-length VCX-A protein (lane 1 and lane 2, respectively) and His-tagged N-terminal 78 amino acids of VCX-A (lane 3) or the C-terminal 105 amino acids of VCX-A (lane 4) was resolved by SDS-PAGE and transferred to nitrocellulose. 32P-uniform-labeled pcP RNA was used as an RNA substrate. Protein size markers are denoted on the left. The protein sizes correspond to the migration of each protein on SDS-PAGE gel stained by Coo-massie blue. The anomalous migration of the VCX-AN78 on SDS-PAGE gel is most likely a consequence of the variable charged nature of VCX-A. Where the N terminus is highly positively charged and the C terminus is highly negatively charged, the N terminus alone migrates slower than would be expected.
(B) VCX-A preferentially binds the cap of a capped RNA. UV crosslinking was carried out with three different substrates, all containing the 32P phosphate at the same position indicated by an asterisk. Twenty picomoles GST-eIF4E, 2 pmol His-VCX-A, and 20 pmol His-αCP1 recombinant proteins were incubated with cap-labeled pcP RNA (lanes 1–3), uncapped 5′ end-labeled pcP RNA (lanes 4–6), or 32P-labeled cap analog (lanes 7–9) at room temperature for 15 min. Reactions were subjected to UV irradiation for 10 min on ice with a 15 W germicidal lamp, subsequently treated with RNase cocktail, and resolved on SDS-PAGE gel.
Since the association of PABP with the cap can lead to inhibition of decapping, we asked whether VCX-A can function as a decapping inhibitor. As shown in Figure 3C, a marked reduction in recombinant hDcp2 decapping activity was detected upon addition of VCX-A (lanes 2–5). No inhibition was detected by the addition of control RNA-binding proteins mDAZL, αCP1, and FMR1 (lanes 6–8). The fact that hDcp2 decapping was refractory to three different RNA-binding proteins with distinct RNA-binding domains (mDAZL, RRM type; αCP1, KH domain; FMR1, KH and RGG box) indicates that the inhibitory function of VCX-A is not a generic RNA-binding property and that VCX-A can function as a decapping inhibitor.
Figure 3. VCX-A Is a Decapping Inhibitor.
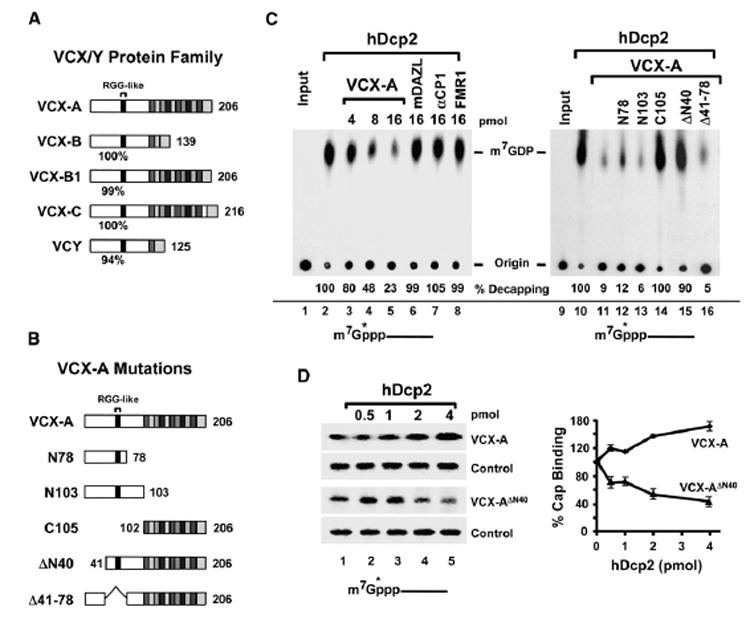
(A) Schematic of VCX/Y gene family proteins. VCX/Y protein sequences were aligned by CLUSTALW software. The N terminus is highly conserved, and the percent identity of each protein relative to VCX-A is denoted under each protein. The RGG-like domain in the N terminus is indicated. The 10 amino acid tandem repeated units are shown at the C terminus of each protein. The number of amino acids in each protein is shown on the right.
(B) VCX-A protein truncations. Labeling is as in (A) above, except that the amino-terminal amino acid of each protein is denoted on the left.
(C) VCX-A inhibits hDcp2 decapping activity in vitro. Decapping assays were carried out with 4 pmol His-hDcp2 in the presence of the indicated concentration of each protein. The percent decapping relative to lanes containing only hDcp2 is presented. Decapping reactions and labeling are as indicated in the legend to Figure 1.
(D) Requirement of the VCX-A first 40 amino acids for hDcp2-resistant association with the cap. Two picomoles of His-VCX-A or His-VCX-A lacking the N-terminal 40 amino acids (VCX-AΔN40) was incubated with cap-labeled pcP RNA at room temperature for 15 min. Following the initial incubation, an increasing concentration of hDcp2 was added to the reaction on ice and incubated an additional 10 min prior to UV crosslinking. Reactions were terminated with SDS sample buffer spiked with 32P-labeled eIF4E protein as an internal loading control prior to resolution of protein on SDS-PAGE gel. The reactions were carried out in a buffer containing Mg+2 as the divalent cation but lacking Mn+2 to minimize hDcp2 catalytic activity. Quantitations of three independent experiments are plotted on the right, with ±standard deviation (SD) denoted by the error bars.
The Amino-Terminal 40 Amino Acids of VCX-A Are Necessary for hDcp2 Decapping Inhibition
VCX-A is a member of a family of proteins encoded on the X and Y chromosomes (VCX and VCY, respectively). VCX consists of four genes at Xp22.31 encoding VCX-A, VCX-B1, VCX-B, and VCX-C; and VCY consists of two genes at Yq11.2 encoding VCY-D and VCY-E (Fukami et al., 2000; Lahn and Page, 2000). The proteins encoded by the different VCX/Y genes are shown schematically in Figure 3A and have a greater than 94% identity at the N-terminal half of the protein, primarily differing in the C terminus by the number of related 10 amino acid repeats (Fukami et al., 2000; Lahn and Page, 2000). The extensive conservation at the N-terminal segment of all the VCX/Y proteins suggests that all members of this family of proteins are RNA-binding proteins.
In order to identify the critical region in VCX-A necessary for hDcp2 decapping inhibition, a series of VCX-A truncations was generated and tested. The truncated proteins are represented schematically in Figure 3B. Equal molar concentrations of each protein were incubated with hDcp2, and their ability to inhibit decapping was analyzed (Figure 3C, lanes 11–16). Removal of the C-terminal repeated region had no effect on decapping inhibition (lanes 12 and 13), while removal of the N-terminal half of VCX-A ablated the ability of the truncated protein to inhibit hDcp2 decapping (lane 14). The region essential for decapping inhibition was further narrowed to the first 40 amino acids, where its removal resulted in a truncated protein incapable of inhibiting decapping (lane 15). However, an internal truncation that removed amino acids 41–78 still retained the ability to inhibit hDcp2 decapping (lane 11 and 16). These data demonstrate that the first 40 amino acids of VCX-A are critical for the inhibition of hDcp2 decapping.
Consistent with a requirement of the first 40 amino acids for decapping inhibition, the VCX-A ΔN40 protein that can still crosslink to the 5′ cap (Figure 3D, third panel) was readily displaced from the 5′ end of a capped RNA by hDcp2 while the full-length VCX was not (first panel). These data demonstrate that the N-terminal 40 amino acids facilitate a more efficient Dcp2 resistant association of VCX-A with the cap and are essential for the inhibition of hDcp2. Interestingly, not only was the full-length VCX-A not displaced by hDcp2, the ability of VCX-A to associate with the 5′ cap was enhanced by the addition of hDcp2. These data imply a potential for protein-protein interactions between VCX-A and hDcp2.
Protein-Protein Interaction of hDcp2 and VCX-A
The potential for hDcp2 and VCX-A protein-protein interactions was next tested. An interaction could be detected between glutathione S-transferase (GST)-VCX-A and 35S-methionine-labeled rabbit reticulocyte in vitro-translated hDcp2. As seen in Figure 4A, full-length hDcp2 was efficiently retained on the GST-VCX-A column, while the catalytically active N-terminal 257 amino acids of hDcp2 (hDcp21–257) were not retained on the affinity matrix (lane 2). Similar results were also obtained when using extract from 293T cells expressing epitope-tagged hDcp2 or a truncated hDcp2 consisting of the minimal hDcp2 region that contains decapping activity, hDcp294–257 (Piccirillo et al., 2003). Myc-hDcp2 interacted with and was retained by GST-VCX-A, but not a control column containing immobilized GST-mDAZL (lane 4 and lane 5, respectively). As expected, hDcp294–257, lacking the C-terminal 163 amino acids, was not retained by either column.
Figure 4. VCX-A Inhibition of Decapping Is Enhanced When It Can Interact with hDcp2.
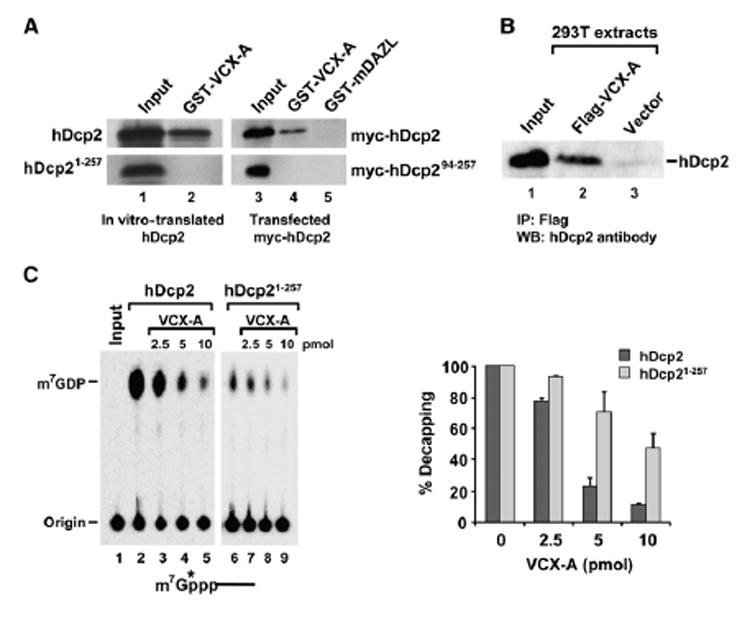
(A) Protein-protein interactions of VCX-A and hDcp2. GST-VCX-A can interact with 35S-methionine in vitro-translated hDcp2 (lane 2, top), but not a truncated hDcp2 consisting of the first 257 amino acids (lane 2, bottom). Forty percent of the sample used in the reaction is shown in lane 1. Interaction with epitope-tagged Myc-hDcp2 or Myc-hDcp2 containing the minimal decapping competent protein (amino acids 94–257) is shown in lane 4. Lane 5 contains the GST-mDAZL control. Lanes 3–5 were detected by western analysis using an anti-Myc antibody.
(B) Endogenous hDcp2 coimmunopurifying with Flag-VCX-A expressed in 293T cells with an anti-Flag M2 column is shown. hDcp2 was detected with affinity-purified anti-hDcp2 antisera.
(C) Protein-protein interaction enhances VCX-A decapping inhibition. Five picomoles each of recombinant His-hDcp2 or truncated His-hDcp21–257 was incubated with a titration of His-VCX-A at 37°C for 30 min in an in vitro decapping assay. The generated 32P-labeled m7GDP products were resolved on PEI-TLC in 0.75 M LiCl. Labeling and quantitations are as described in the legend to Figure 1. The mean of three independent decapping assays is shown on the right bar graph ±SD.
The hDcp2 and VCX-A interactions are not restricted to recombinant proteins and were also detected with epitope-tagged VCX-A expressed in cells and endogenous hDcp2. 293T human embryonic kidney cells were transfected with a vector expressing Flag-VCX-A. Anti-Flag antibody specifically coimmunopurified endogenous hDcp2, while immunoprecipitations from cells containing the vector alone did not (Figure 4B). These data demonstrate that an interaction between hDcp2 and VCX-A can also occur in cells. As all the interaction analyses above were carried out with extract treated with micrococcal nuclease, the detected associations are not a consequence of RNA tethering. These data demonstrate that hDcp2 and VCX-A can interact with one another and that the hDcp2 interaction domain is contained within the C-terminal 163 amino acid noncatalytic domain.
To assess the significance of the detected hDcp2 and VCX-A protein-protein interaction, we tested the ability of VCX-A to inhibit hDcp2 that it can or cannot interact with. Consistent with the premise that the major mode of inhibition by VCX-A is its cap-binding property, decapping inhibition was detected with both the wildtype and truncated hDcp2 proteins (Figure 4C). However, a consistently greater level of inhibition was observed with the full-length protein compared to the hDcp21–257 truncated protein unable to interact with VCX-A. These data demonstrate that, at least with recombinant protein, protein-protein interaction further augments the ability of VCX-A to inhibit hDcp2 decapping.
Inhibition of Endogenous hDcp2 Decapping In Vitro
The above data demonstrate that VCX-A is capable of inhibiting recombinant hDcp2 decapping. We next set out to assess whether endogenous hDcp2 decapping activity can be inhibited by VCX-A. We previously demonstrated that trace amounts of endogenous decapping activity can be detected in the 130,000 × g high-speed cytoplasmic pellet fraction (Wang et al., 2002). We have subsequently observed that maximal endogenous hDcp2 decapping is contained within the 50,000 × g cytoplasmic pellet (P50, see Figure S1 in the Supplemental Data available with this article online). Decapping detected in the P50 fraction was a function of hDcp2, as immunodepletion of hDcp2 from this fraction significantly reduced the level of m7GDP decapping product while decapping was readily detected in the preimmune control sera depletion (Figure 5A, lanes 3 and 4).
Figure 5. VCX-A Inhibits Endogenous hDcp2 Decapping Activity in Extract.
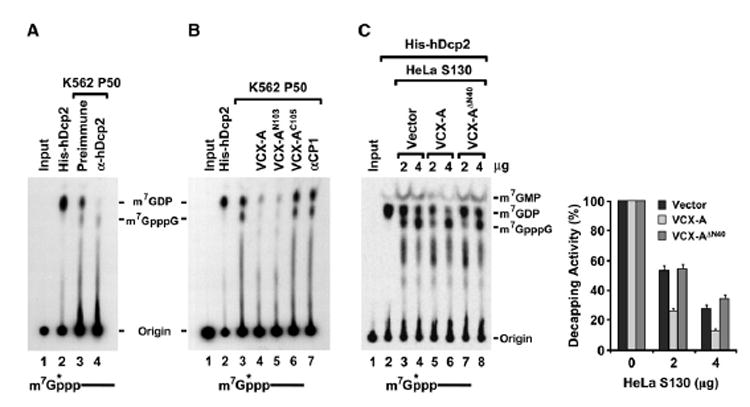
(A) hDcp2 is responsible for the decapping activity detected in the P50 fraction. In vitro decapping assays were carried out with K562 cell P50 fraction either immunodepleted with preimmune sera or hDcp2-specific antisera as indicated. Decapping by recombinant hDcp2 is shown in lane 2.
(B) Recombinant VCX-A inhibits endogenous hDcp2 decapping activity. Eighty picomoles of each indicated histidine-tagged protein was added to 50 μg K562 P50 in an in vitro decapping assay, and products were resolved by TLC in 0.45 M (NH4)2SO4.
(C) VCX-A expressed in cells inhibits recombinant hDcp2 decapping. In vitro decapping assays were carried out with 4 pmol of His-hDcp2 in the presence of 2 and 4 μg HeLa cell S130 extracts expressing Myc-VCX-A, Myc-VCX-A ΔN40, or empty vector as indicated. Quantitation of three independent experiments and corresponding SD denoted by the error bars are shown on the right. Labeling is as described in the legend to Figure 1. Cold cap analog (100 μM) was included in all reactions to inhibit endogenous DcpS activity, and products were resolved as in (B).
The abilities of VCX-A and its truncation derivatives to inhibit endogenous hDcp2 decapping activity were next tested. Consistent with the results of recombinant hDcp2, VCX-A and the N-terminal segment of VCX-A (VCX-AN103) both efficiently inhibited endogenous hDcp2 decapping activity within the P50 fraction derived from K562 cells (Figure 5B, lanes 4 and 5). Decapping was not affected by the C-terminal fragment of VCX-A (VCX-AC105), nor was it hindered by an unrelated control RNA-binding protein, αCP1 (lane 6 and lane 7, respectively). The observed inhibition was not due to the eIF4E cap-binding protein preventing hDcp2 access to the cap, since all the reactions contained 100 μM competitor cap analog to compete for cap-binding proteins and prevent hydrolysis of m7GDP to m7GMP by DcpS (van Dijk et al., 2003).
Inhibition of hDcp2 is also observed with VCX-A expressed in cells. Consistent with a role for VCX-A in the inhibition of hDcp2 decapping, epitope-tagged VCX-A is not present in the P50 fraction that contains hDcp2 decapping activity (data not shown). Its absence from P50 precludes us from testing whether VCX-A expressed in cells is competent to inhibit endogenous hDcp2 decapping activity in vitro. We therefore tested the ability of cellular extract overexpressing VCX-A to inhibit recombinant hDcp2 activity. In vitro decapping assays were carried out with hDcp2 in the presence of HeLa cell extract expressing Myc-tagged VCX-A or VCX-A lacking the N-terminal 40 amino acids (VCX-A ΔN40). We reasoned that, although cell extract can inhibit hDcp2 activity as shown in Figure 1, use of low concentrations of extract should enable the detection of enough decapping to distinguish whether expression of exogenous VCX-A can result in an increased inhibition. With the concentration of extract used, greater inhibition of hDcp2 activity was detected with extract from cells expressing VCX-A (Figure 5C, lanes 5 and 6) relative to extract from cells expressing either the empty vector (lanes 3 and 4) or the VCX-A ΔN40 N-terminal truncation unable to inhibit decapping (lanes 7 and 8). Collectively, the above data demonstrate that endogenous hDcp2 decapping activity can be inhibited by VCX-A in vitro.
VCX-A Can Inhibit Decapping and Stabilize mRNA in Cells
To address whether VCX-A can inhibit decapping in cells, we tested whether decapping of a cap-labeled RNA transfected into cells expressing VCX-A can be stabilized. A stably transformed 293T cell line expressing VCX-A was established since the VCX/Y genes are testis specific (Fukami et al., 2000; Lahn and Page, 2000), and endogenous VCX-A protein is not expected in these cells. A chimeric pcP RNA consisting of the pcDNA3 polylinker sequence with a 16 guanosine G track at the 3′ end to block 3′ to 5′ exonucleolytic decay (Wang and Kiledjian, 2001) was utilized. 32P-cap-labeled RNA was transfected into cells expressing either VCX-A or empty vector, and the demise of the mRNA followed over time. As seen in Figure 6A, the transfected RNA was more stable in cells expressing VCX-A. The RNA decayed with an initial lag in the presence of VCX-A and had an overall 2-fold greater half-life relative to control cells (1.5 hr versus 3.25 hr half-life). The observed stability was a function of decapping, as the RNA was cap labeled and degraded primarily from the 5′ end due to the presence of the 3′ G track (Wang and Kiledjian, 2001). These results demonstrate that VCX-A can inhibit decapping of a chimeric RNA introduced into cells.
Figure 6. VCX-A Inhibits Decapping and Stabilizes mRNA in Cells.
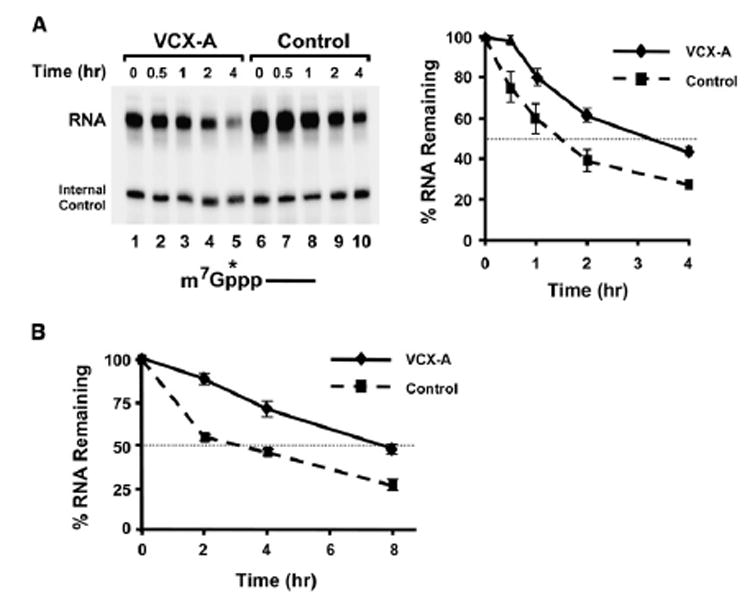
(A) 293T cells stably expressing VCX-A (lanes 1–5) or the empty vector (lanes 6–10) were transfected with 32P-cap-labeled RNA, and the decay was followed for the indicated times. An end-labeled control DNA oligonucleotide was included in the stop buffer as a loading control for normalizations. Quantitations from three independent experiments presented relative to time zero are plotted on the right, with the error bars representing the SD.
(B) Plasmids expressing the VCX-A or the hnRNP U protein RNA-binding domain (Control) were cotransfected with a luciferase expression vector into 293T cells. RNA was isolated from cells following the indicated times after actinomycin D treatment, and the abundance of luciferase mRNA normalized to the endogenous GAPDH mRNA was determined by quantitative real-time PCR. Values are presented relative to time zero and were derived from three independent experiments carried out in triplicate ± SD. The half-life of luciferase mRNA was determined to be 3 hr in the presence of the control protein and 8 hr when coexpressed with VCX-A.
The ability of VCX-A to inhibit decapping indicates that its expression should lead to the stabilization of mRNAs expressed in cells. To test whether VCX-A can stabilize mRNA in cells, Myc-tagged VCX-A was coexpressed with a luciferase reporter gene in 293T cells. Twenty-four hours posttransfection, global transcription was inhibited with actinomycin D and RNA was isolated and analyzed for up to 8 hr following the treatment. As seen in Figure 6B, the luciferase mRNA was more stable when cotransfected with VCX-A compared to coexpression of a control RNA-binding protein consisting of the hnRNP U RNA-binding domain (Kiledjian and Dreyfuss, 1992). The half-life of the luciferase mRNA was increased 2.7-fold from 3 hr in the presence of the control protein to 8 hr when VCX-A was coexpressed. These data demonstrate that VCX-A can inhibit decapping and stabilize mRNA in cells.
Discussion
In this report we address the apparent paradox of the lack of detectable robust hDcp2-mediated decapping in mammalian extract and demonstrate that this is at least in part a consequence of decapping inhibitor(s). Furthermore, we demonstrate that a testis-specific protein of previously unknown function implicated in mental retardation, VCX-A, can function to inhibit decapping. VCX-A is a capped RNA-binding protein that preferentially associates with the 5′ end of mRNA and stabilizes the mRNA. The decapping inhibition requires the first 40 amino acids of VCX-A, implying that all members of the VCX and VCY family of proteins, which shares at least 94% identity in the amino-terminal half of the protein, can also function as decapping inhibitors. The testis specificity of the VCX/Y proteins indicates that proteins of this family are tissue-specific regulators of mRNA decapping and stability.
The observed inhibition of recombinant hDcp2 decapping by the addition of mammalian K562 cell extract could either be due to a trans factor that influences the hDcp2 decapping protein activity or a covalent modification of hDcp2. However, we cannot rule out the possibility that posttranslational modifications could regulate hDcp2 decapping; yet the in-gel decapping assay of Figure 1B strongly suggests that the detected inhibition in our assay parameters was a consequence of a trans factor. Although we have yet to identify the specific trans factor(s) in K562 cells responsible for the inhibition observed in Figure 1, we have independently identified the VCX-A protein as an inhibitor of decapping.
Our data indicate that at least one mechanism by which VCX-A inhibits hDcp2 decapping is by providing a physical impediment at the cap. This conclusion is supported by the demonstration that VCX-A can efficiently crosslink to the cap and is not displaced by hDcp2. In contrast, although the truncated VCX-AΔN40 can still crosslink to the cap, it is readily displaced by hDcp2 (Figure 3). These results demonstrate that the cap association of VCX-A is essential for its ability to inhibit decapping and that the first 40 amino acids are necessary for the hDcp2-resistant association with the cap.
Interestingly, the cap-binding property of VCX-A is analogous to the hDcp2 decapping enzyme and another decapping inhibitor, PABP. All three proteins can only bind the cap when it is linked to an RNA moiety, but they do not recognize the cap structure alone (Figure 2; Khanna and Kiledjian, 2004; Piccirillo et al., 2003). All three appear to utilize a dual recognition mechanism to simultaneously bind the cap and RNA. We propose that this mode of cap recognition defines a novel class of noncanonical cap-binding proteins that bind the 5′ end of capped RNA. A similar cap-binding property has also been reported for the YB-1 protein (Evdokimova et al., 2001), suggesting additional members of this growing noncanonical cap-binding protein family may exist. Future structural analysis of these proteins with capped oligonucleotide will better illustrate their dual binding capability.
Examination of Figure 5 reveals that VCX-A might also influence 3′ to 5′ exonuclease activity. Curiously, in the decapping reactions using P50 pellet fraction, VCX-A reproducibly inhibited both the m7GDP hDcp2 decapping product and generation of cap structure (Figure 5B). The m7GpppG cap structure is generated by 3′ to 5′ exonuclease activity (Wang et al., 1999). These data suggest that the reduced level of exonuclease activity present in the P50 fraction is efficiently inhibited by VCX-A. It is not clear whether the observed inhibition is a simple consequence of the general RNA-binding property of VCX-A or a more active role. In support of the latter hypothesis, a similar inhibition was not detected with the αCP1 control protein (Figure 5B) that has the potential of impeding 3′ to 5′ exonuclease activity (Rodgers et al., 2002). However, it should be noted that a similar inhibition was not detected in the presence of the more robust exonuclease activity present in the soluble cytoplasmic fraction (Figure 5C). Further studies are necessary to determine whether VCX-A has functions in addition to the inhibition of decapping.
Although our results demonstrate that VCX can function to inhibit hDcp2 decapping, it is not the only decapping inhibitor in cells. In addition to their tissue-restricted expression, VCX/Y genes are only present in higher primates (Fukami et al., 2000). We have observed that extracts from primary human tissues, mouse cells, and yeast cells, which should be devoid of VCX proteins, are all capable of inhibiting recombinant hDcp2 decapping (X.J. and M.K., unpublished data). These data suggest that there are additional and most likely multiple decapping inhibitors distinct from VCX/Y that are present in eukaryotic cells potentially controlling decapping.
Although the VCX family members were initially identified as testis-specific genes (Fukami et al., 2000; Lahn and Page, 2000), VCX-A nevertheless correlated with MRX. Analysis of 15 individuals with complex deletions on the terminal end of the X chromosome identified a 15 kb region encompassing the VCX-A gene as the defining interval for the MRX phenotype (Fukami et al., 2000). Curiously, despite the high degree of similarity between the different VCX genes, only VCX-A was found to be a critical determinant of MRX while absence of VCX-B, -B1, and -C did not correlate with MRX (Fukami et al., 2000). These data indicate that VCX-A could uniquely regulate a particular substrate that is critical for cognitive function. Consistent with a role for VCX-A in MRX, a more recent study of four unrelated X-linked ichthyosis patients with MRX found that all four patients contained a disrupted VCX-A gene (Van Esch et al., 2005). However, it is likely that the potential role of the VCX proteins in MRX is more complex and involves a cumulative VCX/Y dosage effect rather than one particular gene (Van Esch et al., 2005). Consistent with this hypothesis, another study reported two siblings with a deletion of both VCX-A and VCX-B1 with only one containing an MRX phenotype (Lesca et al., 2005).
It is not obvious how the VCX/Y proteins could be involved in MRX, considering they are only expressed in male germ cells. It is possible that the VCX genes could be expressed in nongerm cells at specific times in development critical for cognitive development. Our ability to reverse transcribe and clone the VCX-A cDNA from K562 cells indicates that VCX-A could at least be aberrantly expressed in some cell lines. Furthermore, the underlying mechanism of how the VCX/Y protein could influence cognitive function is not clear, although our data indicate it could be by the regulation of mRNA decapping and stability. However, the cap-binding property of VCX-A is also suggestive of a potential regulatory role for VCX-A in mRNA translation. Immunofluorescence of epitope-tagged VCX-A indicates that this protein is localized primarily in the nucleus (Zou et al., 2003). Considering that the decapping enzymes can also exist in the nucleus (Liu et al., 2004), whether the primary function of VCX-A on the decapping enzyme is in the nucleus or cytoplasm remains to be determined. More detailed analysis of the individual endogenous VCX/Y protein localization in germ cells and development will begin to address a potential role of the VCX/Y proteins in mRNA decapping and cognitive function.
Experimental Procedures
Plasmids and Expression of Recombinant Protein
The pET28a-hDcp2, pET28a-hDcp21–257, and pET28a-hDcp294–257 plasmids have previously been described (Piccirillo et al., 2003; Wang et al., 2002), as has the pSV2ALΔ5′ plasmid (de Wet et al., 1987). The coding region of VCX-A was cloned from an oligo d(T)-primed reverse-transcribed product of human K562 cell total RNA by PCR amplification with a 5′ primer (5′-GAATTCGGATCCATGA GTCCAAAGCCGAGAGCCTC-3′) and a 3′ primer (5′-CTCGAGTCGA CCTACACACTCGGTAGTTCTTCC-3′). The PCR product was inserted into the BamHI and XhoI sites of pGEX-6p-1 (GE Healthcare, Piscataway, New Jersey) and pET28a (Novagen, San Diego, California) vectors to generate pGEX-6p-VCX-A and pET28a-VCX-A, respectively. All VCX-A truncations were generated by PCR and subcloned into pET28a vector at the same sites. Primers can be provided upon request. pCMV-VCX-A, pCMV-hDcp2, and pCMV-hDcp294–257 were generated by excision of the insert with BamHI and XhoI from the corresponding pET28a cDNA plasmids and insertion into the same sites in the pCMV-Tag 3B Myc-tag vector (Stratagene, La Jolla, California). pcDNA3-Flag-VCX-A was generated by replacing the hDcp2 cDNA from pcDNA3-Flag-hDcp2 plasmid (kindly provided by J. Lykke-Andersen, University of Colorado; Lykke-Andersen, 2002) at the BamHI and ApaI sites with the VCX-A open reading frame. pIRESpuro3-VCX-A was constructed by excision of the VCX-A open reading frame from pCMV-VCX-A with SacII and XhoI. The ends of the VCX-A fragment were filled and inserted into the EcoRV and AgeI sites within the pIRESpuro3 plasmid (Clontech, Mountain View, California).
GST fusion protein and His-tagged proteins were expressed and purified according to the manufacturer (GE Healthcare, Piscataway, New Jersey; and Novagen, San Diego, California, respectively), except that a final concentration of 300 mM NaCl and 300 mM urea was included in the wash buffer.
Cell Transfection and Extract Preparation
Transfection into human HeLa epithelial carcinoma cells and 293T embryonic kidney cells was carried out with Lipofectamine 2000 reagent (Invitrogen) according to the protocol of the manufacturer. 293T cells were stably transformed with the plasmid pIRESpuro3-VCX-A expressing the VCX-A gene and the puromycin resistance gene and selected with puromycin (Sigma-Aldrich, St. Louis, Missouri) at a concentration of 3 μg/ml. A clonal cell line expressing VCX-A was subsequently isolated (293T-VCX-A). 32P-cap-labeled pcP RNA was transfected into 293T-VCX-A using TransMessenger transfection reagent (QIAGEN, Valencia, California) according to the manufacturer’s suggested protocol. Thirty minutes posttransfection, cells were collected with two washes in PBS and resuspended in PBS containing 200 U/ml micrococcal nuclease (USB Corporation, Cleveland, Ohio) and 5 mM CaCl2 for 15 min at 37°C to degrade residual RNA remaining on the cell surface. Following the incubation, cells were transferred to culture media and an aliquot was removed for time zero; subsequent aliquots were isolated at the indicated time points. RNA was isolated using Trizol Reagent (Invitrogen, Carlsbad, California) and resolved by denaturing polyacrylamide gel electrophoresis. For mRNA half-life determinations, cells were treated with 10 μg/ml actinomycin D (Sigma-Aldrich, St. Louis, Missouri) 16 hr posttransfection and RNA was isolated and treated with RQ1 DNase (Promega, Madison, Wisconsin) according to the manufacturer prior to use for reverse transcription reactions.
For protein extract preparations, cells were harvested 24 hr posttransfection, and cell extracts were prepared by sonication as previously described (Wang et al., 1999). K562 cell extract was prepared by low salt buffer lysis (Wang et al., 1999), and the P50 fraction was derived from the pellet of cytoplasmic extract centrifuged at 50,000 × g. All K562 extract-derived decapping assays in Figure 5 were carried out with freshly prepared extract containing 10 mM Tris (pH 7.5), 100 mM KAc, 2 mM MgAc, and 1 mM DTT. Decapping activity was lost upon freeze thawing.
RNA Production
The pcDNA3 polylinker was amplified by PCR with the SP6 promoter primer and a T7 promoter primer containing 16 cytosines at the 5′ end as previously described (Wang et al., 2002), and was used as template to transcribe the pcP RNA with SP6 RNA polymerase (Promega, Madison, Wisconsin). The resulting RNA contains 16 guanosines at the 3′ end to stabilize the 3′ termini. 32P-cap-labeled pcP RNAs were generated with the vaccinia virus capping enzyme as described previously (Wang et al., 1999) to generate a transcript with the label at the first phosphate following the methylated guanosine (m7G*pppN). Uniform-labeled pcP RNAs or 5′ end-labeled pcP RNAs were generated similarly to pcP RNA in in vitro transcription, except that α-32P-UTP or γ-32P-GTP were included in the respective reactions. The 32P-labeled cap analog was generated by hydrolysis of 32P-cap-labeled RNA with 1 U of Nuclease P1 (Roche Diagnostics, Pleasanton, California) for 1 hr at 37°C as described (Wang and Kiledjian, 2001). All RNAs were gel purified by denaturing polyacrylamide gel electrophoresis as described (Wang and Kiledjian, 2001).
In Vitro Decapping Assays
The indicated amount of extract or recombinant protein was incubated with 32P-cap-labeled pcP RNA in decapping buffer as previously described (Piccirillo et al., 2003). Cold cap analog (100 μM) was included in the decapping assays when cell extract was used to inhibit endogenous DcpS decapping activity. Decapping products were resolved by polyethyleneimine-cellulose TLC (PEI-TLC) plates developed in either 0.75 M LiCl or 0.45 M (NH4)2SO4 at room temperature (Liu et al., 2002; Wang et al., 2002), as indicated in the figure legends, and exposed to a PhosphorImager. Quantifications were carried out using a Molecular Dynamics PhosphorImager (Storm860) using ImageQuant-5 software.
In-Gel Decapping Assay
The in-gel decapping assay was carried out with 32P-cap-labeled pcP embedded in the gel as previously described (Wang et al., 2002) with 1 μg of each indicated recombinant protein with or without 20 μg of K562 cell extract.
Northwestern RNA-Binding Assays and Ultraviolet Crosslinking
Northwestern analysis was carried out as described previously (Kiledjian et al., 1999; Piccirillo et al., 2003). Ten picomoles of GST-VCX-A, His-VCX-A, or histidine-tagged truncated VCX-A proteins was resolved on 15% SDS-PAGE and transferred onto nitrocellulose membrane. The proteins were subsequently renatured and probed with 32P-uniform-labeled pcP RNA lacking a cap. Following three washes, the membrane was exposed to X-ray film and detected by autoradiography.
Ultraviolet (UV) crosslinking assays were carried out as described previously (Khanna and Kiledjian, 2004) with 10 pmol of 5′ end 32P-labeled pcP RNA with or without a cap or 32P-labeled cap structure.
GST Copurifications and Coimmunoprecipitations
Purified GST fusion protein (20 μg) was immobilized on glutathione-agarose beads (50 μl) in PBS and washed with PBS/0.25% Triton X-100 before incubation with 2.5 mg of 293T cell extracts or 35S-methionine-labeled in vitro-translated protein (5 μl) at 4°C for 4 hr in PBS on a rocker. After extensive washing with PBS/0.05% NP-40, bound proteins were released with SDS-PAGE gel loading buffer and fractionated by SDS-PAGE for western analysis. 35S-labeled proteins were detected by autoradiography.
Anti-Flag monoclonal antibody M2 agarose beads (Sigma-Aldrich, St. Louis, Missouri) were used for coimmunoprecipitation assays. Flag-VCX-A was transiently overexpressed in 293T cells. Anti-Flag M2 agarose beads (30 μl) were incubated with 2.5 mg cell extract expressing Flag-VCX-A at 4°C overnight in PBS with moderate shaking. After extensive washing with PBS/0.05% NP-40, bound proteins were eluted with protein elution buffer (0.1 M glycine [pH 3.5] and 100 mM NaCl) and separated by SDS-PAGE gel for immunoblotting with affinity-purified polyclonal hDcp2 as in Wang et al. (Wang et al., 2002).
Quantitative Real-Time PCR
mRNA expression levels were quantified from oligo d(T) M-MLV Reverse Transcriptase (Invitrogen) reverse-transcribed cDNA by real-time PCR using SYBR green PCR core reagent (Applied Biosystems, Foster City, California). Luciferase mRNA abundance was quantitated using the standard curve method according to the recommendations of the manufacturer. Values were normalized to the stable glyceraldehyde-3-phosphate dehydrogenase (GAPDH). Each gene was amplified using the appropriate specific primers: forward primer 5′-ATGACAACAGCCTCAAGATCA-3′ and reverse primer 5′-TCCTTCCACGATACCAAAGTT-3′ for the GAPDH cDNA; and forward primer 5′-CTCACTGAGACTACATCAGC-3′ and reverse primer 5′-TCCAGATCCACAACCTTCGC-3′ for the luciferase cDNA. Real-time PCR was carried out with an ABI Prism 7900HT sequence detection system, and the specificity of the amplified PCR products was assessed by a melting curve analysis after the last cycle by the manufacturer’s suggested program.
Acknowledgments
We thank G. Rappold (University of Heidelberg), R. Parker (University of Arizona), and members of the Kiledjian lab for helpful discussions. X.J. was supported in part by the Cooley’s Anemia Foundation, and this work was supported by NIH grant GM67005 to M.K.
Footnotes
Supplemental Data: Supplemental Data include one figure, Supplemental Experimental Procedures, and supplemental text and can be found with this article online at http://www.molecule.org/cgi/content/full/24/5/713/DC1/.
References
- Caponigro G, Parker R. Multiple functions for the poly(A)-binding protein in mRNA decapping and deadenylation in yeast. Genes Dev. 1995;9:2421–2432. doi: 10.1101/gad.9.19.2421. [DOI] [PubMed] [Google Scholar]
- Coller J, Parker R. Eukaryotic mRNA decapping. Annu Rev Biochem. 2004;73:861–890. doi: 10.1146/annurev.biochem.73.011303.074032. [DOI] [PubMed] [Google Scholar]
- de Wet JR, Wood KV, DeLuca M, Helinski DR, Subramani S. Firefly luciferase gene: structure and expression in mammalian cells. Mol Cell Biol. 1987;7:725–737. doi: 10.1128/mcb.7.2.725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evdokimova V, Ruzanov P, Imataka H, Raught B, Svitkin Y, Ovchinnikov LP, Sonenberg N. The major mRNA-associated protein YB-1 is a potent 5′ cap-dependent mRNA stabilizer. EMBO J. 2001;20:5491–5502. doi: 10.1093/emboj/20.19.5491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenger-Gron M, Fillman C, Norrild B, Lykke-Andersen J. Multiple processing body factors and the ARE binding protein TTP activate mRNA decapping. Mol Cell. 2005;20:905–915. doi: 10.1016/j.molcel.2005.10.031. [DOI] [PubMed] [Google Scholar]
- Fukami M, Kirsch S, Schiller S, Richter A, Benes V, Franco B, Muroya K, Rao E, Merker S, Niesler B, et al. A member of a gene family on Xp22.3, VCX-A, is deleted in patients with X-linked nonspecific mental retardation. Am J Hum Genet. 2000;67:563–573. doi: 10.1086/303047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao M, Wilusz CJ, Peltz SW, Wilusz J. A novel mRNA-decapping activity in HeLa cytoplasmic extracts is regulated by AU-rich elements. EMBO J. 2001;20:1134–1143. doi: 10.1093/emboj/20.5.1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grudzien E, Kalek M, Jemielity J, Darzynkiewicz E, Rhoads RE. Differential inhibition of mRNA degradation pathways by novel cap analogs. J Biol Chem. 2006;281:1857–1867. doi: 10.1074/jbc.M509121200. Published online October 28, 2005. [DOI] [PubMed] [Google Scholar]
- Khanna R, Kiledjian M. Poly(A)-binding-protein-mediated regulation of hDcp2 decapping in vitro. EMBO J. 2004;23:1968–1976. doi: 10.1038/sj.emboj.7600213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiledjian M, Day N, Trifillis P. Purification and RNA binding properties of the polycytidylate-binding proteins alphaCP1 and alphaCP2. Methods. 1999;17:84–91. doi: 10.1006/meth.1998.0710. [DOI] [PubMed] [Google Scholar]
- Kiledjian M, Dreyfuss G. Primary structure and binding activity of the hnRNP U protein: binding RNA through RGG box. EMBO J. 1992;11:2655–2664. doi: 10.1002/j.1460-2075.1992.tb05331.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahn BT, Page DC. A human sex-chromosomal gene family expressed in male germ cells and encoding variably charged proteins. Hum Mol Genet. 2000;9:311–319. doi: 10.1093/hmg/9.2.311. [DOI] [PubMed] [Google Scholar]
- Lesca G, Sinilnikova O, Theuil G, Blanc J, Edery P, Till M. Xp22.3 microdeletion including VCX-A and VCX-B1 genes in an X-linked ichthyosis family: no difference in deletion size for patients with and without mental retardation. Clin Genet. 2005;67:367–368. doi: 10.1111/j.1399-0004.2005.00417.x. [DOI] [PubMed] [Google Scholar]
- Liu H, Rodgers ND, Jiao X, Kiledjian M. The scavenger mRNA decapping enzyme DcpS is a member of the HIT family of pyrophosphatases. EMBO J. 2002;21:4699–4708. doi: 10.1093/emboj/cdf448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu SW, Jiao X, Liu H, Gu M, Lima CD, Kiledjian M. Functional analysis of mRNA scavenger decapping enzymes. RNA. 2004;10:1412–1422. doi: 10.1261/rna.7660804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lykke-Andersen J. Identification of a human decapping complex associated with hUpf proteins in nonsense-mediated decay. Mol Cell Biol. 2002;22:8114–8121. doi: 10.1128/MCB.22.23.8114-8121.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lykke-Andersen J, Wagner E. Recruitment and activation of mRNA decay enzymes by two ARE-mediated decay activation domains in the proteins TTP and BRF-1. Genes Dev. 2005;19:351–361. doi: 10.1101/gad.1282305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piccirillo C, Khanna R, Kiledjian M. Functional characterization of the mammalian mRNA decapping enzyme hDcp2. RNA. 2003;9:1138–1147. doi: 10.1261/rna.5690503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez CV, Vilela C, Berthelot K, McCarthy JE. Modulation of eukaryotic mRNA stability via the cap-binding translation complex eIF4F. J Mol Biol. 2002;318:951–962. doi: 10.1016/S0022-2836(02)00162-6. [DOI] [PubMed] [Google Scholar]
- Rodgers ND, Wang Z, Kiledjian M. Regulated alpha-globin mRNA decay is a cytoplasmic event proceeding through 3′-to-5′ exosome-dependent decapping. RNA. 2002;8:1526–1537. [PMC free article] [PubMed] [Google Scholar]
- Schwartz DC, Parker R. Mutations in translation initiation factors lead to increased rates of deadenylation and decapping of mRNAs in Saccharomyces cerevisiae. Mol Cell Biol. 1999;19:5247–5256. doi: 10.1128/mcb.19.8.5247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz DC, Parker R. mRNA decapping in yeast requires dissociation of the cap binding protein, eukaryotic translation initiation factor 4E. Mol Cell Biol. 2000;20:7933–7942. doi: 10.1128/mcb.20.21.7933-7942.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- She M, Decker CJ, Sundramurthy K, Liu Y, Chen N, Parker R, Song H. Crystal structure of Dcp1p and its functional implications in mRNA decapping. Nat Struct Mol Biol. 2004;11:249–256. doi: 10.1038/nsmb730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon E, Camier S, Sèraphin B. New insights into the control of mRNA decapping. Trends Biochem Sci. 2006;31:241–243. doi: 10.1016/j.tibs.2006.03.001. Published online March 31, 2006. [DOI] [PubMed] [Google Scholar]
- Steiger M, Carr-Schmid A, Schwartz DC, Kiledjian M, Parker R. Analysis of recombinant yeast decapping enzyme. RNA. 2003;9:231–238. doi: 10.1261/rna.2151403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoecklin G, Mayo T, Anderson P. ARE-mRNA degradation requires the 5′-3′ decay pathway. EMBO Rep. 2006;7:72–77. doi: 10.1038/sj.embor.7400572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Dijk E, Le Hir H, Sèraphin B. DcpS can act in the 5′-3′ mRNA decay pathway in addition to the 3′-5′ pathway. Proc Natl Acad Sci USA. 2003;100:12081–12086. doi: 10.1073/pnas.1635192100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Esch H, Hollanders K, Badisco L, Melotte C, Van Hummelen P, Vermeesch JR, Devriendt K, Fryns JP, Marynen P, Froyen G. Deletion of VCX-A due to NAHR plays a major role in the occurrence of mental retardation in patients with X-linked ichthyosis. Hum Mol Genet. 2005;14:1795–1803. doi: 10.1093/hmg/ddi186. [DOI] [PubMed] [Google Scholar]
- Vasudevan S, Peltz SW. Regulated ARE-mediated mRNA decay in Saccharomyces cerevisiae. Mol Cell. 2001;7:1191–1200. doi: 10.1016/s1097-2765(01)00279-9. [DOI] [PubMed] [Google Scholar]
- Wang Z, Day N, Trifillis P, Kiledjian M. An mRNA stability complex functions with poly(A)-binding protein to stabilize mRNA in vitro. Mol Cell Biol. 1999;19:4552–4560. doi: 10.1128/mcb.19.7.4552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Jiao X, Carr-Schmid A, Kiledjian M. The hDcp2 protein is a mammalian mRNA decapping enzyme. Proc Natl Acad Sci USA. 2002;99:12663–12668. doi: 10.1073/pnas.192445599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Kiledjian M. Functional link between the mammalian exosome and mRNA decapping. Cell. 2001;107:751–762. doi: 10.1016/s0092-8674(01)00592-x. [DOI] [PubMed] [Google Scholar]
- Wells SE, Hillner PE, Vale RD, Sachs AB. Circularization of mRNA by eukaryotic translation initiation factors. Mol Cell. 1998;2:135–140. doi: 10.1016/s1097-2765(00)80122-7. [DOI] [PubMed] [Google Scholar]
- Wilusz CJ, Gao M, Jones CL, Wilusz J, Peltz SW. Poly(A)-binding proteins regulate both mRNA deadenylation and decapping in yeast cytoplasmic extracts. RNA. 2001a;7:1416–1424. [PMC free article] [PubMed] [Google Scholar]
- Wilusz CJ, Wormington M, Peltz SW. The cap-to-tail guide to mRNA turnover. Nat Rev Mol Cell Biol. 2001b;2:237–246. doi: 10.1038/35067025. [DOI] [PubMed] [Google Scholar]
- Zou SW, Zhang JC, Zhang XD, Miao SY, Zong SD, Sheng Q, Wang LF. Expression and localization of VCX/Y proteins and their possible involvement in regulation of ribosome assembly during spermatogenesis. Cell Res. 2003;13:171–177. doi: 10.1038/sj.cr.7290161. [DOI] [PubMed] [Google Scholar]