

Abstract
mus301 was identified independently in two genetic screens, one for mutants hypersensitive to chemical mutagens and another for maternal mutants with eggshell defects. mus301 is required for the proper specification of the oocyte and for progression through meiosis in the Drosophila ovary. We have cloned mus301 and show that it is a member of the Mus308 subfamily of ATP-dependent helicases and the closest homolog of human and mouse HEL308. Functional analyses demonstrate that Mus301 is involved in chromosome segregation in meiosis and in the repair of double-strand-DNA breaks in both meiotic and mitotic cells. Most of the oogenesis defects of mus301 mutants are suppressed by mutants in the checkpoint kinase Mei41 and in MeiW68, the Spo11 homolog that is thought to generate the dsDNA breaks that initiate recombination, indicating that these phenotypes are caused by activation of the DNA damage checkpoint in response to unrepaired Mei-W68-induced dsDNA breaks. However, neither mei-W68 nor mei-41 rescue the defects in oocyte specification of mus301 mutants, suggesting that this helicase has another function in oocyte selection that is independent from its role in meiotic recombination.
CELLS need to transmit an intact genome to ensure proper development, survival, and reproduction. The accurate replication of their genome requires both monitoring of DNA integrity and repairing of damages to DNA. Double-strand breaks (DSBs) in the DNA arise spontaneously during development or can be produced by ionizing radiations or by mechanical stress. The repair of DSBs is essential for genome stability and tumor suppression, as interactions between the ends of different DSBs can give rise to tumorigenic chromosome translocations (Elliot and Jasin 2002; Adams et al. 2003; Shivji and Venkitaraman 2004). In eukaryotes, checkpoints are in place to monitor the integrity of the DNA and to avoid the propagation of genomic defects. These checkpoints ensure that a subsequent step in the cell cycle is not initiated in the presence of damaged DNA, allowing additional time for the cell to correct the damage and stimulate the activity of highly conserved repair mechanisms. The DNA damage response in normal cells involves a series of signaling events that include sensors, transducers, and effectors. Central components of these checkpoints in mammals are the ATM/ATR family of phosphatidylinositol-3-OH-kinase-like serine/threonine kinases and their identified targets the checkpoint-1 (Chk1) and checkpoint-2 (Chk2) kinases (Kurz and Lees-Miller 2004). Homologs of these exist in other eukaryotes where they play similar roles.
Recombination normally occurs during prophase I of meiosis and plays a critical role in homolog segregation and in the formation of viable gametes. Current models for meiotic recombination are based on the double-strand break repair model (Szostak et al. 1983; Blanton and Sekelsky 2004). In budding yeast, recombination starts with the formation of double-strand DNA (dsDNA) breaks catalyzed by the type II DNA topoisomerase Spo11, the homolog of mei-W68 in Drosophila (Cao et al. 1990; McKim and Hayashi-Hagihara 1998). Since the lack of function of mei-W68 abolishes meiotic crossing over and gene conversion, it is likely that recombination in the Drosophila ovary is also initiated by the occurrence of DSBs (McKim et al. 1998). In Drosophila, the repair of these dsDNA breaks is monitored by a meiotic checkpoint that involves the activity of the Mei-41 kinase (ATR homolog) (Hari et al. 1995) and of the Drosophila Chk2 homolog maternal nuclear kinase (mnk) (Oishi et al. 1998; Abdu et al. 2002). Its activation results in the modification of two effector proteins, Vasa and Wee1. As a consequence, the meiotic cell cycle is regulated and the efficient translation of gurken (grk) mRNA is prevented (Ghabrial and Schüpbach 1999; Abdu et al. 2002). Since Grk is required for the proper establishment of axial polarity in oogenesis, mutants unable to repair DSBs during recombination lay eggs harboring polarity defects. In addition, oocytes with unrepaired DSBs show an abnormal chromatin condensation in the oocyte nucleus, a phenotype that most probably reflects an arrest in meiotic prophase I (González-Reyes et al. 1997). Interestingly, since both the polarity defects and the abnormal nuclear morphology are rescued in double mutants with mei-W68 and mei-41, progression through meiosis is coupled to specific patterning events during oogenesis (González-Reyes 1999).
Several genes in Drosophila with a known role in progression through meiosis and in early events of oocyte selection and patterning have been identified. Most of the spindle-group of genes were initially identified as maternal-effect mutants with altered eggshell morphology (Tearle and Nüsslein-Volhard 1987; Schüpbach and Wieschaus 1989). Members of this group include spindle (spn)-A, spn-B, spn-C, spn-D, and spn-E and vasa, maelstrom, aubergine, and okra. In spite of the pattern defects and the abnormal nuclear morphology of vasa, aubergine, and maelstrom mutants, these genes seem to be involved only indirectly in meiotic progression. aubergine and maelstrom are required for translational silencing mediated by RNA interference and microRNAs (Wilson et al. 1996; Kennerdell et al. 2002; Findley et al. 2003), whereas vasa is a target of the mei-41-dependent checkpoint pathway that occurs in response to dsDNA breaks and acts as a translational regulator of several maternally provided mRNAs, including grk mRNA (Styhler et al. 1998; Tomancak et al. 1998; Ghabrial and Schüpbach 1999).
A detailed analysis of the mutant phenotypes of spn-A, spn-B, spn-C, spn-D; and spn-E demonstrated the involvement of these genes in the four symmetry-breaking steps that lead to the polarization of the two main body axes of Drosophila (González-Reyes et al. 1997). The loss of function of the spindle genes in the germline produces defects consistent with their role in the selection of the oocyte, the posterior positioning of the oocyte within the egg chamber, and the polarization of the anterior–posterior and dorsal–ventral axes of the follicle (Gillespie and Berg 1995; González-Reyes et al. 1997; Ghabrial et al. 1998; Abdu et al. 2003; Staeva-Vieira et al. 2003). The cloning of okra, spn-A, spn-B, and spn-D established a clear link between these genes and dsDNA break repair. They are all members of the Rad52 epistasis group, a series of genes isolated originally in Saccharomyces cerevisiae because of their role in the response to irradiation damage and that were subsequently found to be deficient in meiotic recombination as well (Symington 2002; Richardson et al. 2004). okra encodes the Drosophila homolog of the yeast DNA-repair protein Rad54, a chromatin-remodeling dsDNA-dependent ATPase with a known function in DSB metabolism; Spn-A is homologous to yeast Rad51, a protein with an essential role in DNA repair and meiotic checkpoint control; spn-B is another Drosophila homolog of Rad51 and has been shown to participate in the repair of meiotic DSBs; finally, spn-D encodes a Rad51C-like protein required exclusively during meiosis. Furthermore, since the patterning defects of okra, spn-A, spn-B, and spn-D mutants can be suppressed by mutations in mei-W68 and in mei-41, the primary defect in these spn mutations seems to be a failure to repair DSBs (Ghabrial et al. 1998; Abdu et al. 2003; Staeva-Vieira et al. 2003; Romeijn et al. 2005).
Mutations in the mutagen-sensitive 301 (mus301) gene were recovered in a screen for mutants hypersensitive to chemical mutagens (Boyd et al. 1981). During the analysis of the contribution of spn-C to the repair of DSBs in the female germline, it was found that spn-C was allelic to mus301 (Ghabrial and Schüpbach 1999). (We have adopted the nomenclature proposed for this locus and its alleles by FlyBase; therefore, we have renamed the spn-C gene mus301.) Since mus301 mutants are associated with high sensitivity to chemical damage and regulate progression through meiosis via the mei-41 checkpoint, it is likely that mus301 participates in the repair of DSBs in mitotic and meiotic cells. Here we report the identification of mus301 as a member of the Mus308 subfamily of ATP-dependent helicases and demonstrate its involvement in DSB repair. We also show that the activation of the DNA damage checkpoint triggered in mus301 mutants requires the function of the checkpoint-2 kinase Mnk. Finally, we find that mus301 has a role in oocyte selection independent of its requirement for mei-W68-induced DSB repair and unrelated to the activation of the mei-41 DNA damage meiotic checkpoint.
MATERIALS AND METHODS
Fly stocks:
Following the nomenclature by FlyBase, the spn-C094, spn-C422, and spn-C660 alleles have been renamed mus301094, mus301422, and mus301660, respectively. These alleles had been described as antimorphs because their phenotype in homozygous or trans-heterozygous conditions is much stronger than any in trans to a deficiency (González-Reyes et al. 1997). Throughout the course of this work, it was discovered that the original mus301094, mus301422, and mus301660 chromosomes carried an enhancer of the spindle phenotype that mapped to the right of mus301. Cleaned-up mus301094, mus301422, or mus301660 chromosomes were obtained by recombining the mutations onto a new background and were used in our phenotypic analyses. Since all cleaned-up chromosomes in trans to each other or to a deficiency show a similar penetrance of the mutant phenotypes, they all classify as genetic nulls. The mus301D1, mus301D2, and mus301D4 mutant alleles have been reported elsewhere (Boyd et al. 1981). mus3012255, mus3013198, and mus3014875 are from the Zuker EMS collection (Koundakjian et al. 2004; Laurençon et al. 2004) and were kindly provided for by A. Laurençon. spn-D349, spn-D150, spn-E616, and spn-E653 are described in detail elsewhere (Tearle and Nüsslein-Volhard 1987; González-Reyes et al. 1997). mei-W681 is a strong allele that eliminates meiotic recombination (McKim et al. 1998). mei-41D3, grpfsA4, and mnkP6 are amorphic alleles (Fogarty et al. 1997; Sibon et al. 1999; Brodsky et al. 2004). Df(2R)LL5 is a deficiency for mei-W68. Df(2L)pr65 is a deficiency for mnk. Df(3L)66C-G28 and Df(3L)ZP3 (66A9-12; 66B5-11, a gift from P. Maróy) are deficiencies that uncover mus301.
Rescue construct:
An ∼11.2-kb SmaI fragment from cosmid 195B2 (European Drosophila Genome Project numbering) was cloned into pBluescript and then digested with XhoI and religated to give an ∼6.2-kb XhoI–SmaI fragment in pBluescript. This fragment contained the ∼4.6 kb of CG7972 with ∼700 bp upstream of the transcriptional start site and ∼900 bp downstream of the poly(A) site. This ∼6.2-kb fragment was cloned directionally into a P-element transformation vector to test for rescue of mus301094 mutants.
Sequencing of mus301 mutant alleles:
Twelve primer pairs were designed to enable PCR amplification of the gene plus ∼200 bp upstream of the transcriptional start and ∼400 bp after the translational stop. Genomic DNA was prepared from homozygous or hemizygous mutant females and used as template in PCR reactions that, after purification, were sequenced directly using the PCR primers. At least two independent PCR reactions were sequenced for each primer pair and both strands were sequenced for each of the mutations found.
MMS mutagenesis:
MMS mutagenesis was performed as described (Ghabrial et al. 1998). Sensitivity to MMS was expressed as a fraction of the percentage expected in the treated vial vs. the percentage expected in the control:
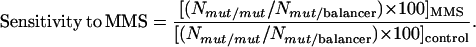 |
X chromosome nondisjunction:
To determine the frequency of X chromosome nondisjunction, yw;+/+ males were crossed to +/w; mus301/Df virgin females and F1 larvae were scored for the presence of y. If X chromosome nondisjunction occurs, exceptional yellow X0 larvae will be obtained, having inherited the X chromosome from the male. The percentage of nondisjunction was calculated as
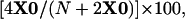 |
where X0 is the number of y larvae obtained and N is the total number of larvae scored. (The number of X0 larvae is multiplied by four to account for the Y0, the XXX, and the XXY progeny that are lethal or cannot be scored. Twice the number of X0 larvae are added to N to account for the lethal Y0 and XXX progeny that would not be counted as part of N.)
Staining procedures:
Antibody, DNA, and rhodamine–phalloidin stainings were performed according to standard procedures. Detailed protocols are available upon request. DNA was counterstained with DAPI (5 mg/ml; Sigma, St. Louis). Antibodies were used at the following concentrations: mouse monoclonal α-Grk (Ghabrial and Schüpbach 1999), 1/10; rabbit α-γHIS2AV, 1/500 (a gift from K. McKim); mouse α-Orb monoclonal antibodies 4H8 and 6H4 (Lantz et al. 1994) from the Developmental Studies Hybridoma Bank (University of Iowa), 1/200 each; guinea pig α-C(3)G (Page and Hawley 2001), 1/500; FITC, Cy2-, Cy3-, and Cy5-conjugated secondary antibodies (Jackson Laboratories, West Grove, PA), 1/200.
RESULTS
mus301 is required for oocyte selection and progression through meiosis:
The predominant phenotype of mus301 mutant females is the production of ventralized egg shells, with phenotypes ranging from fused dorsal appendages to fully ventralized eggs with no dorsal appendages (Figure 1B; Table 1 shows a numerical representation of the mutant phenotypes in Figure 1). This ventralization of the egg shell has been shown to be a consequence of defects in the translation of grk mRNA (González-Reyes et al. 1997; Ghabrial and Schüpbach 1999). In fact, a significant proportion of mutant mus301 egg chambers from stage (S) 8 to S10 of oogenesis present a strong reduction or complete absence of Grk protein (Figure 1D). The early and late expression of Grk protein appear to be regulated independently, because early Grk expression is not disrupted in mus301 mutant females (data not shown). This is also the case in spn-B mutants (Ghabrial et al. 1998). In a low percentage of mus301 mutant egg chambers, the oocyte is not localized posterior to the nurse cells (Figure 1F), most probably as a consequence of a delay in oocyte selection (González-Reyes and St Johnston 1998). In addition, a low proportion of mutant oocytes fail to reach a wild-type size (Figure 1H), indicating that the defect in oocyte selection observed in mus301 mutants may prevent the normal growth of this cell by affecting the directional transport of cytoplasmic constituents from the nurse cells into the oocyte. The most penetrant phenotype of mus301 mutant egg chambers is the lack of a karyosome, a solid sphere of compacted chromatin. Nearly all of the mutant oocytes fail to compact their chromatin into a karyosome. Instead, the chromatin fibers of mutant oocytes adopt a fragmented, thread-like appearance (Figure 1J). Since the fragmented karyosome phenotype is rescued by mutations that prevent recombination or the activation of the meiotic DNA damage checkpoint, it has been suggested that mus301 plays a role in the regulation of meiosis (Ghabrial and Schüpbach 1999).
Figure 1.—

Phenotypes displayed by mus301 mutants. (A) Wild-type egg shell. (B) Fully ventralized egg shell. (C) Wild-type S9 egg chamber showing Gurken protein localization. (D) Mutant egg chamber stained and imaged under the same conditions as in C. No Gurken can be detected. (E and G) Wild-type egg chambers stained with rhodamine–phalloidin to visualize the morphology of cells. Both oocytes (oo) are located at the posterior of the egg chamber. (F and H) Mutant egg chambers labeled with rhodamine–phalloidin showing a misplaced oocyte (F) and a small, misplaced oocyte (H). (I) Wild-type follicle with the oocyte's chromatin condensed into a karyosome (open arrowhead). (J) Mutant karyosome. (B and D) mus301094/Df(3L)66C-G28. (F) mus301422/mus301660. (H) mus301D1/mus301D2. (J) mus301660/Df(3L)66C-G28.
TABLE 1.
Phenotypes of mus301 eggs and egg chambers
| Maternal genotype | % egg chambers with a mutant karyosome (n)a | % egg chambers with mutant Gurken levels (n)b | % egg chambers with misplaced oocytes (n) | % ventralized eggs (n) | % egg chambers with small oocytes (n) |
|---|---|---|---|---|---|
| w | 0 (56) | 2 (25) | 0 (151) | 0 (123) | 0 (151) |
| mus301094/Df(3L)66C-G28 | 100 (161) | ND | 2 (296) | 49 (553) | ND |
| mus301422/Df(3L)66C-G28 | 100 (41) | 30 (86) | 1 (77) | 37 (921) | ND |
| mus301660/Df(3L)66C-G28 | 100 (167) | 20 (95) | 4 (259) | 30 (1251) | 4 (123) |
| mus301D1/Df(3L)66C-G28 | 100 (122) | ND | 3 (219) | 36 (1784) | ND |
| mus301D2/Df(3L)66C-G28 | 100 (68) | ND | 4 (164) | 67 (1209) | ND |
| mus301D4/Df(3L)66C-G28 | 100 (98) | ND | 1 (193) | 69 (675) | ND |
| mus301094/mus301D1 | 100 (168) | ND | 2 (263) | 21 (1667) | ND |
| mus301422/mus301D1 | 100 (203) | ND | 2 (223) | 57 (650) | ND |
| mus301660/mus301D1 | 98 (167) | ND | 0 (198) | 45 (1675) | ND |
| mus301D2/mus301D4 | 99 (154) | ND | 15 (189) | 21 (220) | 12 (189) |
ND, not determined.
Karyosomes were scored in S3–S7 egg chambers.
The levels of Gurken protein were found wild type in hemizygous mutant cysts up to S6; the strong reduction or complete absence of Gurken protein was scored in S8–S10 egg chambers.
The mus301 gene encodes a new member of the Mus308 subfamily of helicases:
mus301 had been mapped to 66B8-11, a region uncovered by two overlapping deficiencies, Df(3L)ZP3 and Df(3L)66C-G28. The breakpoints of the above deficiencies were mapped onto a contig of P1 clones from the Berkeley Drosophila Genome Project using the available sequence-tagged sites (STSs). Next, two P elements inserted in the vicinity of 66B were mapped onto the P1 walk, P{lacW}0903/14 and P{lacW}0898/11 (Deak et al. 1997), in the area deleted by deficiencies Df(3L)ZP3 and Df(3L)66C-G28. Male-mediated recombination (Preston and Engels 1996) placed mus301 distal to the P{lacW}0903/14 insertion point and proximal to the Df(3L)ZP deficiency breakpoint. We then mapped a cosmid contig from the European Drosophila Genome Project (Siden-Kiamos et al. 1990), spanning mus301 onto the P1 contig using cosmid STSs and thereby defining the area in which mus301 must lie to a >120-kb region. Meiotic mapping, combined with the use of two restriction fragment length polymorphisms discovered in the region, placed mus301 within an ∼50-kb fragment. One of the open reading frames predicted to lie in this region (CG7972) encodes a polypeptide very similar to the C-terminal domain of mus308, a helicase involved in DNA crosslink repair (Harris et al. 1996). Taking into account the role of mus301 in DNA repair (Ghabrial and Schüpbach 1999), we tested the possibility that mus301 was an allele of CG7972. We first demonstrated that a genomic construct containing CG7972 is able to rescue the egg shell and karyosome phenotypes of the mus301094 allele (not shown). Second, the sequencing of six mutant alleles of mus301 revealed missense mutations within the coding region of CG7972 (Figure 2 A and Table 2). From these observations we conclude that the CG7972 gene encodes Mus301, a finding in agreement with recent results that identified mus301 as being allelic to CG7972 (Laurençon et al. 2004).
Figure 2.—

The SF2 helicase Mus301 is a member of the Mus308 subfamily. (A) The genomic structure of mus301 (CG7972); boxes represent exons; shaded boxes indicate conserved motifs of SF2 helicases. Mus301 possesses nine conserved motifs characteristic of SF2 helicases, and motif IVa, present in the Mus308 subfamily of helicases. The Q motif is split between exons 1 and 2. The rest of the motifs are contained in exons 2, 3, and 4. The molecular lesions identified in six mus301 mutant alleles are shown. Missense amino acid substitutions or base changes and allele numbers (in parentheses) are indicated. The solid box represents the 3′-UTR. (B) Alignment of the helicase domains of D. melanogaster Mus301 and Mus308 and human HEL308. Open arrowheads indicate the conserved G and Q residues of the Q motif; asterisks denote the unique amino acid variations found in the Mus308 subfamily of helicases.
TABLE 2.
Molecular characterization of some mus301 alleles
| Allele no. | Base change (+1ATG) | aa change | Maps to |
|---|---|---|---|
| 094 | G2101A | G637Q | Fourth exon |
| 422 | G3652A | Fifth intron | |
| 660 | T2185A | M665K | Fourth exon |
| 2255 | T1479G | Y455D | Third exon |
| G3143T | Q811H | Fifth exon | |
| 3198 | A3658G | Fifth intron | |
| 4875 | A1038G | S237G | Second exon |
| T1479G | Y455D | Third exon |
The 094, 422, and 660 alleles were isolated in the Tübingen mutagenesis (Tearle and Nüsslein-Volhard 1987); the 2255, 3198, and 4875 alleles are from the Zuker EMS collection (Koundakjian et al. 2004).
mus301 is predicted to encode a 1051-amino-acid (117-kDa) protein with a strong sequence similarity to Drosophila Mus308. Mus308 is a 229-kDa protein involved in the repair of DNA crosslinks whose amino terminal domain contains the seven motifs characteristic of DNA and RNA helicases, and the carboxy terminal domain shares similarity with the polymerase domains of prokaryotic DNA polymerase I-like enzymes (Harris et al. 1996). Like Mus308, Mus301 contains a series of conserved motifs characteristic of ATP-dependent DNA/RNA helicases belonging to the superfamily 2 (SF2). These regions of Mus301 are mostly distributed along exons 2, 3, and 4 and include the nine best-conserved motifs in SF2 helicases, the Q box, and domains I, Ia, Ib, II, III, IV, V, and VI. In addition, Mus301 also contains both the IVa motif—an amino acid stretch present in the Mus308 subfamily—and a subset of invariant residues that characterize the Mus308 subfamily but that are unusual among other DEAD-box helicases (Figure 2) (Gorbalenya et al. 1989; Harris et al. 1996; Tanner et al. 2003; Tuteja and Tuteja 2004). Thus, Mus301 is a new member of the Mus308 subfamily of SF2 helicases.
The analysis of the molecular lesions in several mus301 mutant alleles is shown in Figure 2 and Table 2. The mus301094 allele is a missense mutation in the highly conserved G637 of motif VI and behaves as a genetic null. Like mus301094, the mus301660, mus3012255, and mus3014875 alleles map to exons and affect positions conserved in Drosophila melanogaster Mus308 and in human, mouse, and Caenorhabditis elegans homologs of mus301 (Figure 2B and data not shown). Of the remaining sequenced alleles, mus301422 introduces a G-to-A change in the conserved splice donor site +5 of intron 5; mus3012255 contains a second missense mutation in exon 5 and mus3013198 harbors a base substitution in the fifth intron (Table 2). Since three of the strongest mus301 mutant alleles, mus301094, mus301422, and mus301660, are viable over a deficiency for the locus, or in trans-heterozygous combinations, we conclude that mus301 is not an essential gene.
Mus301 is the Drosophila ortholog of human HEL308:
Dm Mus301 is 20% similar and 11% identical at the amino acid level over its full length to Dm Mus308. This low similarity is due to the fact that Dm Mus301 lacks the C-terminal polymerase domain of Dm Mus308 and is considerably shorter (1052 aa of Mus301 vs. 2059 aa of Mus308). The helicase domains of Mus301 and Mus308 are more highly conserved (57% similar and 36% identical). In an attempt to isolate mammalian proteins involved in DNA crosslink repair, a human and a mouse homolog of Mus308 were identified (Marini and Wood 2002). A BLAST search shows that Homo sapiens HEL308 and Mus musculus HEL308 are more similar to Dm Mus301 than to Mus308 (Figure 3, A and B). In fact, the analysis of the phylogenetic tree of the helicase domains of Dm Mus301, Dm Mus308, Hs HEL308, Mm HEL308, and C. elegans Y55B1AL shows a branched clade in which Dm Mus301 and Ce Y55B1AL are more closely related to their mammalian counterparts than Dm Mus308 (Figure 3C). On the basis of sequence similarity, we conclude that Dm Mus301 is the structural ortholog of HEL308 in mammals.
Figure 3.—

Sequence comparisons of Drosophila Mus301 with other Mus308 subfamily helicases. (A) The complete sequence of D. melanogaster Mus301 was compared to that of Dm Mus308, human HEL308, mouse HEL308, and C. elegans Y55B1AL.3a. The percentages of similarity and identity (in parentheses) are indicated. (B) Percentages of similarity and identity (in parentheses) in the helicase region of Dm Mus301 and that of other homologs. Solid boxes represent the SF2 helicase motifs present in Dm Mus301. Dm Mus308 is not drawn to scale. (C) Phylogenetic analysis of Dm Mus301 and related proteins. The tree-building method was “neighbor joining,” using the ‘best tree’ mode; numbers represent uncorrected distances. The Drosophila helicase L(2)35Df is a divergent member of the Mus308 subfamily and is shown for comparison.
mus301 is required for chromosome segregation in oogenesis:
To assess a possible role for mus301 in meiosis, we analyzed whether the mutants affect the disjunction of the X chromosome. mus301 hemizygotes were used as they lay some wild-type eggs that give rise to viable larvae. All three mutant alleles analyzed show a high level of X chromosome nondisjunction compared to the wild type (w) control (Table 3). The percentage of X chromosome nondisjunction observed in our experimental conditions is significantly larger than that reported in the initial isolation of the mus301 alleles (Boyd et al. 1981). This difference could be due to the hemizygous conditions utilized in our experiments or to the fact that previous studies measured nondisjunction of only viable adult progeny, which could introduce a bias for progeny that has disjoined properly. From our observations, we conclude that failure to undergo meiotic recombination in mus301 mutants (see below) most probably causes a disruption of chromosome segregation during meiosis. Similarly, mutations in spn-A, spn-B, spn-D, and okra also impair meiotic recombination and they have been shown to have high levels of X chromosome nondisjunction (Ghabrial et al. 1998; Staeva-Vieira et al. 2003).
TABLE 3.
X chromosome nondisjunction in mus301 hemizygous females
| Genotype of mothers | Total no. larvae (N) | No. of wild-type larvae | No. of y larvae (X0 progeny) | % X chromosome nondisjunction |
|---|---|---|---|---|
| w | 3305 | 3304 | 1 | 0.12 |
| mus301094/Df | 328 | 313 | 15 | 17 |
| mus301422/Df | 1600 | 1495 | 105 | 23 |
| mus301660/Df | 202 | 176 | 26 | 41 |
| mus301094/Df(3L)ZP3 | 303 | 291 | 12 | 15 |
| mus301422/Df(3L)ZP3 | 148 | 138 | 10 | 24 |
| mus301660/Df(3L)ZP3 | 188 | 163 | 25 | 42 |
Females of a given genotype were crossed to y males and the presence of y larvae was scored, indicating that X chromosome nondisjunction had occurred. To avoid unspecific effects due to genetic background, the experiment was repeated with a second deficiency for mus301, Df(3L)ZP3.
mus301 is required for mitotic dsDNA break repair:
Given the phenotypic similarities between mus301 and okra, spn-A, spn-B, and spn-D and the involvement of the latter group of genes in the repair of dsDNA breaks, it seems possible that mus301 also encodes a protein required for DSB repair. To test if this is the case, mutant larvae were assayed for sensitivity to MMS, a mutagen that induces dsDNA breaks. Crosses producing different combinations of mus301 mutant larvae were fed a solution of 0.8% MMS and the survival of the treated larvae was compared to that of untreated controls. In agreement with the characterization of the original mus301 alleles (Boyd et al. 1981), we found that mus301 mutants are sensitive to MMS, suggesting that mus301 is involved in mitotic DSB repair, similar to spn-A, spn-B, and okra (Figure 4). Further confirmation of a role for mus301 in DSB repair comes from the fact that mus301 mutants are sensitive to X-rays, which, like MMS, also induce DSBs (Oliveri et al. 1990).
Figure 4.—

An essential role for mus301 in chromosome segregation during meiosis and for DSB repair in mitotic cells. MMS sensitivity of mus301, spindle-D, and spindle-E mutant combinations. MMS sensitivity was expressed as a fraction of the percentage expected in the treated vial vs. the percentage expected in the control. spn-D and spn-E are not required for DSB repair in mitotic cells and are shown for comparison. Df, Df(3L)66C-G28.
mus301 mutants are defective in meiotic dsDNA break repair:
During oogenesis, the oocyte is specified in a stepwise manner from a cyst of 16 sibling cells, which are interconnected through actin-rich cytoplasmic bridges called ring canals. Initially, two cells, each containing four ring canals, behave like “pro-oocytes” as they accumulate oocyte-specific markers. Later on, one of them is selected as the oocyte, while the remaining 15 cells of the cyst acquire a nurse-cell fate. In 16-cell cysts of region 2a of the germarium, the two pro-oocytes and several nurse cells enter meiosis and form synaptonemal complexes (SC), which are proteinaceous structures that connect aligned homologous chromosomes that can be visualized by staining for the SC component C(3)G (Carpenter 1979; Page and Hawley 2001). In region 2b, only the oocyte and an adjacent nurse cell show a distinguishable SC. As the cyst matures, meiosis is restricted to the presumptive oocyte and the SC is found in this cell only in germarial region 3 cysts. By S5–S6 of oogenesis, the SC is disassembled (Figure 5A and data not shown). To determine if the meiotic defects seen in mus301 mutants are due to a failure in SC formation, we stained mutant germaria with α-C(3)G. As in the wild-type, C(3)G is visible in four cells of region 2a cysts. In contrast to the gradual reduction in the number of C(3)G-positive cells in wild-type cysts, the C(3)G staining persists in several cells per cyst until S2 (Figure 5B and data not shown). Later on, the C(3)G signal is restricted to one cell and disappears at ∼S5–S6. These results indicate that the SC forms normally in mus301 mutant cysts. There is, however, a delay in the restriction of the SC to a single cell, most probably reflecting the late selection of the oocyte characteristic of this mutant. A similar abnormal distribution of the SC was reported for mus301 mutant cysts using an antibody against an unknown component of the SC (Huynh and St Johnston 2000).
Figure 5.—

A role for mus301 in recombinational DSB repair. Wild-type (A) and mutant (B and C) germaria showing the distribution of γ-HIS2AV (red), C(3)G (green), and Orb (blue) proteins. (A) One cyst each from region 2b, region 2b/3, and region 3 is shown (see Figure 6 for a schematic of these germarial stages). γ-HIS2AV foci are abundant in region 2b oocytes, but they are lost in successive stages and are barely detectable in region 3 oocytes. (B and C) γ-HIS2AV foci are visible in region 2b/3 and region 3 mutant oocytes. The sibling nurse cells also possess increased γ-HIS2AV staining compared to wild type. All images are projections of several confocal sections. Dashed lines delineate individual cysts. Arrowheads point toward oocytes. (B and C) mus301094/Df(3L)66C-G28. Bar, 10 μm.
Double-strand breaks in the DNA are produced during meiotic recombination in germline cells of early cysts. One of the earliest known responses to DSB in mammals and S. cerevisiae is the phosphorylation of H2A histone variants in the nucleosomes situated in the vicinity of the break. In Drosophila, the phosphorylation of the single histone variant HIS2AV can be detected as soon as 1 min after DSB induction in somatic cells and is removed after 3 hr of exposure to the DSB-inducing agent, thus making this variant histone an excellent marker to study the dynamics of DSB repair (Madigan et al. 2002). To monitor the presence of DSBs in germline cells of both wild-type and mus301 mutant cysts, we made use of an antibody that recognizes a phosphorylated form of HIS2AV, γ-HIS2AV. In wild-type female meiosis, γ-HIS2AV staining appears in region 2a of the germarium after the initiation of SC formation. As meiosis proceeds and the DSBs are repaired, the γ-HIS2AV-positive foci disappear, and by germarial region 3 there is no detectable signal in the oocyte (Figure 5A) (Jang et al. 2003). Consistent with a role for Mus301 in DSB repair, a large proportion of mutant oocytes show a dramatic accumulation of γ-HIS2AV foci in their nuclei compared to wild-type controls. In addition, these foci persist after region 3 until S3–S4, when they disappear prior to SC disassembly (Figure 5, B and C). A similar increase in the number and persistence of foci can be detected in nurse-cell nuclei. Since the phosphorylation of HIS2AV indicates the presence of DSBs (Madigan et al. 2002; Jang et al. 2003), our observations strongly suggest that DSBs are not processed efficiently in mus301 mutant oocytes.
mus301 mutant oocytes activate a Mei-41/Chk2-dependent checkpoint:
The cell cycle and patterning defects observed in okra, spn-A, spn-B, and spn-D mutants result from the activation of the checkpoint kinase Mei-41 (Ghabrial and Schüpbach 1999), but there are conflicting data on the identity of the downstream kinases that transduce this signal. On the one hand, it has been shown that Mei-41 acts through the Drosophila Chk2 homolog Mnk1, since mnk1 mutants suppress the ventralization of okra, spn-A, spn-B, and spn-D mutants (Abdu et al. 2002; Staeva-Vieira et al. 2003). On the other hand, Masrouha et al. (2003) have observed that mnk1 does not suppress the ventralization of spn-B or mus301 mutant alleles. We therefore reexamined this issue by generating mnk1; mus301 double mutants. Consistent with the results of Abdu et al. (2002), we find that double-mutant egg chambers for mnk and mus301 show nearly wild-type levels of Grk protein and a normal karyosome (Table 4), strongly suggesting that, as in the case of okra, spn-A, spn-B, and spn-D, Dm Chk2 is a transducer of the meiotic checkpoint activated in mus301 mutant oocytes.
TABLE 4.
Phenotypes of double-mutant combinations of mus301 with grp/DmChk1 or mnk/DmChk2
| Maternal genotype | % egg chambers with mutant karyosomes (n) | % egg chambers with mutant Gurken levels (n) |
|---|---|---|
| grpFSA4 | 0 (107) | ND |
| mus301660/Df(3L)66C-G28 | 100 (105) | ND |
| grpFSA4; mus301660/Df(3L)66C-G28 | 100 (193) | ND |
| mnkP6/CyO; mus301660/TM3 | 0 (103) | 3 (18) |
| mnkP6/Df(2L)pr65; mus301660/TM3 | 1 (96) | 3 (36) |
| mnkP6/CyO; mus301660/Df(3L)66C-G28 | 100 (35) | 28 (18) |
| mnkP6/Df(2L)pr65; mus301660/Df(3L)66C-G28 | 4 (23) | 7 (14) |
Karyosomes were scored in S3–S7 egg chambers. The levels of Gurken protein were scored in S9 and S10 egg chambers. grp; mus301 double mutants lay ventralized eggs with a similar frequency to mus301 single mutants (not shown). ND, not determined.
Similarly, we analyzed the role of grapes (grp), a putative serine/threonine kinase with extensive homology to Chk1 kinase in Schizosaccharomyces pombe (Fogarty et al. 1997; Sibon et al. 1999) in the Mei-41 checkpoint activated in mus301 mutants. Since grp; mus301 mutants lay a large proportion of ventralized eggs (<70%, n = 196), produce a low frequency of misplaced oocytes (3%, n = 363), and fail to form a normal karyosome (Table 4), the abnormal patterning and the defective meiosis of mus301 mutant egg chambers most probably do not involve the activity of Dm Chk1. This result adds to the finding that Grp is not the transducer of the pachytene checkpoint in okra, spn-D, or spn-B mutants (Abdu et al. 2002).
mus301 is required for oocyte specification independently of Mei-W68 and of Mei-41 checkpoint activation:
During meiosis, DSBs are made to enable recombination to take place. Since a defect in the initial stages of meiosis is likely to occur prior to the defects in pattern formation observed in mus301 mutant ovaries, we decided to investigate if the latter were a consequence of a failure to proceed through meiosis correctly by analyzing double mutants for mei-W68 or mei-41 and mus301. In agreement with previous results (Ghabrial and Schüpbach 1999), we find that mei-W68; mus301, and mei-41; mus301 double mutants possess essentially wild-type karyosomes and Grk protein levels (data not shown). In contrast, the analysis of double mutants shows that the delay in oocyte selection observed in mus301 mutants is not a consequence of unrepaired DSBs. A fraction of mus301 mutant cysts accumulate oocyte-specific markers such as Orb protein in the two pro-oocytes until germarial region 3 when compared to wild-type controls (Figure 6). This delay in oocyte selection most probably accounts for the occurrence of misplaced oocytes in older mutant egg chambers (Table 1) (González-Reyes and St Johnston 1998). mei-W68; mus301 mutants behave in this regard like single mus301 mutants and still show a delay in choosing one pro-oocyte to become the oocyte and display a low percentage of misplaced oocytes (Figure 6; 4% of double-mutant region 2b/3 and region 3 cysts show misplaced oocytes; n =135). Similarly, mei-41; mus301 cysts also present a delay in Orb accumulation in a single cell (Figure 6D). Thus, the delay in oocyte selection characteristic of mus301 mutants is not a consequence of the activation of the mei-41 checkpoint. Altogether, our results suggest a role for the Mus301 helicase in oocyte specification independent of the initiation of meiotic recombination by Mei-W68 and the DNA damage checkpoint.
Figure 6.—

mei-W68 does not rescue the two-oocyte phenotype of mus301 mutant cysts. (A) Scheme of a wild-type germarium to show the arrangement of region 2b, region 2b/3, and region 3 cysts. (B–E) Germaria double stained to visualize filamentous actin and Orb protein. (B) Wild-type germarium showing Orb accumulated in a single cell in region 2b and region 3 cysts. (C) Mutant germarium carrying region 2b and region 3 cysts, each containing two cells that accumulate Orb. (D and E) Double-mutant germaria showing region 3 cysts with two cells containing high levels of Orb protein. (F) Double-mutant egg chambers stained with anti-Orb to show a misplaced oocyte. The white bars mark the anterior–posterior axes of the follicles. (G) Germaria of different genetic combinations were analyzed and the number of region 2b/3 and region 3 cysts containing two cells with increased levels of the oocyte marker Orb and each possessing four4 ring -canals (as visualized with rRhodamine–-pPhalloidin) were counted as “2× oocyte” cysts. (B) Wild type. (C) mus301094/Df(3L)66C-G28. (D) mei-41D3/Df(2R)LL5; mus301094/Df(3L)66C-G28. (E and F) mei-W681; mus301094/Df(3L)66C-G28. Asterisks label the cells with highest Orb contents. Bar, 10 μm.
DISCUSSION
Several helicases are involved in the repair of dsDNA breaks. One of the best characterized is the RecBCD complex of eubacteria, which possesses a bipolar helicase activity with a defined role in the homologous recombination repair pathway. The RecBCD recombinase is able to process DNA ends using a combination of helicase and nuclease activities (Anderson and Kowalczykowski 1997; Singleton et al. 2004). Also in Escherichia coli, RecG helicase has been reported to promote DSB repair subsequent to the activity of RecBCD and RecA (Meddows et al. 2004). In eukaryotic cells, defects in DSB repair have been associated with cancer predisposition and genomic instability. For instance, Bloom's syndrome is a rare disorder in humans caused by mutations in the RecQ helicase BLM that results in a predisposition to cancers of all types (Hickson 2003). Evidence for a role of this helicase in DSB metabolism comes from its involvement in homologous recombination-dependent repair of damaged replication forks (Wu and Hickson 2003) and from the fact that mending of dsDNA breaks in the absence of BLM results in defective products with large deletions (Runger and Kraemer 1989; Gaymes et al. 2002). The ortholog of BLM in Drosophila is encoded by mus309 (Kusano et al. 2001), a gene identified in a screen for hypersensitivity to chemical DNA-damaging agents such as nitrogen mustard, a mutagen that induces interstrand DNA crosslinks, and MMS (Boyd et al. 1981). In this screen, a total of 11 complementation groups were isolated, including mus301 and mus308. In contrast to mus308, mus301 is strongly sensitive to both mutagens, implicating Mus301 in the repair of interstrand crosslinks and double-strand breaks. We have identified Mus301 as a new member of the Mus308 subfamily of ATP-dependent helicases and present evidence for a role of Mus301 in the repair of the dsDNA breaks that arise during recombination.
A role for mus301 in DSB repair and oocyte specification:
Recombination begins with the occurrence of dsDNA breaks on one chromatid, catalyzed in budding yeast by the Spo11 protein. Subsequently, the DSB is resected to produce an intermediate with a 3′-overhanging single-strand DNA (ssDNA) tail. Rad51 and Dmc1 proteins then bind to this ssDNA to form a filamentous structure that promotes a search for homologous, nonsister DNA to prime repair DNA synthesis. The requirement of the strand-invasion protein Rad51 in dsDNA break repair is demonstrated by the hypersensitivity to ionizing radiation conferred by mutations in Rad51 (Symington 2002). The Drosophila genome has five Rad51 family members (spn-A, spn-B, spn-D, CG2412, and CG6318) (Staeva-Vieira et al. 2003) and mutations in three of these have confirmed their role in the early aspects of DSB repair, as the absence of Spn-A, Spn-B, or Spn-D function leads to the activation of a meiotic checkpoint triggered by unrepaired dsDNA breaks or unresolved recombination intermediates. Furthermore, some of these genes are partially redundant, as double-mutant combinations display stronger phenotypes than single mutants alone (González-Reyes et al. 1997). In this context, our finding that, like spn-A, mus301 mutants do not process DSBs efficiently suggests that Mus301 acts in the same step of DSB repair as Spn-A. Moreover, since mus301 and several of the Rad51-like genes interact genetically, as demonstrated by the enhancement of the mutant phenotypes observed in the double mutants mus301 spn-A or mus301 spn-B (González-Reyes et al. 1997), it is likely that mus301- and rad51-like genes do not act in a linear pathway. Rather, they seem to collaborate in the formation of stable recombination intermediates necessary for efficient DSB repair. In such a scenario, it is interesting to note that mus301 is the Drosophila ortholog of Hs HEL308, a single-stranded DNA-dependent ATPase and DNA helicase of unknown function that in vitro is able to translocate on DNA with 3′–5′ polarity and to displace 20- to 40-mer duplex oligonucleotides (Marini and Wood 2002). The considerable sequence similarity (73%) between the helicase domain of Mus301 and that of Hs HEL308 raises the possibility that mus301 possesses a DNA-unwinding activity with 3′–5′ polarity. In support of our model, budding yeast mer3 encodes an ATP-dependent DNA helicase that unwinds dsDNA with a 3′–5′ polarity and that stimulates 3′–5′ heteroduplex extension by Rad51 in crossover recombination (Mazina et al. 2004). Finally, okra, a gene required for the repair of DSB after P-element excision and for DNA repair during oogenesis (Kooistra et al. 1997; Ghabrial et al. 1998; Kooistra et al. 1999; Romeijn et al. 2005), is the Drosophila homolog of yeast Rad54, a SWI2/SNF2 chromatin-remodeling dsDNA-dependent ATPase that binds Rad51 directly and that stimulates DSB repair in both meiotic and mitotic cells (Mazin et al. 2000; Krogh and Symington 2004). Since Okra, Mus301, Spn-A, and, to a lesser extent, Spn-B are also involved in the repair of DNA damage caused by MMS treatment and ionizing radiation in mitotic cells (this work; Oliveri et al. 1990; Staeva-Vieira et al. 2003), it is likely that the Mus301-Rad51-Okra interaction is also maintained in DSB repair in the soma.
In contrast to the situation in S. cerevisiae, the rad52-group genes okra, spn-A, spn-B, and spn-D were isolated because of their role in egg chamber polarization in oogenesis. Their molecular characterization allowed the establishment of a clear link between DNA repair and pattern formation in the female germline in Drosophila. Additional experiments that involved double-mutant combinations defined the realm of action of the spindle genes. They act after the induction of dsDNA breaks by Mei-W68 and are necessary for DSB repair, as demonstrated by the rescue of the meiotic phenotypes of okra, spn-A, spn-B, and spn-D mutants in the absence of mei-W68 or mei-41. The phenotypic analysis of mus301 places this gene at the same level as these spindle genes in the recombination pathway. Lack of function of mus301 prevents the efficient processing of DSBs, thus triggering the activation of the meiotic checkpoint, which in turn induces a Chk2-dependent cell cycle delay similar to the situation in spn-A, spn-B, and spn-D mutants (Abdu et al. 2002; Staeva-Vieira et al. 2003). In addition, our experiments involving double-mutant combinations for mei-W68 or mei-41 and mus301 have revealed a novel role for mus301 in oocyte selection independent of initiation of recombination and of DNA damage checkpoint activation during cyst formation. Although the mechanisms by which mus301 regulates oocyte development independently of mei-W68 or mei-41 are unclear, they most probably involve Mus301 helicase activity, since the mus301 allele used in these experiments carries a missense mutation in a conserved glycine of helicase domain VI and there are not other recognizable domains in the protein. Since there are no detectable meiotic DSBs in the absence of Mei-W68 activity (McKim et al. 1998; Jang et al. 2003), our observation suggests a new function for Mus301 unrelated to DSB repair in oocyte specification. In this regard, it would be interesting to know if mutations in other genes involved in recombinational DNA repair and in oocyte selection, such as okra, spn-A, spn-B, and spn-D, also affect oocyte specification independently of mei-W68 and mei-41.
Acknowledgments
We thank Kim McKim, Anne Laurençon, Trudi Schüpbach, Scott Page, Peter Maróy, Jeanette Natzle, the Developmental Studies Hybridoma Bank (University of Iowa), and the Bloomington Stock Center for fly stocks and reagents. Financial support from the European Community Marie Curie Programme to R.M.; from the Medical Research Council (UK), the Spanish Ministerio de Ciencia y Tecnología (BMC2003-01512), and the Junta de Andalucía (CVI-280) to A.G.-R.; and from a Wellcome Trust (UK) Principal Research Fellowship to D.StJ. is acknowledged.
References
- Abdu, U., M. Brodsky and T. Schüpbach, 2002. Activation of a meiotic checkpoint during Drosophila oogenesis regulates the translation of gurken through Chk2/Mnk. Curr. Biol. 12: 1645–1651. [DOI] [PubMed] [Google Scholar]
- Abdu, U., A. González-Reyes, A. Ghabrial and T. Schupbach, 2003. The Drosophila spn-D gene encodes a Rad51C-like protein that is required exclusively during meiosis. Genetics 165: 197–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams, M., M. McVey and J. Sekelsky, 2003. Drosophila BLM in double-strand break repair by synthesis-dependent strand annealing. Science 299: 265–267. [DOI] [PubMed] [Google Scholar]
- Anderson, D. G., and S. C. Kowalczykowski, 1997. The translocating RecBCD enzyme stimulates recombination by directing RecA protein onto ssDNA in a chi-regulated manner. Cell 90: 77–86. [DOI] [PubMed] [Google Scholar]
- Blanton, H., and J. Sekelsky, 2004. Unique invasions and resolutions: DNA repair proteins in mitotic recombination in Drosophila melanogaster. Cytogenet. Genome Res. 107: 172–179. [DOI] [PubMed] [Google Scholar]
- Boyd, J., M. Golino, K. Shaw, C. Osgood and M. Green, 1981. Third-chromosome mutagen sensitive mutants of Drosophila melanogaster. Genetics 97: 607–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brodsky, M. H., B. T. Weinert, G. Tsang, Y. S. Rong, N. M. McGinnis et al., 2004. Drosophila melanogaster MNK/Chk2 and p53 regulate multiple DNA repair and apoptotic pathways following DNA damage. Mol. Cell. Biol. 24: 1219–1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao, L., E. Alani and N. Kleckner, 1990. A pathway for generation and processing of double-strand breaks during meiotic recombination in S. cerevisiae. Cell 61: 1089–1101. [DOI] [PubMed] [Google Scholar]
- Carpenter, A., 1979. Synaptonemal complex and recombination nodules in wild-type Drosophila melanogaster females. Genetics 92: 511–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deak, P., M. Omar, R. Saunders, M. Pál, O. Komonyi et al., 1997. P-element insertion alleles of essential genes on the third chromosome of Drosophila melanogaster: correlation of physical and cytogenetic maps in chromosomal region 86E–87F. Genetics 147: 1697–1722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliot, B., and M. Jasin, 2002. Human genome and diseases: review. Double strand breaks and translocations in cancer. Cell. Mol. Life Sci. 59: 373–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Findley, S., M. Tamanaha, N. Clegg and H. Ruohola-Baker, 2003. maelstrom, a Drosophila spindle-class gene, encodes a protein that colocalizes with Vasa and RDE1/AGO1 homolog, Aubergine, in nuage. Development 130: 859–871. [DOI] [PubMed] [Google Scholar]
- Fogarty, P., S. Campbell, R. Abu-Shumays, B. de Saint Phalle, K. Yu et al., 1997. The Drosophila grapes gene is related to checkpoint gene chk1/rad27 and is required for late syncytial division fidelity. Curr. Biol. 7: 418–426. [DOI] [PubMed] [Google Scholar]
- Gaymes, T. J., P. S. North, N. Brady, I. D. Hickson, G. J. Mufti et al., 2002. Increased error-prone non homologous DNA end-joining—a proposed mechanism of chromosomal instability in Bloom's syndrome. Oncogene 21: 2525–2533. [DOI] [PubMed] [Google Scholar]
- Ghabrial, A., and T. Schüpbach, 1999. Activation of a meiotic checkpoint regulates translation of Gurken during Drosophila oogenesis. Nat. Cell Biol. 1: 354–357. [DOI] [PubMed] [Google Scholar]
- Ghabrial, A., R. Ray and T. Schüpbach, 1998. okra and spindle-B encode components of the RAD52 DNA repair pathway and affect meiosis and patterning in Drosophila oogenesis. Genes Dev. 12: 2711–2723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillespie, D., and C. Berg, 1995. homeless is required for RNA localization in Drosophila oogenesis and encodes a new member of the DE-H family of RNA-dependent ATPases. Genes Dev. 9: 2495–2508. [DOI] [PubMed] [Google Scholar]
- González-Reyes, A., 1999. DNA repair and pattern formation come together. Nat. Cell Biol. 1: E150–E152. [DOI] [PubMed] [Google Scholar]
- González-Reyes, A., and D. St Johnston, 1998. The Drosophila AP axis is polarised by the cadherin-mediated positioning of the oocyte. Development 125: 3635–3644. [DOI] [PubMed] [Google Scholar]
- González-Reyes, A., H. Elliott and D. St Johnston, 1997. Oocyte determination and the origin of polarity in Drosophila: the role of the spindle genes. Development 124: 4927–4937. [DOI] [PubMed] [Google Scholar]
- Gorbalenya, A. E., E. V. Koonin, A. P. Donchenko and V. M. Blinov, 1989. Two related superfamilies of putative helicases involved in replication, recombination, repair and expression of DNA and RNA genomes. Nucleic Acids Res. 17: 4713–4730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hari, K., A. Santerre, J. Sekelsky, K. McKim, J. Boyd et al., 1995. The mei-41 gene of D. melanogaster is a structural and functional homolog of the human ataxia telangiectasia gene. Cell 82: 815–821. [DOI] [PubMed] [Google Scholar]
- Harris, P., O. Mazina, E. Leonhardt, R. Case, J. Boyd et al., 1996. Molecular cloning of Drosophila mus308, a gene involved in DNA cross-link repair with homlogy to prokaryotic DNA polymerase I genes. Mol. Cell. Biol. 16: 5764–5771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickson, I. D., 2003. RecQ helicases: caretakers of the genome. Nat. Rev. Cancer 3: 169–178. [DOI] [PubMed] [Google Scholar]
- Huynh, J.-R., and D. St Johnston, 2000. The role of BicD, Egl, Orb and the microtubules in the restriction of meiosis to the Drosphila oocyte. Development 127: 2785–2794. [DOI] [PubMed] [Google Scholar]
- Jang, J. K., D. E. Sherizen, R. Bhagat, E. A. Manheim and K. S. McKim, 2003. Relationship of DNA double-strand breaks to synapsis in Drosophila. J. Cell Sci. 116: 3069–3077. [DOI] [PubMed] [Google Scholar]
- Kennerdell, J., S. Yamaguchi and R. Carthew, 2002. RNAi is activated during Drosophila oocyte maturation in a manner dependent on aubergine and spindle-E. Genes Dev. 16: 1884–1889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kooistra, R., K. Vreeken, J. Zonneveld, A. de Jong, J. Eekne et al., 1997. The Drosophila melanogaster RAD54 homolog, DmRAD54, is involved in the repair of radiation damage and recombination. Mol. Cell. Biol. 17: 6097–6104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kooistra, R., A. Pastink, J. Zonneveld, P. Lohman and J. Eeken, 1999. The Drosophila melanogaster DmRAD54 gene plays a crucial role in double-strand break repair after P-element excision and acts synergistically with Ku70 in the repair of X-ray damage. Mol. Cell. Biol. 19: 6269–6275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koundakjian, E., D. Cowan, R. Hardy and A. Becker, 2004. The Zuker collection: a resource for the analysis of autosomal gene function in Drosophila melanogaster. Genetics 167: 203–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krogh, B. O., and L. S. Symington, 2004. Recombination proteins in yeast. Annu. Rev. Genet. 38: 233–271. [DOI] [PubMed] [Google Scholar]
- Kurz, E., and S. Lees-Miller, 2004. DNA damage-induced activation of ATM and ATM-dependent signalling pathways. DNA Rep. 3: 889–900. [DOI] [PubMed] [Google Scholar]
- Kusano, K., D. M. Johnson-Schlitz and W. R. Engels, 2001. Sterility of Drosophila with mutations in the Bloom syndrome gene–complementation by Ku70. Science 291: 2600–2602. [DOI] [PubMed] [Google Scholar]
- Lantz, V., J. Chang, J. Horabin, D. Bopp and P. Schedl, 1994. The Drosophila orb RNA-binding protein is required for the formation of the egg chamber and establishment of polarity. Genes Dev. 8: 598–613. [DOI] [PubMed] [Google Scholar]
- Laurençon, A., C. Orme, H. Peters, C. Boulton, E. Vladar et al., 2004. A large-scale screen for mutagen sensitive loci in Drosophila. Genetics 167: 217–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madigan, J. P., H. L. Chotkowski and R. L. Glaser, 2002. DNA double-strand break-induced phosphorylation of Drosophila histone variant H2Av helps prevent radiation-induced apoptosis. Nucleic Acids Res. 30: 3698–3705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marini, F., and R. Wood, 2002. A human DNA helicase homologous to the DNA cross-link sensitivity protein Mus308. J. Biol. Chem. 277: 8716–8723. [DOI] [PubMed] [Google Scholar]
- Masrouha, N., L. Yang, S. Hijal, S. Larochelle and B. Suter, 2003. The Drosophila chk2 gene loki is essential for embryonic DNA double-strand-break checkpoints induced in S phase or G2. Genetics 163: 974–981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazin, A., C. Bornarth, J. Solinger, W.-D. Heyer and S. Kowalczykowski, 2000. Rad54 protein is targeted to pairing loci by the Rad51 nucleoprotein filament. Mol. Cell 6: 583–592. [DOI] [PubMed] [Google Scholar]
- Mazina, O. M., A. V. Mazin, T. Nakagawa, R. D. Kolodner and S. C. Kowalczykowski, 2004. Saccharomyces cerevisiae Mer3 helicase stimulates 3′-5′ heteroduplex extension by Rad51: implications for crossover control in meiotic recombination. Cell 117: 47–56. [DOI] [PubMed] [Google Scholar]
- McKim, K., and A. Hayashi-Hagihara, 1998. mei-W68 in Drosophila melanogaster encodes a Spo11 homolog: evidence that the mechanism for initiating meiotic recombination is conserved. Genes Dev. 12: 2932–2942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKim, K., B. Green-Marroquin, J. Sekelsky, G. Chin, C. Steinberg et al., 1998. Meiotic synapsis in the absence of recombination. Science 279: 876–878. [DOI] [PubMed] [Google Scholar]
- Meddows, T. R., A. P. Savory and R. G. Lloyd, 2004. RecG helicase promotes DNA double-strand break repair. Mol. Microbiol. 52: 119–132. [DOI] [PubMed] [Google Scholar]
- Oishi, I., S. Sugiyama, H. Otani, H. Yamamura, Y. Nishida et al., 1998. A novel Drosophila nuclear protein serine/threonine kinase expressed in the germline during its establishment. Mech. Dev. 71: 49–63. [DOI] [PubMed] [Google Scholar]
- Oliveri, D., P. Harris and J. Boyd, 1990. X-ray sensitivity and single-strand DNA break repair in mutagen-sensitive mutants of Drosophila melanogaster. Mutat. Res. 235: 25–31. [DOI] [PubMed] [Google Scholar]
- Page, S., and R. Hawley, 2001. c(3)G encodes a Drosophila synaptonemal complex protein. Genes Dev. 15: 3130–3143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preston, C., and W. Engels, 1996. P-element-induced male recombination and gene conversion in Drosophila. Genetics 144: 1611–1622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson, C., N. Horikoshi and T. K. Pandita, 2004. The role of the DNA double-strand break response network in meiosis. DNA Rep. 3: 1149–1164. [DOI] [PubMed] [Google Scholar]
- Romeijn, R. J., M. M. Gorski, M. A. van Schie, J. N. Noordermeer, L. H. Mullenders et al., 2005. Lig4 and rad54 are required for repair of DNA double-strand breaks induced by P-element excision in Drosophila. Genetics 169: 795–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Runger, T. M., and K. H. Kraemer, 1989. Joining of linear plasmid DNA is reduced and error-prone in Bloom's syndrome cells. EMBO J. 8: 1419–1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schüpbach, T., and E. Wieschaus, 1989. Female sterile mutations on the second chromosome of Drosophila melanogaster. I. Maternal effect mutations. Genetics 121: 101–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shivji, M., and A. Venkitaraman, 2004. DNA recombination, chromosomal stability and carcinogenesis: insights into the role of BRCA2. DNA Rep. 3: 835–843. [DOI] [PubMed] [Google Scholar]
- Sibon, O., A. Laurençon, R. Hawley and W. Theurkauf, 1999. The Drosophila ATM homologue Mei-41 has an essential checkpoint function at the midblastula transition. Curr. Biol. 9: 302–312. [DOI] [PubMed] [Google Scholar]
- Siden-Kiamos, I., R. Saunders, L. Spanos, T. Majerus, J. Treanear et al., 1990. Towards a physical map of the Drosophila melanogaster genome: mapping of cosmid clones within defined genomic divisions. Nucleic Acids Res. 18: 6261–6270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singleton, M. R., M. S. Dillingham, M. Gaudier, S. C. Kowalczykowski and D. B. Wigley, 2004. Crystal structure of RecBCD enzyme reveals a machine for processing DNA breaks. Nature 432: 187–193. [DOI] [PubMed] [Google Scholar]
- Staeva-Vieira, E., S. Yoo and R. Lehmann, 2003. An essential role of DmRad51/SpnA in DNA repair and meiotic checkpoint control. EMBO J. 22: 5863–5874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Styhler, S., A. Nakamura, A. Swan, B. Suter and P. Lasko, 1998. vasa is required for Gurken accumulation in the oocyte, and is involved in oocyte differentiation and germline cyst development. Development 125: 1569–1578. [DOI] [PubMed] [Google Scholar]
- Symington, L. S., 2002. Role of RAD52 epistasis group genes in homologous recombination and double-strand break repair. Microbiol. Mol. Biol. Rev. 66: 630–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szostak, J. W., T. L. Orr-Weaver, R. J. Rothstein and F. W. Stahl, 1983. The double-strand-break repair model for recombination. Cell 33: 25–35. [DOI] [PubMed] [Google Scholar]
- Tanner, N. K., O. Cordin, J. Banroques, M. Doere and P. Linder, 2003. The Q motif: a newly identified motif in DEAD box helicases may regulate ATP binding and hydrolysis. Mol. Cell 11: 127–138. [DOI] [PubMed] [Google Scholar]
- Tearle, R., and C. Nüsslein-Volhard, 1987. Tübingen mutants stocklist. Dros. Inf. Serv. 66: 209–226. [Google Scholar]
- Tomancak, P., A. Guichet, P. Zavorsky and A. Ephrussi, 1998. Oocyte polarity depends on regulation of gurken by Vasa. Development 125: 1723–1732. [DOI] [PubMed] [Google Scholar]
- Tuteja, N., and R. Tuteja, 2004. Unraveling DNA helicases. Motif, structure, mechanism and function. Eur. J. Biochem. 271: 1849–1863. [DOI] [PubMed] [Google Scholar]
- Wilson, J., J. Connell and P. Macdonald, 1996. aubergine enhances oskar translation in the Drosophila ovary. Development 122: 1631–1639. [DOI] [PubMed] [Google Scholar]
- Wu, L., and I. D. Hickson, 2003. The Bloom's syndrome helicase suppresses crossing over during homologous recombination. Nature 426: 870–874. [DOI] [PubMed] [Google Scholar]