

Abstract
The factors driving or preventing pathological expansion of tandem repeats remain largely unknown. Here, we assessed the FGF14 (GAA)·(TTC) repeat locus in 2,530 individuals by long-read and Sanger sequencing and identified a common 5′-flanking variant in 70.34% of alleles analyzed (3,463/4,923) that represents the phylogenetically ancestral allele and is present on all major haplotypes. This common sequence variation is present nearly exclusively on nonpathogenic alleles with fewer than 30 GAA-pure triplets and is associated with enhanced stability of the repeat locus upon intergenerational transmission and increased Fiber-seq chromatin accessibility.
Dominantly inherited (GAA)·(TTC) repeat expansions in intron 1 of the fibroblast growth factor 14 gene (FGF14) have recently been shown to cause spinocerebellar ataxia 27B (SCA27B)1,2. Initial observations suggest that intermediate and expanded alleles are unstable during intergenerational transmission1,3, although the underlying mechanisms driving this instability remain unknown. Here, we investigated whether sequence variants flanking the FGF14 repeat locus affect its stability.
We first analyzed the FGF14 repeat locus and its flanking sequences in a set of 541 alleles from 339 individuals (including 32 patients with SCA27B) using Sanger sequencing. We defined the flanking sequences as the 15 nucleotides immediately adjacent to the repeat locus (T2T-CHM13v2.0, chr13:101,377,550–101,377,792) on both 5′ and 3′ ends according to the genomic context. We found that only 166 of the 541 alleles (30.68%) matched the T2T-CHM13 reference 5′-flanking sequence (5′-CAACCAACTTTCTGT), whereas 32 additional alleles (5.91%) carried a single terminal T > A substitution of the reference sequence (5′-CAACCAACTTTCTGA). Although we will hereafter refer to both the T2T-CHM13 reference 5′-flanking sequence and this variant carrying a single substitution as the reference 5′-flanking sequence (5′-RFS), it is not the major allele. The majority of the 541 alleles (343, 63.40%) contained a sequence variant (5′-CAACCAACTTTCTGTTAGTCATAGTACCCCAA) immediately flanking the repeat locus (Fig. 1a,b). Remarkably, this common 5′-flanking variant (5′-CFV) was exclusively observed in alleles carrying fewer than 30 GAA-pure repeats, whereas the 5′-RFS was only present in alleles with more than 30 repeats (Fig. 1b). Alleles shorter than 30 repeats were perfectly separated from longer alleles by the presence of the 5′-CFV, such that none of the pathogenic (GAA)≥250 expansions carried the 5′-CFV.
Fig. 1 |. A common 5′-flanking sequence variant is associated with smaller FGF14 GAA repeat sizes.
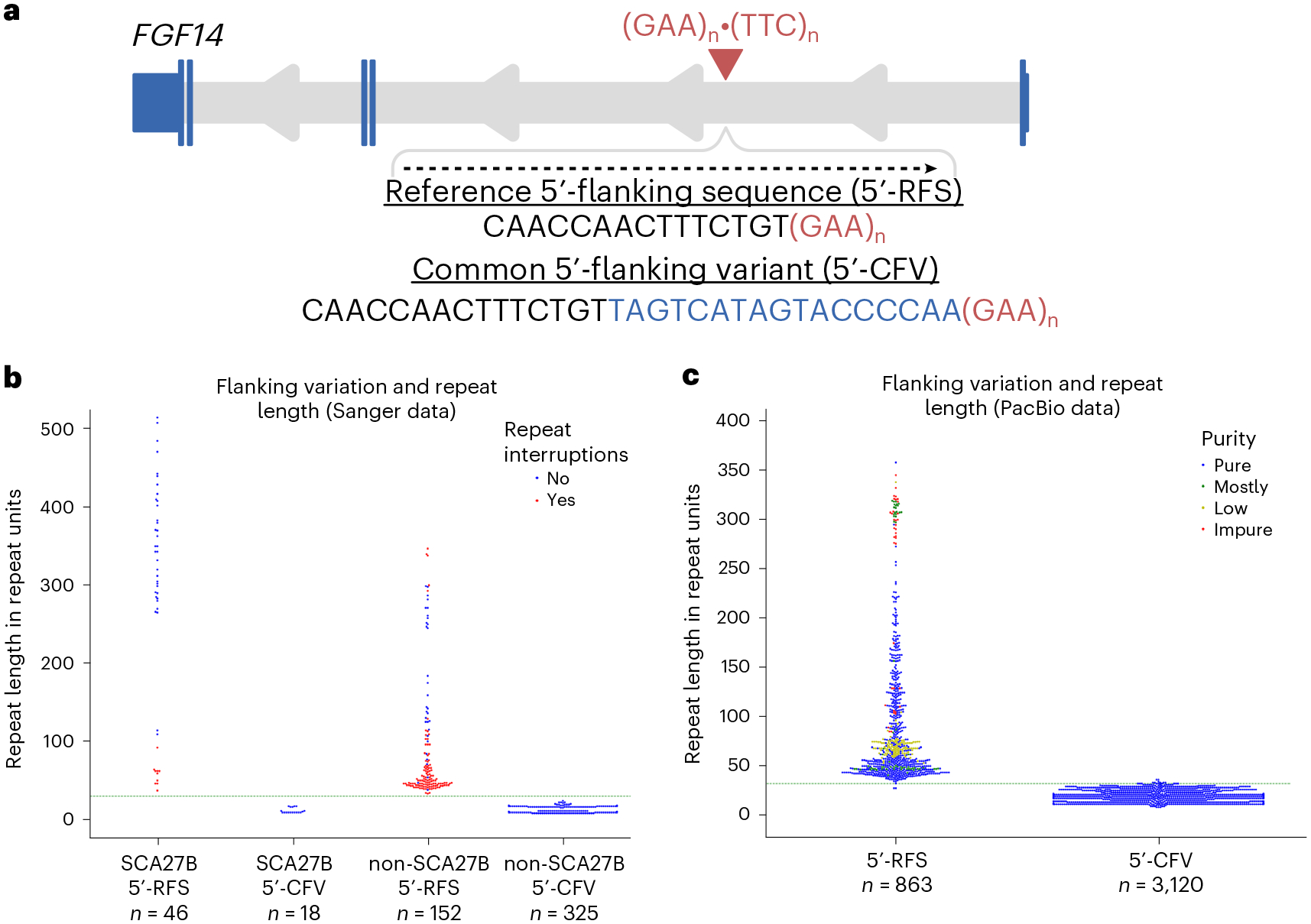
a, Schematic representation of the FGF14 gene, isoform 1b with the location of the (GAA)n•(TTC)n repeat locus in the first intron. The sequences of the reference 5′-flanking sequence (5′-RFS) and the common 5′-flanking variant (5′-CFV, in blue) are shown. The sequences are presented relative to the positive strand (genomic context). b, Swarmplot of repeat lengths as estimated by Sanger sequencing for 541 alleles shows that the 5′-CFV is consistently associated with alleles containing fewer than 30 GAA repeats, whereas the 5′-RFS is associated with larger alleles, including pathogenic ones. Each of the two alleles of patients with SCA27B is shown in either the 5′-RFS or 5′-CFV categories, even though only (GAA)≥250 alleles are pathogenic (all carrying the 5′-RFS). Blue dots represent uninterrupted GAA repeats, whereas red dots represent interrupted GAA repeats. c, Swarmplot of repeat lengths as estimated by PacBio HiFi sequencing for 3,983 alleles (from the Genomic Answers for Kids, Care4Rare-SOLVE and All of Us cohorts) shows a similar pattern. Alleles possessing any other flanking sequences and two alleles of over 800 repeat units carrying the 5′-RFS were omitted for clarity (Extended Data Fig. 1). The color of the data points corresponds to the GAA repeat motif purity (a color legend is shown in the top right corner of the plot). In b and c, the green dashed horizontal line indicates 30 GAA repeats. Abbreviations: 5′-CFV, common 5′-flanking variant; 5′-RFS, reference 5′-flanking sequence; SCA27B, spinocerebellar ataxia 27B.
We next sought to replicate these findings using PacBio HiFi sequencing in 2,191 individuals (4,382 alleles). We found that 863 alleles (19.69%) carried the 5′-RFS (Supplementary Note and Supplementary Fig. 1). The 5′-CFV was observed in 3,120 alleles (71.20%), which consistently had smaller repeat lengths than the alleles containing the 5′-RFS. Specifically, 3,103 (99.46%) of the 3,120 alleles containing the 5′-CFV had fewer than 30 GAA repeats and 3,120 (100.00%) of them were GAA-pure, whereas 861 (99.77%) of the 863 alleles containing the 5′-RFS had more than 30 triplets and 644 (74.62%) of them were GAA-pure (Fig. 1c). These results show that the 5′-CFV is strongly associated with alleles carrying fewer than 30 GAA repeats compared to the 5′-RFS. Finally, the 5′-flanking sequence in the remaining 399 alleles (9.11%) not carrying the 5′-CFV nor the 5′-RFS displayed a range of variations.
These variations mainly consisted of differences in the number of cytosines (Cs) and/or adenines (As) in the final six nucleotides of the 5′-CFV. We defined these groups as: C1 (5′-TAGTCATAGTACAA), C2 (5′-TAGTCATAGTACCAA), C3 (5′-TAGTCATAGTACCCAA) and C5 (5′-TAGTCATAGTACCCCCAA) by the number of terminal cytosines (Extended Data Fig. 1a). The 5′-CFV corresponds to the C4 group (5′-TAGTCATAGTACCCCAA). We observed 30 alleles with the C5 (0.68% of 4,382 alleles), 24 alleles with the C3 (0.55%), 116 alleles with the C2 (2.65%) and 168 alleles with the C1 (3.83%) variant. The alleles harboring the C3, C4 or C5 variants generally possessed fewer than 30 GAA repeats (3,159/3,177; 99.43%), whereas alleles with the C1 or C2 variants all possessed more than 30 GAA repeats (284/284; 100%) (Extended Data Fig. 1b). Many of these 5′-flanking sequences also included variants with a single or double terminal adenine. Three C4 alleles not counted as part of the 5′-CFV group were observed with a single terminal adenine. Analyzing the variants by their number of terminal adenines revealed that the only C3 sequence over 30 GAA repeats had a single terminal A (Extended Data Fig. 1b). In addition to those groups, ten alleles (0.23%) had a short 5′-flanking sequence (5′-CAACCAACTTTCT), all of which carried more than 30 GAA repeats. Finally, 48 alleles (1.10%) had a 5′-flanking sequence with other configurations. Together, these data indicate that the 5′-CFV, and any 5′-flanking sequence within one nucleotide variation, were overwhelmingly associated with shorter alleles containing fewer than 30 GAA repeats (3,159/3,176; 99.46%). Conversely, 5′-flanking variants with two or more nucleotide variations from the 5′-CFV were associated with sequences longer than 30 repeats (1,184/1,206; 98.18%).
In a further analysis, we observed no relationship between variants in the 3′-flanking sequence and the GAA repeat length, suggesting that only variants in the 5′-flanking region impact the FGF14 repeat locus stability (Supplementary Note and Extended Data Fig. 2). Furthermore, since Friedreich ataxia is the only other known disease caused by intronic (GAA)·(TTC) repeat expansions, we analyzed the flanking sequences surrounding the FXN repeat locus in 1,027 individuals and found no clear segregation of allele sizes by 5′- or 3′-flanking variants (Supplementary Fig. 2).
We next studied the intergenerational transmission of the FGF14 repeat in 478 intergenerational events by Sanger sequencing (67 events) and PacBio sequencing (411 events) (Fig. 2, Extended Data Fig. 3 and Supplementary Fig. 3). Allowing for ‘off-by-one’ triplet call errors in the PacBio dataset4, we observed that 295 of 297 alleles (99.33%) with the 5′-CFV were stably transmitted (Fig. 2). All of these 297 alleles were GAA-pure. The two nonstably transmitted alleles with the 5′-CFV each differed in size by two triplets in the offspring compared to the parent. We did not observe a single event involving deletion of part or all of the 5′-CFV upon transmission. In comparison, we found that alleles carrying the 5′-RFS exhibited an increasing degree of instability upon intergenerational transmission proportional to their length and GAA purity (Fig. 2 and Supplementary Note). To disambiguate the effects of flanking sequences on intergenerational stability from those of repeat length and purity, we used a regression model and found that the 5′-CFV was a significant predictor of stability (estimate = 0.560, standard error = 0.037, t = 14.94, P = 0) when controlling for repeat length and purity. This model explained 58% of the variance of intergenerational stability. Together, these data show that the 5′-CFV is associated with greater intergenerational stability than the 5′-RFS. Our data also confirm and extend previous findings showing that GAA repeat length and purity are major factors contributing to intergenerational instability5.
Fig. 2 |. Intergenerational instability of the FGF14 repeat locus.
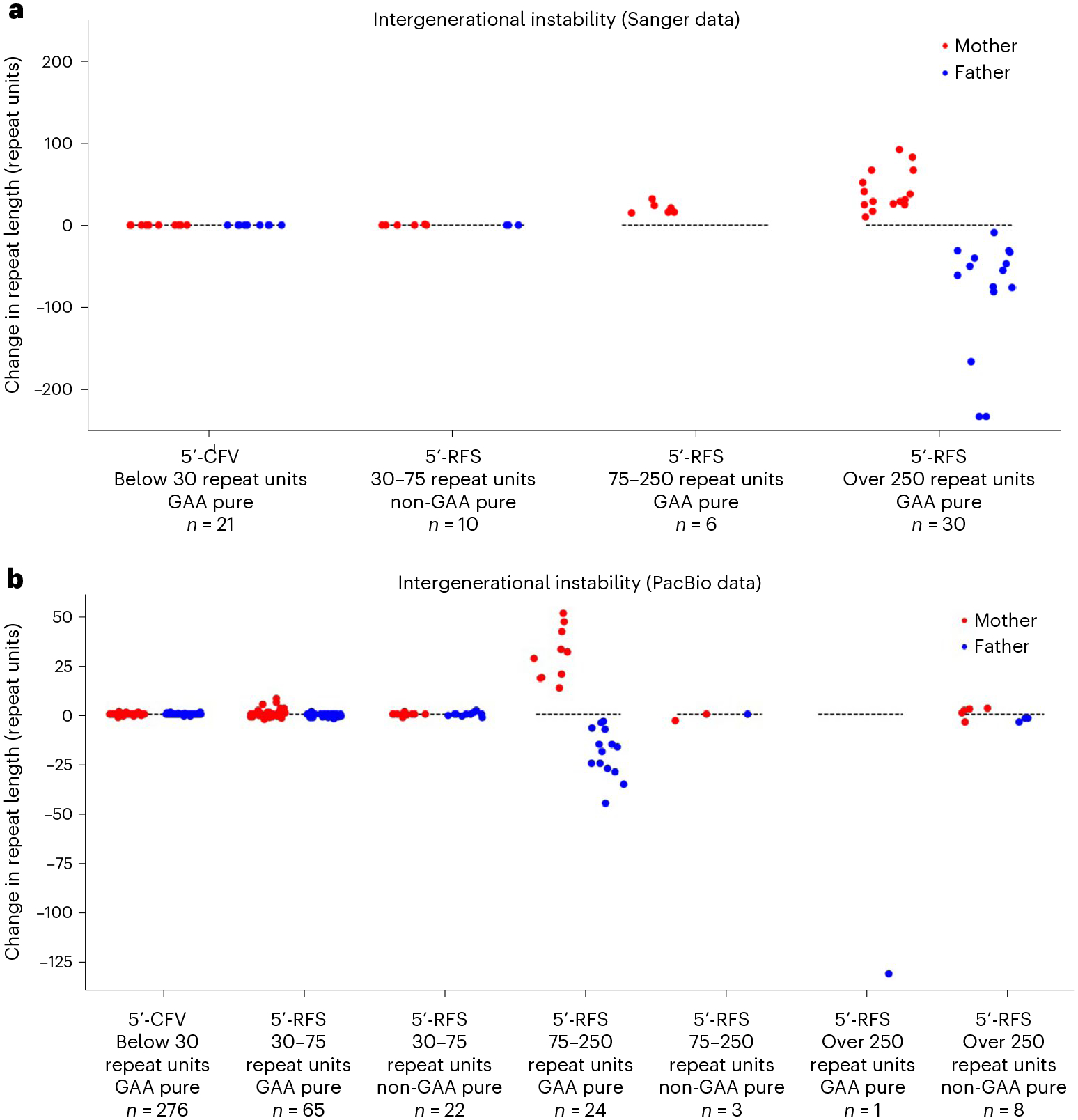
a,b, Change in GAA repeat length across intergenerational events, grouped by parental allele size, 5′-flanking sequence group (5′-RFS or 5′-CFV), and repeat purity as estimated by Sanger sequencing for 67 intergenerational events (a) and PacBio sequencing for 399 intergenerational events (from the Genomic Answers for Kids cohort) (b). The y axis shows the change in repeat length from parent to child. Contractions are plotted below the dashed lines, whereas expansions are plotted above them. Random noise was applied across the x axis within each category to improve data visualization. Twelve alleles with flanks other than the 5′-RFS or 5′-CFV were omitted for clarity (Extended Data Fig. 3). Red dots are alleles transmitted from mother to child, whereas blue dots represent alleles transmitted from father to child. Abbreviations: 5′-CFV, common 5′-flanking variant; 5′-RFS, reference 5′-flanking sequence.
We observed that the 5′-CFV occurred on all major haplotype groups defined using the 1000 Genomes and Human Genome Diversity Project cohort6 (Extended Data Fig. 4, Supplementary Note and Supplementary Fig. 4). The observation of the FGF14 repeat locus flanked by sequences within a few nucleotide variations of the human 5′-CFV in the reference genome of anthropoid primates (Supplementary Fig. 5) further suggests that the 5′-CFV represents the ancestral allele. Moreover, all hominoids, except Pongo abelii (Sumatran orangutan), carry a 5′-flanking sequence identical to the human 5′-CFV sequence. These results from reference genomes were replicated using individual genomes from 79 great apes (Supplementary Note and Supplementary Fig. 6). In contrast, the 5′-RFS is present on six of the ten major human haplotype groups studied (Extended Data Fig. 4), suggesting that it is a recurrent variant. Additional long-read data from seven unrelated patients with SCA27B revealed that the pathogenic expansion has arisen on at least three of those six haplotypes containing the 5′-RFS, suggesting that the repeat expansion can independently arise on different haplotypes (Supplementary Fig. 7).
Seeking to understand how the 5′-CFV may exert an effect on repeat stability, we analyzed single-molecule chromatin architectures surrounding the FGF14 repeat locus by Fiber-seq7 in postmortem cortical neuronal (NeuN+) and non-neuronal (NeuN−) nuclei. Neurons and glia exhibit high and low levels of FGF14 expression, respectively8. We detected increased chromatin accessibility on both flanks of the repeat locus, as measured by 6-methyl adenine (m6A) methylation, on fibers bearing the 5′-CFV sequence in NeuN+ nuclei relative to fibers carrying the 5′-RFS sequence in NeuN+ nuclei or fibers carrying the 5′-CFV sequence in NeuN− nuclei (Supplementary Fig. 8). Fibers carrying the C2A1 degenerate sequence showed a pattern of chromatin accessibility intermediate to fibers carrying the 5′-CFV and the 5′-RFS.
In summary, the current reference 5′-flanking sequence is in fact a minor allele at the FGF14 repeat locus and appears to confer a risk of developing SCA27B. Conversely, the 5′-CFV appears to stabilize the repeat and represents the major and phylogenetically ancestral allele despite being absent from reference assemblies. Our data showing near-perfect separation of short (<30 GAA repeats) from long (>30 GAA repeats) alleles by the presence of the 5′-CFV suggest that deletion of this variant is likely to lead to further expansion, rather than its deletion resulting from expansion of the repeat locus. The Fiber-seq data revealed increased chromatin accessibility surrounding the locus in the presence of the 5′-CFV, suggesting that this flanking sequence-dependent chromatin configuration may facilitate the recruitment of factors that promote tandem repeat stability. The four terminal cytosines of the 5′-CFV seem necessary for this process, as shown by the population-level data of GAA length variation among alleles with only one or two cytosines in that sequence as well as the Fiber-seq data demonstrating an altered chromatin state in a C2A1 allele. Irrespective of the mechanism, the 5′-CFV may represent a protective genomic element insulating the highly mutagenic (and likely functionally important) FGF14 repeat locus. Finally, identification of additional similar variants across the genome will likely yield further insight into the mechanisms protecting against tandem repeat expansion.
Methods
Institutional review board approval
The institutional review board of the Montreal Neurological Hospital, Montreal (MPE-CUSM-15–915); the Centre Hospitalier de l’Université de Montréal, Montreal (ND02.045); the Clinical Trials Ontario (REB# 1577 CTO); the Children’s Mercy Kansas City (Study #11120514); the Centre Hospitalier Régional Universitaire de Nancy, Nancy (2020PI220); the Center for Neurology, Tübingen (598/2011BO1); the University of Western Australia, Perth (RA/4/20/1008); and the Xiangya Hospital, Central South University, China (202310206) approved this study. We obtained written informed consent from all the participants in this study or their legal guardians.
Sanger sequencing
Genomic DNA isolated from peripheral blood using standard methods was used for all genetic analyses performed in this study. PCR reactions were performed in a 30 μl volume using the Phusion Flash High-Fidelity PCR Master Mix 2X (catalog no. F548L, Thermo Fisher Scientific) with 1 μM forward and reverse primers (forward primer, 5′-TGCAAATGAAGGAAAACTCTT-3′; reverse primer, 5′-CAATGATGAATTAAGCAGTTCC-3′) and 120 ng genomic DNA. Thermal cycling conditions were 98 °C for 3 min, 12 cycles of 98 °C for 10 s, 65 °C for 15 s (decreasing the annealing temperature by 1 °C every two cycles), 72 °C for 3 min, 20 cycles of 98 °C for 10 s, 59 °C for 15 s, 72 °C for 3 min and a final extension at 72 °C for 5 min. Sanger sequencing of PCR amplification products9 was performed at the Centre d’Expertise et de Services Génome Québec using the ABI 3730xl DNA Analyzer (Applied Biosystems) and in the Laboratoire de Génétique du Centre Hospitalier Régional Universitaire de Nancy using the ABI 3130xl DNA Analyzer (Applied Biosystems). Sequences were analyzed using Snap-Gene v.5.0.8 software (Dotmatics). A total of 339 samples (146 controls and 193 patients with late-onset ataxia, including 32 patients with SCA27B) were sequenced. The flanking sequences and repeat of 541 of the 678 alleles could be determined and were kept for downstream analysis. Alleles were assessed for the presence of sequence variants in the 5′- and 3′-flanking regions and repeat interruptions.
Sizing of expanded FGF14 alleles
We also measured the size of expanded FGF14 alleles by capillary electrophoresis of FAM-labeled long-range PCR amplification products9. The following primers were used: forward primer, 5′−6-FAM-TGCAAATGAAGGAAAACTCTT-3′; reverse primer, 5′-CAATGATGAATTAAGCAGTTCC-3′. Amplification products were analyzed on an ABI 3730xl DNA Analyzer (Applied Biosystems) with a 50-cm POP-7 capillary using the GeneScan 1200 Liz Dye Size Standard (catalog no. 4379950, Applied Biosystems). Results were analyzed using the GeneMapper software using the built-in microsatellite default settings (version 6.0, Applied Biosystems).
Targeted nanopore sequencing
Results of Sanger sequencing for samples carrying a large FGF14 allele were confirmed by means of targeted long-read Oxford Nanopore Technologies (ONT) sequencing. ONT sequencing was performed on a total of 47 individuals, 32 of whom had SCA27B. PCR amplification products were selected for molecular size >400 bp using SPRIselect paramagnetic beads for DNA size selection following manufacturer’s protocol (catalog no. B23319, Beckman Coulter Life Sciences). Presequencing size selection was performed to increase coverage depth of larger alleles. Amplicons were normalized to 150 ng μl−1 and then multiplexed using native barcoding expansion PCR-free library preparation kits and the SQK-LSK109 sequencing kit as per manufacturer’s instructions (Oxford Nanopore Technologies), multiplexed and sequenced on the MinION or PromethION platform using the R9.4.1 flow cell (Oxford Nanopore Technologies). Each run included a libprep negative control. Reads were base-called and demultiplexed with stringent barcodes_both_ends setting using Guppy 5.0.13. Sequences were aligned to the GRCh37 reference human genome using minimap2 (v2.22)10 with the predefined settings for nanopore data. STRique11-v0.4.2 was then used to count the number of repeated units observed for each read spanning the FGF14 tandem repeat site. Motif purity was assessed for each sequencing read and calculated as the number of GAA units observed in the portion of the repeat locus-spanning segment of the read divided by the STRique estimation of the total number of repeat units for that read.
Whole-genome nanopore sequencing
Whole-genome ONT sequencing was performed on three unrelated patients with SCA27B within the Garvan Institute Sequencing Platform (Sydney, Australia). Briefly, genomic DNA was sheared to ~15–20 kb fragment size using Covaris g-tubes (catalog no. 520079, Covaris), and libraries were prepared from ~1 to 2 μg DNA using a ligation kit (SQK-LSK114, ONT). Each library was loaded onto an ONT PromethION flow cell (R10.4.1) and sequenced on a P48 device. Samples were run for a maximum duration of 72 h, with one to three nuclease flushes and reloads performed during the run, where necessary, to maximize sequencing yield. Raw ONT sequencing data were converted from FAST5 to the more compact BLOW5 format12 in real time during sequencing using slow5tools (v0.3.0)13. Data were base-called with Guppy (v6; HAC model) using the Buttery-eel wrapper for BLOW5 input14. FASTQ reads passing quality control were aligned to the GRCh38 reference genome using minimap2 (v2.22)10 with the following optional parameters: -x map-ont -a–secondary=no–MD.
Pacific Biosciences high-fidelity sequencing
The 2,191 samples whose whole-genome long-read sequencing data were used in this study were drawn from three sources: 1,126 samples spanning 525 families from the Children’s Mercy Research Institute’s Genomic Answers for Kids program, 1,027 unrelated samples from the All of Us Program Long Read Data release CDRv7 (April 2023: https://support.researchallofus.org/hc/en-us/articles/14769699298324-v7-Curated-Data-Repository-CDR-Release-Notes-2022Q4R9-versions) and 38 samples spanning 12 families from the Care4Rare Canada research program’s Care4Rare-SOLVE study. The Genomic Answers for Kids dataset included 224 trios, from which 411 intergenerational transmission events were able to be confidently resolved. The Genomic Answers for Kids dataset included persons from a wide range of self-reported geographic origins (European, 85.23%; Admixed American, 9.38%; African, 3.09%; South Asian, 1.50%; East Asian, 0.80%) and largely focused on children with rare diseases and their unaffected family members. The All of Us Program Long Read Data released in CDRv7 was composed of adults unrelated to each other and not known to have a rare disease. All persons in that dataset self-reported as black or African American. The Care4Rare Canada dataset was composed of individuals of self-reported European geographic origin with various unsolved rare genetic diseases (no patient with SCA27B was included in this dataset) and their relatives, who were often unaffected. Samples from the Genomic Answers for Kids program were sequenced to a coverage of approximately 8–25× using one (most parents) to three (most probands) SMRT cells per sample on a Sequel IIe platform at Children’s Mercy Hospital. The samples comprising the All of Us Program Long Read Data released in CDRv7 were sequenced to a coverage of approximately 8× using a single SMRT cell per sample on a Sequel IIe platform at the HudsonAlpha Institute for Biotechnology. The Care4Rare Canada samples were sequenced to a coverage of approximately 30× for affected individuals (n = 26) using three SMRT cells per sample and a minimum of 10× for unaffected family members (n = 12) using a single SMRT cell per sample on a Sequel IIe platform at the Pacific Biosciences Applications lab in Menlo Park, CA.
All samples were aligned to the GRCh38 build of the human genome. TRGT v0.3.3 or v0.3.4 software4 was then run on each sample using default parameters to resolve the flanking and repeat sequences of the two alleles in each person. The repeat specification given to TRGT was for the genomic region chr13:102161544–102161756, which includes the FGF14 GAA repeat locus along with 25 bp of flanking sequence on each side. The alleles called by TRGT were then analyzed for variants in the flanking regions as well as the sequence content of the repetitive region. While the genomic data were aligned to GRCh38, the subsequent analyses using the sequences generated by TRGT were done comparing to the T2T-CHM13 reference assembly, as the reference 5′-flanking sequence in the T2T-CHM13 assembly at this locus is more prevalent compared to the corresponding sequence in GRCh38 (Supplementary Note). The repetitive regions were segmented based on fuzzy matching of the repeat motifs, such that up to one off-pattern nucleotide was tolerated every 12 bp. Then, the GAA purity of each allele was calculated as the proportion of the allele (excluding the flanking sequence) that was spanned by segmentations carrying the GAA motif. The following ordinal scale was used to classify motif purity: impure (non-GAA motif), low purity (<75% GAA motif), mostly pure (75–95% GAA motif) and pure (≥95% GAA motif). The threshold for an allele to be considered GAA-pure was set at 95% purity.
An additional cohort of two unrelated patients with SCA27B of self-reported French-Canadian descent was recruited through the Care4Rare-SOLVE study. These samples were prepared and processed in the same way as the controls from that cohort described above.
An additional cohort of two unrelated patients with SCA27B of self-reported Han Chinese descent was recruited. Library preparation utilized the SMRTbell Express Template Prep Kit 2.0 (catalog no. 100-938-900, Pacific Biosciences), following the manufacturer’s protocol. These samples were sequenced to a coverage exceeding 100× for target region using a single SMRT cell per sample on a Sequel IIe platform. Subsequently, PacBio subreads were converted into HiFi reads through circular consensus sequencing (CCS) with default parameters. Following sequencing, the HiFi reads were aligned to GRCh38 using minimap2 v2.24. TRGT was used to determine the flanking sequence and repeat count of the FGF14 repeat locus, as described above.
For haplotype analysis, we collected stable population variants from a recent analysis of the 1000 Genomes and Human Genome Diversity Project datasets6. This dataset had been statistically phased and represented a broad array of ancestries. From it, we collected 15 biallelic variants with a minor allele frequency >0.2 which were within 10 kb of the FGF14 repeat locus. We then fitted a k-means clustering model so that it could assign a haplotype group label to new alleles, given phased calls of these 15 biallelic variants. We chose ten clusters for this analysis to define ten ‘major haplotype groups’ from this diverse set of short-read data. PacBio genomes had single nucleotide variants (SNVs) and insertion/deletion polymorphisms (indels) called with PEPPER-Margin-DeepVariant v0.8 (ref. 15), structural variants called with PBSV v2.9.0 (https://github.com/PacificBiosciences/pbsv) and their FGF14 repeat locus and flanking variant evaluated with TRGT4. The VCF files from these three methods were then input, along with the BAM file, to HiPhase v1.0.0 (ref. 16), which returned physical phase blocks. Samples where physical phasing could not be established for at least 10 kb on either side of the FGF14 repeat were excluded, leaving 1,674 samples for the haplotype analysis. We then used the fitted k-means clustering model to assign haplotype group IDs to each of the 3,348 haplotypes so that the labels on the long-read samples matched those on the short-read samples.
The population descriptors included here did not factor in any way into the sample selection used for this manuscript. An exception was received for this manuscript to the Data and Statistics Dissemination Policy from the All of Us Resource Access Board.
Phylogenetic analysis
FASTQ files of the 79 great apes’ whole genomes generated by Prado-Martinez et al.17 were downloaded from the Sequence Read Archive under the accession number PRJNA189439. The FASTQ files were aligned to the GRCh38 reference genome with BWA v0.7.17 (ref. 18). The aligned genomic data were then processed with Expansion-Hunter v5.0.0 (ref. 19) (configured to anticipate the presence of the human common 5′-flanking variant as the reference sequence) to determine the size of the repeat region and REViewer v0.2.7 (ref. 20) to determine the status of the flanking variant and check for quality. From this dataset, we determined the repeat length and phased flanking variant for both alleles from each of the 79 genome sequences. Analysis of the reference genomes of the human and 14 non-human primate species (Pan troglodytes, panTro6 assembly; Pan paniscus, panPan3 assembly; Gorilla gorilla gorilla, gorGor6 assembly; Nomascus leucogenys, nomLeu3 assembly; Pongo abelii, ponAbe3 assembly; Chlorocebus sabaeus, chlSab2 assembly; Macaca fascicularis, macFas5 assembly; Macaca mulatta, rheMac10 assembly; Papio anubis, papAnu4 assembly; Callithrix jacchus, calJac4 assembly; Saimiri boliviensis, saiBol1 assembly; Tarsius syrichta, tarSyr2 assembly; Microcebus murinus, micMur2 assembly; Otolemur garnettii, otoGar3 assembly) was performed using the UCSC genome browser. Multiple sequence alignment of the reference genomes of human and non-human primate species was performed using Jalview (version 2)21,22.
Fiber-seq
Single molecular chromatin architectures were assessed utilizing minor modifications to Fiber-seq protocol7. All human samples used for Fiber-seq were collected and used de-identified and in accordance with the policies of Icahn School of Medicine at Mount Sinai and in regulation with its institutional review board. Anonymized postmortem brain tissue samples from neurologically healthy individuals are initially homogenized in hypotonic lysis buffer (0.32 M sucrose, 5 mM CaCl2, 3 mM magnesium acetate, 0.1 mM EDTA, 10 mM Tris-HCl pH 8, 1 mM dithiothreitol and 0.1% Triton X-100). A sucrose gradient (1.8 M sucrose, 3 mM magnesium acetate, 1 mM dithiothreitol and 10 mM Tris-HCl, pH 8) was layered under the homogenate and nuclei were pelleted via sucrose gradient ultracentrifugation at 110,000 × g for 60 min at 4 °C in a swinging bucket rotor. Nuclei are then collected and labeled with NeuN Ab (Anti-NeuN Alexa 488 antibody, catalog no. MAB377X, EMD Millipore) rotating for 1 h at 4 °C protected from ambient light. Fluorescence-activated nuclei sorting (as described in Girdhar et al.23) was used to sort 1.5 million nuclei from 1 g frozen samples of cortex. 4,6-Diamidino-2-phenylindole was added at a concentration of 5 μg ml−1 just before fluorescence-activated nuclei sorting. NeuN+ and NeuN− nuclei were sorted using BD Biosciences FACSAria II. Nuclei were then incubated with Hia5 bacterial m6A-methyltransferase (m6A-MTase) (generated in-house) for selective tagging of nucleosome-free accessible linker DNA. We incubated up to 1 × 106 intact nuclei with 1 U Hia5 per 20 μg DNA. Libraries were prepared using the SMRTbell Prep Kit 3.0 (catalog no. 102-182-700, Pacific Biosciences), following the manufacturer’s protocol, and sequenced on a Sequel IIe platform.
Raw PacBio sequenced reads are output from the sequencer in CCS format with polymerase kinetics (hifi-kinetics) information per long read (read). If sequencing information is in raw subread format, CCS with hifi-kinetics can be generated using pbccs with the –hifi-kinetics flag (ccs.how). Kinetic information shows the average interpulse duration of polymerase pausing per base pair sequenced, where methylated adenines have longer interpulse durations than unmodified bases. CCS reads with hifi-kinetics are then processed using the fibertools predict-m6a command and PacBio primrose3 to add m6A and 5-methyl cytosine (5mC) information to the reads, respectively24. Next, we predict locations of methyltransferase-sensitive patches on each fiber using a hidden Markov model on the m6A information by running the fibertools add-nucleosomes command. A methyltransferase-sensitive patch is created by combining multiple m6A positions that the model identifies as a single patch of accessibility by the methyltransferase. These patches and their inverse, which are called nucleosomes, are added to the BAM file as well. The reads with base-modification information are then aligned to the T2T-CHM13 human genome release using pbmm2 align (version 1.13.1).
Statistical analysis
We used a linear regression model to evaluate the effect of the flanking sequence on intergenerational stability correcting for repeat length and purity. We analyzed the data in Python version 3.10.9. P < 0.05 was considered significant. All analyses were two sided.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Extended Data
Extended Data Fig. 1 |. Relationship between 5′-flanking sequences and FGF14 GAA repeat lengths.
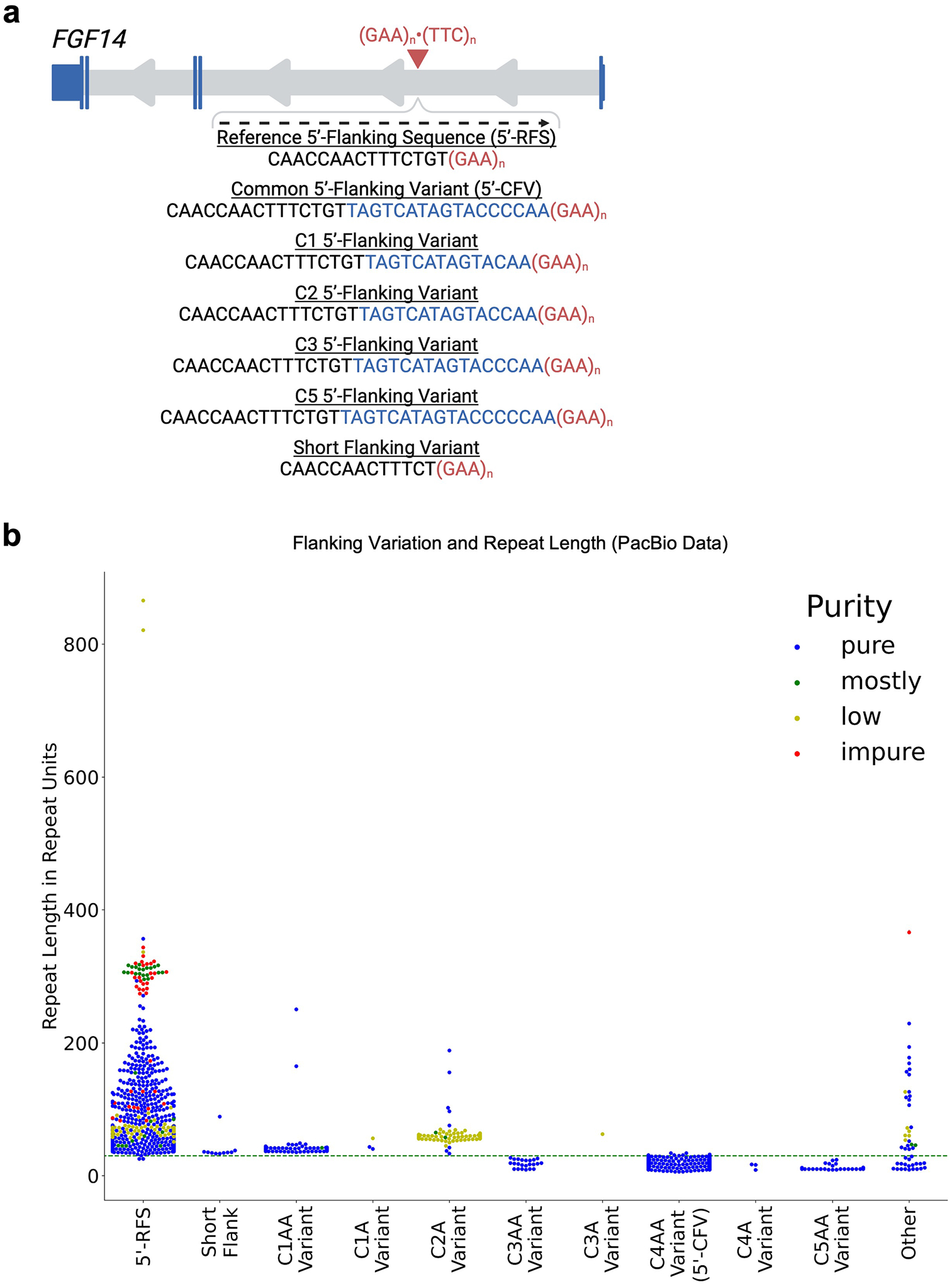
a, Schematic representation of the FGF14 gene, isoform 1b with the location of the (GAA)n·(TTC)n repeat locus in the first intron. The sequences of the reference 5′-flanking sequence (5′-RFS), the common 5′-flanking variant (5′-CFV; C4 variant), and the C1, C2, C3, C5, and short flanking variant sequences are shown. The sequences of the C1 through C5 variants are highlighted in blue. The sequences are presented relative to the positive strand (genomic context). b, Swarmplot related to Fig. 1c showing repeat lengths as estimated by PacBio HiFi sequencing for 4,382 alleles (from the Genomic Answers for Kids, Care4Rare-SOLVE, and All of Us cohorts) including each of the C1 through C5 sequence variants, separated into subgroups based on the presence of a single terminal adenine (A) or dual terminal adenines (AA). No alleles with C2AA or C5A 5′-flanking variants were found. Three C4 alleles not counted as part of the 5′-CFV group were observed with a single terminal adenine. This plot also extends the y-axis to show the two alleles of over 800 repeat units carrying the 5′-RFS that were not plotted in Fig. 1c for visual clarity. The color of the data points corresponds to the GAA repeat motif purity (a color legend is shown in the top right corner of the plot). The green dashed horizontal line indicates 30 GAA repeats. Abbreviations: 5′-CFV, common 5′-flanking variant; 5′-RFS, reference 5′-flanking sequence.
Extended Data Fig. 2 |. Relationship between 3′-flanking sequences and FGF14 GAA repeat lengths.
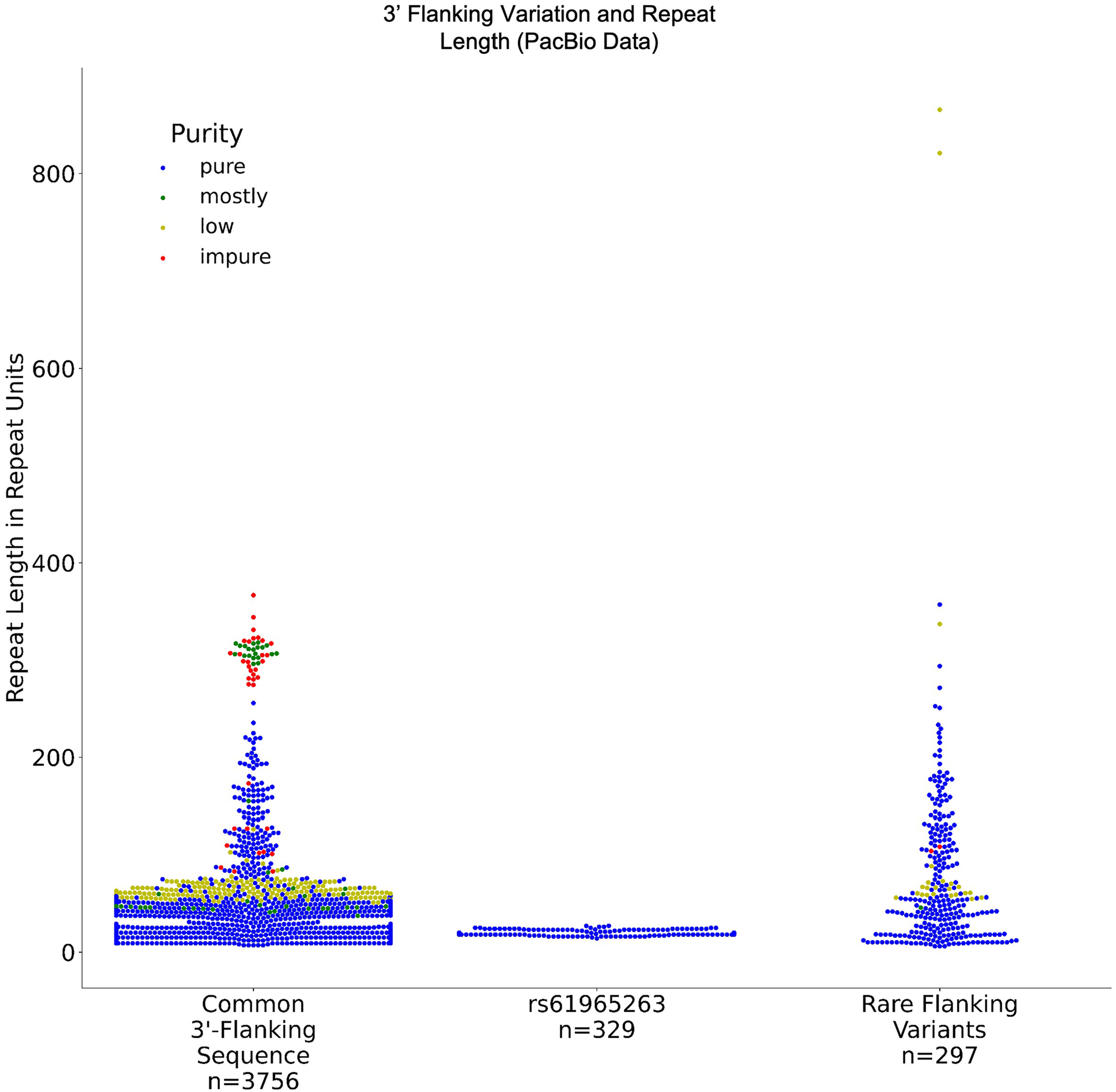
Distribution of repeat lengths estimated by PacBio HiFi sequencing for 4,382 alleles (data from the Genomic Answers for Kids, Care4Rare-SOLVE, and All of Us cohorts) in relation to variations observed in the 3′-flanking sequence of the FGF14 repeat locus. The distribution of FGF14 GAA repeat lengths in alleles with the common 3′-flanking sequence, alleles with the single nucleotide variation rs61965263, and alleles with other rare flanking variants is illustrated. The color of the data points corresponds to the GAA repeat motif purity (a color legend is shown in the top right corner of the plot).
Extended Data Fig. 3 |. Analysis of parent-offspring transmission of the FGF14 repeat according to the 5′-flanking sequence variant.
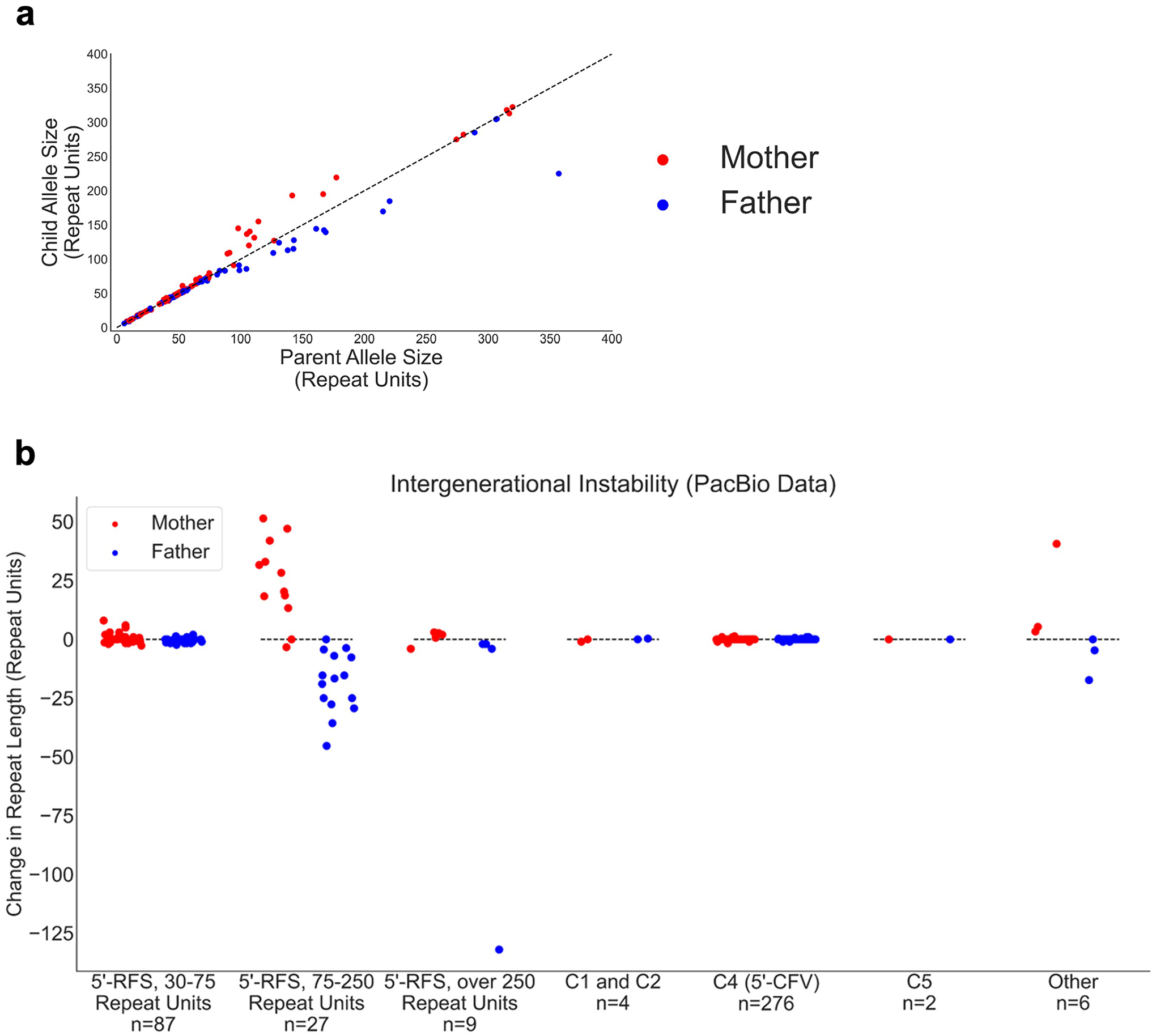
a, Analysis of GAA repeat size changes across 411 intergenerational transmissions (from the Genomic Answers for Kids cohort) as estimated by PacBio HiFi sequencing. Contractions are plotted below the dashed identity line while expansions are plotted above that line. b, Change in GAA repeat length across 411 intergeneration transmission (from the Genomic Answers for Kids cohort) as measured by PacBio HiFi sequencing separated by flanking variant group and parental allele size. The number of intergenerational transmission events in each group is indicated below the x-axis. The y-axis shows the change in repeat length from parent to child. Contractions are plotted below the dashed lines while expansions are plotted above them. Random noise was applied across the x-axis within each category to maximize data visualization. This panel extends Fig. 2b by plotting the 12 additional intergenerational events involving alleles carrying a C1, C2, C5, or other rare 5′-flanking sequence variant. In a and b, red dots are alleles passed from mother to child, while blue dots represent alleles passed from father to child. Abbreviations: 5′-CFV, common 5′-flanking variant; 5′-RFS, reference 5′-flanking sequence.
Extended Data Fig. 4 |. Haplotype analysis of the FGF14 flanking sequence variants.
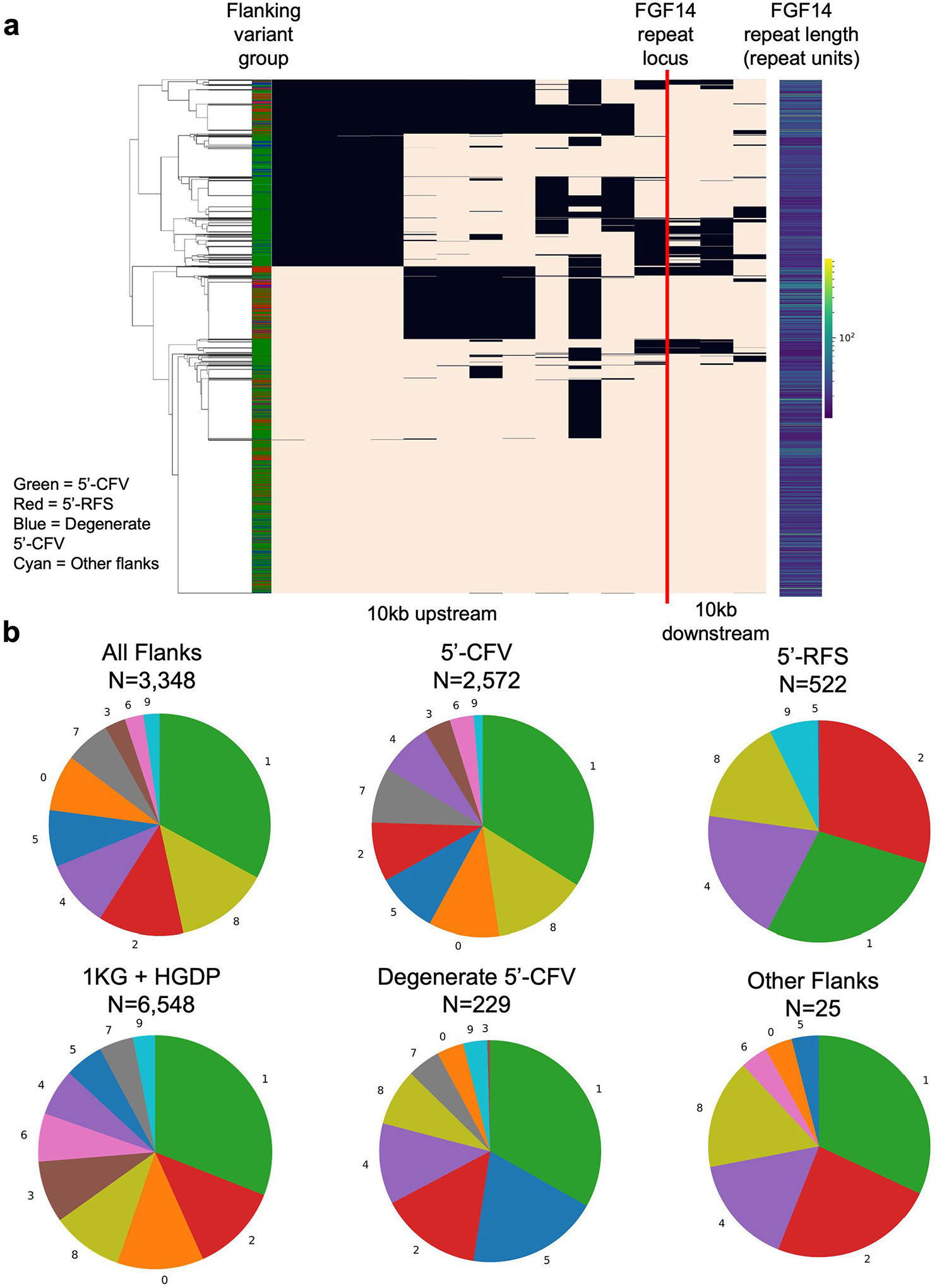
Haplotype analysis of the FGF14 flanking sequence variants for 1,674 individuals (from the Genomic Answers for Kids and All of Us cohorts). a, Visualization of haplotypes for 1,674 individuals physically phased through the FGF14 repeat locus. Each row represents one of two alleles per individual. A color-coded legend adjacent to the dendrogram indicates the flanking variant group on each allele: green for 5′-CFV, red for 5′-RFS, blue for degenerate 5′-CFV sequences, and cyan for other flanking sequences. The 15 columns in the plot represent the variant status of each allele for 15 common SNVs derived from the 1000 Genomes and Human Genome Diversity Project (HGDP) dataset, with black indicating reference genotype and tan showing alternate genotype. A vertical red line marks the location of the GAA repeat locus. The heatmap on the right side of the plot displays the GAA repeat length of each allele, with yellow indicating larger repeats and dark blue smaller ones. b, Frequency distribution of the 10 major haplotype groups for each flanking sequence variant. The lower-left pie chart shows the distribution of the 10 major haplotype groups in the 1000 Genomes and HGDP dataset. The number of alleles plotted in each pie chart is given above the chart. Abbreviations: 1KG, 1000 Genomes; 5′-CFV, common 5′-flanking variant; 5′-RFS, reference 5′-flanking sequence; HGDP, Human Genome Diversity Project.
Supplementary Material
Acknowledgements
We thank all the individuals who participated in this study. We gratefully acknowledge All of Us participants for their contributions, without whom this research would not have been possible. We also thank the National Institutes of Health’s All of Us Research Program for making available the participant data examined in this study. We thank the Centre d’Expertise et de Services Génome Québec for assistance with Sanger sequencing. We thank Pacific Biosciences Applications lab in Menlo Park, CA, and Pacific Biosciences bioinformatics team for HiFi sequencing and alignment of the Care4Rare Canada dataset. This work was supported by the National Institutes of Health (NIH) National Institute of Neurological Disorders and Stroke (grant 2R01NS072248-11A1 to S.Z.), the NIH National Human Genome Research Institute (grant R21HG013397 to M.C.D. and S.Z.), the All of Us Data and Research Center through support of the long-read demo projects (to M.C.D. and S.Z.), the Fondation Groupe Monaco (to B.B.), the Canadian Institutes of Health Research (grant 189963 to B.B.) and the Care4Rare Canada Consortium, funded in part by Genome Canada and the Ontario Genomics Institute (grant OGI-147 to K.M.B.), the Canadian Institutes of Health Research (grant GP1-155867 to K.M.B.), Ontario Research Fund, Genome Quebec and the Children’s Hospital of Eastern Ontario Foundation. This work was also supported by the European Joint Programme on Rare Diseases, under the EJP RD COFUND-EJP 825575 via the Deutsche Forschungsgemeinschaft grant 441409627 as part of the PROSPAX consortium (to M.S. and B.B.), and the National Key R&D Program of China (grant 2021YFA0805200 to H.J.). This work was supported in part by the Bioinformatics for Next Generation Sequencing shared resource facility within the Tisch Cancer Institute at the Icahn School of Medicine at Mount Sinai, which is partially supported by NIH grant P30CA196521. This work was also supported in part through the computational resources and staff expertise provided by Scientific Computing at the Icahn School of Medicine at Mount Sinai and supported by the Clinical and Translational Science Awards grant UL1TR004419 from the National Center for Advancing Translational Sciences. Research reported in this paper was supported by the Office of Research Infrastructure of the National Institutes of Health under award numbers S10OD026880 and S10OD030463. This work was also supported by the Australian Medical Research Future Fund Genomics Health Futures Mission Grants 2007681 and 2023126 (to I.W.D.). N.M.T. is supported by the National Institute on Drug Abuse (grant RF1DA048810) and the National Institute of Neurological Disorders and Stroke (grant R01NS106229). H.H. is supported by the Wellcome Trust, the UK Medical Research Council and the UCLH/UCL Biomedical Research Centre. The Nussenzweig laboratory is supported by the Intramural Research Program of the NIH funded in part with federal funds from the National Cancer Institute under contract HHSN261201500003. G.R. is supported by an EL2 Investigator Grant (APP2007769) from the Australian National Health and Medical Research Council (NHMRC). S.A. is supported by the National Institute on Drug Abuse (grant DP1DA056018). We thank generous donors to Genomic Answers for Kids program (to T.P.) at Children’s Mercy Kansas City. D.P. and G.F.D.G. hold fellowship awards from the Canadian Institutes of Health Research. The funders had no role in the conduct of this study.
Footnotes
Online content
Any methods, additional references, Nature Portfolio reporting summaries, source data, extended data, supplementary information, acknowledgements, peer review information; details of author contributions and competing interests; and statements of data and code availability are available at https://doi.org/10.1038/s41588-024-01808-5.
Code availability
Code for running TRGT and relevant data analysis is available through our manuscript companion repository: https://github.com/ZuchnerLab/FGF14FlankingVariant and https://doi.org/10.5281/zenodo.11239003 (ref. 25).
Competing interests
E.D. is an employee of Pacific Biosciences. M.S. has received consultancy honoraria from Ionis, Prevail, Orphazyme, Servier, Reata, GenOrph and AviadoBio, all unrelated to the present manuscript. M.A.E. is an employee of Pacific Biosciences. S.Z. received consultancy honoraria from Neurogene, Aeglea BioTherapeutics and Applied Therapeutics and is an unpaid officer of the TGP Foundation, all unrelated to the present manuscript. The other authors declare no competing interests.
Extended data is available for this paper at https://doi.org/10.1038/s41588-024-01808-5.
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s41588-024-01808-5.
Contributor Information
All of Us Research Program Long Read Working Group:
Emily Bateman, Chelsea Berngruber, Fabio Cunial, Colleen P. Davis, Huyen Dinh, Harsha Doddapaneni, Kim Doheny, Shannon Dugan-Perez, Tara Dutka, Evan E. Eichler, Philip Empey, Sarah Fazal, Chris Frazar, Kiran Garimella, Jessica Gearhart, Richard Gibbs, Jane Grimwood, Namrata Gupta, Salina K. Hall, Yi Han, William T. Harvey, Jess Hosea, PingHsun Hsieh, Jianhong Hu, Yongqing Huang, James Hwang, Michal Izydorczyk, Hyeonsoo Jeong, Ziad Khan, Sarah Kirkpatrick, Michelle Kokosinski, Sam Kovaka, Edibe Nehir Kurtas, Rebecca Lakatos, Emily LaPlante, Samuel K. Lee, Niall Lennon, Shawn Levy, Qiuhui Li, Lee Lichtenstein, Glennis A. Logsdon, Chris Lord, Ryan Lorig-Roach, Medhat Madmoud, Anant Maheshwari, Beth Marosy, Heer Mehta, Ginger Metcalf, David Mohr, Carolina Montano, Luke Morina, Yulia Mostovoy, Anjene Musick, Donna Muzny, Shane Neph, Justin Paschall, Karynne Patterson, Arianna Pionzio, David Porubsky, Nripesh Prasad, Allison N. Rozanski, Alba Sanchis-Juan, Michael C. Schatz, Sophie Schwartz, Alan Scott, Adriana Sedeno-Cortes, Fritz Sedlazeck, Tristan Shaffer, Hua Shen, Beri Shifaw, Joshua D. Smith, Natthapon Soisangwan, Andrew Stergachis, Hang Su, Michael Talkowski, Winston Timp, Vanesa Vee, Evie Wan, Yuanyuan Wang, George Weissenberger, Julie Wertz, Marsha Wheeler, Christopher Whelan, DongAhn Yoo, Shadi Zaheri, Xinchang Zheng, Yiming Zhu, and Michelle Zilka
Data availability
The data created through the All of Us Program Long Read Data release CDRv7 (April 2023: https://support.researchallofus.org/hc/en-us/articles/14769699298324-v7-Curated-Data-Repository-CDR-Release-Notes-2022Q4R9-versions) are available through the All of Us Research Program researcher workbench (https://researchallofus.org/). The Care4Rare-SOLVE data are available through Genomics4RD (https://www.genomics4rd.ca) via controlled access requests to genomics4rd@cheo.on.ca. The data created as part of Genomic Answers for Kids are available through NIH/NCBI dbGaP (https://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs002206.v4). Genome sequences of the 79 great apes generated by Prado-Martinez et al.17 were downloaded from the Sequence Read Archive under the accession number PRJNA189439. Patient-level whole-genome sequencing data are not publicly available, as they could compromise privacy and have not been consented for open sharing. Data from Sanger sequencing have not been consented for sharing. Additional data that support the findings of this study are available on request from the corresponding author (M.C.D.).
References
- 1.Pellerin D et al. Deep intronic FGF14 GAA repeat expansion in late-onset cerebellar ataxia. N. Engl. J. Med 388, 128–141 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rafehi H et al. An intronic GAA repeat expansion in FGF14 causes the autosomal-dominant adult-onset ataxia SCA27B/ATX-FGF14. Am. J. Hum. Genet 110, 1018 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Méreaux JL et al. Clinical and genetic keys to cerebellar ataxia due to FGF14 GAA expansions. EBioMedicine 99, 104931 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dolzhenko E et al. Characterization and visualization of tandem repeats at genome scale. Nat. Biotechnol 10.1038/s41587-023-02057-3 (2024). [DOI] [PubMed] [Google Scholar]
- 5.Sakamoto N et al. GGA*TCC-interrupted triplets in long GAA*TTC repeats inhibit the formation of triplex and sticky DNA structures, alleviate transcription inhibition, and reduce genetic instabilities. J. Biol. Chem 276, 27178–27187 (2001). [DOI] [PubMed] [Google Scholar]
- 6.Koenig Z et al. A harmonized public resource of deeply sequenced diverse human genomes. Genome Res 10.1101/gr.278378.123 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stergachis AB, Debo BM, Haugen E, Churchman LS & Stamatoyannopoulos JA Single-molecule regulatory architectures captured by chromatin fiber sequencing. Science 368, 1449–1454 (2020). [DOI] [PubMed] [Google Scholar]
- 8.Duffy MF, et al. Divergent patterns of healthy aging across human brain regions at single-cell resolution reveal links to neurodegenerative disease. Preprint at bioRxiv 10.1101/2023.07.31.551097 (2023). [DOI] [Google Scholar]
- 9.Bonnet C et al. Optimized testing strategy for the diagnosis of GAA-FGF14 ataxia/spinocerebellar ataxia 27B. Sci. Rep 13, 9737 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li H Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Giesselmann P et al. Analysis of short tandem repeat expansions and their methylation state with nanopore sequencing. Nat. Biotechnol 37, 1478–1481 (2019). [DOI] [PubMed] [Google Scholar]
- 12.Gamaarachchi H et al. Fast nanopore sequencing data analysis with SLOW5. Nat. Biotechnol 40, 1026–1029 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Samarakoon H et al. Flexible and efficient handling of nanopore sequencing signal data with slow5tools. Genome Biol 24, 69 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Samarakoon H, Ferguson JM, Gamaarachchi H & Deveson IW Accelerated nanopore basecalling with SLOW5 data format. Bioinformatics 39, btad352 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shafin K et al. Haplotype-aware variant calling with PEPPER-Margin-DeepVariant enables high accuracy in nanopore long-reads. Nat. Methods 18, 1322–1332 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Holt JM et al. HiPhase: jointly phasing small, structural, and tandem repeat variants from HiFi sequencing. Bioinformatics 40, btae042 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Prado-Martinez J et al. Great ape genetic diversity and population history. Nature 499, 471–475 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li H Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM Preprint at https://ar5iv.labs.arxiv.org/html/1303.3997 (2013).
- 19.Dolzhenko E et al. ExpansionHunter: a sequence-graph-based tool to analyze variation in short tandem repeat regions. Bioinformatics 35, 4754–4756 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dolzhenko E et al. REViewer: haplotype-resolved visualization of read alignments in and around tandem repeats. Genome Med 14, 84 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Waterhouse AM, Procter JB, Martin DM, Clamp M & Barton GJ Jalview Version 2—a multiple sequence alignment editor and analysis workbench. Bioinformatics 25, 1189–1191 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Goujon M et al. A new bioinformatics analysis tools framework at EMBL-EBI. Nucleic Acids Res 38, W695–W699 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Girdhar K et al. Chromatin domain alterations linked to 3D genome organization in a large cohort of schizophrenia and bipolar disorder brains. Nat. Neurosci 25, 474–483 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jha A et al. DNA-m6A calling and integrated long-read epigenetic and genetic analysis with fibertools. Preprint at bioRxiv 10.1101/2023.04.20.537673 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Danzi M FGF14 Flanking Variant Manuscript Codebase. Zenodo 10.5281/zenodo.11239003 (2024). [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data created through the All of Us Program Long Read Data release CDRv7 (April 2023: https://support.researchallofus.org/hc/en-us/articles/14769699298324-v7-Curated-Data-Repository-CDR-Release-Notes-2022Q4R9-versions) are available through the All of Us Research Program researcher workbench (https://researchallofus.org/). The Care4Rare-SOLVE data are available through Genomics4RD (https://www.genomics4rd.ca) via controlled access requests to genomics4rd@cheo.on.ca. The data created as part of Genomic Answers for Kids are available through NIH/NCBI dbGaP (https://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs002206.v4). Genome sequences of the 79 great apes generated by Prado-Martinez et al.17 were downloaded from the Sequence Read Archive under the accession number PRJNA189439. Patient-level whole-genome sequencing data are not publicly available, as they could compromise privacy and have not been consented for open sharing. Data from Sanger sequencing have not been consented for sharing. Additional data that support the findings of this study are available on request from the corresponding author (M.C.D.).