

Abstract
Objective
IRF2BPL mutation has been associated with a rare neurodevelopmental disorder with abnormal movements, including dystonia. However, the role of IRF2BPL in dystonia remains elusive. We aimed to investigate IRF2BPL mutations in a Taiwanese dystonia cohort.
Methods
A total of 300 unrelated patients with molecularly unassigned isolated (n = 256) or combined dystonia (n = 44) were enrolled between January 2015 and July 2023. The IRF2BPL variants were analyzed based on whole exome sequencing. The in silico prediction of the identified potential pathogenic variant was performed to predict its pathogenicity. We also compared the clinical and genetic features to previous literature reports.
Results
We identified one adolescent patient carrying a de novo heterozygous pathogenic variant of IRF2BPL, c.379C>T (p.Gln127Ter), who presented with generalized dystonia, developmental regression, and epilepsy (0.33% of our dystonia cohort). This variant resides within the polyglutamine (poly Q) domain before the first PEST sequence block of the IRF2BPL protein, remarkably truncating the protein structure. Combined with other patients with IRF2BPL mutations in the literature (n = 60), patients with variants in the poly Q domain have a higher rate of nonsense mutations (p < 0.001) and epilepsy (p = 0.008) than patients with variants in other domains. Furthermore, as our index patient, carriers with substitutions before the first PEST sequence block have significantly older age of onset (p < 0.01) and higher non‐epilepsy symptoms, including generalized dystonia (p = 0.003), and ataxia (p = 0.003).
Interpretation
IRF2BPL mutation is a rare cause of dystonia in our population. Mutations in different domains of IRF2BPL exhibit different phenotypes.
Introduction
Dystonia is a clinically and genetically diverse movement disorder marked by sustained or intermittent muscle contractions, resulting in abnormal, frequently repetitive movements or postures. It may manifest as isolated dystonia, in which dystonia represents the sole motor symptom, or as combined dystonia, which encompasses dystonia occurring alongside other neurological manifestations, such as parkinsonism, myoclonus, epilepsy, ataxia, or developmental delay. 1 Currently, more than 30 dystonia‐causative genes have been identified. 1 , 2 The co‐occurrence of dystonia combined with neurodevelopmental delay implicates causative genetic variants related to neurodevelopment, with a pathogenic substitution frequency of up to 45% in complex dystonia syndrome. 3 These genetic discoveries shed light on the molecular mechanisms involved in dystonia, including endoplasmic reticulum function and protein trafficking, presynaptic and postsynaptic neurotransmitter signaling, and gene transcription modulation during neurodevelopment. 4 The recognition that multiple dystonia‐related genes coalesce in shared biological pathways is a crucial advance in understanding dystonia and will aid in the development of mechanism‐targeted therapeutic strategies.
Rare heterozygous pathogenic variants in the interferon regulatory factor 2‐binding protein‐like gene (IRF2BPL) have been associated with a progressive neurodevelopmental disorder characterized by severe regression, speech abnormalities, epilepsy, ataxia, and a variety of movement disorders. 5 , 6 These movement disorders mainly encompass dystonia and ataxia, though some patients may present with a pure epileptic encephalopathy without movement presentations, with symptoms mainly developing between childhood and adolescence. 5 , 6 The expression of IRF2BPL is ubiquitous, including in the central nervous system. Although its precise functions remain largely unclear, various studies have shown its potential involvement in neuronal development and maintenance, 7 transcription of gonadotropin‐releasing hormone, 8 , 9 modulation of the ubiquitin‐proteasome system in the nucleus, 10 , 11 and acting as an E3 ubiquitin ligase that targets ‐catenin for proteasomal degradation. 6 , 10 , 12 These reports have drawn attention to the possibility that IRF2BPL may contribute to dystonia, especially combined dystonia. However, the exploration of IRF2BPL mutation in patients with dystonia is limited. To date, the literature has documented several cases with IRF2BPL mutation, mostly in the presence of epilepsy and developmental delay. Here, we investigated IRF2BPL mutations in a Taiwanese dystonia cohort with molecularly unidentified dystonia to elucidate the frequency and genotype–phenotype correlation of IRF2BPL mutations, focusing on non‐epilepsy symptoms.
Methods
Participants
A total of 300 unrelated patients with molecularly unassigned isolated or combined dystonia from the movement disorder clinics of National Taiwan University Hospital and Kaohsiung Chang Gung Memorial Hospital were enrolled in this study between January 2015 and July 2023. For patients with combined dystonia, dystonia would be the main feature alongside other movement disorder features. Patients with secondary causes, such as neuroleptic agent‐induced tardive dystonia, cerebral palsy, posttraumatic dystonic syndrome, and structural brain lesions with gadolinium contrast enhancement on brain MRIs (including brain tumor, demyelinating lesions, and infectious or autoimmune encephalitis), were excluded. We excluded patients with mutations in known causative genes related to familial dystonia or parkinsonism listed in our previous panel sequencing. 13 We also excluded patients having pathogenic variants in other known dystonia genes based on the current exome data (Table S1).
Among the 300 recruited patients, 37 had an age at onset of symptom <20 years, and 61 patients had a family history of dystonia Of these latter, 23 had a family inheritance pattern compatible with autosomal‐dominant inheritance and the remaining 38 patients had an autosomal recessive inheritance pattern with affected siblings or at least one other first‐ and/or second‐degree relative with dystonia. None of the patients were from a consanguineous family. All participants provided written informed consent, and the study was approved by the institutional ethics review board of National Taiwan University Hospital and Kaohsiung Chang Gung Memorial Hospital.
Whole exome sequencing
Genomic DNA was isolated from 10 ml of venous blood obtained from all participants following a standard protocol. 13 Whole exome sequencing was performed for all enrolled patients on the Illumina NovaSeq 6000 sequencer. 14 The paired‐end reads were trimmed of the sequencing adaptor sequences and low‐quality bases (Q < 30, Phred scale). After trimming, the sequences were aligned to the human reference genome (GRCh37/hg19). Average on‐target coverage of at least 30X was obtained for all included samples.
Variant analysis
We excluded genetic variants that did not alter coding sequences and variants with a minor allele frequency >0.001 in control subjects in one or more reference databases, specifically the Single‐Nucleotide Polymorphism Database (dbSNP144; https://www.ncbi.nlm.nih.gov/snp), 1000 Genomes Project, 15 Exome Aggregation Consortium (ExAC version 0.3), The Genome Aggregation Database (gnomAD, n = 123,136 exomes and 15,496 whole‐genome sequences; http://gnomad.broadinstitute.org/), and The Taiwan biobank (https://taiwanview.twbiobank.org.tw/index, n = 1517 exomes). 16 We analyzed variants that were annotated as missense, splice donor, splice acceptor, start‐lost, stop‐gained, stop‐loss, or frameshift substitution by ANNOVAR software. To predict the function of the variant in the coding region, we used PROVEAN, 17 SIFT, 18 PolyPhen‐2, 19 MutationTaster, 20 and CADD 21 to predict the possible impact of the variant on the protein structure. Missense variants with PolyPhen‐2 scores <0.95 and SIFT scores >0.05 were excluded. The Varsome version 10.0 tool platform (Varsome, https://varsome.com) was applied to classify pathogenicity based on the American College of Medical Genetics and Genomics interpretation criteria. 22 Variants meeting the criteria were then subjected to Sanger sequencing to confirm the nucleotide change.
Sanger sequencing
The potentially pathogenic variants were ascertained by Sanger sequencing. Genomic DNA samples from the index patients were sequenced as described previously. 6
Systematic review of previous reports of patients with IRF2BPL mutation
We searched the PubMed database for all published studies in English that contained the terms “IRF2BPL mutation,” “IRF2BPL pathogenic variant,” “IRF2BPL likely pathogenic variant,” “C14orf4 mutation,” “C14orf4 pathogenic variant,” and “C14orf4 likely pathogenic variant.” We selected case reports, case series, or original articles of patients with genetically confirmed IRF2BPL‐related mitochondrial complex I deficiency published in 2000–2023. Next, we summarized the clinical, genetic, and imaging characteristics of different genotypes and compared them to the patients enrolled in this study to delineate the genotype and phenotype correlations of this rare disorder.
Statistical analysis
Data were expressed as mean ± standard error of the mean (SEM) and nominal variables as numbers and percentages. Continuous variables were compared by the Mann–Whitney U‐test, whereas categorical data were examined using Fisher's exact test due to small sample number in some compassion groups. Multiple comparisons were adjusted using Bonferroni's correction for the significance level of p value. The threshold p value for statistical significance in the multiple clinical feature comparisons was set at 0.009. Baseline characteristics were compared between different genotype groups using analysis of variance or the Kruskal–Wallis test when appropriate. All analyses were performed in Stata software (StataCorp LLC, College Station, TX).
In silico modeling
Both the wild‐type sequence and mutated variant of IRF2BPL were obtained from the SWISS MODEL (https://swissmodel.expasy.org/). Subsequently, we performed an in silico comparison of these two models to predict potential alterations in the three‐dimensional protein structure after mutation.
Results
The mean age at onset among the 300 enrolled patients was 43.42 ± 18.57 years, and 39.67% were men. A total of 256 (85.33%) patients presented with isolated dystonia, and the remaining 44 (14.67%) patients had combined dystonia with concomitant myoclonus [n = 12 (27.27%)], parkinsonism [n = 25 (56.82%)], ataxia [n = 4 (9.09%)], or epilepsy and developmental delay [n = 3 (6.82%)]. Notably, 3 patients had two non‐dystonia features, that is, epilepsy and developmental delay. For patients with combined dystonia, the dystonia was mostly generalized in distribution.
Among the 300 enrolled participants, 1 patient presenting with generalized dystonia combined with epilepsy and neurodevelopment regression was found to carry a de novo heterozygous nonsense c.379C>T (p.Gln127Ter) mutation in IRF2BPL. None of the patients with non‐epileptic dystonia (n = 297) had a pathogenic variant in IRF2BPL. This index patient is an 18‐year‐old female, the only child born from non‐consanguineous healthy parents, and had no family history of neurological diseases. Initially, she achieved motor development milestones well, though she exhibited speech delay. At the age of 4 years, she gradually developed strabismus and regressed in motor skills. Starting at the age of 13 years, she experienced ataxic gait, generalized dystonia, and generalized tonic–clonic seizures. At 18 years of age, a neurological examination revealed reduced cognitive function, left eye esotropia, generalized dystonia, bilateral lower limb myoclonus (more severe on the right side), and a mixed dystonic and ataxic gait. Brain MRI revealed thinning of the corpus callosum without signal changes or structural lesions in the bilateral basal ganglia (Fig. 1A). Awake electroencephalography (EEG) showed generalized epileptic discharges mixed with focal epileptic activity (Fig. 1B). A 99mTc‐TRODAT single‐photon emission computed tomography scan did not show reduced dopamine transporter uptake at the bilateral basal ganglia (Fig. 1C).
Figure 1.
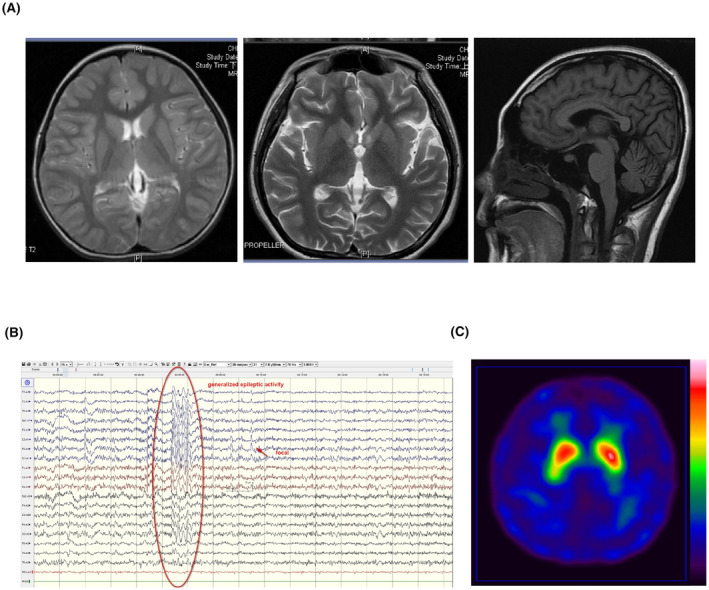
Neuroimaging and electroencephalogram studies of the index patient with IRF2BPL c.379C>T (p.Gln127Ter) mutation. (A) T2‐weighted axial view and T1‐weighted sagittal view of brain MRI. (B) electroencephalogram. (C) 99mTc‐TRODAT single‐photon emission computed tomography scan.
The patient underwent laboratory testing, including a complete blood count, liver function test, renal function tests, ceruloplasmin, autoimmune profiles, urine organic acid analysis and plasma tandem mass analysis of metabolites, which were all unremarkable. We then performed whole exome sequencing for the index patient and identified a pathogenic nonsense variant in IRF2BPL, c.379C>T (p.Gln127Ter), in a heterozygous state (Fig. 2A). Direct Sanger sequencing verified this substitution in the index patient but not in the proband's asymptomatic parents (Fig. 2B). The variant is absent in the population database gnomAD and is classified as pathogenic according to ACMG guidelines. 22 Given the deleterious nature of the c.379C>T (p.Gln127Ter) variant and its absence in population databases, it was considered a de novo likely pathogenic variant for the index patient.
Figure 2.
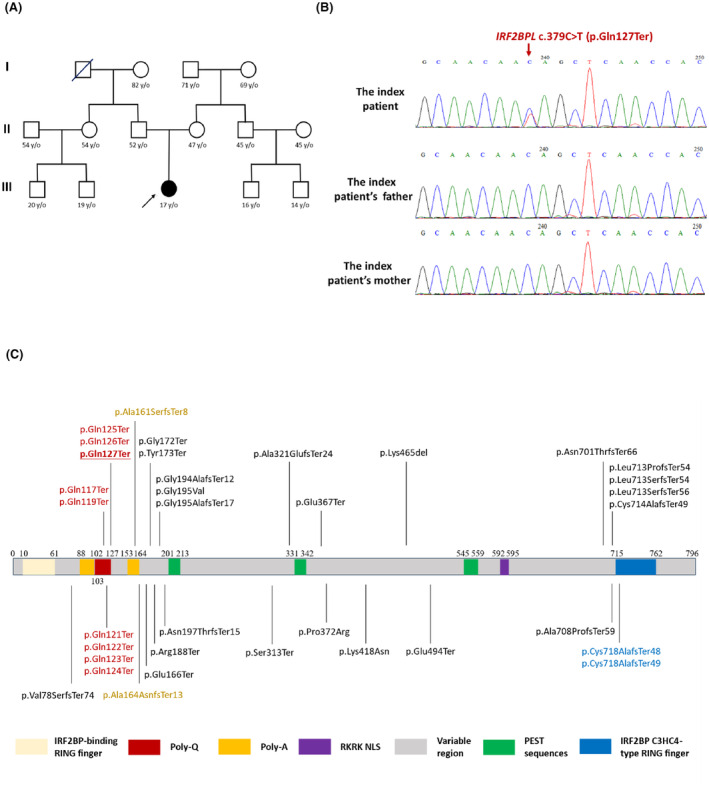
Family pedigree and genetic analysis of the index patient. (A) Pedigree of the index patient. Open symbol: unaffected; filled symbol: affected; arrow: proband. (B) Sanger sequencing traces confirm the de novo heterozygous c.379C > T (p.Gln127Ter) mutation in IRF2BPL. (C) Reported pathogenic variant, including the reported p.Gln127Ter variant (underlined) identified in the current study on the individual protein domain of IRF2BPL.
The mutated nucleotide is located within the N‐terminus polyglutamine (poly Q) domain of IRF2BPL (Fig. 2C), which is known to form the protein's coiled‐coil structure. IRF2BPL contains two highly conserved domains, including a coiled‐coil DNA‐binding domain (IRF2BP zinc finger domain) at the N‐terminus and a C3HC4‐type RING finger domain at the C‐terminus. 7 Between the two conserved domains is a variable region that contains a potential nuclear targeting signal, as well as poly Q and polyalanine tracts. IRF2BPL also contains three putative PEST (proline, glutamic acid, serine, and threonine‐rich) sequences, suggesting that this protein is post‐translationally regulated. 23 Based on the in silico computational protein modeling, nucleotide 127 is predicted to be located on the poly Q domain, and the nonsense substitution markedly truncates the protein (Fig. 3A,B). The c.379C>T (p.Gln127Ter) variant is predicted to cause premature termination of the protein at amino acid 127/796, resulting in loss of the nuclear localization sequence and the C‐terminal C3HC4 type ring finger domain, which is predicted to affect the cellular localization and function of IRF2BPL protein.
Figure 3.
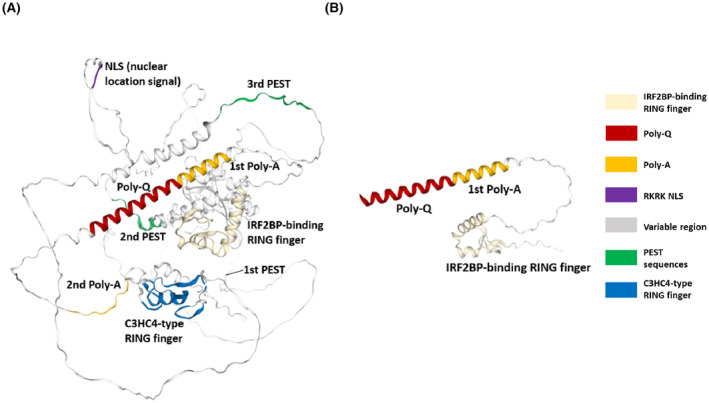
In silico protein structures of wild‐type and mutant IRF2BPL. (A) The mutated nucleotide of p.Gln127 is located within the N‐terminal coiled‐coilstructure of the poly Q domain of the IRF2BPL protein. (B) The truncated mutant IRF2BPL protein harboring the nonsense p.Gln127Ter mutation.
To further delineate the potential genotype and phenotype correlations of the rare mutations in IRF2BPL, we combined our data with results from previous studies to clarify whether dystonia or other non‐epilepsy clinical characteristics are observed in patients having mutations in the poly Q domain of IRF2BPL. Combining 60 patients with IRF2BPL mutations previously described in the literature and the patient reported in the present study (Table S2), 5 , 6 , 7 , 9 , 10 , 11 , 24 , 25 , 26 , 27 , 28 , 29 , 30 , 31 , 32 , 33 , 34 , 35 , 36 , 37 , 38 we created four subgroups (Table 1) based on the locations of the identified mutations in the protein: the N‐terminal poly Q domain (n = 29), poly A domain (n = 2), variable region (n = 28), and the C‐terminal C3HC4 type ring finger domain (n = 2). Among these patients, only four individuals were documented to have affected parents with heterozygous variant following an autosomal‐dominant inheritance trait, indicating a high rate of de novo mutation in this gene. Of note, the majority of patients have nonsense variants or small deletions causing frameshift mutations, while only two patients possess missense pathogenic variants (Table S2). Since a previous study has demonstrated no association between either the neurological features or the MRI findings with mutation type, 10 we thus categorized patients based on the locations of the identified mutations in the protein structure of IRF2BPL, despite the varying mutation types. Compared with the other three groups, patients with mutations in the poly Q domain all had nonsense mutations, whereas mutations within the poly A and C3HC4 domains were exclusively frameshift mutations (p < 0.001, Table 1). Moreover, patients with pathogenic variants in the poly Q domain had a higher frequency of epilepsy (poly Q domain: 13/14 = 92.86% vs. poly A domain: 1/2 = 50% vs. variable region: 12/25 = 48.0% vs. C3HC4 domain: 1/2 = 50%, p = 0.008). Within the variable region, patients with mutations clustering in the N‐terminus before the first PEST (proline, glutamic acid, serine, and threonine‐rich) sequence block had a later age of onset (p = 0.002) and lower initial developmental delay (p = 0.04) but higher occurrence of motor regression (p < 0.001), dystonia (p = 0.003), and ataxia (p = 0.003) compared to mutations located after the first PEST sequence block (Table 2). These observations highlight the crucial role of the N‐terminus of IRF2BPL, especially the poly Q domain and sequences before the first PEST sequence block.
Table 1.
Clinical and genetic features in patients with IRF2BPL mutations in different protein domain derived from previous literature and the current study.
| Poly Q domain (N = 29) | Poly A domain (N = 2) | Variable region (N = 28) | C3HC4 domain (N = 2) | p Value | |
|---|---|---|---|---|---|
| Age of onset, years | 9.51 ± 8.14 | 7.75 ± 5.25 | 7.27 ± 8.73 | 3.25 ± 1.75 | 0.06 |
| Male sex (n, %) | 6 (46.1%, n = 13) | 1 (50%) | 13 (46.43%) | 1 (50%) | 0.99 |
| Ethnicity | |||||
| Asian | 1 (3.45%) | 0 | 4 (14.29%) | 0 | 0.43 |
| Western | 28 (96.55%) | 2 (100%) | 23 (82.14%) | 2 (100%) | 0.41 |
| Unknown | 0 | 0 | 1 (3.57%) | 0 | 0.52 |
| Mutation type | |||||
| Frameshift | 0 | 2 (100%) | 13 (46.43%) | 2 (100%) | <0.001*** |
| Nonsense | 29 (100%) | 0 | 13 (46.43%) | 0 | <0.001*** |
| Missense | 0 | 0 | 2 (7.14%) | 0 | 0.33 |
| Main clinical features | |||||
| Initial global developmental delay | 6 (27.27%, n = 22) | 0 | 11 (42.31%, n = 26) | 1 (50%) | 0.48 |
| Motor regression | 5 (22.73%, n = 22) | 2 (100%) | 10 (38.46%, n = 26) | 0 | 0.09 |
| Language regression | 2 (10%, n = 20) | 0 | 7 (26.92%, n = 26) | 0 | 0.44 |
| Dystonia | 11 (39.29%, n = 28) | 1 (50%) | 8 (34.78%, n = 23) | 0 | 0.82 |
| Ataxia | 25 (86.21%) | 1 (50%) | 14 (50%, n = 24) | 1 (50%) | 0.04* |
| Pyramidal signs | 16 (55.17%) | 1 (50%) | 12 (50%, n = 24) | 1 (50%) | 0.94 |
| Epilepsy | 13 (92.86%, n = 14) | 1 (50%) | 12 (48%, n = 25) | 1 (50%) | 0.008** |
| Brain MRI findings | |||||
| Normal | 4 (28.57%, n = 14) | 2 (100%) | 11 (52.38%, n = 21) | 1 (50%) | 0.22 |
| Cerebral or cerebellar or brainstem atrophy | 5 (35.71%, n = 14) | 0 | 9 (42.86%, n = 21) | 1 (50%) | 0.78 |
| Corpus callosum atrophy | 4 (28.57%, n = 14) | 0 | 1 (4.76%, n = 21) | 0 | 0.25 |
p < 0.05;
p < 0.01.
p < 0.001.
Table 2.
Clinical and genetic characteristics in patients with IRF2BPL mutations located before and after first PEST region on the variable domain.
| Variable region (N = 28) | p Value | ||
|---|---|---|---|
| Before first PEST region (N = 15) | After first PEST region (N = 13) | ||
| Age of onset, years | 11.11 ± 10.05 | 2.40 ± 1.48 | 0.002** |
| Male sex (n, %) | 6 (40%) | 7 (53.85%) | 0.71 |
| Ethnicity | |||
| Asian | 3 (20%) | 1 (7.69%) | 0.60 |
| Western | 12 (80%) | 11 (84.62%) | 0.99 |
| Unknown | 0 | 1 (7.69%) | 0.46 |
| Mutation type | |||
| Frameshift | 6 (40%) | 7 (53.85%) | 0.71 |
| Nonsense | 9 (60%) | 4 (30.77%)) | 0.15 |
| Missense | 0 | 2 (15.38%) | 0.21 |
| Main clinical features | |||
| Initial global developmental delay | 3 (21.43%, n = 14) | 8 (66.67%, n = 12) | 0.04* |
| Motor regression | 10 (71.43%, n = 14) | 0 (n = 12) | <0.001** |
| Language regression | 6 (42.86%, n = 14) | 1 (8.33%, n = 12) | 0.08 |
| Dystonia | 8 (61.54%, n = 13) | 0 (n = 10) | 0.003** |
| Ataxia | 12 (85.7%, n = 14) | 2 (20%, n = 10) | 0.003** |
| Pyramidal signs | 9 (64.29%, n = 14) | 3 (30%, n = 10) | 0.21 |
| Epilepsy | 4 (28.57%, n = 14) | 8 (72.73%, n = 11) | 0.05 |
| Brain MRI findings | |||
| Normal | 5 (41.67%, n = 12) | 6 (66.67%, n = 9) | 0.39 |
| Cerebral or cerebellar or brainstem atrophy | 7 (58.33%, n = 12) | 2 (22.22%, n = 9) | 0.18 |
| Abnormal corpus callosum | 1 (8.33%, n = 12) | 0 (n = 9) | 0.98 |
Data are the number (%) or the mean ± SD. p‐Values that compare individual characteristics between four groups with different genotypes were evaluated with Fisher's exact test. Variables without a normal distribution were compared with the Kruskal–Wallis test.
p < 0.05;
p < 0.01.
Discussion
We described the screening of IRF2BPL in a large Taiwanese dystonia cohort. The low prevalence within the cohort suggests that IRF2BPL mutations are a rare cause of dystonia in our population, even in the subgroup with combined dystonia. Compared with prior reports, our study revealed that mutations across various domains of IRF2BPL exhibit distinct phenotypic spectrum. N‐terminal pathogenic variants, mostly nonsense mutations, are associated with later age at onset and non‐epileptic symptoms, encompassing dystonia and ataxia. Our results extend the current knowledge and provide further insights into the phenotype–genotype correlation of rare mutations in IRF2BPL.
IRF2BPL mutations are a rare cause of dystonia in our population. The simultaneous presence of dystonia and neurodevelopmental delay or regression has long been acknowledged to involve causative genetic variants associated with neurodevelopment, with a pathogenic substitution rate of up to nearly half of complex dystonia syndrome. 3 The causative genes related to neurodevelopment involved in dystonia combined with neurodevelopmental delay include IRF2BPL. 3 , 7 In this study, we did not identify any likely pathogenic variants in the aforementioned genes but one de novo mutation in IRF2BPL. However, IRF2BPL is not a major contributing genetic factor in our dystonia patients, even those with combined dystonia. Due to the rarity of IRF2BPL‐related disorder, the information from previously reported cases implicated IRF2BPL in a severe pediatric phenotype characterized by developmental delay and epilepsy in patients with de novo mutations, although inherited IRF2BPL variants have also been reported in patients with late‐onset progressive dystonic and ataxic syndrome. 5 , 6 , 7 Consistent with our observation, one documented case with the same de novo mutation as our index patient, c.379C>T (p.Gln127Ter) variant, has been reported. 7 Both our index patient and the previously described case exhibited initially normal neurodevelopment, followed by motor regression, occurrence of ataxia, dystonia, and epilepsy. Notably, ataxia or dystonia preceded the onset of epilepsy in both cases. With the high occurrence of de novo mutations, the absence of a family history in dystonia patients should not preclude the molecular diagnosis of IRF2BPL mutations.
Pathogenic variants have been reported throughout IRF2BPL. One study has proposed that nonsense variants are associated with the phenotypes of severe developmental delay or regression and movement disorders, whereas missense substitutions are associated with epilepsy, 7 but conflicting results exist. 10 Another study revealed that the mutation type (nonsense or missense variations) is not related to either clinical presentation or MRI findings. 10 In the current study, we characterized the clinical presentations based on the protein domains in which the mutations were located. Our analysis revealed that patients carrying pathogenic variants in the poly Q domain exhibited a higher frequency of epilepsy compared to those harboring mutations in other domains. Notably, all mutations identified within the poly Q domain were nonsense mutations leading to a truncated IRF2BPL protein. Furthermore, mutations located before the first PEST block had significantly later onset, less developmental delay, and more frequent non‐epilepsy features of dystonia and ataxia compared to mutations close to the C‐terminus. We hypothesized that mutations close to the N‐terminus in the poly Q domain or before the first PEST block will result in a truncated protein with loss of the nuclear localization signal and C‐terminal C3HC4 RING domain, which will lead to neuronal impairment. The C‐terminal C3HC4 RING domain belongs to the E3 ligase family and is associated with protein ubiquitination and degradation. 11 , 39 Dysregulation of the ubiquitin system not only contributes to neurodegenerative diseases, but also results in delayed neurodevelopmental processes. 11 , 40 The c.379C>T (p.Gln127Ter) mutation in our patient is situated within the poly Q domain of IRF2BPL. Computational modeling revealed alterations in the tertiary structure of the protein, supporting our hypothesis and suggesting an impact on the function of IRF2BPL. Several studies have investigated potential mechanisms associated with IRF2BPL mutations. One study demonstrated that cells with the truncated IRF2BPL mutation had decreased levels of ubiquitinated proteins. 11 Other research utilized astrocytes reprogrammed from patient skin fibroblasts and found partial mis‐localization of IRF2BPL to the cytoplasm, along with reduced astrocyte‐mediated neuronal support and abnormal energy metabolism. 41 Recently, loss of IRF2BPL in in vivo Drosophila and zebrafish models revealed increased WNT transcription. 7 Another study using RNA‐Seq data showed that some WNT ligands, including WNT5A, WNT16, WNT9B, and WNT10B, are upregulated in astrocytes differentiated from IRF2BPL mutated patients' induced pluripotent stem cells. 41 The Wnt canonical pathway controls multiple biological processes throughout development and adult life. Growing evidence suggests that deregulation of the Wnt canonical pathway is involved in the pathogenesis of neurodegenerative diseases. Further functional studies are required to determine whether loss of IRF2BPL transcriptional regulation activity directly causes differential expression of neurodevelopmental genes.
In conclusion, IRF2BPL mutation is a rare cause of dystonia in our population. Mutations in different domains of IRF2BPL result in a phenotypic spectrum. Further functional studies are needed to delineate the pathogenicity of mutant IRF2BPL in neuron dysfunction.
Funding Information
This work was supported by grants from National Science and Technology Council (MOST 110‐2314‐B‐002‐150‐MY3) and National Taiwan University Hospital (113‐N0070).
Author Conributions
Study concept and design: PS Chen, YF Chen, and CH Lin. Acquisition of data: PS Chen, YF Chen, JY Chiu, MC Wu, CH Tai, YY Chang, MY Lan, NC Lee, and CH Lin. Analysis and interpretation of data: PS Chen and CH Lin. Drafting of the manuscript: PS Chen. Critical revision of the manuscript for important intellectual content: CH Lin. Obtained funding: CH Lin. Study supervision: CH Lin.
Conflict of Interest
All authors report no conflict of interests.
Supporting information
Table S1.
Acknowledgments
We thank the patients for participating in this study. We also thank the staff of the Second and the fifth Core Lab, Department of Medical Research, National Taiwan University Hospital, for technical support during the study.
Funding Statement
This work was funded by National Taiwan University Hospital grant 113‐N0070; National Science and Technology Council grant MOST 110‐2314‐B‐002‐150‐MY3.
Data Availability Statement
The data supporting the findings of this study are available on request from the corresponding author.
References
- 1. Vidailhet M, Meneret A, Roze E. Dystonia: genetics, phenomenology, and pathophysiology. Lancet Neurol. 2020;19:881‐882. [DOI] [PubMed] [Google Scholar]
- 2. Lange LM, Junker J, Loens S, et al. Genotype‐phenotype relations for isolated dystonia genes: MDSGene systematic review. Mov Disord. 2021;36:1086‐1103. [DOI] [PubMed] [Google Scholar]
- 3. Zech M, Jech R, Boesch S, et al. Monogenic variants in dystonia: an exome‐wide sequencing study. Lancet Neurol. 2020;19:908‐918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Gonzalez‐Latapi P, Marotta N, Mencacci NE. Emerging and converging molecular mechanisms in dystonia. J Neural Transm (Vienna). 2021;128:483‐498. [DOI] [PubMed] [Google Scholar]
- 5. Tran Mau‐Them F, Guibaud L, Duplomb L, et al. De novo truncating variants in the intronless IRF2BPL are responsible for developmental epileptic encephalopathy. Genet Med. 2019;21:1008‐1014. [DOI] [PubMed] [Google Scholar]
- 6. Shelkowitz E, Singh JK, Larson A, Elias ER. IRF2BPL gene mutation: expanding on neurologic phenotypes. Am J Med Genet A. 2019;179:2263‐2271. [DOI] [PubMed] [Google Scholar]
- 7. Marcogliese PC, Shashi V, Spillmann RC, et al. IRF2BPL is associated with neurological phenotypes. Am J Hum Genet. 2018;103:245‐260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Heger S, Mastronardi C, Dissen GA, et al. Enhanced at puberty 1 (EAP1) is a new transcriptional regulator of the female neuroendocrine reproductive axis. J Clin Invest. 2007;117:2145‐2154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Khan WJ, Maqsood H, Younus S. Novel IRF2BPL gene mutation manifesting as a broad spectrum of neurological disorders: a case report. BMJ Neurol Open. 2023;5:e000459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Pisano S, Melis M, Figorilli M, et al. Neurological phenomenology of the IRF2BPL mutation syndrome: analysis of a new case and systematic review of the literature. Seizure. 2022;99:12‐15. [DOI] [PubMed] [Google Scholar]
- 11. Qian XH, Liu XY, Zhu ZY, et al. Neurodevelopmental disorder caused by a truncating de novo variant of IRF2BPL. Seizure. 2021;84:47‐52. [DOI] [PubMed] [Google Scholar]
- 12. Higashimori A, Dong Y, Zhang Y, et al. Forkhead box F2 suppresses gastric cancer through a novel FOXF2‐IRF2BPL‐β‐catenin signaling Axis. Cancer Res. 2018;78:1643‐1656. [DOI] [PubMed] [Google Scholar]
- 13. Wu MC, Chang YY, Lan MY, et al. A clinical and integrated genetic study of isolated and combined dystonia in Taiwan. J Mol Diagn. 2022;24:262‐273. [DOI] [PubMed] [Google Scholar]
- 14. Fan SP, Lee NC, Lin CH. Novel phenotype of 6p25 deletion syndrome presenting juvenile parkinsonism and brain calcification. Mov Disord. 2020;35:1457‐1462. [DOI] [PubMed] [Google Scholar]
- 15. Genomes Project C , Abecasis GR, Auton A, et al. An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491:56‐65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lin JC, Fan CT, Liao CC, Chen YS. Taiwan biobank: making cross‐database convergence possible in the big data era. Gigascience. 2018;7:1‐4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Choi Y, Chan AP. PROVEAN web server: a tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics. 2015;31:2745‐2747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Vaser R, Adusumalli S, Leng SN, Sikic M, Ng PC. SIFT missense predictions for genomes. Nat Protoc. 2016;11:1‐9. [DOI] [PubMed] [Google Scholar]
- 19. Adzhubei IA, Schmidt S, Peshkin L, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248‐249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Schwarz JM, Cooper DN, Schuelke M, Seelow D. MutationTaster2: mutation prediction for the deep‐sequencing age. Nat Methods. 2014;11:361‐362. [DOI] [PubMed] [Google Scholar]
- 21. Kircher M, Witten DM, Jain P, O'Roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. 2014;46:310‐315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405‐424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Rogers S, Wells R, Rechsteiner M. Amino acid sequences common to rapidly degraded proteins: the PEST hypothesis. Science. 1986;234:364‐368. [DOI] [PubMed] [Google Scholar]
- 24. Heide S, Davoine CS, Cunha P, et al. IRF2BPL causes mild intellectual disability followed by late‐onset ataxia. Neurol Genet. 2023;9:e200096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Costa C, Oliver KL, Calvello C, et al. IRF2BPL: a new genotype for progressive myoclonus epilepsies. Epilepsia. 2023;64:e164‐e169. [DOI] [PubMed] [Google Scholar]
- 26. Ganos C, Zittel S, Hidding U, Funke C, Biskup S, Bhatia KP. IRF2BPL mutations cause autosomal dominant dystonia with anarthria, slow saccades and seizures. Parkinsonism Relat Disord. 2019;68:57‐59. [DOI] [PubMed] [Google Scholar]
- 27. Gardella E, Michelucci R, Christensen HM, et al. IRF2BPL as a novel causative gene for progressive myoclonus epilepsy. Epilepsia. 2023;64:e170‐e176. [DOI] [PubMed] [Google Scholar]
- 28. Spagnoli C, Rizzi S, Salerno GG, Frattini D, Fusco C. IRF2BPL gene variants: one new case. Am J Med Genet A. 2020;182:255‐256. [DOI] [PubMed] [Google Scholar]
- 29. Skorvanek M, Dusek P, Rydzanicz M, et al. Neurodevelopmental disorder associated with IRF2BPL gene mutation: expanding the phenotype? Parkinsonism Relat Disord. 2019;62:239‐241. [DOI] [PubMed] [Google Scholar]
- 30. Horovitz DDG, de Faria Domingues de Lima MA , Pires LC, et al. Neurological phenotypes of IRF2BPL gene variants: a report of four novel variants. J Cent Nerv Syst Dis 2023;15:11795735231181467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ginevrino M, Battini R, Nuovo S, et al. A novel IRF2BPL truncating variant is associated with endolysosomal storage. Mol Biol Rep. 2020;47:711‐714. [DOI] [PubMed] [Google Scholar]
- 32. Prilop L, Buchert R, Woerz S, Gerloff C, Haack TB, Zittel S. IRF2BPL mutation causes nigrostriatal degeneration presenting with dystonia, spasticity and keratoconus. Parkinsonism Relat Disord. 2020;79:141‐143. [DOI] [PubMed] [Google Scholar]
- 33. Sainio MT, Aaltio J, Hyttinen V, et al. Effectiveness of clinical exome sequencing in adult patients with difficult‐to‐diagnose neurological disorders. Acta Neurol Scand. 2022;145:63‐72. [DOI] [PubMed] [Google Scholar]
- 34. Antonelli F, Grieco G, Cavallieri F, Casella A, Valente EM. Adult onset familiar dystonia‐plus syndrome: a novel presentation of IRF2BPL‐associated neurodegeneration. Parkinsonism Relat Disord. 2022;94:22‐24. [DOI] [PubMed] [Google Scholar]
- 35. Johannesen KM, Nikanorova N, Marjanovic D, et al. Utility of genetic testing for therapeutic decision‐making in adults with epilepsy. Epilepsia. 2020;61:1234‐1239. [DOI] [PubMed] [Google Scholar]
- 36. Mahdiannasser M, Rashidi‐Nezhad A, Badv RS, Akrami SM. Exploring the genetic etiology of drug‐resistant epilepsy: incorporation of exome sequencing into practice. Acta Neurol Belg. 2022;122:1457‐1468. [DOI] [PubMed] [Google Scholar]
- 37. Yang F, Li H, Dai Y, Zhang R, Zhang JT. IRF2BPL gene variants with dystonia: one new Chinese case report. BMC Neurol. 2023;23:32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hong SY, Yang JJ, Li SY, Lee IC. A wide Spectrum of genetic disorders causing severe childhood epilepsy in Taiwan: a case series of Ultrarare genetic cause and novel mutation analysis in a pilot study. J Pers Med. 2020;10:281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Budhidarmo R, Nakatani Y, Day CL. RINGs hold the key to ubiquitin transfer. Trends Biochem Sci. 2012;37:58‐65. [DOI] [PubMed] [Google Scholar]
- 40. Zheng Q, Huang T, Zhang L, et al. Dysregulation of ubiquitin‐proteasome system in neurodegenerative diseases. Front Aging Neurosci. 2016;8:303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sinha Ray S, Dutta D, Dennys C, et al. Mechanisms of IRF2BPL‐related disorders and identification of a potential therapeutic strategy. Cell Rep. 2022;41:111751. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1.
Data Availability Statement
The data supporting the findings of this study are available on request from the corresponding author.