

Abstract
Adult T‐cell leukemia (ATL) is an aggressive type of leukemia, originating from T‐cells infected with human T‐cell leukemia virus type 1. Accumulating evidence suggests the aberrant activation of NF‐κB to be a causative factor mediating the abnormal proliferation of leukemic cells, thus resulting in the development of ATL. A rearranged NF‐κB2/p100 gene was isolated from an ATL‐derived cell line, which was generated by a chromosomal translocation. The isolated NF‐κB2 mutant is fused with the with no (lysine) deficient protein kinase 1 gene, coding for a 58 kDa protein that retains the DNA binding Rel homology domain, but it lacks the entire ankyrin repeat inhibitory domain, thus suggesting its constitutive activation. This rearranged NF‐κB2 gene product (p58) was localized in the nucleus, and formed a complex with NF‐κB p65 or RelB. Moreover, a T‐cell line expressing p58 increased the amount of an NF‐κB2‐inducible gene, NF‐κB2/p100 by itself. These results suggest that such NF‐κB2 gene rearrangement may therefore be a factor in the constitutive activation of NF‐κB in ATL, and thereby playing a role in the ATL pathogenesis. (Cancer Sci 2008; 99: 792–798)
Adult T‐cell leukemia (ATL) is a highly aggressive leukemia, characterized by the clonal proliferation of CD4 positive T‐cells infected with human T‐cell leukemia virus type 1 (HTLV‐1).( 1 , 2 , 3 , 4 , 5 ) HTLV‐1 establishes a life‐long persistent infection through the immortalization of infected T‐cells.( 6 , 7 ) This immortalization is, however, not sufficient for ATL development, since only a minority of HTLV‐1 infected individuals (approximately 5%) develop ATL after a long latency period (approximately 60 years).( 4 , 5 ) Accumulating evidence indicates that genetic and epigenetic changes of the host genome and HTLV‐1‐associated deterioration of host immunity may therefore be major factors in ATL development.( 4 , 5 )
Primary leukemic cells in the patients, as well as ATL‐derived cell lines demonstrate the constitutive activation of transcription factor NF‐κB.( 8 , 9 , 10 , 11 ) Moreover, agents which block the NF‐κB activity induce apoptosis in these ATL cells.( 12 , 13 , 14 ) Although HTLV‐1 Tax protein potently activates NF‐κB, primary leukemic cells as well as ATL derived cell lines express little, if any, tax mRNA. This indicates that the NF‐κB activation in ATL cells is caused by genetic and/or epigenetic changes in the host genome.( 4 , 10 , 15 , 16 ) However, precisely how NF‐κB is deregulated in ATL cells still remains poorly elucidated.
NF‐κB plays a crucial role in cell proliferation, apoptosis, as well as inflammation, differentiation and development, by regulating the transcription of numerous genes.( 17 , 18 ) NF‐κB is a family of factors containing the DNA binding Rel homology domain including p50, p65, c‐Rel, RelB, and p52. NF‐κB is normally sequestered in the cytoplasm through physical interaction with ankyrin‐repeat‐containing inhibitor proteins, including IκBα, IκBβ, IκBɛ, and NF‐κB2/p100. NF‐κB is activated by either the canonical or non‐canonical pathway. In the canonical pathway, the p50‐p65 complex forms a ternary complex with IκBα, IκBβ or IκBɛ and then it is sequestered in the cytoplasm. An activating stimulus induces the phosphorylation and degradation of the IκBs, and free p50‐p65 moves into the nucleus where it thereafter activates transcription. On the other hand, the non‐canonical NF‐κB pathway is triggered through the signal‐dependent processing of NF‐κB2/p100, thereby generating active p52‐RelB.
Accumulating evidence suggests that the NF‐κB2 gene is associated with certain lymphoid malignancies.( 11 , 19 ) For instance, NF‐κB2 rearrangements are found in 1–2% of B‐cell lymphoma, chronic lymphocytic leukemia and multiple myeloma, and more frequently (around 14%) in mature CD4 positive T‐cell malignancies such as Sezary syndrome.( 20 ) These NF‐κB2 rearrangements always produce a protein containing an intact Rel homology domain with the deletion of most of the ankyrin domain. Moreover, these tumor‐associated NF‐κB2 mutants lose their inhibitory activity against p65, and activate transcription without the Bcl‐3 coactivator.( 21 , 22 )
In this study, a rearranged NF‐κB2/p100 gene (NF‐κB2/p58) was isolated from an ATL‐derived cell line. NF‐κB2/p58 is a 540 amino acid protein, containing a Rel homology DNA binding domain but lacking the entire ankyrin‐repeat inhibitory domain. Like other tumor‐associated NF‐κB2 mutants, NF‐κB2/p58 was constitutively localized in the nucleus, and it induced the expression of an NF‐κB‐regulated gene in a T‐cell line. These results suggest that NF‐κB2 gene rearrangement is potentially responsible for the constitutive activation of NF‐κB in ATL cells, and thereby playing a role in the development of ATL.
Materials and Methods
Cells and culture conditions. TL‐OmI, MT1, KK1, KOB1, and ST1 are ATL derived cell lines.( 23 , 24 , 25 , 26 ) HUT102, SLB1, C5/MJ, and MT4 are HTLV‐1‐infected human T‐cell lines.( 2 , 27 ) Jurkat, HUT78, and MOLT4 are HTLV‐1‐negative human T‐cell lines. CTLL‐2 is an interleukin(IL)‐2‐dependent mouse T‐cell line.( 28 ) All cell lines were cultured in RPMI1640 medium supplemented with 10% heat inactivated fetal calf serum (FCS) (RPMI/10%FCS), 55 µM 2‐mercaptoethanol and antibiotics. In addition, recombinant human IL‐2 (500 pM) was added to the culture medium for KK1, KOB1, ST1 and CTLL‐2. 293T is a human embryonic kidney cell line which was cultured in Dulbecco's modified Eagle's medium supplemented with 10% FCS and antibiotics.
Isolation of NF‐κB2 cDNA and genomic DNA. The TL‐OmI cDNA library in a pMX plasmid( 26 ) was used as a template to isolate NF‐κB2 cDNA from TL‐OmI cells using the polymerase chain reaction (PCR). Two nested primers were designed corresponding to the Rel homology domain of human NF‐κB2 gene as well as one primer corresponding to the sequence downstream of the cDNA cloning site in pMX. The primer sequences were 5′‐ATTCTGGGAAGCAGAACCTG‐3′ for the 1st NF‐κB2 primer, 5′‐GAGACATGGAGAGTTGCTAC‐3′ for the 2nd NF‐κB2 primer, and 5′‐CCCCTTTTTCTGGAGAC TAAAT‐3′ for pMX. To isolate a rearranged NF‐κB2 genomic DNA of TL‐OmI, genomic DNA was extracted from TL‐OmI and Jurkat cells by using the DNeasy Tissue kit (Qiagen). The primers used were 5′‐TGCGGGGTGGAGATGAAGTTTA‐3′ for human NF‐κB2 and 5′‐TCTCGGGCCCAATTGTCTACCA‐3′ for human with no K (lysine) lysine deficient protein kinase 1. The PCR products were cloned into pCR‐Blunt II‐TOPO (Invitrogen), and then were sequenced using the Big Dye Terminator v1.1 Cycle Sequencing kit (Applied Biosystems).
Plasmids. κB‐Luc is a luciferase expression plasmid regulated by the NF‐κB elements of the IL‐2 receptor α‐chain gene and the minimal HTLV‐1 long‐terminal repeat promoter.( 29 ) pGK/β‐gal expresses β‐galactosidase under the control of the phosphoglycerate kinase promoter and it is used to normalize the transfection efficiency.( 30 ) pEFneo‐p100 is an NF‐κB2/p100 expression vector regulated by the elongation factor α promoter, constructed by inserting a HindIII‐NotI fragment from pBluescript‐p100( 31 ) into the BamHI and NotI sites of pEFneo( 32 ) by blunt end ligation. pSG‐p65 is an NF‐κB/p65 expression plasmid.( 33 ) The lentiviral expression vectors, CSII‐EF‐MCS and CSII‐CMV‐MCS‐IRES2‐Bsd, were kindly provided by Dr H. Miyoshi (RIKEN Tsukuba Institute, Tsukuba, Japan). CSII‐EF‐IB is a bicistronic blasticidin resistance gene expression lentivirus vector, constructed by inserting an IRES‐Bsd (blasticidin S deaminase) fragment from CSII‐CMV‐MCS‐IRES2‐Bsd into the EcoRI and XbaI sites of CSII‐EF‐MCS. The following plasmids were constructed using the Gateway recombination system (Invitrogen). The Gateway destination vectors, CS‐EF‐IB‐RfA and pEFneo‐RfA, were constructed by inserting the Gateway reading frame cassette A (Invitrogen) into the EcoRI site of CSII‐EF‐IB and the EcoRV site of pEFneo, respectively. To construct the Gateway entry vector, pENTR‐p52, NF‐κB2/p52 sequence was PCR‐amplified by using the following primers, 5′‐CACCATGGAGAGTTGCTACAACCC‐3′ and 5′‐TTACCCGCCCCCGCCTCCCGGGTAGCA‐3′, and the PCR product was cloned into pENTR/D‐TOPO (Invitrogen). To construct pENTR‐p58, a NF‐κB2/p58 fragment was excised from pCR‐NF‐κB2/p58 (described above) by EcoRI and then was inserted into the EcoRI sites of pENTR2B (Invitrogen). To construct the entry vectors for green fluorescent protein (GFP) and Tax1, PCR fragments derived from pEGFP‐N3 (Clontech) and pHβPr‐1‐neoTax1( 34 ) were inserted into pENTR/D‐TOPO. These cDNAs in the entry vectors were then transferred to CS‐EF‐IB‐RfA and pEFneo‐RfA by an LR recombination reaction using LR clonase (Invitrogen), and designated as CS‐EF‐IB‐p52, CS‐EF‐IB‐p58, CS‐EF‐IB‐GFP, CS‐EF‐IB‐Tax1, pEFneo‐p52, and pEFneo‐p58, respectively. The expression vector pCMV‐HAp58 encoding hemagglutinin (HA)‐tagged p58 was constructed by inserting a PCR‐amplified p58 fragment into the EcoRI and NotI sites of pCMV‐HA (Clontech). To construct pCMV‐HAp100 and pCMV‐HAp52, BglII‐NotI fragments from pEFneo‐p100 and pEFneo‐p52 were inserted into the BglII and NotI sites of pCMV‐HAp58 (the BglII site is located in common coding sequences of p52 and p58 while the NotI site is located downstream from their stop codons).
Immunoprecipitation and Western blotting. To prepare total cell extracts, human T‐cell lines (2 × 106) were lyzed in sodium dodecyl sulfate (SDS) sample buffer (2% SDS, 62.5 mM Tris‐HCl, pH 6.8, 20% glycerol, 0.01% bromophenol blue, 50 mM dithiothreitol), sonicated, and heated at 95°C for 3 min. The cell lysates (30 µg) were cleared by centrifugation, then were separated on 8% polyacrylamide gel containing SDS, and electronically transferred to polyvinylidene difluoride‐membranes. The membranes were incubated with anti‐NF‐κB2/p100 (C‐5 from Santa Cruz Biotechnology), Tax (Taxy‐7), HA (HA‐7 from Sigma) or Tubulin antibody (DM1A from Calbiochem), followed by visualization using an enhanced chemiluminescence Western blotting detection system (GE health science). For the immunopreipitation assays, 293T cells were transfected either with pCMV‐HAp100, pCMV‐HAp58 or pCMV‐HAp52 by using the FuGENE 6 (Roche). The transfected cells were lyzed in ice cold lysis buffer (1% Nondiet P‐40, 25 mM Tris‐HCl, pH 7.2, 150 mM NaCl, 1 mM EDTA, 1 mM phenylmethylsulfonyl fluoride, 20 µg/mL aprotinin). The cell lysates were immunoprecipitated with anti‐HA antibody (HA‐7). The precipitated proteins were then analyzed by Western blotting using anti‐RelB (C‐19 from Santa Cruz Biotechnology), anti‐p65 (F‐6 from Santa Cruz Biotechnology), or anti‐HA antibody as described above.
Immunostaining. 293T cells (1 × 105) were cultured on a coverslip in a 6‐well culture plate for 24 h, and the cells were transfected either with pEFneo‐p58, pEFneo‐p100 or pEFneo using FuGENE 6. 48 h after the transfection, the cells were washed with phosphate buffered saline (PBS), and fixed with methanol for 10 min. The fixed cells were treated with blocking buffer (1% bovine serum albumin in PBS) for 1 h, incubated with anti‐NF‐κB2/p100 antibody for 1 h, and further incubated with Alexa 594‐labeled anti‐mouse immunoglobulin antibody (Invitrogen) for 1 h. After mounting on slides, the cells were examined by fluorescent microscopy (BZ‐8000, Keyence).
Luciferase assay. Jurkat cells (2 × 105 cells) were transfected with pEFneo‐p58, pEFneo‐p52, or pEFneo‐p100, together with κB‐Luc and pGK/β‐gal with or without pSG‐p65 using TransFectin (Bio‐Rad). At 48 h after the transfection, the cell lysates were prepared, and then the luciferase and β‐galactosidase activities in the lysates were measured using the Luciferase Assay System (Promega) and Galacto‐Light System (Applied Biosystems), respectively.
Establishment of stable NF‐κB2 expressing cell lines by lentiviruses. Recombinant lentiviruses were generated by transfecting pCAG‐HIVgp, pCMV‐VSV‐G‐RSV‐Rev (provided by Dr H. Miyoshi) and the respective lentiviral vectors (CS‐EF‐IB‐GFP, CS‐EF‐IB‐p58, CS‐EF‐IB‐p52, and CS‐EF‐IB‐Tax1) into 293T cells by using FuGENE 6. The resultant lentiviruses were added to CTLL‐2 culture (4 × 105 cells) in a final volume of 2 mL of RPMI/10%FCS containing 8 µg/mL polybrene and 500 pM IL‐2. After 48 h, they were then cultured in the selection medium containing 14 µg/mL blasticidin for more than two weeks.
IL‐2‐independent growth assay. CTLL‐2 and its derivatives were washed with PBS twice and then were cultured in the absence of IL‐2 for 1–4 days. Viable cell numbers were counted by a trypan blue exclusion assay under light microcopy.
Results
Expression of NF‐κB2 in human T‐cell lines. To examine whether NF‐κB2 plays a role in the ATL pathogenesis, NF‐κB2 protein was characterized in ATL‐derived cell lines (Fig. 1). Five ATL‐derived cell lines used here were previously characterized to be derived from leukemic cells of ATL patients.( 23 , 24 , 25 ) SLB‐1, C5/MJ, and MT‐4 are human T‐cell lines transformed by HTLV‐1 in vitro. HUT102 was established from the peripheral blood of a mycosis fungoides patient( 2 ) but this cell line was classified as an HTLV‐1‐transformed T‐cell line, since it is unclear whether HUT102 originated from the leukemic cells of the patient or normal T‐cells infected with HTLV‐1 during the establishment. Cell lysates were prepared from these human T‐cell lines, and the NF‐κB2 gene product in the cell lysates was examined by a Western blot analysis using anti‐p100/p52 antibody which recognizes the N‐terminal peptide of NF‐κB2. HTLV‐1‐negative HUT78 cells expressed aberrant NF‐κB2 proteins (p84/p85) as well as their processed product p52 as observed in a previous report (Fig. 1).( 21 ) On the other hand, Jurkat expressed p100 but not p52 and MOLT‐4 expressed neither p100 nor p52, thus indicating that the NF‐κB2 pathway is not activated in these two HTLV‐1‐neagtive T‐cell lines. Of the five ATL‐derived cell lines, TL‐OmI expressed an aberrant NF‐κB2 product, around 58 kDa, as well as p52. The NF‐κB2 gene in TL‐OmI was further characterized below. In addition to TL‐OmI, another ATL‐derived cell line (MT‐1) expressed p52 as well as p100, whereas the three other ATL‐derived cell lines expressed p100 but not p52. These results suggested that some ATL‐derived cell lines (TL‐OmI, MT‐1), but not all, possess constitutive NF‐κB2 activation. Although the HTLV‐1 Tax protein activates the NF‐κB2 pathway( 35 ) MT‐1 expressed undetectable amount of Tax protein (Fig. 1), thus indicating that the activation of the NF‐κB2 pathway in MT‐1 is not mediated by Tax. All the HTLV‐1‐transformed IL‐2‐independent cell lines expressed p100 as well as p52, indicating an activation of the NF‐κB2 pathways in HTLV‐1 mediated T‐cell transformation, and these findings were consistent with those of previous reports.( 35 )
Figure 1.
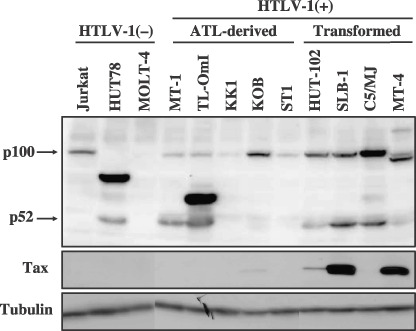
Expression of NF‐κB2 in human T‐cell lines. Cell lysates prepared from indicated human T‐cell lines were characterized by a Western blot analysis using anti‐NF‐κB2/p100, anti‐Tax, or anti‐tubulin antibody. The positions of NF‐κB2/p100 and p52 are indicated. The three adult T‐cell leukemia (ATL)‐derived cell lines KK1, KOB, and ST1 are IL‐2‐dependent cell lines, while the others including TL‐OmI and MT‐1 are IL‐2‐independent lines. HTLV‐1, human T‐cell leukemia virus type 1.
Isolation of a rearranged NF‐κB2 cDNA from TL‐OmI cDNA library. To isolate cDNA encoding an aberrant NF‐κB2 protein in TL‐OmI, the nested PCR using the TL‐OmI cDNA library( 26 ) as a template was performed. The nucleotide sequence of one cDNA isolated from TL‐OmI mRNA encoded a fusion protein between N‐terminal Rel homology domain of NF‐κB2 without ankyrin repeat and C‐terminal partial fragment of a WNK lysine deficient protein kinase 1 (WNK1) (Fig. 2).( 36 ) The expected molecular weight of that protein was 58 kDa, and this cDNA expressed a protein detectable by anti‐p100/p52 in 293T or CTLL‐2 cells, the size of which was identical to that of the aberrant NF‐κB2 protein in TL‐OmI. These results indicated that the aberrant NF‐κB2 gene in TL‐OmI encodes a fusion protein of N‐terminal Rel homology domain of NF‐κB2 with a C‐terminal fragment of WNK1. The aberrant NF‐κB2 gene in TL‐OmI is designated as NF‐κB2/p58.
Figure 2.
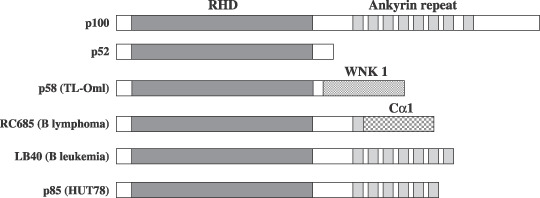
Structure of tumor‐associated NF‐κB2 mutants. The positions of the Rel homology domain (RHD) and ankyrin repeat are shown. The RC685, LB40 and p85 (HUT78) are previously characterized tumor‐associated NF‐κB2 mutants.( 19 , 21 ) WNK1, with no K (lysine) lysine deficient protein kinase 1.
Characterization of genomic structure of NF‐κB2/p58 gene. To characterize the genomic DNA structure of NF‐κB2/p58, a genomic DNA fragment encompassing the boundary region of p58 between NF‐κB2 and WNK1 was amplified by PCR using primers corresponding to the respective boundary sequences in NF‐κB2/p58 cDNA (Fig. 3). The amplified genomic DNA sequence confirmed that NF‐κB2/p58 in TL‐OmI is derived from a genomic DNA rearrangement of two genes (Fig. 3c). The chromosomal translocations occurred at exon 12 of NF‐κB2 and at the intron between the exon 26 and the exon 27 of WNK1. It should be noted that PCR using the same primers did not amplify any specific fragment from Jurkat genomic DNA, thus demonstrating that the NF‐κB2 cDNA isolated from TL‐OmI is not an artifact of cDNA cloning (Fig. 3b).
Figure 3.
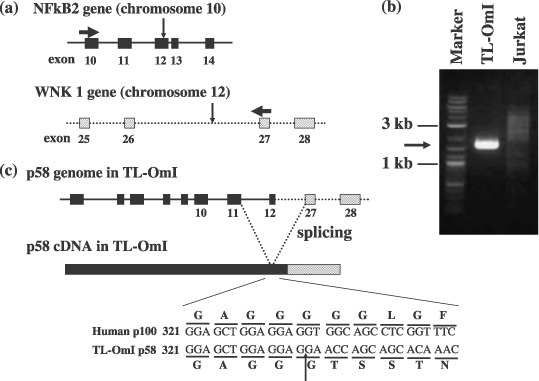
Genomic and cDNA structures of NF‐κB2/p58 in TL‐OmI. (a) Horizontal and vertical arrows indicate the positions of the primer sequences to amplify genomic NF‐κB2 DNA and the chromosomal breakpoints, respectively. (b) Genomic DNA was extracted from TL‐OmI and Jurkat cells, and NF‐κB2 gene was amplified by polymerase chain reaction. The specific amplification from TL‐OmI is indicated by an arrow. (c) The expected genomic DNA and cDNA structures of NF‐κB2/p58 in TL‐OmI are shown. The nucleotide and amino acid sequences encompassing the boundary region between p100 and WNK1 in p58 are shown.
Subcellular localization of NF‐κB2/p58. Next, the subcellular localization of NF‐κB2/p58 was characterized by immunostaining (Fig. 4). The full‐length p100 was exclusively localized in the cytoplasm, whereas p58 was localized in the nucleus. This is consistent with the findings of a previous report which showed the tumor‐associated NF‐κB2 proteins to be constitutively localized in the nucleus.( 22 )
Figure 4.
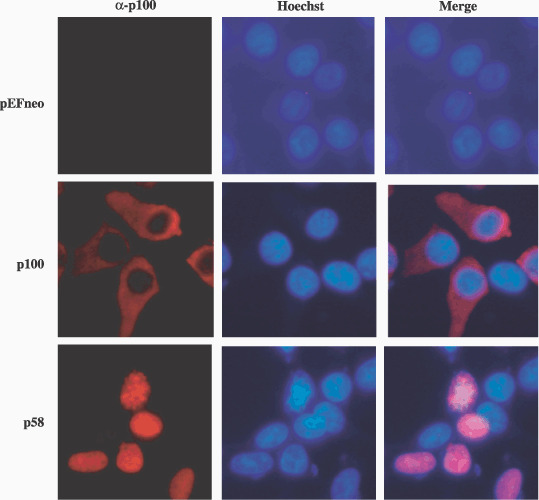
Subcellular localization of NF‐κB2/p58 and p100 in 293T cells. 293T cells were transfected with either the pEFneo‐p58, pEFneo‐p100 or pEFneo plasmid. The cells were then stained with anti‐p100 (red) and with Hoechst 33258 (blue) for nuclear staining. The stained cells were examined using fluorescent light microscopy.
NF‐κB2/p58 induces NF‐κB2/p100 expression in a mouse T cell line CTLL‐2. CTLL‐2 is a mouse IL‐2‐dependent T‐cell line. To explore the role of NF‐κB2/p58 in T‐cells, CTLL‐2 cells expressing p58 were established using a lentivirus vector. The transformants were selected using blasticidine, because the lentivirus vector confers resistance to the antibiotic. CTLL‐2 cells infected with p58‐virus showed a significant level of cell death in comparison to the cells infected with the control (GFP) or p52‐virus, and it took a longer length of time to establish p58 expressing cells than the others (data not shown). This indicates that NF‐κB2/p58 over‐expression has a growth inhibitory activity toward CTLL‐2. A Western blot analysis showed that the established CTLL‐2 cells expressed either p58 or p52, although the amount of p58 was much less than that in TL‐OmI (Fig. 5a). Interestingly, p58 as well as p52‐transduced cells expressed p100 more than control cells. This phenotype was observed in another p58‐transducing cell line that was established independently (Fig. 5b). Tax‐transduced cells also showed augmented expression of p100, and the amount of p100 was equivalent to that in p58‐transduced cells (Fig. 5a). These results suggest that NF‐κB2/p58 can induce p100 expression in CTLL‐2 cells as much as Tax. To examine the effect of p58 expression on either cell growth or apoptosis, the CTLL‐2 cells expressing p58, p52 or Tax, were cultured in the absence of IL‐2, and the number of viable cells was thus counted over four days (Fig. 5c). CTLL‐2 cells expressing Tax continued to grow in the absence of IL‐2, and established IL‐2‐independent cells, consistent with the previous report.( 28 ) On the other hand, p58‐cells as well as p52‐cells died while demonstrating kinetics which was closely similar to that of the control cells, thus indicating that p58 does not have the capacity to induce IL‐2‐independent growth of CTLL‐2.
Figure 5.
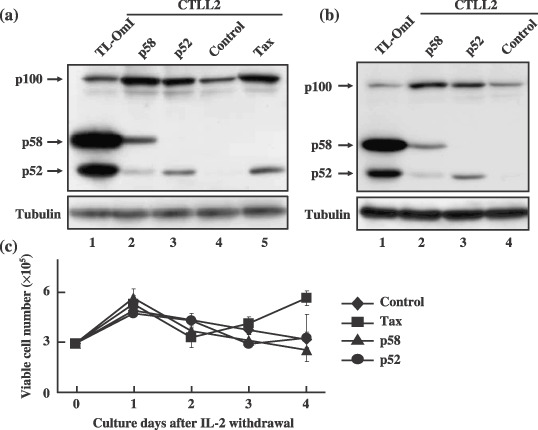
NF‐κB2/p58 activates the endogenous NF‐κB2/p100 expression in a T‐cell line. (a, b) The cell lysates were prepared from TL‐OmI (lane 1), and CTLL‐2 infected with either p58‐virus (lane 2), p52‐virus (lane 3), control virus (lane 4) or Tax‐virus (lane 5). NF‐κB2 proteins in cell lysates were analyzed by a Western blot analysis using an anti‐p100 antibody. The CTLL‐2 cells expressing p58 or p52 in (a) and (b) were independently established. (c) The CTLL‐2 cells characterized above were cultured in the absence of IL‐2, and viable cell numbers were counted using the trypan blue exclusion assay under microscopy.
NF‐κB2/p58 forms a heterodimer with p65 and RelB. NF‐κB2/p52 as well as p100 forms a hetero‐dimmer with other NF‐κB family members through the Rel homology domain. Thereafter, we next examined whether p58 interacts with other NF‐κB subunits. 293T cells were transfected with the expression plasmids encoding either hemagglutinin (HA)‐tagged p100 (HA‐p100), HA‐p58 or HA‐p52, and cultured for 2 days. The cell lysates were then immunoprecipitated with anti‐HA antibody and the immunoprecipitated proteins were analyzed by a Western blot analysis either with anti‐RelB or anti‐p65 antibody (Fig. 6). Not only p100 and p52 but also p58 coimmunoprecipitated RelB and p65, thus indicating that p58 also has a capacity to form a complex with these NF‐κB subunits. We noticed that p58 and p52 coimmunoprecipitated more RelB proteins than p100. This is likely to be due to the relative abundance of RelB in the lysates of p58 and p52 transfected cells, thus suggesting that the RelB expression is transcriptionally activated by p58 and p52. In addition, it should be noted that 293T cells transfected with the HA‐p58 plasmid expressed around 52 kDa protein recognized by anti‐HA antibody, thus indicating that HA‐p58 was cleaved to produce p52 kDa protein (Fig. 6).
Figure 6.
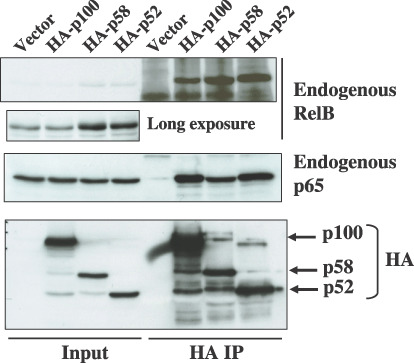
The interaction of NF‐κB2/p58 with other NF‐κB subunits. The cell lysates were prepared from 293T cells transfected either with pCMV‐HA‐p100, pCMV‐HA‐p52, or pCMV‐HA‐p58, and they were immunoprecipitated with anti‐hemagglutinin(HA) antibody. The total cell lysates (Input) and immunoprecipitates (HA IP) were then characterized by a Western blot analysis by using either anti‐RelB, anti‐p65, or anti‐HA antibody.
Reduced IκB like activity of NF‐κB2/p58. The activity of p58 to induce a luciferase reporter containing five copies of the NF‐κB binding site from IL‐2 receptor α‐chain gene was examined. A transient transfection experiment showed that p58 reproducibly but minimally stimulated luciferase expression in a human T‐cell line Jurkat, and the activity was reproducibly less than that by p52 (Fig. 7b). In addition, while p100 inhibited the p65‐mediated activation of NF‐κB‐reporter, the inhibitory activity of p58 to p65 greatly decreased, and p58, even at a low dose, slightly augmented the activation (Fig. 7c). These results indicated that p58, but not p100, is an activator of NF‐κB‐dependent transcription.
Figure 7.
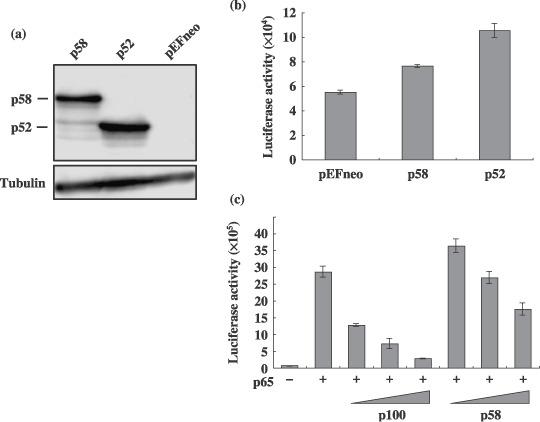
Transcriptional activity of NF‐κB2/p58 in a T‐cell line. (a) Cell lysates were prepared from 293T cells transfected with either the pEFneo‐p58, pEFneo‐p100 or pEFneo plasmid (2 µg), and the amount of NF‐κB2 protein in each lysate was measured by a Western blot analysis using an antip100 antibody. The arrows indicate the p58 and p52 recognized by the antibody. (b, c) Jurkat cells were transfected with either the pEFneo‐p58, pEFneo‐p100 or pEFneo plasmid (0.1 µg) together with the luciferase plasmid (0.5 µg) regulated by the NF‐κB element (kB‐Luc) and the β‐galactosidase plasmid (0.1 µg). The pSG‐p65 plasmid (0.1 µg) was cotransfected with increasing amount of pEFneo‐p100 or pEFneo‐p58 (0.05, 0.1, and 0.2 µg) into Jurkat cells as indicated in (c). Cell lysates were prepared from transfected cells, and the luciferase and β‐galactosidase activities were determined. The luciferase activity normalized by the β‐galactosidase activity was shown as the average with standard deviations. Three independent experiments were carried out to confirm reproducibility.
Discussion
In this study, a mutant NF‐κB2 gene (NF‐κB2/p58) was isolated from an ATL‐derived cell line. Like other tumor‐associated NF‐κB2 mutants, p58 is constitutively localized in the nucleus (Fig. 4), and it induced the expression of an endogenous NF‐κB‐regulated gene, NF‐κB2/p100 by itself, in a T‐cell line (Fig. 5). These results suggest that NF‐κB2/p58 plays a role in the activation of the NF‐κB pathway in TL‐OmI, and thereby, in the malignant growth.
NF‐κB2/p58 is the smallest of the tumor‐associated NF‐κB2 mutants reported so far, and it does not contain any ankyrin repeat sequence (Fig. 2). This indicates that the Rel homology DNA binding domain is sufficient for the aberrant constitutive activation of NF‐κB2 in tumors. It should be noted that p52 was consistently detected in the cells expressing NF‐κB2/p58 or HA‐p58, although NF‐κB2/p58 did not contain the authentic proteolytic cleavage site downstream of Rel homology domain (Fig. 2). Heusch et al. showed the authentic proteolytic cleavage site in p100 is dispensable for the cleavage of p100, instead such cleavage needs a glycine‐rich region (GRR) upstream of the cleavage site.( 37 ) Since NF‐κB2/p58 keeps GRR but not the authentic cleavage site, around p52 kDa protein in the cells expressing NF‐κB2/p58 is likely to be generated from p58 through cleavage at the site distinct from the authentic one.
NF‐κB2/p58 induced the expression of endogenous NF‐κB2/p100, a well‐known NF‐κB‐inducible gene, in CTLL‐2 cells, and the induction was equivalent to that of Tax (Fig. 5). These results suggest that NF‐κB2/p58 acts as a transcriptional activator through NF‐κB in T‐cells. In contrast, NF‐κB2/p58 demonstrated only a minimal stimulation of the NF‐κB activity in a transient assay using Jurkat cells. It should be noted that the same reporter was strongly stimulated by Tax.( 38 ) Although it is unclear why NF‐κB2/p58 is a weak activator in a transient assay, this may be because NF‐κB2/p58 and Tax have distinct specificities to NF‐κB‐regulated promoters, dependent on NF‐κB binding sequences or other enhancer sequences in promoters, since the NF‐κB luciferase reporter used here has five copies of the NF‐κB binding site derived from IL‐2 receptor α‐chain but not NF‐κB2/p100 gene. It is therefore considered important to identify which genes are regulated by NF‐κB2/p58 in TL‐OmI.
NF‐κB2/p58 efficiently interacted with RelB and p65 in 293T cells (Fig. 6). While the interaction of p65 with p100 inhibited the p65‐mediated transcriptional activation through the NF‐κB‐site, the interaction with p58 slightly augmented the activity. Moreover, the p58‐transduced cells expressed increased amounts of RelB and p100 proteins (5, 6). Taken together, these results indicated that NF‐κB2/p58 acts as a transcriptional activator by forming a homo‐ and/or hetero‐dimmer. Although it is unclear whether p58 needs to be cleaved to p52 to activate the transcription, we assume that p58 by itself has the ability to stimulate the transcription, since the amount of p52 processed from p58 in CTLL‐2 or 293T cells is much less than that of p58 (5, 7).
NF‐κB2/p58 contains the C‐terminal portion of WNK1 ( Fig. 2 ). Although all tumor‐associated NF‐κB2 mutants have deletions of ankyrin sequences, the amino acid sequences of their C‐terminus varied with each other (Fig. 2). For instance, NF‐κB2/p85 in HUT78 contains a repetitive sequence derived from Alu and Line‐1 in human genome (Fig. 2), and thus it is unlikely that such a C‐terminal peptide in this NF‐κB2 mutant plays a role in transcriptional regulation.( 21 ) We therefore speculate that the C‐terminal WNK1 peptide in p58 does not play a significant role in transcriptional regulation.
Intriguingly, NF‐κB2 activation was observed in IL‐2‐independent ATL‐derived cell lines (MT‐1, TL‐OmI) but not IL‐2‐dependent ones (KK1, KOB, ST1) (Fig. 1). Hironaka et al. also showed that all three IL‐2‐independent ATL‐derived cell lines have NF‐κB2 activation, although they did not characterize any IL‐2‐dependent lines.( 39 ) It should be noted that NF‐κB2 activation by Tax is required but not sufficient for IL‐2‐independent growth transformation of CTLL‐2.( 40 ) Therefore, these results suggest that NF‐κB2 activation may be somehow associated with the IL‐2‐independent growth of ATL‐derived cell lines.
We tried to knockdown NF‐κB/p58 in TL‐OmI using RNA interference multiple times in order to investigate its functional role, but only to fail to significantly reduce the expression. This may be due to the high amount of NF‐κB2/p58 mRNA in TL‐OmI. Therefore, further analyzes are required to elucidate the biological significance of NF‐κB2 activation in TL‐OmI as well as the ATL pathogenesis.
Acknowledgments
We thank Dr Yasuaki Yamada for providing the KK1, KOB1, and ST1 cell lines, and Dr Hiroyuki Miyoshi at RIKEN Tsukuba Institute for the pCAG‐HIVgp, pCMV‐VSV‐G‐RSV‐Rev, CSII‐EF‐MCS, and CSII‐CMV‐MCS‐IRES2‐Bsd plasmids. We also thank the Takeda Pharmaceutical Company for providing recombinant human IL‐2. We would like to express our gratitude to Chika Yamamoto and Misako Tobimatsu for their excellent technical assistances. This work was supported in part by a Grant‐in‐Aid for Scientific Research on Priority Areas and for Scientific Research (C) of Japan, as well as Grant for Promotion of Niigata University Research Projects.
References
- 1. Uchiyama T, Yodoi J, Sagawa K et al . Adult T‐cell leukemia: clinical and hematologic features of 16 cases. Blood 1977; 50: 481–92. [PubMed] [Google Scholar]
- 2. Poiesz BJ, Ruscetti FW, Gazdar AF et al . Detection and isolation of type C retrovirus particles from fresh and cultured lymphocytes of a patient with cutaneous T‐cell lymphoma. Proc Natl Acad Sci USA 1980; 77: 7415–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hinuma Y, Nagata K, Hanaoka M et al . Adult T‐cell leukemia: antigen in an ATL cell line and detection of antibodies to the antigen in human sera. Proc Natl Acad Sci USA 1981; 78: 6476–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Matsuoka M, Jeang KT. Human T‐cell leukaemia virus type 1 (HTLV‐1) infectivity and cellular transformation. Nat Rev Cancer 2007; 7: 270–80. [DOI] [PubMed] [Google Scholar]
- 5. Giam CZ, Jeang KT. HTLV‐1 Tax and adult T‐cell leukemia. Front Biosci 2007; 12: 1496–507. [DOI] [PubMed] [Google Scholar]
- 6. Miyoshi I, Kubonishi I, Yoshimoto S et al . Type C virus particles in a cord T‐cell line derived by co‐cultivating normal human cord leukocytes and human leukaemic T cells. Nature 1981; 294: 770–1. [DOI] [PubMed] [Google Scholar]
- 7. Yamamoto N, Okada M, Koyanagi Y et al . Transformation of human leukocytes by cocultivation with an adult T cell leukemia virus producer cell line. Science 1982; 217: 737–9. [DOI] [PubMed] [Google Scholar]
- 8. Mori N, Fujii M, Ikeda S et al . Constitutive activation of NF‐kappaB in primary adult T‐cell leukemia cells. Blood 1999; 93: 2360–8. [PubMed] [Google Scholar]
- 9. Arima N, Matsushita K, Obata H et al . NF‐kappaB involvement in the activation of primary adult T‐cell leukemia cells and its clinical implications. Exp Hematol 1999; 27: 1168–75. [DOI] [PubMed] [Google Scholar]
- 10. Sun SC, Yamaoka S. Activation of NF‐kappaB by HTLV‐I and implications for cell transformation. Oncogene 2005; 24: 5952–64. [DOI] [PubMed] [Google Scholar]
- 11. Courtois G, Gilmore TD. Mutations in the NF‐kappaB signaling pathway: implications for human disease. Oncogene 2006; 25: 6831–43. [DOI] [PubMed] [Google Scholar]
- 12. Mori N, Yamada Y, Ikeda S et al . Bay 11‐7082 inhibits transcription factor NF‐kappaB and induces apoptosis of HTLV‐I‐infected T‐cell lines and primary adult T‐cell leukemia cells. Blood 2002; 100: 1828–34. [DOI] [PubMed] [Google Scholar]
- 13. Satou Y, Nosaka K, Koya Y et al . Proteasome inhibitor, bortezomib, potently inhibits the growth of adult T‐cell leukemia cells both in vivo and in vitro . Leukemia 2004; 18: 1357–63. [DOI] [PubMed] [Google Scholar]
- 14. Watanabe M, Ohsugi T, Shoda M et al . Dual targeting of transformed and untransformed HTLV‐1‐infected T cells by DHMEQ, a potent and selective inhibitor of NF‐kappaB, as a strategy for chemoprevention and therapy of adult T‐cell leukemia. Blood 2005; 106: 2462–71. [DOI] [PubMed] [Google Scholar]
- 15. Furukawa Y, Kubota R, Tara M et al . Existence of escape mutant in HTLV‐I tax during the development of adult T‐cell leukemia. Blood 2001; 97: 987–93. [DOI] [PubMed] [Google Scholar]
- 16. Okazaki S, Moriuchi R, Yosizuka N et al . HTLV‐1 proviruses encoding non‐functional TAX in adult T‐cell leukemia. Virus Genes 2001; 23: 123–35. [DOI] [PubMed] [Google Scholar]
- 17. Hayden MS, Ghosh S. Signaling to NF‐kappaB. Genes Dev 2004; 18: 2195–224. [DOI] [PubMed] [Google Scholar]
- 18. Karin M. Nuclear factor‐kappaB in cancer development and progression. Nature 2006; 441: 431–6. [DOI] [PubMed] [Google Scholar]
- 19. Neri A, Chang CC, Lombardi L et al . B cell lymphoma‐associated chromosomal translocation involves candidate oncogene lyt‐10, homologous to NF‐kappa B p50. Cell 1991; 67: 1075–87. [DOI] [PubMed] [Google Scholar]
- 20. Neri A, Fracchiolla NS, Migliazza A et al . The involvement of the candidate proto‐oncogene NFKB2/lyt‐10 in lymphoid malignancies. Leuk Lymphoma 1996; 23: 43–8. [DOI] [PubMed] [Google Scholar]
- 21. Zhang J, Chang CC, Lombardi L, Dalla‐Favera R. Rearranged NFKB2 gene in the HUT78 T‐lymphoma cell line codes for a constitutively nuclear factor lacking transcriptional repressor functions. Oncogene 1994; 9: 1931–7. [PubMed] [Google Scholar]
- 22. Chang CC, Zhang J, Lombardi L et al . Rearranged NFKB‐2 genes in lymphoid neoplasms code for constitutively active nuclear transactivators. Mol Cell Biol 1995; 15: 5180–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Miyoshi I, Kubonishi I, Sumida M et al . A novel T‐cell line derived from adult T‐cell leukemia. Gann 1980; 71: 155–6. [PubMed] [Google Scholar]
- 24. Sugamura K, Fujii M, Kannagi M et al . Cell surface phenotypes and expression of viral antigens of various human cell lines carrying human T‐cell leukemia virus. Int J Cancer 1984; 34: 221–8. [DOI] [PubMed] [Google Scholar]
- 25. Yamada Y, Ohmoto Y, Hata T et al . Features of the cytokines secreted by adult T cell leukemia (ATL) cells. Leuk Lymphoma 1996; 21: 443–7. [DOI] [PubMed] [Google Scholar]
- 26. Higuchi M, Matsuda T, Mori N et al . Elevated expression of CD30 in adult T‐cell leukemia cell lines: possible role in constitutive NF‐kappaB activation. Retrovirology 2005; 2: 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. De Vecchis L, Graziani G, Macchi B et al . Decline of natural cytotoxicity of human lymphocytes following infection with human T‐cell leukemia/lymphoma virus (HTLV). Leuk Res 1985; 9: 349–55. [DOI] [PubMed] [Google Scholar]
- 28. Iwanaga Y, Tsukahara T, Ohashi T et al . Human T‐cell leukemia virus type 1 tax protein abrogates interleukin‐2 dependence in a mouse T‐cell line. J Virol 1999; 73: 1271–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Endo K, Hirata A, Iwai K et al . Human T‐cell leukemia virus type 2 (HTLV‐2) Tax protein transforms a rat fibroblast cell line but less efficiently than HTLV‐1 Tax. J Virol 2002; 76: 2648–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Devergne O, Cahir McFarland ED, Mosialos G et al . Role of the TRAF binding site and NF‐kappaB activation in Epstein−Barr virus latent membrane protein 1‐induced cell gene expression. J Virol 1998; 72: 7900–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bours V, Burd PR, Brown K et al . A novel mitogen‐inducible gene product related to p50/p105‐NF‐kappa B participates in transactivation through a kappa B site. Mol Cell Biol 1992; 12: 685–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Asao H, Fu XY. Interferon‐gamma has dual potentials in inhibiting or promoting cell proliferation. J Biol Chem 2000; 275: 867–74. [DOI] [PubMed] [Google Scholar]
- 33. Ruben SM, Dillon PJ, Schreck R et al . Isolation of a rel‐related human cDNA that potentially encodes the 65‐kD subunit of NF‐kappa B. Science 1991; 254: 11. [DOI] [PubMed] [Google Scholar]
- 34. Matsumoto K, Shibata H, Fujisawa JI et al . Human T‐cell leukemia virus type 1 Tax protein transforms rat fibroblasts via two distinct pathways. J Virol 1997; 71: 4445–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Xiao G, Cvijic ME, Fong A et al . Retroviral oncoprotein Tax induces processing of NF‐kappaB2/p100 in T cells: evidence for the involvement of IKKalpha. EMBOJ 2001; 20: 6805–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Xu B, English JM, Wilsbacher JL et al . WNK1, a novel mammalian serine/threonine protein kinase lacking the catalytic lysine in subdomain II. J Biol Chem 2000; 275: 16795–801. [DOI] [PubMed] [Google Scholar]
- 37. Heusch M, Lin L, Geleziunas R, Greene WC. The generation of nfkb2 p52: mechanism and efficiency. Oncogene 1999; 18: 6201–8. [DOI] [PubMed] [Google Scholar]
- 38. Niinuma A, Higuchi M, Takahashi M et al . Aberrant activation of the interleukin‐2 autocrine loop through the nuclear factor of activated T cells by nonleukemogenic human T‐cell leukemia virus type 2 but not by leukemogenic type 1 virus. J Virol 2005; 79: 11925–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hironaka N, Mochida K, Mori N et al . Tax‐independent constitutive IkappaB kinase activation in adult T‐cell leukemia cells. Neoplasia 2004; 6: 266–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Higuchi M, Tsubata C, Kondo R et al . Cooperation of NF‐kappaB2/p100 activation and the PDZ domain binding motif signal in human T‐cell leukemia virus type 1 (HTLV‐1) Tax1 but not HTLV‐2 Tax2 is crucial for interleukin‐2‐independent growth transformation of a T‐cell line. J Virol 2007; 81: 11900–7. [DOI] [PMC free article] [PubMed] [Google Scholar]