

Abstract
In this study, we found that in the adipose tissue of wildtype animals, insulin and TGF‐β signalling converge via a BMP antagonist short gastrulation (sog) to regulate ECM remodelling. In tumour bearing animals, Sog also modulates TGF‐β signalling to regulate ECM accumulation in the fat body. TGF‐β signalling causes ECM retention in the fat body and subsequently depletes muscles of fat body‐derived ECM proteins. Activation of insulin signalling, inhibition of TGF‐β signalling, or modulation of ECM levels via SPARC, Rab10 or Collagen IV in the fat body, is able to rescue tissue wasting in the presence of tumour. Together, our study highlights the importance of adipose ECM remodelling in the context of cancer cachexia.
Keywords: cachexia, Drosophila, ECM, insulin, TGF‐β
Subject Categories: Cancer, Metabolism, Signal Transduction
During cancer cachexia, insulin and TGF‐β signalling converge in the fat body via Sog to hinder ECM secretion, which in turn affects muscle integrity.
Introduction
Cachexia is a metabolic wasting syndrome characterised by the loss of adipose tissue and muscle. Cachexia occurs in approximately 80% of advanced‐stage cancer patients and is ultimately responsible for approximately 30% of cancer mortalities. Despite the clinical significance of cachexia in cancer and diseases such as HIV or other chronic diseases, there is no gold standard for its treatment (Baracos et al, 2018). Cachexia is often resolved when the tumour can be resected in early disease; however, there are few strategies to treat cachexia in late stage or metastatic patients, where removal of the tumour is not an option. Therefore, understanding the mechanisms that drive wasting in peripheral tissues and how to target these deregulations can offer new therapeutic avenues to treat cachexia.
Drosophila melanogaster is emerging as an excellent model to identify tumour‐secreted factors that drive cancer cachexia. Adult and larval tumour models that induce cachectic phenotypes have been established in Drosophila, and these models have revealed several different mechanisms by which tumours induce these cachectic phenotypes (Figueroa‐Clarevega & Bilder, 2015; Kwon et al, 2015; Song et al, 2019; Newton et al, 2020; Ding et al, 2021; Hodgson et al, 2021; Khezri et al, 2021; Lee et al, 2021; Lodge et al, 2021; Santabárbara‐Ruiz & Léopold, 2021). We recently demonstrated that eye imaginal disc tumours, caused by the expression of constitutively activate Ras (Ras V12 ) and an RNAi against the polarity protein disc‐large 1 (dlg1 RNAi ), secrete two cachectic factors: ImpL2 and Matrix metalloproteinase 1 (Mmp1) (Lodge et al, 2021). ImpL2 is a secreted protein that induces cachexia by binding to circulating Insulin‐like peptides (Ilps) to prevent them from activating Insulin‐like receptor (InR) in host tissues, thus causing host tissue wasting (Honegger et al, 2008). This mechanism is likely conserved, as Insulin Growth Factor Binding Protein‐2 (IGFBP‐2), which also antagonises insulin signalling, is correlated with muscle atrophy in pancreatic ductal adenocarcinoma (PDAC) patients (Dong et al, 2021). In addition, Mmp1 drives cachexia by increasing the amount of the Transforming growth factor‐β (TGF‐β) ligand Glass bottom boat (Gbb). Gbb subsequently induces an elevation of TGF‐β signalling in the Drosophila adipose tissue (called the fat body), to induce muscle detachment. Together, our findings highlighted the importance of fat body signalling in promoting muscle degradation during cachexia. However, what occurs downstream of these signals to drive fat body and muscle disruption remains unclear.
In this study, we demonstrate that tumour‐derived ImpL2 and Gbb facilitate muscle degradation during cachexia via two main mechanisms. Firstly, ImpL2 mediates a reduction in fat body insulin signalling, which in turn activates fat body TGF‐β signalling by upregulating short gastrulation (sog), a TGF‐β antagonist. As Gbb also activates fat body TGF‐β signalling, our findings reveal that ImpL2 and Gbb converge on this activation. We further show that fat body TGF‐β signalling activation subsequently causes an aberrant upregulation of ECM proteins in the fat body. As muscle ECM proteins are mostly fat body‐derived (Dai et al, 2017, 2018), this in turn causes a reduction in muscle ECM to promote muscle detachment. Modulating SPARC, a collagen binding protein (Shahab et al, 2015) or Rab10, a regulator of basement membrane fibril formation (Isabella & Horne‐Badovinac, 2016) can ameliorate ECM accumulation in the fat body in tumour bearing animals, and in turn improve muscle integrity. Secondly, tumour secreted ImpL2 causes a reduction in muscle insulin signalling, which contributes towards reduced translation and increased muscle atrophy. Enhanced activation of insulin in the muscle can specifically improve muscle atrophy (but not ECM). Together, the two mechanisms attribute towards the muscle detachment phenotype we have observed. Therefore, our data demonstrate that in addition to targeting tumour secreted factors, modulating key signalling and ECM regulators in peripheral tissues could be an effective strategy to treat cachexia.
Results
Reduced insulin signalling promotes the activation of TGF‐β signalling in the cachectic fat body
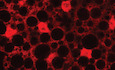
In this study, we utilise two Drosophila larval tumour models to study cachexia. In the first model, the tumour is induced via the GAL4‐UAS mediated overexpression of Ras V12 and Discs Large 1 (dlg1) RNAi in the eye (Fig 1A). Using this system, we can knockdown or overexpress candidate genes in the tumour. In the second model, the tumour is induced via the QF2‐QUAS mediated overexpression of Ras V12 and scrib RNAi (Fig 1A), allowing us to knockdown or overexpress genes of interest in the fat body of tumour‐bearing animals using drivers such as R4‐GAL4. We previously showed that Ras V12 , dlg1 RNAi eye imaginal disc tumours express elevated levels of the dIlp antagonist ImpL2, consequently leading to a reduction in fat body insulin signalling (Lodge et al, 2021). In parallel, tumour secreted Gbb accounts for the upregulation of TGF‐β signalling. Here, we show that tumours induced by the overexpression of Ras V12 and an RNAi against the polarity protein Scribble (scrib RNAi ) using the QF/QUAS system, caused a downregulation of insulin signalling (pAkt, Fig 1B–D), and an upregulation of TGF‐β signalling (pMad, Fig 1E–G) in the fat body.
Figure 1. Insulin signalling in the fat body negatively inhibits TGFß signalling in cachexia.
-
ASchematic depicting the two tumour models utilised in this study. The GAL4‐UAS induced Ras V12 ;dlg1 RNAi tumour, or the QF2‐QUAS induced Ras V12 scrib RNAi tumour.
-
B–CFat body stained for pAkt from control (r4>mCherry RNAi ) and QRas V12 scrib RNAi ; r4>mCherry RNAi tumour‐bearing animals, counter stained in (B′–C′) for DAPI.
-
DQuantification of normalised (to tumour) fat body pAkt intensity in (B–C). r4>mCherry RNAi (n = 28), QRas V12 scrib RNAi ; r4>mCherry RNAi (n = 25).
-
E–FFat body stained for pMad from control (w 1118 ) and QRas V12 scrib RNAi ; r4>mCherry RNAi tumour‐bearing animals, counter stained in (E'–F′) for DAPI.
-
GQuantification of normalised (to tumour) fat body pMad intensity in (E–F). w 1118 (n = 32), QRas V12 scrib RNAi ; r4>mCherry RNAi (n = 31).
-
H–IFat body of Ras V12 dlg1 RNAi tumour‐bearing animals where mCherry RNAi or ImpL2 RNAi was expressed in the tumour, stained for pMad, counter stained in (H′–I′) for DAPI.
-
JQuantification of fat body pMad intensity in (H–I). mCherry RNAi (n = 29), ImpL2 RNAi (n = 37).
Data information: Scale bar is 50 μm for fat body pAkt and pMad staining, fat body is stained at 6 days after tumour induction. Graphs are represented as Mean ± SEM, n = the number of samples. (****) P < 0.0001, two‐tailed unpaired student's t‐tests were used to test for significant differences. The Welch's correction was applied in cases of unequal variances.
Source data are available online for this figure.
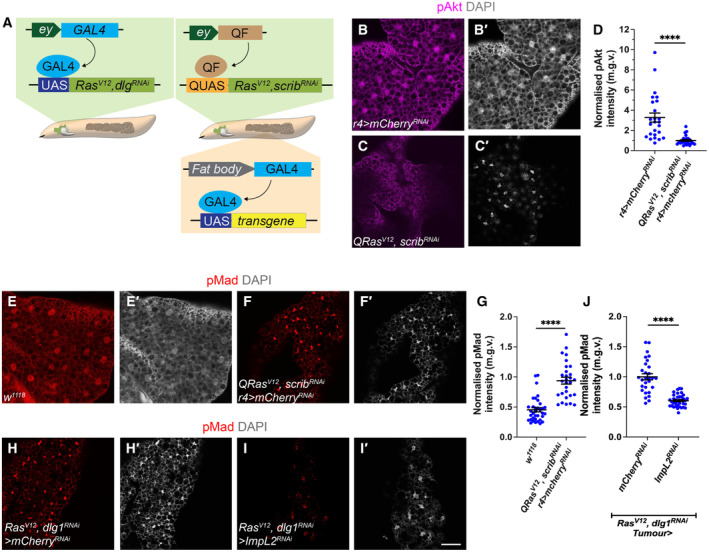
Tumour‐specific ImpL2 inhibition was sufficient to elevate fat body pAkt levels (Lodge et al, 2021) and ameliorate tumour‐induced muscle disruption (Honegger et al, 2008; Figueroa‐Clarevega & Bilder, 2015; Kwon et al, 2015; Lee et al, 2021). Surprisingly, we found that the knockdown of ImpL2 in the tumour (Ras V12 , dlg1 RNAi ) caused a reduction in fat body pMad staining (Fig 1H–J), suggesting that there may be signalling crosstalk between the insulin/PI3K and TGF‐β pathways in the fat body. Furthermore, expression of Akt overexpression in the fat body of tumour animals (QRas V12 , scrib RNAi ), caused an upregulation of pAkt (Fig 2A–C), but an unexpected downregulation of the TGF‐β signalling readout pMad (Fig 2D–F), indicating that an upregulation of insulin/PI3K signalling maybe correlated with a downregulation of TGF‐β signalling. However, conversely, TGF‐β signalling does not seem to affect the activation of PI3K signalling pathway. Upon the expression of the TGF‐β inhibitor sog (Biehs et al, 1996) in the fat body (r4‐GAL4) of tumour bearing animals (QRas V12 , scrib RNAi ), we observed a significant reduction in fat body pMad levels (Fig 2G–I) but no significant effects on pAkt levels (Fig 2J–L).
Figure 2. Insulin signalling negatively inhibits TGFß signalling, but TGFß signalling does not affect insulin signalling in the fat bodies of cachectic animals.
-
A–BFat body stained for pAkt of QRas V12 scrib RNAi tumour‐bearing animals where mCherry RNAi or Akt was expressed in the fat body (r4‐GAL4), counter stained in (A′–B′) for DAPI.
-
CQuantification of normalised fat body pAkt intensity in (A–B). QRas V12 scrib RNAi ; r4>mCherry RNAi (n = 14), QRas V12 scrib RNAi ; r4>Akt (n = 14).
-
D–EFat body of QRas V12 scrib RNAi tumour‐bearing animals where mCherry RNAi or Akt was expressed in the fat body (r4‐GAL4), with TGF‐ß signalling activation indicated by pMad staining, counter stained in (D′–E′) for DAPI.
-
FQuantification of normalised fat body pMad intensity in (D–E). mCherry RNAi (n = 30), Akt (n = 30).
-
G–HFat body from animals expressing mCherry RNAi or sog under the control of r4‐GAL4 in QRas V12 scrib RNAi tumour‐bearing animals stained with pMad, counter stained in (G–H′) for DAPI.
-
IQuantification of normalised fat body pMad intensity in (G–H). mCherry RNAi (n = 30), sog (n = 20).
-
J–KFat body from animals expressing mCherry RNAi or sog under the control of r4‐GAL4 in QRas V12 scrib RNAi tumour‐bearing animals stained with pAkt, counter stained in (J′–K′) for DAPI.
-
LQuantification of normalised fat body pAkt intensity in (J–K). mCherry RNAi (n = 40), sog (n = 17).
Data information: Scale bar is 50 μm for fat body pAkt and pMad staining, fat body is stained at 6 days after tumour induction. Graphs are represented as Mean ± SEM, n = the number of samples. (****) P < 0.0001, (ns) P > 0.05), two‐tailed unpaired student's t‐tests were used to test for significant differences. The Welch's correction was applied in cases of unequal variances.
Source data are available online for this figure.
Enhancing insulin signalling in the fat body improves muscle integrity in cachectic animals
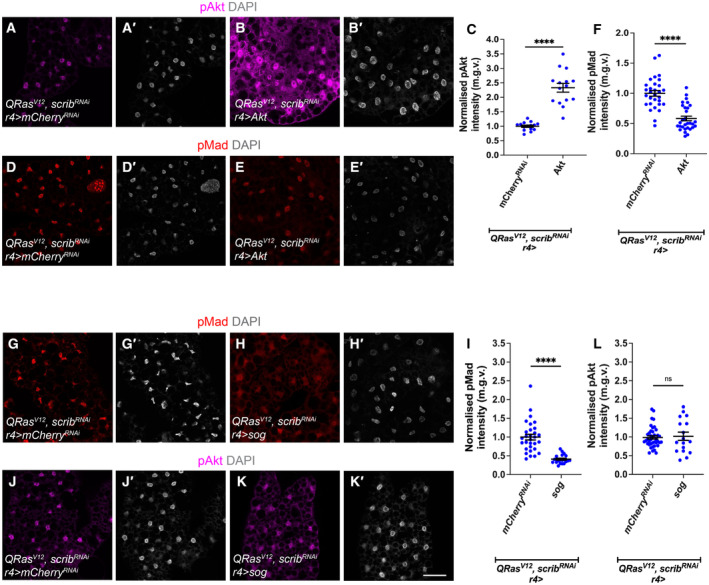
Next, we tested whether fat body‐specific changes in Insulin signalling influences muscle breakdown during cachexia. Similar to GAL4‐induced Ras V12 , scrib RNAi tumour, QF2 induced Ras V12 , scrib RNAi tumours were able to induce a 40% reduction in muscle/cuticle attachment (Lodge et al, 2021; Dark et al, 2022) (Fig 3A–E). Upon overexpression of Akt in the fat body (r4‐GAL4) of tumour bearing animals (QRas V12 , scrib RNAi ), we found there was a significant improvement in muscle morphology (Fig 3F–H). Similarly, overexpression of InR CA in the fat body of tumour bearing animals (QRas V12 , scrib RNAi ) also improved muscle integrity (Fig 3I–K). Together, our data indicate that the elevation of fat body insulin signalling level can improve muscle integrity in the context of cachexia.
Figure 3. Insulin signalling in the fat body improves muscle integrity in cachectic animals.
-
A, BSchematic depicting intact muscle fillet in control animals, versus deteriorated muscle fillets in tumour animals.
-
C, DMuscle fillets stained with phalloidin (Actin) from w 1118 or QRas V12 scrib RNAi tumour‐bearing animals where mCherry RNAi was expressed in the fat body (r4‐GAL4). Detachments are marked with yellow arrows.
-
EQuantification of normalised muscle detachment in (C, D). w 1118 (n = 3), QRas V12 scrib RNAi (n = 10).
-
F, GMuscle fillets stained with phalloidin (Actin) from QRas V12 scrib RNAi tumour‐bearing animals raised at 25°C, where in the fat body, either mCherry RNAi , or Akt was overexpressed.
-
HQuantification of normalised muscle detachment in (F, G), r4>mCherry RNAi (n = 3). QRas V12 scrib RNAi ; r4>mCherry RNAi (n = 5), QRas V12 scrib RNAi ; r4>Akt (n = 8).
-
I, JMuscle fillets stained with phalloidin (Actin) from QRas V12 scrib RNAi tumour‐bearing animals raised at 25°C, where in the fat body, either lacZ;mCherry RNAi , or InR CA ; mCherry RNAi was overexpressed.
-
KQuantification of normalised muscle detachment in (I, J), r4>mCherry RNAi (n = 3). QRas V12 scrib RNAi ; r4>lacZ;mCherry RNAi (n = 3), QRas V12 scrib RNAi ; r4>InR CA ; mCherry RNAi (n = 9).
-
L–NMuscle fillets of QRas V12 scrib RNAi tumour‐bearing animals raised at 25°C where InR CA ; mCherry RNAi or mad RNAi ; LacZ RNAi or InR CA ; mad RNAi was expressed in the fat body (r4‐GAL4).
-
OQuantification of muscle detachment in (L–N). w 1118 (n = 10), InR CA ; mCherry RNAi (n = 8) or mad RNAi ; LacZ RNAi (n = 14) or InR CA ; mad RNAi (n = 9).
Data information: Scale bar is 200 μm for muscle fillet staining carried out at 7 days after tumour induction. Graphs are represented as Mean ± SEM, n = the number of samples. (***) P < 0.001, (****) P < 0.0001, (ns) P > 0.05. For experiments with two genotypes, two‐tailed unpaired student's t‐tests were used to test for significant differences. The Welch's correction was applied in cases of unequal variances. For experiments with more than two genotypes, significant differences between specific genotypes were tested using a one‐way ANOVA and a subsequent Šidák post‐hoc test.
Source data are available online for this figure.
To test if fat body insulin signalling facilitates cachexia progression via TGF‐β signalling, we next activated insulin signalling via fat body expression of InR CA and simultaneously knocked down TGF‐β signalling via fat body expression of mad RNAi (Fig 3L–O) in tumour bearing animals (QRas V12 , scrib RNAi ). We have previously shown that mad RNAi was sufficient to rescue muscle integrity (Lodge et al, 2021) and here found that InR CA (InR CA ;mCherry RNAi ) was similarly able to rescue muscle integrity (Fig 3I–K). Co‐expression of InR CA and mad RNAi gave a similar rescue as either InR CA (InR CA ;mCherry RNAi ) or mad RNAi (mad RNAi ;LacZ RNAi ) alone, suggesting that TGF‐β signalling likely acts downstream of insulin signalling in the fat body of tumour‐bearing animals.
Fat body insulin signalling has been known to modulate the overall size of the animal by altering systemic ecdysone levels and thus we considered whether the insulin signalling pathway may influence cachexia via this growth hormone (Caldwell et al, 2005; Colombani et al, 2005; Mirth et al, 2005). We have previously shown that decreasing systemic ecdysone levels, via prothoracic gland (PG)‐specific expression of an RNAi against torso, increased overall body size, but was not sufficient to cause muscle detachment (Lodge et al, 2021). Here we show that a reduction in body size by increasing global ecdysone levels through PG‐specific expression of Ras V12 (Caldwell et al, 2005) failed to alter the level of muscle detachment in tumour bearing animals (Appendix Fig S1A–C, QRas V12 , scrib RNAi ). Finally, in tumour bearing animals fed a sterol‐free diet, that underwent a prolonged 3rd instar stage due to reduced ecdysone levels (Parkin & Burnet, 1986), we activated insulin signalling in the fat body via Akt overexpression (QRas V12 , scrib RNAi ). We found that this manipulation caused a significant decrease in pMad levels in the fat body (Appendix Fig S1G–I) and a rescue of muscle detachment (Appendix Fig S1D–F), similar to animals fed a standard diet (Fig 3F–H). Together, our data suggest that systemic ecdysone levels are unlikely to be involved in mediating the effects of fat body Insulin/TGF‐β signalling on muscle detachment.
Tumour secreted ImpL2 and Gbb act additively to affect muscle integrity
Although our data suggest that insulin and TGF‐β signalling act in a linear pathway in the fat body to facilitate cachexia, it is unclear how the tumour‐secreted proteins ImpL2 and Gbb interact with each other to facilitate the progression of cachexia. To determine if the two proteins interact, we first assessed whether ImpL2 regulates TGF‐β signalling by influencing gbb expression in the tumour. Upon the knockdown of Impl2, we found that tumour gbb was not significantly altered (Fig EV2A). Next, to determine whether ImpL2 and Gbb acted synergistically to affect muscle integrity, we knocked down both gbb and ImpL2 in the tumour (Ras V12 , dlg1 RNAi ). Inhibition of both ligands resulted in a greater rescue in muscle integrity than knockdown of either ligand alone (Fig EV1A–E). We suspected that these additive effects of the proteins were because each protein rescued different aspects of cachexia and thus tested their effects on two important aspects of cachexia: protein synthesis and ECM accumulation. Protein translation (measured using the O‐propargyl‐puromycin incorporation assay, OPP assay) is significantly downregulated during cachexia (Fig EV1F–H), and ECM proteins such as Nidogen accumulated in the fat body of tumour bearing animals (Lodge et al, 2021, Fig EV1N–P). We next examined whether the knockdown of gbb and ImpL2, either alone or together in the tumour, rescued protein synthesis or ECM accumulation. Knockdown of gbb alone in the tumour did not significantly rescue protein synthesis in the fat body, however, ImpL2 RNAi alone or ImpL2 RNAi and gbb RNAi together rescued protein synthesis (Fig EV1I–M). Conversely, knockdown of gbb alone or knockdown of gbb together with ImpL2 significantly rescued the Nidogen overaccumulation defects observed at the plasma membrane of fat body from tumour‐bearing animals, while ImpL2 RNAi alone did not (Fig EV1Q–U). Finally, the knockdown of ImpL2 alone or the co‐knockdown of gbb and ImpL2 in the tumour significantly rescued the reduction in OPP levels observed in the muscles of tumour‐bearing animals (Fig EV2B–E). Whereas the knockdown of gbb alone or knockdown of gbb together with ImpL2 significantly rescued the reduction in Nidogen levels in the muscles of tumour‐bearing animals (Fig EV2F–I). Altogether, our data indicate that ImpL2 RNAi and gbb RNAi rescue different aspects of cachexia to additively rescue muscle degradation.
Figure EV2. Knockdown of tumour‐derived Gbb and ImpL2 rescues muscle OPP and ECM protein Nidogen.
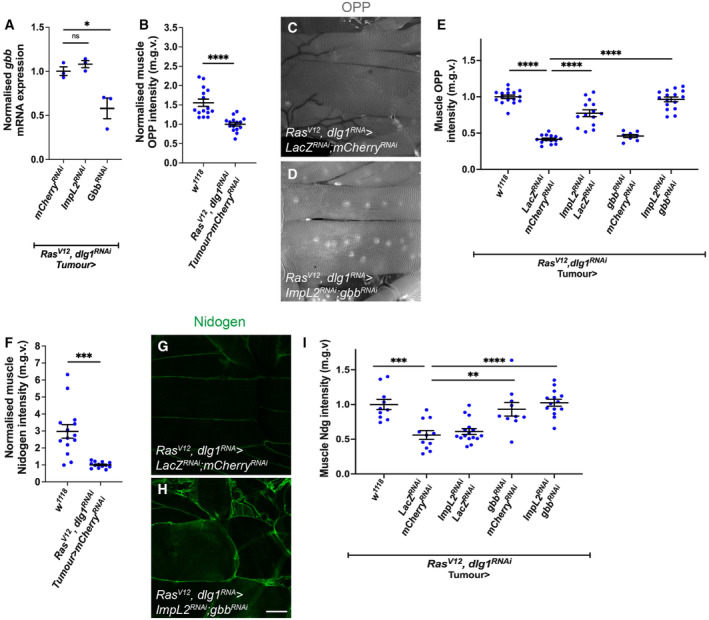
-
ATumour qPCRs showing mRNA expression levels of gbb in Ras V12 dlg1 RNAi larvae expressing mCherry RNAi , ImpL2 RNAi , Gbb RNAi in the tumour (n = 3).
-
BQuantification of normalised muscle OPP intensity in w 1118 and Ras V12 dlg1 RNAi tumour‐bearing animals, where mCherry RNAi is specifically expressed in the tumour. w 1118 (n = 15) and Ras V12 dlg1 RNAi (n = 15).
-
C, DOPP staining detecting protein translation in the muscles of Ras V12 dlg1 RNAi tumour‐bearing animals that express lacZ RNAi ; mCherry RNAi or ImpL2 RNAi ; gbb RNAi .
-
EQuantification of OPP in the muscles of wild type w 1118 (n = 18) and tumour‐bearing animals expressing LacZ RNAi ;mCherry RNAi (n = 16), ImpL2 RNAi ; lacZ RNAi (n = 14), gbb RNAi ; mCherry RNAi (n = 8), ImpL2 RNAi ; gbb RNAi (n = 15).
-
FQuantification of normalised muscle Nidogen intensity in w 1118 and Ras V12 dlg1 RNAi tumour‐bearing animals, where mcherry RNAi is specifically expressed in the tumour. w 1118 (n = 14) and Ras V12 dlg1 RNAi (n = 13)
-
G, HNidogen staining detecting ECM localisation in the muscles of Ras V12 dlg1 RNAi tumour‐bearing animals that express lacZ RNAi ; mCherry RNAi or ImpL2 RNAi ;gbb RNAi .
-
IQuantification of Nidogen in wild type w 1118 (n = 10) and tumour‐bearing animals expressing LacZ RNAi ;mCherry RNAi (n = 11), ImpL2 RNAi ; lacZ RNAi (n = 16), gbb RNAi ; mCherry RNAi (n = 10), ImpL2 RNAi ; gbb RNAi (n = 14).
Data information: Scale bar is 50 μm. Graphs are represented as Mean ± SEM, n = the number of samples. (*) P < 0.05 (**) P < 0.01, (***) P < 0.001, (****) P < 0.0001, (ns) P > 0.05. For experiments with two genotypes, two‐tailed unpaired student's t‐tests were used to test for significant differences. The Welch's correction was applied in cases of unequal variances. For experiments with more than two genotypes, significant differences between specific genotypes were tested using a one‐way ANOVA and a subsequent Šidák post‐hoc test.
Figure EV1. Tumour secreted Gbb and ImpL2 rescue muscle integrity additively.
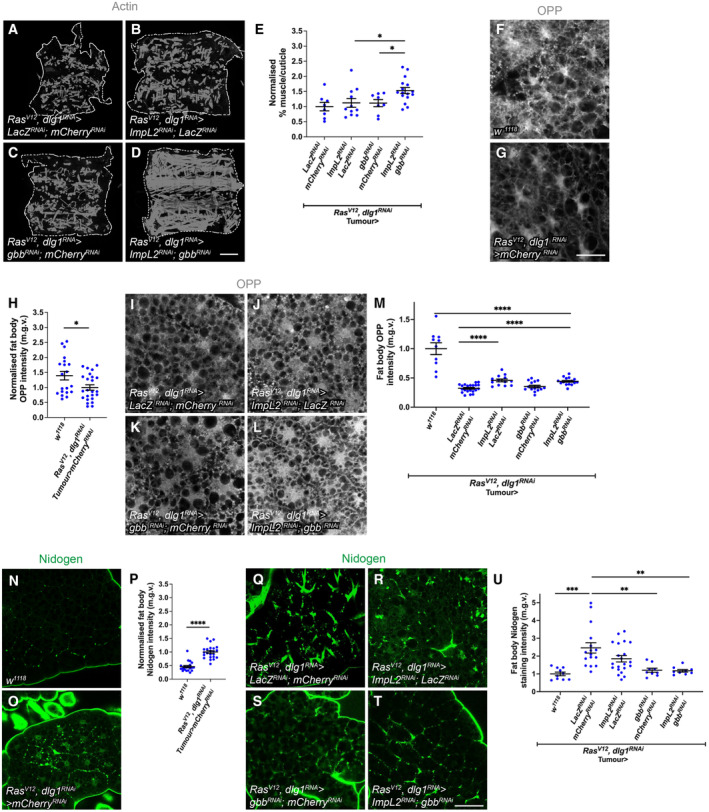
-
A–DMuscle fillets of Ras V12 dlg1 RNAi tumour‐bearing animals that express in the tumour lacZ RNAi ; mCherry RNAi or ImpL2 RNAi ; lacZ RNAi , or gbb RNAi ; mCherry RNAi or ImpL2 RNAi ; gbb RNAi .
-
EQuantification of muscle detachment in (A–D). lacZ RNAi ; mCherry RNAi (n = 8), ImpL2 RNAi ; lacZ RNAi (n = 11), gbb RNAi ; mCherry RNAi (n = 8), ImpL2 RNAi ; gbb RNAi (n = 16).
-
F, GOPP staining detecting protein translation in the fat body of w 1118 and Ras V12 dlg RNAi tumour‐bearing animals.
-
HQuantification of OPP in (F, G). w 1118 (n = 19) Ras V12 dlg1 RNAi (n = 26)
-
I–LOPP staining detecting protein translation in the fat body of Ras V12 dlg1 RNAi tumour‐bearing animals, that express in the tumour lacZ RNAi ; mCherry RNAi or ImpL2 RNAi ; lacZ RNAi , or gbb RNAi ; mCherry RNAi or ImpL2 RNAi ; gbb RNAi .
-
MQuantification of OPP in (I–L). w 1118 (n = 10); LacZ RNAi ; mCherry RNAi (n = 22), ImpL2 RNAi ; lacZ RNAi (n = 13), gbb RNAi ; mCherry RNAi (n = 15), ImpL2 RNAi ; gbb RNAi (n = 14).
-
N, ONidogen staining detecting ECM localisation in the fat body of w 1118 and Ras V12 dlg1 RNAi animals.
-
PQuantification of nidogen staining in (N, O). w 1118 (n = 23) Ras V12 dlg1 RNAi (n = 20).
-
Q–TRas V12 dlg1 RNAi tumour‐bearing animals that express lacZ RNAi ; mCherry RNAi or ImpL2 RNAi ; lacZ RNAi , or gbb RNAi ; mCherry RNAi or ImpL2 RNAi ; gbb RNAi .
-
UQuantification of Nidogen in (Q, T). w 1118 (n = 9); LacZ RNAi ; mCherry RNAi (n = 17), ImpL2 RNAi ; lacZ RNAi (n = 22), gbb RNAi ; mCherry RNAi (n = 9), ImpL2 RNAi ; gbb RNAi (n = 9).
Data information: Scale bar is 50 μm for fat body staining (F, G, I–L, N, O, Q–T) and 500 μm for muscle fillets (A–D). Graphs are represented as Mean ± SEM, n = the number of samples. (*) P < 0.05 (**) P < 0.01, (***) P < 0.001, (****) P < 0.0001. For experiments with two genotypes, two‐tailed unpaired student's t‐tests were used to test for significant differences. The Welch's correction was applied in cases of unequal variances. For experiments with more than two genotypes, significant differences between specific genotypes were tested using a one‐way ANOVA and a subsequent Šidák post‐hoc test.
Muscle insulin signalling regulates atrophy and muscle integrity in cachexia
A key feature of cancer‐induced wasting is the reduction in myofiber size, termed atrophy. This reduction in muscle size can be measured as a ratio of muscle width/length of the VL 3 muscle (Lodge et al, 2021; Graca et al, 2022). Since tumour‐specific ImpL2 inhibition significantly rescued muscle translation, we suspected that insulin signalling in the muscle may play a role in modulating muscle integrity. To test this, we activated insulin signalling in the muscle of tumour bearing animals (QRas V12 , scrib RNAi ) by overexpressing Akt with the muscle‐specific driver MHC‐GAL4. This manipulation significantly rescued muscle integrity (Fig EV3A–C), muscle width/length ratio (Fig EV3D–F), as well as muscle protein translation as indicated by OPP (Fig EV3P). Surprisingly, the muscle ECM levels was further reduced by muscle‐specific Akt overexpression (Fig EV3G–I). This suggests that muscle insulin signalling predominantly regulate translation and atrophy. Interestingly, while muscle pMad levels are elevated in tumour‐bearing animals and that tumour‐specific ImpL2 inhibition was able to reduce muscle TGF‐β signalling in tumour‐bearing animals (Fig EV3Q–S); muscle‐specific expression of mad RNAi in tumour bearing animals (QRas V12 , scrib RNAi ) using MHC‐GAL4 (Fig EV3T) was not able to improve muscle integrity. This suggests that although muscle TGF‐β signalling is responsive to circulating insulin levels, muscle TGF‐β signalling is functionally dispensable in facilitating muscle degradation during cachexia.
Figure EV3. Muscle overexpression of Akt (but not fat body Akt or mad RNAi overexpression) rescues atrophy in cachectic animals, and TGF‐ß signalling in the muscle is responsive to modulation in tumour insulin signalling but is not required for muscle integrity in cachexia.
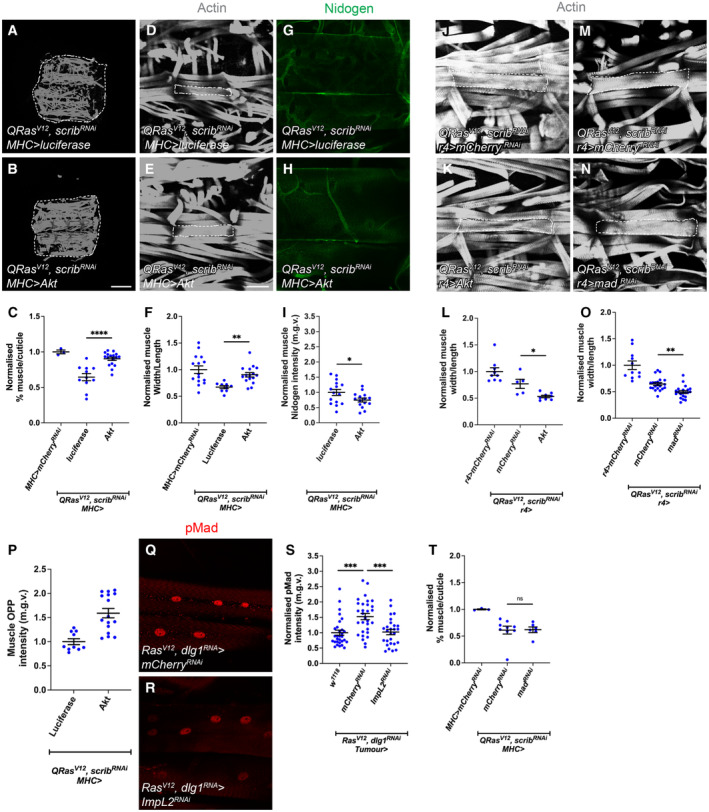
-
A, BMuscle fillets stained with phalloidin (Actin) where luciferase or Akt was expressed under the control of MHC‐GAL4 in QRas V12 scrib RNAi tumour‐bearing animals.
-
CQuantification of normalised muscle detachment in (A, B). Wilt type control (MHC>mCherry RNAi , n = 3), UAS‐luciferase (n = 10), Akt (n = 17).
-
D, EMuscle segment (outlined) where luciferase or Akt was expressed under the control of MHC‐GAL4 in QRas V12 scrib RNAi tumour‐bearing animals.
-
FQuantification of normalised muscle width/length in (D, E). Wild type control (n = 15), luciferase (n = 9), Akt (n = 16).
-
G, HMuscle Nidogen staining where luciferase or Akt was expressed under the control of MHC‐GAL4 in QRas V12 scrib RNAi tumour‐bearing animals.
-
IQuantification of normalised muscle Nidogen staining in (G, H). luciferase (n = 16), Akt (n = 15).
-
J, KMuscle segment from QRas V12 scrib RNAi tumour‐bearing animals, where mCherry RNAi or Akt was expressed in the fat body (r4‐GAL4).
-
LQuantification of normalised muscle width/length in (J, K). r4>mCherry RNAi (n = 9), QRas V12 scrib RNAi ;r4>mCherry RNAi (n = 5), QRas V12 scrib RNAi ;r4>Akt (n = 8).
-
M, NMuscle segment from QRas V12 scrib RNAi tumour‐bearing animals, where mCherry RNAi or mad RNAi was expressed in the fat body (r4‐GAL4).
-
OQuantification of normalised muscle width/length in (M, N). r4>mCherry RNAi (n = 11), QRas V12 scrib RNAi ; r4>mCherry RNAi (n = 18), QRas V12 scrib RNAi ; r4>mad RNAi (n = 15).
-
PQuantification of normalised muscle OPP intensity where luciferase (n = 10) or Akt (n = 16) was expressed under the control of MHC‐GAL4 in QRas V12 scrib RNAi tumour‐bearing animals.
-
Q, RMuscle from Ras V12 dlg1 RNAi tumour‐bearing animals expressing either mCherry RNAi or ImpL2 RNAi in the tumour, where TGF‐ß signalling activation is indicated by pMad staining.
-
SQuantification of normalised muscle pMad intensity in (Q, R). w 1118 (n = 30), mCherry RNAi (n = 30), ImpL2 RNAi (n = 26).
-
TQuantification of normalised muscle detachment where MHC>mCherry RNAi (n = 5) or mCherry RNAi (n = 9) or lacZ RNAi ; mad RNAi (n = 6) was expressed under the control of MHC‐GAL4 in QRas V12 scrib RNAi tumour‐bearing animals.
Data information: Scale bar is 500 μm for muscle fillets (A, B), 100 μm for muscle segment atrophy measurements (D, E, J, K, M, N) and 50 μm for muscle Nidogen and pMad staining (G, H, Q, R). Muscles in tumour bearing animals were dissected at day 6 after tumour induction. Graphs are represented as Mean ± SEM, n = the number of samples. (*) P < 0.05 (**) P < 0.01, (***) P < 0.001, (****) P < 0.0001, (ns) P > 0.05. For experiments with two genotypes, two‐tailed unpaired student's t‐tests were used to test for significant differences. The Welch's correction was applied in cases of unequal variances. For experiments with more than two genotypes, significant differences between specific genotypes were tested using a one‐way ANOVA and a subsequent Šidák post‐hoc test.
Fat body TGF‐β signalling, Rab10 and SPARC regulate ECM accumulation and muscle integrity in cachexia
Next, we examined whether modulating fat body insulin or TGF‐β signalling can improve muscle integrity. We found that fat body specific Akt expression was not able to rescue muscle atrophy caused by QRas V12 Scrib RNAi tumours (Fig EV3J–L). As TGF‐β signalling acts downstream of insulin signalling in the fat body of tumour bearing animals, we next asked if inhibition of TGF‐β signalling in the fat body via the expression of an RNAi against mad improved muscle atrophy in tumour bearing animals (QRas V12 Scrib RNAi). Interestingly, although fat body mad RNAi expression was able to improve overall muscle integrity (Lodge et al, 2021), this manipulation did not significantly improve muscle atrophy (Fig EV3M–O). It however was able to significantly reduced ECM accumulation in the fat body (Fig 4A–B and E) which consequently led to increased muscle Nidogen levels (Fig 4C, D and F). Together, these data suggest that fat body insulin and TGF‐β signalling converge to specify ECM levels in the fat body and in turn, the muscle during cachexia.
Figure 4. TGF‐ß signalling in the fat body rescues muscle detachment via regulating fat body ECM secretion.
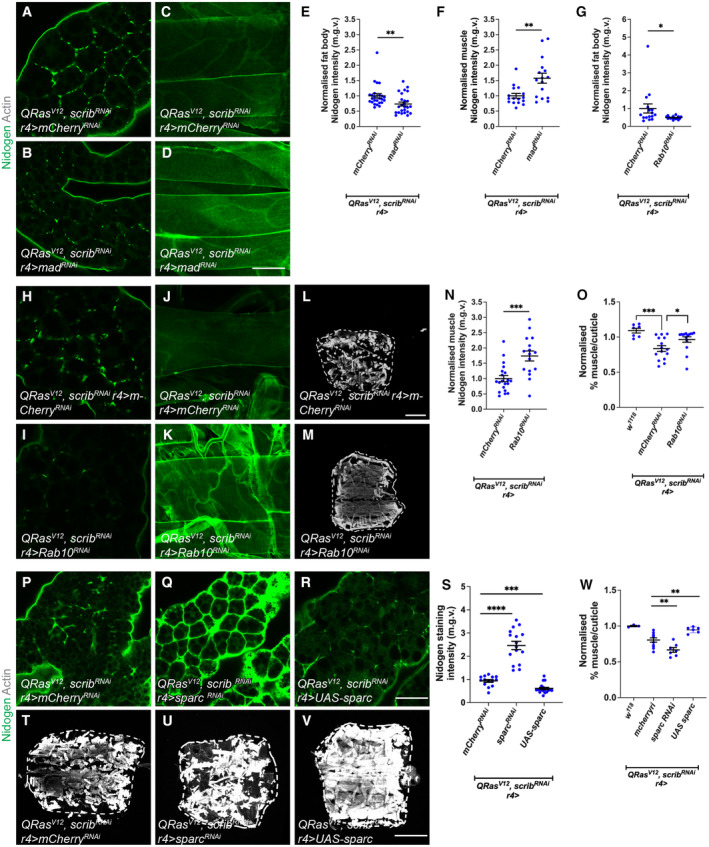
-
A, BFat body stained with the ECM protein Nidogen from QRas V12 scrib RNAi tumour‐bearing animals, where mCherry RNAi or mad RNAi was expressed in the fat body (r4‐GAL4).
-
C, DMuscles stained with Nidogen from QRas V12 scrib RNAi tumour‐bearing animals, where mCherry RNAi or mad RNAi was expressed in the fat body (r4‐GAL4).
-
EQuantification of fat body Nidogen staining in (A, B). mCherry RNAi (n = 25), mad RNAi (n = 25).
-
FQuantification of muscle Nidogen in (C, D). mCherry RNAi (n = 16), mad RNAi (n = 16).
-
GQuantification of normalised fat body Nidogen staining in (H, I). mCherry RNAi (n = 16), Rab10 RNAi (n = 16).
-
H, IFat body stained with the ECM protein Nidogen from QRas V12 scrib RNAi tumour‐bearing animals, where mCherry RNAi or Rab10 RNAi was expressed in the fat body (r4‐GAL4).
-
J, KMuscles stained with Nidogen from QRas V12 scrib RNAi tumour‐bearing animals, where mCherry RNAi or Rab10 RNAi was expressed in the fat body (r4‐GAL4).
-
L, MMuscle fillets stained with phalloidin (Actin) from QRas V12 scrib RNAi tumour‐bearing animals, where mCherry RNAi or Rab10 RNAi was expressed in the fat body (r4‐GAL4). Day 6 muscles used here.
-
NQuantification of normalised muscle Nidogen staining in (J, K). mCherry RNAi (n = 20), Rab10 RNAi (n = 16).
-
OQuantification of normalised muscle detachment in (L, M). w 1118 (n = 5), QRas V12 scrib RNAi ; r4>mCherry RNAi (n = 11), QRas V12 scrib RNAi ;r4>Rab10 RNAi (n = 11).
-
P–RFat body stained with the ECM protein nidogen from QRas V12 scrib RNAi tumour‐bearing animals, where mCherry RNAi or sparc RNAi or UAS‐sparc was expressed in the fat body (r4‐GAL4).
-
SQuantification of normalised fat body nidogen staining in (P–R). mCherry RNAi (n = 12), sparc RNAi (n = 16), UAS‐sparc (n = 10).
-
T–VMuscle fillets stained with phalloidin (Actin) from QRas V12 scrib RNAi tumour‐bearing animals, where mCherry RNAi or sparc RNAi or UAS‐sparc was expressed in the fat body (r4‐GAL4).
-
WQuantification of normalised muscle fillet in (T–V). w 1118 (n = 3), QRas V12 scrib RNAi ; r4>mCherry RNAi (n = 10), QRas V12 scrib RNAi ; r4>sparc RNAi (n = 6), QRas V12 scrib RNAi ; r4>UAS‐sparc (n = 6).
Data information: Scale bar is 200 μm for muscle fillet (L, M, T–V) and 50 μm for muscle Viking staining done at day 7 after tumour induction (C, D, J, K), and scale bar is 50 μm for fat body staining (A, B, H, I, P–R) done at day 6 after tumour induction. Graphs are represented as Mean ± SEM, n = the number of samples. (*) P < 0.05 (**) P < 0.01, (***) P < 0.001, (****) P < 0.0001. For experiments with two genotypes, two‐tailed unpaired student's t‐tests were used to test for significant differences. The Welch's correction was applied in cases of unequal variances. For experiments with more than two genotypes, significant differences between specific genotypes were tested using a one‐way ANOVA and a subsequent Šidák post‐hoc test.
Source data are available online for this figure.
It has previously been shown that muscle ECM proteins are mostly derived from the fat body and blood cells (Dai et al, 2018); furthermore, in the context of cachexia, we observed a correlation between ECM accumulation in the fat body, and ECM depletion in the muscle. We therefore hypothesised that cachectic fat body may be trapping ECM proteins and preventing ECM secretion to the muscle, causing muscle degradation. We first examined whether this ECM accumulation is intracellular or extracellular. We found that without detergent, we could still detect GFP labelled Viking with an anti‐GFP antibody, suggesting that these accumulations are extracellular (Fig EV4A–A'). To explore whether this may be the case, we blocked endocytosis in the fat body (CG‐GAL4) through expression of a temperature‐sensitive dominant negative form of shibire (shi ts) (Zang et al, 2015). It was previously shown that fat body shibire knockdown causes trapping of outgoing ECM proteins, such as Nidogen, in the fat body membrane (Zang et al, 2015). Consistent with our hypothesis, we found that blocking endocytosis resulted in a significant downregulation of Nidogen in the muscles (Fig EV4B–D) and an increase in muscle detachment (Fig EV4E–G). Together, these data suggest that fat body ECM accumulation may contribute to the muscle ECM deficit and muscle detachment.
Figure EV4. Inhibition of fat body endocytosis causes muscle detachment, increasing Vkg expression in the fat body improves muscle integrity in tumour‐bearing animals.
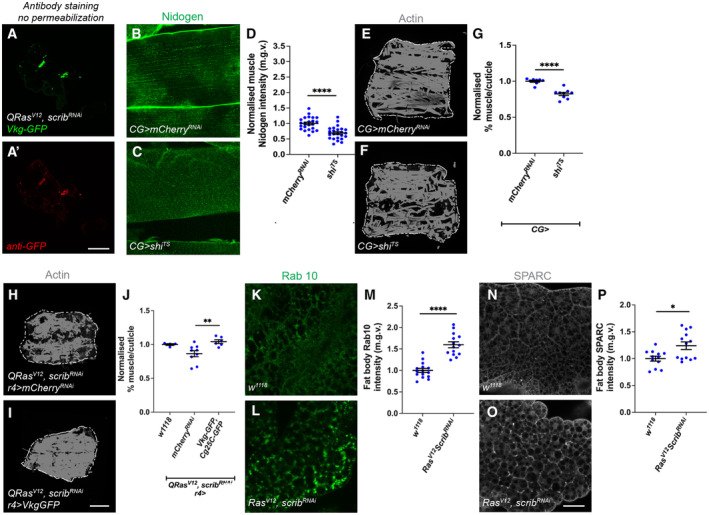
-
A, A'Antibody staining against GFP without permeabilisation can detect Vkg‐GFP.
-
B, CNidogen staining in muscles of animals where mCherry RNAi or shi TS was expressed in the fat body via CG‐GAL4.
-
DQuantification of muscle nidogen staining in (B, C). mCherry RNAi (n = 23), shi TS (n = 24).
-
E, FMuscle fillets stained with phalloidin (Actin) from animals where mCherry RNAi or shi TS was expressed in the fat body via CG‐GAL4.
-
GQuantification of normalised muscle detachment in (E, F). mCherry RNAi (n = 8), shi TS (n = 9).
-
H, IMuscle fillets stained with phalloidin (Actin) from QRas V12 scrib RNAi tumour‐bearing animals, where mCherry RNAi or UAS‐VkgGFP;UAS‐Cg25C‐GFP was expressed in the fat body (r4‐GAL4). Day 6 muscles used here.
-
JQuantification of normalised muscle detachment in (H, I). w 1118 (n = 4), QRas V12 scrib RNAi ;r4>mCherry RNAi (n = 9), QRas V12 scrib RNAi ;r4>UAS‐VkgGFP;UAS‐Cg25C‐GFP (n = 6).
-
K, LFat body staining for Rab10 in w 1118 and QRas V12 scrib RNAi tumour‐bearing animals.
-
MQuantification of normalised Rab10 levels in (K, L). w 1118 (n = 15), QRas V12 scrib RNAi (n = 14).
-
N, OFat body staining for SPARC in w 1118 and QRas V12 scrib RNAi tumour‐bearing animals.
-
PQuantification of normalised SPARC levels in (N, O). w 1118 (n = 12), QRas V12 scrib RNAi (n = 13).
Data information: Scale bar is 500 μm for muscle fillets (E, F, H, I) 50 μm for muscle Nidogen and GFP staining (A, A′, B, C) and 50 μm for fat body staining (K, L, N, O). Muscles in Shi TS animals were dissected at day 5 ALH, muscles and fat body in tumour bearing animals were dissected at day 6 after tumour induction. Graphs are represented as Mean ± SEM, n = the number of samples. (*) P < 0.05 (**) P < 0.01, (****) P < 0.0001. For experiments with two genotypes, two‐tailed unpaired student's t‐tests were used to test for significant differences. The Welch's correction was applied in cases of unequal variances. For experiments with more than two genotypes, significant differences between specific genotypes were tested using a one‐way ANOVA and a subsequent Šidák post‐hoc test.
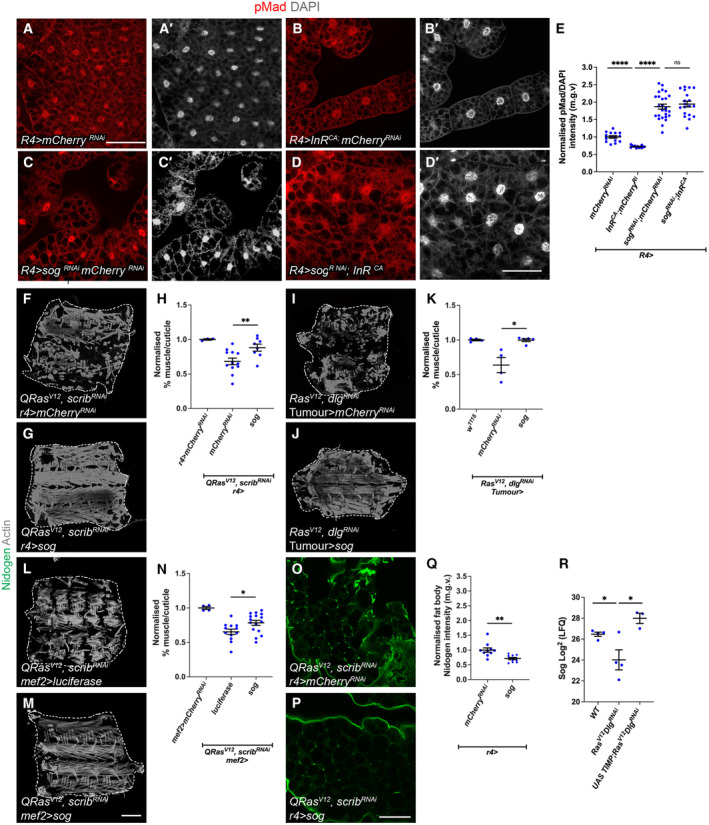
To test whether ECM accumulation in the fat body can affect muscle detachment in the context of cancer cachexia, we next tried to modulate ECM levels in the fat body of tumour bearing animals (QRas V12 Scrib RNAi ). We found that an increase in the expression of Collagen IV (viking [vkg] and Collagen at 25C [Cg25C]) (Pastor‐Pareja & Xu, 2011) in the fat body of tumour bearing animals was sufficient to improve muscle integrity (Fig EV4H–J). It was previously reported that the overexpression of a small GTPase protein called Rab10 can cause an accumulation of ECM proteins (Isabella & Horne‐Badovinac, 2016). We found that Rab10 protein levels was indeed elevated in the fat body of tumour bearing animals (Fig EV4K–M). The overexpression of Rab10 RNAi in the fat body of tumour bearing animals was sufficient to reduce the accumulation of fat body ECM protein Nidogen (Fig 4G–I), increased muscle Nidogen levels (Fig 4J, K and N), and improved muscle attachment (Fig 4L, M and O), suggesting that modulating fat body ECM secretion in cachectic animals can have a direct effect on muscle ECM levels and integrity.
SPARC, the Drosophila homologue of BM40/SPARC/osteonectin, is known to be required for ColIV secretion and has been shown to be a chaperone protein for ColIV both intracellularly and extracellularly (Duncan et al, 2020). Reminiscent of ColIV accumulation in the cachectic fat body, we found that SPARC also accumulated at the cachectic fat body adhesion sites (Fig EV4N–P), suggesting that SPARC together with ColIV might be stuck within the fat body. It has been shown previously that the disruption of SPARC caused the retention of ColIV in the membranes of fat body cells (Pastor‐Pareja & Xu, 2011; Shahab et al, 2015). We found that the expression of a sparc RNAi in the fat body of tumour bearing animals caused further accumulation of Nidogen, and additional deterioration of muscle morphology (Fig 4P–W). Conversely, the overexpression of sparc (Portela et al, 2010), significantly reduced Nidogen accumulation in the fat body, and improved muscle morphology (Fig 4P–W), suggesting that by over‐riding the secretion problems from the fat body, SPARC can help with ECM secretion and muscle integrity. Finally, the activation of TGF‐β signalling pathway, via the overexpression of Mad, caused the transcriptional upregulation of both SPARC and Rab10 (Fig EV5B). This suggests that in the wildtype fat body, TGF‐β signalling can modulate the expression level of these ECM secretion modulators. Together, our data suggest that the activation of TGF‐β signalling pathway can contribute to an accumulation of ECM proteins in the fat body, through the modulation of ECM regulators SPARC and Rab10. In the context of cachexia, this can result in a deficit of ECM proteins in the muscle, causing muscle detachment.
Figure EV5. Modulating fat body TGF‐ß signalling alters sparc and rab10 transcription, and modulating fat body InR alters sog transcription.
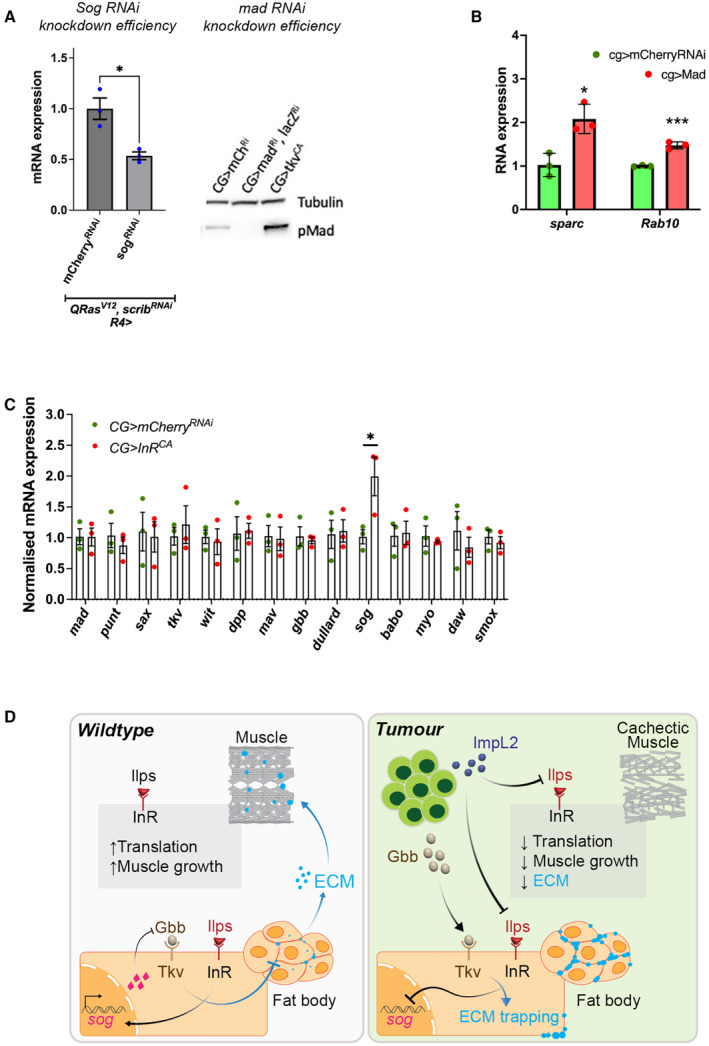
- Knockdown efficiency of sog RNAi and mad RNAi as indicated by qPCR and western blotting respectively. For qPCR, error bars represent SEM, n = 3 for biological replicates.
- Fat body qPCR showing normalised mRNA expression levels of SPARC and Rab10 upon the expression of mCherry RNAi compared to UAS‐Mad. Error bars represent SD, n = 3 for biological replicates.
- Fat body qPCRs showing normalised mRNA expression levels of TGF‐ß receptors and ligands in InR CA or mCherry RNAi larvae (raised at 18°C) with CG‐GAL4 (n = 3, biological replicates). Error bars represent SD.
- Summary diagram—Left: during development, insulin signalling in the fat body activates the transcription of sog, which inhibits Gbb and prevents the activation of TGF‐ß signalling in the fat body. This allows fat body ECM to be secreted to function in the muscle. Insulin signalling in the muscle in parallel enhances translation and muscle growth. Right: in tumour‐bearing/cachectic animals, tumours secrete two ligands: ImpL2 and Gbb. In the fat body, ImpL2 inhibits insulin signalling, preventing the transcription of sog and thus Sog can no longer inhibit Gbb. In addition, tumour secreted Gbb binds to Tkv to activate TGF‐ß signalling in the fat body, resulting in an accumulation of ECM proteins, to prevent ECM transport out of the fat body to reach the muscle. In the muscle, ImpL2 inhibits insulin signalling, which inhibits translation and muscle growth. Fat body in tumour bearing animals were dissected at day 6 after tumour induction. Fat body in non‐tumour bearing animals were dissected at day 5 ALH.
Data information: (*) P < 0.05 (**) P < 0.01, (***) P < 0.001, (****) P < 0.0001. Two‐tailed unpaired student's t‐tests were used to test for significant differences. The Welch's correction was applied in cases of unequal variances.
Insulin signalling can function via TGF‐β signalling in the wildtype fat body
Next, we assessed whether the cross‐regulatory relationship between insulin signalling and TGF‐β signalling observed in the cachectic fat body is relevant in a developmental context. For this, we first starved larvae by subjecting animals to a diet of 1% agarose in PBS for 24 h to reduce their levels of circulating Ilps (Brogiolo et al, 2001). Starved larvae exhibited the expected downregulation in fat body pAkt, and a significant increase in fat body pMad levels (Fig 5A–C) compared to controls fed on a standard laboratory diet. Next, we used CG‐GAL4 to express a constitutive activate form of InR (InR CA ). This caused an upregulation in fat body pAkt and significant reduction in fat body pMad levels (Fig 5D–F). Conversely, when CG‐GAL4 was used to overexpress the gene for the adaptor protein p60 which acts downstream of InR to recruit DP110 and downregulates insulin signalling when overexpressed (Weinkove et al, 1999), we observed an upregulation in fat body pMad levels (Fig 5G–I). Together, our data suggest that the insulin signalling pathway inhibits TGF‐β signalling in the wildtype fat body.
Figure 5. Fat body insulin signalling negatively regulates TGF‐ß signalling.
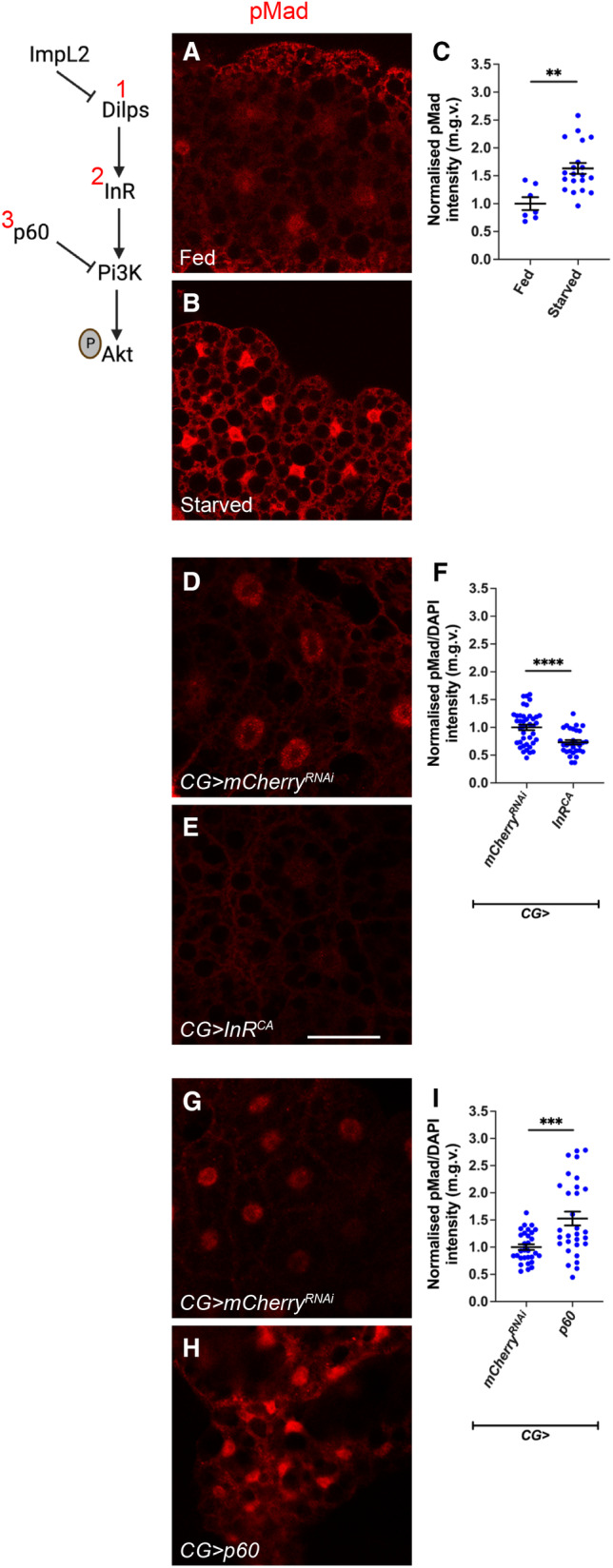
-
A, BFat body from fed and starved animals where Dilps level is reduced (1) with TGF‐ß signalling activation indicated by pMad staining, dissected at day 5 after larval hatching (ALH).
-
CQuantification of normalised pMad staining in (A, B). Fed (n = 7), starved (n = 20).
-
D, EFat body from animals raised at 18°C, dissected at day 7 ALH, that express mCherry RNAi or InR CA (2) under the control of CG‐GAL4, with TGF‐ß signalling activation indicated by pMad staining.
-
FQuantification of normalised pMad staining in (D, E). mCherry RNAi (n = 30), InR CA (n = 40).
-
G, HFat body from animals expressing mCherry RNAi or p60 (3) under the control of CG‐GAL4, with TGF‐ß signalling activation indicated by pMad staining, dissected at day 5 ALH.
-
IQuantification of normalised pMad staining in (G, H). mCherry RNAi (n = 30), p60 (n = 30).
Data information: Scale bar is 50 μm. Graphs are represented as Mean ± SEM, n = the number of samples. (**) P < 0.01, (***) P < 0.001, (****) P < 0.0001, two‐tailed unpaired student's t‐tests were used to test for significant differences. The Welch's correction was applied in cases of unequal variances.
Source data are available online for this figure.
We next tested if reduced Insulin signalling in the fat body phenocopies the Vkg accumulation observed upon fat body activation of TGF‐β signalling. Vkg accumulated in the membranes of fat body upon the overexpression of a dominant negative form of Target of Rapamycin (TOR DN ) using r4‐GAL4 (Fig 6A–C). Similarly, Vkg accumulation was observed upon p60 overexpression (Fig 6E–G). Moreover, as with the activation of Tkv (Lodge et al, 2021), expression of TOR DN did not induce muscle detachment (Fig 6D). This indicates that these signals are indeed functionally equivalent and thus wild‐type fat body insulin signalling may also act through TGF‐β. Interestingly, however, fat body p60 overexpression caused a small but significant reduction in muscle attachment, suggesting that fat body insulin signalling may play additional roles to induce muscle detachment that are independent of TGF‐β (Fig 6H).
Figure 6. Fat body insulin signalling negatively regulates ECM accumulation.
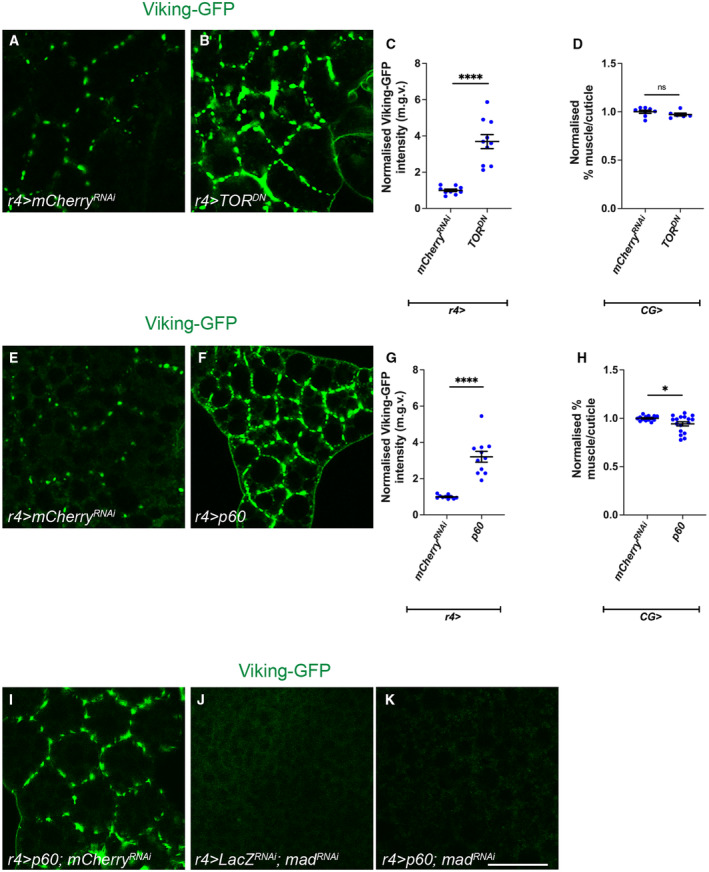
-
A, BViking::GFP localisation in fat body from animals expressing mCherry RNAi or TOR DN under the control of r4‐GAL4.
-
CQuantification of normalised Viking::GFP intensity in (A, B). mCherry RNAi (n = 10) TOR DN (n = 9).
-
DQuantification of normalised muscle detachment caused by the expression of mCherry RNAi or TOR DN under the control of r4‐GAL4. mCherry RNAi (n = 8) TOR DN (n = 7).
-
E, FViking::GFP localisation in fat body from animals expressing mCherry RNAi or p60 under the control of r4‐GAL4.
-
GQuantification of normalised Viking::GFP intensity in (E, F). mCherry RNAi (n = 9), p60 (n = 10).
-
HQuantification of normalised muscle detachment caused by the expression of mCherry RNAi or p60 under the control of r4‐GAL4. mCherry RNAi (n = 12), p60 (n = 16).
-
I–KViking::GFP localisation in fat body from animals expressing p60; mCherry RNAi or lacZ RNAi ; mad RNAi or p60; mad RNAi under the control of r4‐GAL4.
Data information: Scale bar is 50 μm for fat body staining, dissected at day 5 ALH. Graphs are represented as Mean ± SEM, n = the number of samples. (*) P < 0.05 (****) P < 0.0001, (ns) P > 0.05, two‐tailed unpaired student's t‐tests were used to test for significant differences. The Welch's correction was applied in cases of unequal variances.
Source data are available online for this figure.
To determine whether insulin signalling knockdown induces ECM accumulation in the fat body via TGF‐β signalling, we expressed p60 while simultaneously knocking down TGF‐β signalling using mad RNAi . Similar to CG>lacZ RNAi ; mad RNAi , very little Vkg was detected at the plasma membrane of CG>p60; mad RNAi fat bodies (Fig 6I–K). Together, these data suggest that the accumulation of ECM proteins in the fat bodies of CG>p60 or CG>TOR DN larvae is dependent on mad, functionally placing insulin signalling upstream of TGF‐β signalling in the wild‐type fat body.
Insulin signalling affects TGF‐β signalling via TOR and S6K
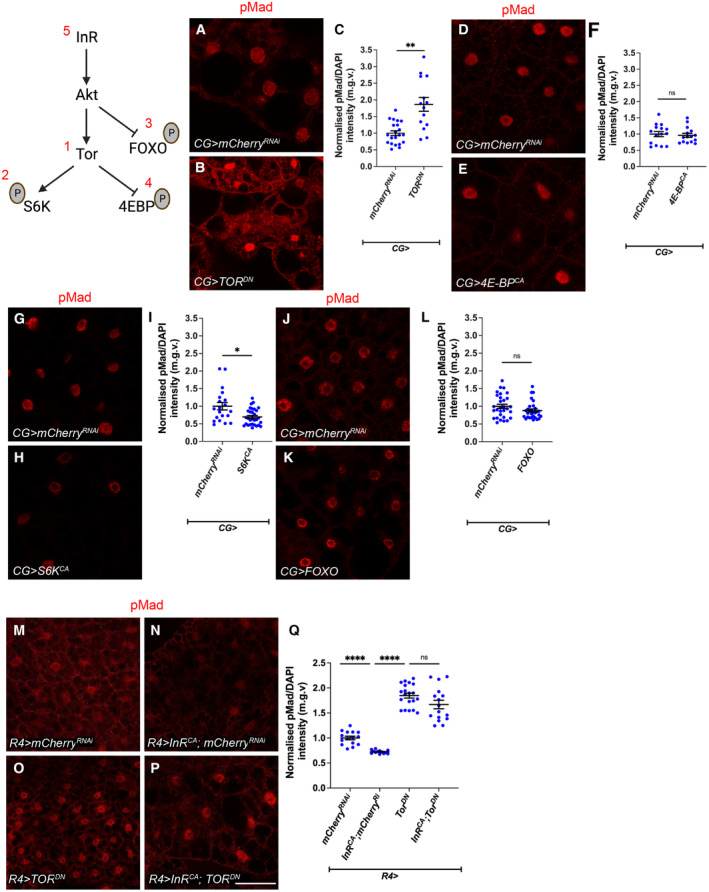
pAkt triggers phosphorylation cascades that result in deactivation of the transcription factor Forkhead box, sub‐group O (FOXO) and activation of TOR. Once activated, TOR activates S6K and deactivates 4E‐BP respectively by inducing their phosphorylation (Zhang et al, 2000). Together, FOXO, TOR, S6K and 4E‐BP make up the most downstream components of the insulin signalling pathway and induce the cellular changes triggered by the activation of the pathway (Giannakou & Partridge, 2007; Semaniuk et al, 2021). To test which of these factors regulate TGF‐β signalling, we used CG‐GAL4 to overexpress FOXO, TOR DN or constitutively activated forms of S6K (S6K CA ) or 4E‐BP (4E‐BP CA ) in the fat body. We found that fat body from CG>4E‐BP CA and CG>FOXO larvae exhibited no change in pMad levels (Fig 7D–F and J–L), whereas fat body from CG>TOR DN larvae exhibited increased pMad levels (Fig 7A–C) and CG>S6K CA fat body exhibited reduced pMad levels (Fig 7G–I). To assess whether InR inhibited TGF‐β signalling in the fat body via TOR, we tested the epistatic relationship between TOR DN and InR CA expression. We found that the co‐expression of TOR DN and InR CA caused elevated pMad, phenocopying knockdown of TOR (TOR DN ) alone, suggesting that insulin signalling modulates fat body TGF‐β signalling via TOR (Fig 7M–Q).
Figure 7. Fat body TOR signalling negatively regulates TGF‐ß signalling.
-
A, BFat body from animals expressing mCherry RNAi or TOR DN (1) under the control of CG‐GAL4, with TGF‐ß signalling activation indicated by pMad staining.
-
CQuantification of normalised pMad (to DAPI) staining in (A, B). mCherry RNAi (n = 21), TOR DN (n = 14).
-
D, EFat body from animals expressing mCherry RNAi or 4E‐BP CA (4) under the control of CG‐GAL4, with TGF‐ß signalling activation indicated by pMad staining.
-
FQuantification of normalised pMad staining in (D, E). mCherry RNAi (n = 15), 4E‐BP CA (n = 15).
-
G, HFat body from animals expressing mCherry RNAi or S6K CA (2) under the control of CG‐GAL4, with TGF‐ß signalling activation indicated by pMad staining.
-
IQuantification of normalised pMad staining in (G, H). mCherry RNAi (n = 20), S6K CA (n = 30).
-
J, KFat body from animals expressing mCherry RNAi or FOXO (3) under the control of CG‐GAL4, with TGF‐ß signalling activation indicated by pMad staining.
-
LQuantification of normalised pMad staining in (J, K). mCherry RNAi (n = 30), FOXO (n = 30).
-
M–PFat body from animals expressing mCherry RNAi or Tor DN (1) or InR CA ; mCherry RNAi (5) or InR CA ;Tor DN (5, 1) under the control of r4‐GAL4, with TGF‐ß signalling activation indicated by pMad staining (the same mCherry RNAi sample was used in Fig 8A, as these experiments were carried out together).
-
QQuantification of normalised pMad staining in (M–P). mCherry RNAi (n = 16), Tor DN (n = 20), InR CA ; mCherry RNAi (n = 12), InR CA ;Tor DN (n = 16). The same mCherry RNAi and InR CA ; mCherry RNAi data points were used as in Fig 8F.
Data information: Scale bar is 50 μm, dissection carried out at 5 days ALH. Graphs are represented as Mean ± SEM, n = the number of samples. (*) P < 0.05 (**) P < 0.01, (****) P < 0.0001, (ns) P > 0.05. For experiments with two genotypes, two‐tailed unpaired student's t‐tests were used to test for significant differences. The Welch's correction was applied in cases of unequal variances. For experiments with more than two genotypes, significant differences between specific genotypes were tested using a one‐way ANOVA and a subsequent Šidák post‐hoc test.
Source data are available online for this figure.
Insulin does not regulate TGF‐β in the muscle or the wing imaginal discs
To determine whether the effect of insulin signalling on TGF‐β signalling is fat body specific, we next assessed whether modulating insulin signalling in the muscle, or the wing discs affects pMad levels in these tissues. We found that overexpression of a constitutively active form of DP110 (DP110 CAAX ) (Leevers et al, 1996) using the muscle driver MHC‐GAL4 did not significantly alter muscle pMad staining (Appendix Fig S2A–C). In the wing imaginal disc, heat shock‐induced clones overexpressing InR CA or p60 exhibited expected changes in clone size (Appendix Fig S2D–E') and pAkt levels. However, these alterations in insulin signalling did not significantly alter TGF‐β signalling (Appendix Fig S2D–E'). Similarly, tkv CA or mad RNAi clones caused altered clone size (Appendix Fig S2F–G') and changes in pMad levels but caused no significant changes in pAkt levels in the wing (Appendix Fig S2F–G'). This indicates that the cross‐regulation of these signalling pathways does not hold in the muscle or the wing imaginal discs.
Insulin signalling influences TGF‐β signalling by modulating fat body sog expression
Since the Bone Morphogenic Protein (BMP) arm of the TGF‐β signalling pathway controls Mad phosphorylation, we suspected that the insulin signalling pathway may modulate TGF‐β signalling levels by directly modulating the transcription of components or regulators of the BMP signalling pathway. To test this, qPCR was conducted on the fat body of CG>InR CA and CG>mCherry RNAi animals (Fig EV5C). We assessed the expression levels of the BMP ligand encoding genes (gbb, decapentaplegic [dpp] and maverick [mav], but not scarecrow as it is only expressed in the CNS during the larval stage, Yoo et al, 2020), the four BMP receptor encoding genes (thickveins [tkv], saxophone [sax], punt and wishful thinking [wit]), the BMP signalling inhibitor sog, the transcription factor mad, and the putative regulator of Mad dephosphorylation dullard (Urrutia et al, 2016). Since non‐BMP components of the TGF‐β signalling pathway have also been observed to influence Mad phosphorylation (Gesualdi & Haerry, 2007; Peterson et al, 2012), we also assessed the transcription of several other TGF‐β signalling pathway genes: babo, myoglianin (myo), dawdle (daw) and smad on X (smox). Strikingly, we found that sog transcription was increased two‐fold in the fat body of CG>InR CA larvae, while none of the other genes tested exhibited any changes in transcription levels (Fig EV5C). To assess whether Sog mediates the downstream effects of insulin signalling on TGF‐β, we expressed sog RNAi in the fat body of larvae expressing InR CA and tested whether sog knockdown inhibits the effects of InR CA on fat body pMad levels (Fig 8A–E). We found that sog knockdown increased pMad level, and the expression of sog RNAi together with InR CA resulted in an increase in pMad level similar to sog RNAi expression alone. These findings suggest that Sog lies downstream of insulin signalling and mediates the effects of insulin signalling on TGF‐β signalling.
Figure 8. Insulin signalling activates the TGF‐ß inhibitor Sog.
-
A–D′Overexpression of mCherry RNAi , InR CA ; mCherry RNAi , Sog RNAi ; mCherry RNAi and Sog RNAi ;InR CA under the control of r4‐GAL4 stained for pMad and DAPI. The same mCherry RNAi sample was used in Fig 7M, as these experiments were carried out together).
-
EQuantification of pMad levels of (A–D′). mCherry RNAi (n = 17), InR CA ; mCherry RNAi (n = 7), Sog RNAi ; mCherry RNAi (n = 26), and Sog RNAi ;InR CA (n = 19), The same mCherry RNAi and InR CA ; mCherry RNAi data points were used as in Fig 6Q.
-
F, GMuscle fillets stained with phalloidin (Actin) from tumour‐bearing (QRas V12 scrib RNAi ) animals where mCherry RNAi or sog was expressed in the fat body using r4‐GAL4.
-
HQuantification of muscle detachment in (F, G). w 1118 (n = 3), mCherry RNAi (n = 10), sog (n = 8).
-
I, JMuscle fillets stained with phalloidin (Actin) from upon tumour‐specific overexpression of mCherry RNAi or Sog in Ras V12 dlg1 RNAi tumour‐bearing animals.
-
KQuantification of muscle detachment in (I, J). w 1118 (n = 3), mCherry RNAi (n = 4), sog (n = 6).
-
L, MMuscle fillets stained with phalloidin (Actin) from tumour‐bearing animals with overexpression of luciferase or sog in the muscle via MEF2‐GAL4.
-
NQuantification of muscle detachment in (L, M). w 1118 (n = 3), luciferase (n = 10), Sog (n = 16).
-
O, PFat body stained with the ECM protein Nidogen from QRas V12 scrib RNAi tumour‐bearing animals, where mCherry RNAi or UAS‐sog was expressed in the fat body (r4‐GAL4).
-
QQuantification of normalised fat body Nidogen staining in (O, P). mCherry RNAi (n = 9), UAS‐sog (n = 10). Scale bar is 50 μm for fat body.
-
RSog levels in the haemolymph of w 1118 , Ras V12 dlg1 RNAi and UAS‐TIMP; Ras V12 dlg1 RNAi tumour‐bearing animals (n = 4,4,3).
Data information: Scale bar is 200 μm for muscle fillet done at day 7 after tumour induction (F, G, I, J, L,M) and scale bar is 50 μm for fat body staining done at day 5 for wildtype fat body (A–D′) and 6 days after tumour induction (O, P). Graphs are represented as Mean ± SEM, n = the number of samples. (*) P < 0.05 (**) P < 0.01, (****) P < 0.0001, (ns) P > 0.05. For experiments with two genotypes, two‐tailed unpaired student's t‐tests were used to test for significant differences. The Welch's correction was applied in cases of unequal variances. For experiments with more than two genotypes, significant differences between specific genotypes were tested using a one‐way ANOVA and a subsequent Šidák post‐hoc test.
Source data are available online for this figure.
Sog is a critical regulator of cachexia
Since insulin signalling likely regulates TGF‐β signalling by modulating sog expression, we assessed whether Sog modulation is relevant in cachectic animals. Analysis of our previously published hemolymph proteomics data (Lodge et al, 2021) showed that Sog is one of the most downregulated circulating proteins in tumour‐bearing animals (Fig 8R). The downregulation of haemolymph Sog appears to be dependent on tumour‐derived Mmps. Upon the inhibition of Mmps in the tumour via the overexpression of TIMP, we found that circulating Sog levels is restored to wildtype levels (Fig 8R). Together, this suggests that circulating Sog levels are low in the tumour bearing animals. Consistent with this, increasing Sog levels specifically in the fat body, muscle or tumour via Sog overexpression, significantly improved tumour induced muscle detachment (Fig 8F–H, I–K and L–N). Furthermore, the overexpression of Sog in the fat body significantly improved Nidogen accumulation in the fat body (Fig 8O–Q), consistent with our model that TGF‐β signalling regulates fat body ECM accumulation. Together, our data show that Sog is a critical regulator of cachexia.
Discussion
It has been shown that cachexia is mediated by humoral factors secreted from the tumour, and the complete removal of cachexia‐associated tumours can reverse cachexia (Ni et al, 2012). However, in late stage/metastatic cancer patients, the removal of the tumour is often not an option. In this study, we demonstrate, using a Drosophila tumour model, that targeting signalling deregulations in the peripheral tissues can be a novel strategy to treat cachexia even when tumours are present, and the signalling status of peripheral tissues can serve as diagnostic biomarker for cachexia. We show that the insulin and TGF‐β signalling pathways are deregulated in the peripheral tissues in tumour bearing animals, their disruption can recapitulate some aspects of tissue wasting, and their modulation can rescue tissue wasting in the presence of a tumour.
We have identified the fat body as a central node in our cachexia model. In both fly and mouse cachexia models, the breakdown of fat precedes that of the muscles (Das et al, 2011; Lodge et al, 2021). Here, we find that tumour secreted proteins ImpL2 and Gbb converge in the fat body to induce the activation of TGF‐β signalling (Fig EV5D). TGF‐β activation then drives an aberrant accumulation of ECM at the inter‐adipocyte junctions in the fat body, which likely prevents ECM secretion and subsequent transport to the muscle. We found that the modulation of ECM secretion via SPARC or Rab10 can ameliorate the cachexia phenotype. It was previously reported that plasma membrane (PM) overgrowth induced by increased inflammation can cause pericellular collagen accumulation in the fat body (Zang et al, 2015). In the cachectic fat body, we have not observed PM thickening, therefore, it is likely this occurs in cachexia via a different mechanism. Aberrant ECM accumulation in the adipose tissue has been observed in human cachexia patients, and this accumulation has been associated with elevated levels of TGF‐β signalling (Alves et al, 2017). Furthermore, reduced ECM levels have been reported in the muscles in a rat cachexia model (Moraes et al, 2017). Thus, altered ECM localisation appears to be a conserved mechanism in cancer cachexia.
In this study, we show that insulin signalling (regulated by tumour‐derived ImpL2) directly affects muscle translation rates and atrophy. This, together with fat body TGF‐β activation/ECM accumulation, appears to contribute towards the regulation of muscle integrity in the context of cachexia (Fig EV5D). Additional tumour‐secreted signals likely exist. Whether these signals also act through the fat body (through regulation of ECM production or via alternative mechanisms), or act directly on the muscle or even via other target tissues, remains to be addressed. Studying these signals, their downstream signalling, as well as identifying additional target tissues where the signalling is relevant, remain important areas of future research.
The insulin signalling pathway plays many important roles in the regulation of growth and metabolism. TGF‐β signalling is best known for roles in development, growth and differentiation. More recently, these pathways have been shown to intersect in metabolic contexts. In the fat body, it has been shown under high fat diet induced obesity, that fat body derived Gbb can cause insulin resistance via negative regulation of the insulin signalling pathway (Hong et al, 2016). Furthermore, in both Drosophila and C. elegans, it has been shown that BMP signalling can regulate Ilps to modulate lipid homeostasis (Ballard et al, 2010; Clark et al, 2021). In the developing muscle, it was recently shown that the Activin branch of the TGF‐β signalling pathway positively regulates the insulin signalling pathway via the regulation of a structural protein, MHC (Kim & O'Connor, 2021). In contrast to these studies, we show that, in the fat body, insulin signalling regulates TGF‐β signalling by its modulation at the extracellular level via the secreted BMP inhibitor Sog. Interestingly, it has been shown in Zebrafish that IGF signalling can influence the expression of BMPs via Chordin (Sog homologue) during embryonic pattering, indicating that this relationship is conserved across distantly related species (Eivers et al, 2004). Although it was previously shown that the insulin signalling effector FOXO can directly bind to the promoter of Sog in adult flies (Birnbaum et al, 2019), this does not seem to be the case in the larval fat body as FOXO overexpression did not significantly alter fat body pMad levels. Instead, our data suggest that insulin signalling regulates sog expression via TOR and S6K. In support of this, TOR has been shown to influence other transcriptional regulators such as Raptor and Yorkie, thus it may regulate sog expression by modulating one of these transcriptional regulators (Parker & Struhl, 2015; Tiebe et al, 2015).
Sog has previously been shown to be important in embryonic patterning as well as at the developing cross‐veins in the pupal wing. Our study also demonstrates that the inhibition of extracellular BMP ligands via Sog is a novel link between insulin and TGF‐β signalling in the fat body. We show that Sog is highly relevant in the context of cachexia, as it is found to be highly downregulated in the haemolymph of tumour‐bearing animals. In addition, tissue specific overexpression of sog in the tumour, fat body and muscle significantly rescued muscle integrity in the tumour bearing animals. Therefore, it appears that Sog levels in the tumour, as well as in peripheral tissues, play a role in mediating cachexia. As RNAseq data from the peripheral tissues of cachexia patients are not readily available through public data bases, we instead examined cBioPortal data which assesses gene expression in the tumour (5,541 samples in 25 pan‐cancer studies) (Cerami et al, 2012; Gao et al, 2013). We found that there is a mild negative correlation between BMP7 expression (52% homology with gbb) and CHORDIN (CHRD, 40% homology with Sog) in cancers commonly associated with cachexia as well as non‐cachectic cancers (Appendix Fig S3), where the correlation is slightly strongly in the cachexia‐related cancers. Together, this suggests that BMP and its regulators are expressed in many types of cancers, and that they may regulate each other's expression. As such, CHRD may also be relevant for the progression of cachexia in humans, thus it would be interesting to explore whether circulating CHRD can serve as a potential biomarker for the diagnosis of cachexia. Together, our data suggest that TGF‐β and insulin signalling crosstalk is relevant in the context of cancer cachexia. Our data also reveal potential therapeutic avenues to treat cachexia in the clinic when resection of the tumour is not an option in late stage/metastatic patients. These include targeting BMP antagonists such as Sog/CHRD, inhibition of TGF‐β signalling, activation of insulin signalling, or enhancement of ECM secretion in the adipose tissue.
Material and Methods
Drosophila stocks and husbandry
The following stocks were used from the Bloomington Drosophila stock centre: CG‐GAL4 (BL7011), Mef2‐GAL4 (BL27391), MHC‐GAL4 (BL55133), r4‐GAL4 (BL33832), UAS‐4E‐BP CA (BL24854), UAS‐Akt (BL8191), UAS‐Dp110 CAAX (BL25908), UAS‐Gbb RNAi#2 (BL34898), UAS‐InR CA (BL8263), UAS‐luciferase (BL64774), UAS‐mad RNAi (BL31316), UAS‐mCherry RNAi (BL35785), UAS‐S6K CA (BL6914), UAS‐Ras V12 (BL64195), UAS‐Shi TS (BL44222), UAS‐FOXO (BL9575), UAS‐tkv CA (BL36536), UAS‐GFP (BL4775), UAS‐Sparc3 (Helena Richardson), ey‐FLP1;act>CD2>GAL4, UAS‐GFP (Lodge et al, 2021), UAS‐lacZ RNAi (BL31562), sog‐lacZ (BL10132). The following stocks were obtained from the Vienna Drosophila Resource Centre: UAS‐Gbb RNAi (v330684), UAS‐Impl2 RNAi (v30931), UAS‐Sparc RNAi (v16677), UAS‐Sog RNAi (v37405) (Hattori et al, 2013). The following stock was obtained from the Kyoto stock centre: Vkg‐GFP (110692). The following stocks were also used: Ey‐FLP1; QUAS‐Ras V12 , QUAS‐scrib RNAi /CyOQS; act>CD2>QF, UAS‐RFP/TMBQS (Lodge et al, 2021) Ey‐FLP1; UAS‐Ras V12 , UAS‐dlg1 RNAi /CyO, GAL80; act>CD2>GAL4, UAS‐GFP (Manent et al, 2017), Phm‐GAL4 (McBrayer et al, 2007), UAS‐p60 (Cheng et al, 2011), UAS‐Sog‐HA (Yu et al, 2000), UAS‐TOR DN (Zhang et al, 2006), UAS‐cg25C;UAS‐Vkg (Van De Bor et al, 2015). The knockdown efficiency of mad RNAi and sog RNAi are shown in Fig EV5.
For the r4>InR CA ; mad RNAi interaction experiment and analysis of atrophy in QRas V12 scrib RNAi larvae carrying r4>mad RNAi larvae were reared at 25°C. For experiments with CG>InR CA larvae were reared at 18°C. For experiments with CG>shi TS , adults were allowed to lay for 24 h at 25°C and progeny transferred to 31°C after an additional 48 h. For all other experiments, adults were allowed to lay for 24 h at 25°C and the progeny then moved to 29°C. Animals were dissected at wandering stage in non‐tumour bearing animals (with the exception of the CG>shi TS experiments, conducted on day 7 animals), and for tumour‐bearing animals, they were dissected on 7 days after egg lay or as indicated throughout.
Immunostaining
For muscle staining, larvae were filleted as previously described (Lodge et al, 2021; Dark et al, 2022), fixed for 30 min in PBS containing 4% formaldehyde and washed with PBS containing 0.3% Triton‐X (PBST‐0.3). Fat body was fixed for 45 min and washed with PBS containing 0.2% Triton‐X (PBST‐0.2). Wing discs were fixed for 20 min and washed with PBS containing 0.2% Triton‐X (PBST‐0.2). Tissues were then stained as per the manufacturer's specifications. Samples were mounted in glycerol and imaged on an Olympus FV3000 confocal microscope. Within a given experiment, all images were acquired using identical settings. Fat body samples were imaged the same day as mounting. To assay for translation in the muscle, we used Click‐iT™ Plus OPP Alexa Fluor™ 488 Protein Synthesis Assay Kit (Thermo Fisher, #C10456). Primary antibodies used: ßgal (1:100, Promega), pMad (1:800, Cell Signalling, #9516, used for fat body staining), pMad (1:100, abcam, ab52903, used for muscle staining) pAkt (Cell signalling, 1:100, #4060), Nidogen (gift of Anne Holtz, 1:500), SPARC (1:500, (Shahab et al, 2015). Secondary donkey antibodies conjugated to Alexa 555 and Alexa 647 (Molecular Probes) were used at 1:200. DAPI (Molecular Probes) was used at 1:10,000, Phalloidin (Molecular Probes) was used at 1:10,000. Actin muscle stains were conducted on day 7 animals when dissected from QRas V12 scrib RNAi animals except when raised at 25°C and on day 8 animals when dissected from Ras V12 dlg1 RNAi . All other muscle and fat body staining were conducted at day 6 when dissected from QRas V12 scrib RNAi animals and at day 7 when dissected from Ras V12 dlg1 RNAi animals (except when specified in the figure legend).
Western blotting
Fatbodies dissected from three wandering stage larvae (or day 6 larvae where tumour‐bearing animals were used) were homogenised in RIPA lysis buffer containing EDTA‐free cOmplete ULTRA protease inhibitor (Roche, #05892791001). Lysate protein concentrations were determined using the DC protein assay (BioRad, #5000112) and protein concentrations in all samples equalised before adding 10 mM DTT and Bolt LDS sample buffer (Novex, #B0007) to samples and then boiling at 70°C for 10 min. Samples were run in 12‐well Bolt 4–12% Bis‐Tris Plus precast gels (Invitrogen, #NW04122BOX) using Bolt MOPS SDS running buffer (Invitrogen, #B0001) and transferred to an Immobilon‐FL PVDF membrane (Sigma‐Aldrich, #IPFL00010) via wet transfer using transfer buffer (50 mM Tris, 38 mM Glycine, 20% ethanol). Membranes were blocked in 5% skim milk (w/v) diluted in TBS containing 0.2% Tween‐20 (TBST), incubated with antibodies as per the manufacturer's specifications, developed using Amersham ECL Prime Western Blotting Reagent (Cytiva, #RPN2236) and imaged on the iBright 1,500 (Invitrogen). Primary antibodies used: pMad (1:1,000, Cell Signalling, #9516). Secondary antibodies used: anti‐Mouse HRP (1:10,000, Jackson ImmunoResearch Labs, #115‐035‐003), anti‐Rabbit HRP (1:10,000, Jackson ImmunoResearch Labs, #111‐035‐003).
Image analysis
All images were quantified using ImageJ. In wildtype fat body and muscle samples pMad intensity was normalised to DAPI, pMad and DAPI levels were quantified by drawing a circle around the nucleus in the DAPI channel, and the mean grey value (m.g.v.) determined for pMad and DAPI channels. In tumour bearing fat body, pMad, pAkt and OPP were not normalised to DAPI levels, as fat body tissue penetrance is altered in the presence of tumour. Here, the ROI circle was drawn around the nucleus in the pMad, pAkt or OPP channel. To measure fluorescence intensity of Vkg‐GFP and Ndg, a line was drawn around membrane of a single cell in the fat body or along the edge of a single muscle segment in fillets on the z‐plane where fluorescence was most intense, the line made to be five pixels wide, and m.g.v. determined along the line. %muscle/cuticle was determined using ImageJ as previously described (Lodge et al, 2021; Dark et al, 2022). For muscle atrophy measurements, muscle length and width measurements were done on the VL3 muscle on tiled 10X images of individual fillets using FIJI software with the line selection tool.
qPCR
For each biological replicate, three to five larvae were selected at day 6 (for fat body samples). Samples were dissected in cold PBS and snap‐frozen in liquid nitrogen. Frozen tissue was then lysed in 300 μL of TRI Reagent (Invitrogen, #10296010) Total RNA was extracted using a Direct‐zol RNA Microprep Kit (Zymo Research, #R2061). cDNA was obtained by reverse transcription of 0.5 μg of total RNA using ProtoScript II First Strand cDNA Synthesis Kit (NEB, #E6560S). Three independent biological replicates were prepared for each genotype. qPCR was performed with the Fast SYBR™ Green PCR Master Mix (Applied Biosystems, #4385612) on a LightCycler 480 (Roche). Gene expression was normalised to the geometric mean of the reference gene rpl32.
TCGA tumour data analyses
mRNA expression data for CHRD and BMP7 were downloaded for 24 TCGA studies from cBioPortal on 3rd August 2022. A total of 9,409 samples had RSEM data that had been batch normalised for both genes, and Spearman correlation was performed.
Statistical analysis
Hemolymph analysis was done using Limma. All other statistical analyses were conducted using GraphPad Prism. At least three animals per genotype were used for all muscle and fat body experiments. For staining intensity quantifications, individual data points represent fluorescence intensity of a single cell. For muscle integrity quantifications, individual data points represent a single larva. Data in Fig 4H was confirmed to be normally distributed using the Shapiro–Wilk test. For experiments with two genotypes or treatments, two‐tailed unpaired student's t‐tests were used to test for significant differences. The Welch's correction was applied in cases of unequal variances. For experiments with more than two genotypes, significant differences between specific genotypes were tested using a one‐way ANOVA and a subsequent Šidák post‐hoc test. The results for all post‐hoc tests conducted in each analysis are shown in graphs. For all graphs, error bars represent SEM. P and adjusted‐P values are reported as follows: P > 0.05, ns (not significant); P < 0.05, *; P < 0.01, **; P < 0.001, ***; P < 0.0001, ****.
Author contributions
Daniel Bakopoulos: Conceptualization; data curation; formal analysis; investigation; methodology; writing – original draft; writing – review and editing. Sofya Golenkina: Conceptualization; data curation; formal analysis; investigation; methodology; writing – review and editing. Callum Dark: Conceptualization; data curation; formal analysis; investigation; methodology; writing – review and editing. Elizabeth L Christie: Data curation. Besaiz J Sánchez‐Sánchez: Resources. Brian M Stramer: Resources. Louise Y Cheng: Conceptualization; resources; data curation; formal analysis; supervision; funding acquisition; investigation; methodology; writing – original draft; project administration; writing – review and editing.
Disclosure and competing interests statement
The authors declare that they have no conflict of interest.
Supporting information
Appendix S1
Expanded View Figures PDF
PDF+
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Source Data for Figure 7
Source Data for Figure 8
Acknowledgements
We are grateful to Kieran Harvey, Helena Richardson, Gary Hime, Anne Holz, Travis Johnson, Christen Mirth, Maurice Ringuette and Ana Traven for generous sharing of fly stocks, yeast stock and antibodies. We would like to thank Bloomington Drosophila Stock Center, Vienna Drosophila Resource Center and Developmental Studies Hybridoma Bank for fly stocks and antibodies. We would like to also thank OZDros for Drosophila quarantine, Peter MacCallum Cancer Institute CAHM for microscopy assistance. We are grateful to Kellie Veen and Khanh Phuong Nguyen for assistance with graphics production. LYC's laboratory is supported by funding from the NHMRC Ideas Grant (APP1182847), and the Peter MacCallum Cancer Foundation (APP2218).
EMBO reports (2023) 24: e57695
Data availability
These data include no data deposited in external repositories.
References
- Alves MJ, Figuerêdo RG, Azevedo FF, Cavallaro DA, Neto NIP, Lima JDC, Matos‐Neto E, Radloff K, Riccardi DM, Camargo RG et al (2017) Adipose tissue fibrosis in human cancer cachexia: the role of TGFβ pathway. BMC Cancer 17: 190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballard SL, Jarolimova J, Wharton KA (2010) Gbb/BMP signaling is required to maintain energy homeostasis in Drosophila . Dev Biol 337: 375–385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baracos VE, Martin L, Korc M, Guttridge DC, Fearon KCH (2018) Cancer‐associated cachexia. Nat Rev Dis Primers 4: 1–18 [DOI] [PubMed] [Google Scholar]
- Biehs B, François V, Bier E (1996) The Drosophila short gastrulation gene prevents Dpp from autoactivating and suppressing neurogenesis in the neuroectoderm. Genes Dev 10: 2922–2934 [DOI] [PubMed] [Google Scholar]
- Birnbaum A, Wu X, Tatar M, Liu N, Bai H (2019) Age‐dependent changes in transcription factor FOXO targeting in female Drosophila . Front Genet 10: 312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brogiolo W, Stocker H, Ikeya T, Rintelen F, Fernandez R, Hafen E (2001) An evolutionarily conserved function of the Drosophila insulin receptor and insulin‐like peptides in growth control. Curr Biol 11: 213–221 [DOI] [PubMed] [Google Scholar]
- Caldwell PE, Walkiewicz M, Stern M (2005) Ras activity in the Drosophila prothoracic gland regulates body size and developmental rate via ecdysone release. Curr Biol 15: 1785–1795 [DOI] [PubMed] [Google Scholar]
- Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E et al (2012) The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov 2: 401–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng LY, Bailey AP, Leevers SJ, Ragan TJ, Driscoll PC, Gould AP (2011) Anaplastic lymphoma kinase spares organ growth during nutrient restriction in Drosophila . Cell 146: 435–447 [DOI] [PubMed] [Google Scholar]
- Clark JF, Ciccarelli EJ, Kayastha P, Ranepura G, Yamamoto KK, Hasan MS, Madaan U, Meléndez A, Savage‐Dunn C (2021) BMP pathway regulation of insulin signaling components promotes lipid storage in Caenorhabditis elegans . PLoS Genet 17: e1009836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colombani J, Bianchini L, Layalle S, Pondeville E, Dauphin‐Villemant C, Antoniewski C, Carré C, Noselli S, Léopold P (2005) Antagonistic actions of ecdysone and insulins determine final size in Drosophila . Science 310: 667–670 [DOI] [PubMed] [Google Scholar]
- Dai J, Ma M, Feng Z, Pastor‐Pareja JC (2017) Inter‐adipocyte adhesion and signaling by collagen IV intercellular concentrations in Drosophila . Curr Biol 27: 2729–2740 [DOI] [PubMed] [Google Scholar]
- Dai J, Estrada B, Jacobs S, Sánchez‐Sánchez BJ, Tang J, Ma M, Magadán‐Corpas P, Pastor‐Pareja JC, Martín‐Bermudo MD (2018) Dissection of nidogen function in Drosophila reveals tissue‐specific mechanisms of basement membrane assembly. PLoS Genet 14: e1007483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dark C, Cheung S, Cheng LY (2022) Analyzing cachectic phenotypes in the muscle and fat body of Drosophila larvae. STAR Protoc 3: 101230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das SK, Eder S, Schauer S, Diwoky C, Temmel H, Guertl B, Gorkiewicz G, Tamilarasan KP, Kumari P, Trauner M et al (2011) Adipose triglyceride lipase contributes to cancer‐associated cachexia. Science 333: 233–238 [DOI] [PubMed] [Google Scholar]
- Ding G, Xiang X, Hu Y, Xiao G, Chen Y, Binari R, Comjean A, Li J, Rushworth E, Fu Z et al (2021) Coordination of tumor growth and host wasting by tumor‐derived Upd3. Cell Rep 36: 109553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong J, Yu J, Li Z, Gao S, Wang H, Yang S, Wu L, Lan C, Zhao T, Gao C et al (2021) Serum insulin‐like growth factor binding protein 2 levels as biomarker for pancreatic ductal adenocarcinoma‐associated malnutrition and muscle wasting. J Cachexia Sarcopenia Muscle 12: 704–716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan S, Delage S, Chioran A, Sirbu O, Brown TJ, Ringuette MJ (2020) The predicted collagen‐binding domains of Drosophila SPARC are essential for survival and for collagen IV distribution and assembly into basement membranes. Dev Biol 461: 197–209 [DOI] [PubMed] [Google Scholar]
- Eivers E, McCarthy K, Glynn C, Nolan CM, Byrnes L (2004) Insulin‐like growth factor (IGF) signalling is required for early dorso‐anterior development of the zebrafish embryo. Int J Dev Biol 48: 1131–1140 [DOI] [PubMed] [Google Scholar]
- Figueroa‐Clarevega A, Bilder D (2015) Malignant Drosophila tumors interrupt insulin signaling to induce cachexia‐like wasting. Dev Cell 33: 47–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E et al (2013) Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal 6: pl1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gesualdi SC, Haerry TE (2007) Distinct signaling of Drosophila activin/TGF‐beta family members. Fly (Austin) 1: 212–221 [DOI] [PubMed] [Google Scholar]
- Giannakou ME, Partridge L (2007) Role of insulin‐like signalling in Drosophila lifespan. Trends Biochem Sci 32: 180–188 [DOI] [PubMed] [Google Scholar]
- Graca FA, Rai M, Hunt LC, Stephan A, Wang Y‐D, Gordon B, Wang R, Quarato G, Xu B, Fan Y et al (2022) The myokine Fibcd1 is an endogenous determinant of myofiber size and mitigates cancer‐induced myofiber atrophy. Nat Commun 13: 2370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hattori Y, Usui T, Satoh D, Moriyama S, Shimono K, Itoh T, Shirahige K, Uemura T (2013) Sensory‐neuron subtype‐specific transcriptional programs controlling dendrite morphogenesis: genome‐wide analysis of Abrupt and Knot/Collier. Dev Cell 27: 530–544 [DOI] [PubMed] [Google Scholar]
- Hodgson JA, Parvy J‐P, Yu Y, Vidal M, Cordero JB (2021) Drosophila larval models of invasive tumorigenesis for in vivo studies on tumour/peripheral host tissue interactions during cancer cachexia. Int J Mol Sci 22: 8317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honegger B, Galic M, Köhler K, Wittwer F, Brogiolo W, Hafen E, Stocker H (2008) Imp‐L2, a putative homolog of vertebrate IGF‐binding protein 7, counteracts insulin signaling in Drosophila and is essential for starvation resistance. J Biol 7: 10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong S‐H, Kang M, Lee K‐S, Yu K (2016) High fat diet‐induced TGF‐β/Gbb signaling provokes insulin resistance through the tribbles expression. Sci Rep 6: 30265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isabella AJ, Horne‐Badovinac S (2016) Rab10‐mediated secretion synergizes with tissue movement to build a polarized basement membrane architecture for organ morphogenesis. Dev Cell 38: 47–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khezri R, Holland P, Schoborg TA, Abramovich I, Takáts S, Dillard C, Jain A, O'Farrell F, Schultz SW, Hagopian WM et al (2021) Host autophagy mediates organ wasting and nutrient mobilization for tumor growth. EMBO J 40: e107336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim M‐J, O'Connor MB (2021) Drosophila Activin signaling promotes muscle growth through InR/TORC1‐dependent and ‐independent processes. Development 148: dev190868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon Y, Song W, Droujinine IA, Hu Y, Asara JM, Perrimon N (2015) Systemic organ wasting induced by localized expression of the secreted insulin/IGF antagonist ImpL2. Dev Cell 33: 36–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Ng KG‐L, Dombek KM, Eom DS, Kwon YV (2021) Tumors overcome the action of the wasting factor ImpL2 by locally elevating Wnt/Wingless. Proc Natl Acad Sci USA 118: e2020120118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leevers SJ, Weinkove D, MacDougall LK, Hafen E, Waterfield MD (1996) The Drosophila phosphoinositide 3‐kinase Dp110 promotes cell growth. EMBO J 15: 6584–6594 [PMC free article] [PubMed] [Google Scholar]
- Lodge W, Zavortink M, Golenkina S, Froldi F, Dark C, Cheung S, Parker BL, Blazev R, Bakopoulos D, Christie EL et al (2021) Tumor‐derived MMPs regulate cachexia in a Drosophila cancer model. Dev Cell 56: 2664–2680 [DOI] [PubMed] [Google Scholar]
- Manent J, Banerjee S, de Matos Simoes R, Zoranovic T, Mitsiades C, Penninger JM, Simpson KJ, Humbert PO, Richardson HE (2017) Autophagy suppresses Ras‐driven epithelial tumourigenesis by limiting the accumulation of reactive oxygen species. Oncogene 36: 5576–5592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McBrayer Z, Ono H, Shimell M, Parvy J‐P, Beckstead RB, Warren JT, Thummel CS, Dauphin‐Villemant C, Gilbert LI, O'Connor MB (2007) Prothoracicotropic hormone regulates developmental timing and body size in Drosophila . Dev Cell 13: 857–871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirth C, Truman JW, Riddiford LM (2005) The role of the prothoracic gland in determining critical weight for metamorphosis in Drosophila melanogaster . Curr Biol 15: 1796–1807 [DOI] [PubMed] [Google Scholar]
- Moraes LN, Fernandez GJ, Vechetti‐Júnior IJ, Freire PP, Souza RWA, Villacis RAR, Rogatto SR, Reis PP, Dal‐Pai‐Silva M, Carvalho RF (2017) Integration of miRNA and mRNA expression profiles reveals microRNA‐regulated networks during muscle wasting in cardiac cachexia. Sci Rep 7: 6998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newton H, Wang Y‐F, Camplese L, Mokochinski JB, Kramer HB, Brown AEX, Fets L, Hirabayashi S (2020) Systemic muscle wasting and coordinated tumour response drive tumourigenesis. Nat Commun 11: 4653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni X, Yang J, Li M (2012) Imaging‐guided curative surgical resection of pancreatic cancer in a xenograft mouse model. Cancer Lett 324: 179–185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker J, Struhl G (2015) Scaling the Drosophila wing: TOR‐dependent target gene access by the Hippo pathway transducer Yorkie. PLoS Biol 13: e1002274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkin CA, Burnet B (1986) Growth arrest of Drosophila melanogaster on erg‐2 and erg‐6 sterol mutant strains of Saccharomyces cerevisiae . J Insect Physiol 32: 463–471 [Google Scholar]
- Pastor‐Pareja JC, Xu T (2011) Shaping cells and organs in Drosophila by opposing roles of fat body‐secreted collagen IV and perlecan. Dev Cell 21: 245–256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson AJ, Jensen PA, Shimell M, Stefancsik R, Wijayatonge R, Herder R, Raftery LA, O'Connor MB (2012) R‐Smad competition controls activin receptor output in Drosophila . PloS One 7: e36548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Portela M, Casas‐Tinto S, Rhiner C, López‐Gay JM, Domínguez O, Soldini D, Moreno E (2010) Drosophila SPARC is a self‐protective signal expressed by loser cells during cell competition. Dev Cell 19: 562–573 [DOI] [PubMed] [Google Scholar]
- Santabárbara‐Ruiz P, Léopold P (2021) An Oatp transporter‐mediated steroid sink promotes tumor‐induced cachexia in Drosophila . Dev Cell 56: 2741–2751 [DOI] [PubMed] [Google Scholar]
- Semaniuk U, Piskovatska V, Strilbytska O, Strutynska T, Burdyliuk N, Vaiserman A, Bubalo V, Storey KB, Lushchak O (2021) Drosophila insulin‐like peptides: from expression to functions – a review. Entomol Exp Appl 169: 195–208 [Google Scholar]
- Shahab J, Baratta C, Scuric B, Godt D, Venken KJT, Ringuette MJ (2015) Loss of SPARC dysregulates basal lamina assembly to disrupt larval fat body homeostasis in Drosophila melanogaster . Dev Dyn 244: 540–552 [DOI] [PubMed] [Google Scholar]
- Song W, Kir S, Hong S, Hu Y, Wang X, Binari R, Tang H‐W, Chung V, Banks AS, Spiegelman B et al (2019) Tumor‐derived ligands trigger tumor growth and host wasting via differential MEK activation. Dev Cell 48: 1–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiebe M, Lutz M, De La Garza A, Buechling T, Boutros M, Teleman AA (2015) REPTOR and REPTOR‐BP regulate organismal metabolism and transcription downstream of TORC1. Dev Cell 33: 272–284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urrutia H, Aleman A, Eivers E (2016) Drosophila Dullard functions as a Mad phosphatase to terminate BMP signaling. Sci Rep 6: 32269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van De Bor V, Zimniak G, Papone L, Cerezo D, Malbouyres M, Juan T, Ruggiero F, Noselli S (2015) Companion blood cells control ovarian stem cell niche microenvironment and homeostasis. Cell Rep 13: 546–560 [DOI] [PubMed] [Google Scholar]
- Weinkove D, Neufeld TP, Twardzik T, Waterfield MD, Leevers SJ (1999) Regulation of imaginal disc cell size, cell number and organ size by Drosophila class IA phosphoinositide 3‐kinase and its adaptor. Curr Biol 9: 1019–1029 [DOI] [PubMed] [Google Scholar]
- Yoo S, Nair S, Kim H, Kim Y, Lee C, Lee G, Park JH (2020) Knock‐in mutations of scarecrow, a Drosophila homolog of mammalian Nkx2.1, reveal a novel function required for development of the optic lobe in Drosophila melanogaster . Dev Biol 461: 145–159 [DOI] [PubMed] [Google Scholar]
- Yu K, Srinivasan S, Shimmi O, Biehs B, Rashka KE, Kimelman D, O'Connor MB, Bier E (2000) Processing of the Drosophila Sog protein creates a novel BMP inhibitory activity. Development 127: 2143–2154 [DOI] [PubMed] [Google Scholar]
- Zang Y, Wan M, Liu M, Ke H, Ma S, Liu L‐P, Ni J‐Q, Carlos Pastor‐Pareja J (2015) Plasma membrane overgrowth causes fibrotic collagen accumulation and immune activation in Drosophila adipocytes. Elife 4: e07187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Stallock JP, Ng JC, Reinhard C, Neufeld TP (2000) Regulation of cellular growth by the Drosophila target of rapamycin dTOR. Genes Dev 14: 2712–2724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Billington CJ, Pan D, Neufeld TP (2006) Drosophila target of rapamycin kinase functions as a multimer. Genetics 172: 355–362 [DOI] [PMC free article] [PubMed] [Google Scholar]