

Abstract
Rationale:
Eosinophilic esophagitis (EoE) is a chronic allergic inflammatory disease. Multiple genetic risk factors linked to EoE have been identified; however, these studies have been primarily focused on populations of European ancestry. There is a lack of studies leveraging the genetic architecture of Black or African American (AA) populations for the identification of loci involved in EoE susceptibility. Herein, we present admixture mapping (AM) and genome-wide association analysis (GWAS) of EoE using the participants of AA populations.
Methods:
We conducted AM and GWAS of EoE using 137 EoE cases and 1465 healthy controls from AA population. Samples were genotyped using the Multi-Ethnic Genotyping Array (MEGA). Genotype imputation was carried out with the CAAPA reference panel using the Michigan Imputation Server. Global and local ancestry inference was carried out using RFMix v2, followed by fine-mapping analysis based on imputed genotypes, and RNAseq analysis. After standard quality control filtering, over 6,000,000 variants were tested by logistic regression adjusted for sex, age, and global ancestry.
Results:
Global African ancestry proportion was found to be significantly lower among cases than controls (0.751 vs. 0.786, p-value = 0.012). Case-only AM identified four significant loci (9p13.3, 12q24.22–23, and 15q11.2) associated with EoE, two of which (12q24.22–23 and 9p13.3) were further replicated in the case-control analysis. At the two loci (12q24.23 and 9p13.3), the associations were observed for excess African ancestry. Fine mapping and multi-omic functional annotations prioritized the variants rs11068264 (FBXW8) and rs7307331 (VSIG10) at 12q24.23 and rs2297879 (ARHGEF39) at 9p13.3. GWAS identified one genome-wide significant locus at chromosome 1p22.3 (rs17131726, p-value = 2.39e-27, DDAH1) and 10 other suggestive loci including FAM179A (rs145050353), SCAND3 (rs56100858), TBC1D13 (rs114834583), MT2 (rs34800257) and PCSK2 (rs75293413) associated with EoE at p-value < 1×10−6. Interestingly, most of the GWAS variants were low frequency African-ancestry specific variants, which suggests the associations were ancestry-specific. RNASeq analysis showed esophageal DDAH1 and VSIG10 were downregulated and ARHGEF39 was upregulated among EoE cases.
Conclusions:
We have identified an African ancestry specific genetic susceptibility locus DDAH1 at 1p22.3, 1p22.3, 9p13.3, and 12q24.23, through GWAS and admixture mapping for EoE in AA, providing evidence of ancestral specific inheritance of EoE. These findings highlight the need of independent genetic studies of different ancestries for EoE.
Keywords: Eosinophilic Esophagitis, Admixture mapping, African American, Genome-wide association, Annotation, Gene score
Graphical Abstract
Capsule Summary
Strides have been made in identifying the genetic underpinnings of eosinophilic esophagitis (EoE) in populations with European ancestry; however, there has been a paucity of studies focused on admixed populations such as African Americans. This is the first EoE genetic etiology study on the African American population.
Background
Eosinophilic esophagitis (EoE) is a chronic inflammatory disease characterized by accumulation of eosinophils in the esophagus. It is associated with symptoms of esophageal dysfunction such as difficulty feeding, dysphagia, chest pain, food refusal, odynophagia, and food impaction1–3. EoE is a global health condition now reported in all continents, with prevalence of 1 per 2,000 individuals2. The majority of individuals with EoE are atopic with a high rate of food allergy, and ~70% of the EoE cases have other atopic diseases such as asthma and atopic dermatitis4, 5. Monozygotic twins have a 41% disease concordance compared with 22% for dizygotic twins indicating a genetic and environmental basis of EoE6.
Many studies have found racial and sex differences in EoE. The prevalence of EoE is found to be higher in the European Americans (EA) as compared to their Black and African American (hereafter referred as African American or AA)7–9. However, there are also reports that do not support these observations10–12. In fact, Weiler et al. noted that a higher proportion of AA patients who underwent endoscopy were diagnosed with EoE than the EA patients12. Sperry et al. found that the proportion of AA EoE patients was very similar to the proportion of AA individuals in the general population in the region10. Studies have shown that AA patients with EoE have an earlier age at diagnosis, are more likely to present with failure-to-thrive, have lower incidence of dysphagia and in general, have a more aggressive form of the disease10, 12, 13. Studies investigating inequities regarding diagnosis delay, biopsy rates, and structural factors that may affect diagnosis in AA individuals are currently lacking in the published literature. More males are affected by EoE than females with male to female ratio of 2.5:1. The racial and sex disparities in EoE could be attributed to race- or racial disparity specific environmental factors (e.g. pollution and segregation), ancestry-specific genetic differences or combination of both as reported in other allergic diseases, and other biopsychosocial factors14, 15. Epigenetic mechanisms are known to mediate environmental influences contributing to development of allergic diseases16, 17 and may also contribute to pathogenesis of EoE18.
Genome wide association studies of EoE using European ancestry has identified multiple loci including 2p23 (encodes Calpain 14, CAPN14 gene), 5q22 (encodes TSLP and WDR36 genes), 8p23 (encodes XK, Kell blood group complex subunit-related family, member 6, XKR6 gene), 11q13 (encodes EMSY), and 16p13 (encodes CLEC16A) as strong candidate regions for EoE susceptibility4, 19–23. Kottyan et al. developed an EoE-Custom single-nucleotide polymorphism (SNP) Chip (EoE-CSC) with 956 candidate EoE risk single-nucleotide polymorphisms (SNPs) and identified associations at 2p23, 5q22, 11q13, and 16p1324. Candidate gene studies found a strong association of a nonsynonymous variants in TSLP receptor (TSLPR), Cytokine receptor-like factor 2, located on a pseudo-autosomal region on Xp22.3 and Yp11.3 among male EoE cases, a susceptibility factor behind the male predominance of EoE19. Candidate gene studies have further found that FLG (encoding filaggrin) and TGFB1 (encoding Transforming growth factor, beta 1) are associated with EoE susceptibility19. Kottyan et al. suggested a model of EoE that relates traditional allergy risk factors with risk factors specific to EoE (thus highlighting the shared molecular and genetic environment across other allergic conditions and EoE), best demonstrated by an EoE specific esophageal response that is driven in part by CAPN14 following upregulation by IL-13, the Th2 cytokine involved in allergic responses20. Recent GWAS meta-analysis also identified multiple loci implicated with allergic disorders including RAD50, RORA, and SMAD3 to be genetically linked with EoE.23. To date, there are 26 independent GWAS risk loci with significance p-value < 5×10−8 reported in the GWAS catalog (https://www.ebi.ac.uk/gwas/home, accessed on 12/09/2021). However, these studies have been primarily focused on populations of European ancestry.
GWAS on AA or Latino/Hispanic are lacking for most of the common complex diseases including EoE15. Given that populations vary in terms of disease-allele frequencies, linkage disequilibrium (LD) patterns, disease prevalence, and effect size, it is informative to investigate the disease risk variants in diverse ancestral populations. Current euro-centric genomics studies in human disease impedes our ability to fully understand the ancestry-specific genetic architecture of common and complex diseases including EoE. In addition, without a diverse population, assuring that genetic research applied to clinical practice will be difficult and may not reflect the full spectrum of genetic and immunologic pathomechanisms for exploring treatment interventions. Hence, in addition to increase participation of diverse populations in genomic studies, there may be a benefit to conducting population-specific assessment of pathogenic variants.
In admixed populations, AM methods identify association between phenotype and locus-specific genomic segments, as they have proportions that are significantly higher or lower from the average ancestry proportion in the admixed population25. The premises of AM is that the risk variants among cases are transmitted in much higher proportion from the risk population than the other25. Compared with GWAS, AM requires fewer ancestral blocks to be tested for ancestry association, resulting in reduced burden for multiple testing correction26, 27. Hence, with relatively small sample sizes, AM offers more statistical power to detect genetic risk factors of EoE compared with GWAS. In addition, admixture mapping enables identification of chromosomal regions associated with disease and enriched with either African or European ancestry loci among African Americans. On the other hand, GWAS has higher resolution than AM and is suitable to detect the genomic regions with shared ancestry28. Derived from the admixture between the African and European descendants in the proportion of approximately 80% and 20%, respectively, the AA mixed genome is expected to vary from the African and European populations26, 28. Accordingly, the AM and GWAS mapping may provide ancestry specific as well as multi-ancestry genetic architecture of EoE in AA population.
In this study, we report both AM and GWAS on EoE in a self-identified AA population using the Multi-Ethnic Global Array (MEGA), a custom array from Illumina that contains SNP sets tailored towards admixed ancestry. This approach provides the optimal coverage of ancestry-specific genetic variants and is thus more suitable to capture the genetic architecture of AA population29. Such high-density genome-wide markers provide increased resolution compared to a sparse ancestry informative markers panel30, 31. To prioritize the target variants from the AM loci, the regions were fine-mapped with imputation followed by functional annotation of SNPs using Combined Annotation Dependent Depletion (CADD)32, RegulomeDB33, ancestry informativeness of markers, and expression quantitative trait loci (eQTL). To further validate potential SNPs/genes, we used the publicly available GWAS results on EoE from the GWAS Catalog34 and the differential gene expression analysis using an esophageal RNASeq case-control EoE dataset35. Herein, we describe the results of the first admixture and association analysis of EoE in AA individuals.
Materials and Methods
Study design and population
Study participants were composed from the local cohort at the Cincinnati Children’s Hospital Medical Center (CCHMC) and partly from external cohorts. The local cohort collected at CCHMC consisted of EoE cases and non-EoE controls from the Cincinnati Center for Eosinophilic Disorders and the Cincinnati Genomic Control Cohort (GCC)22, 36. The local cohort also included 26 samples derived from collaborating institutions (University of Alabama Birmingham, Emory University, and University of North Carolina). The external cohorts were composed of cases from the National Institutes of Health Consortium of Food Allergy Researchers (CoFAR)13, and the Consortium of Eosinophilic Gastrointestinal Disease Researchers (CEGIR)37. All participants were of self-reported “Black or African American” race. Parental informed consent was obtained for all participants under eighteen years of age in the study for the purpose of DNA collection and genotyping, and from patients age 18 and older. Cases were confirmed by the physician to fulfill the diagnostic criteria for EoE. EoE was defined as the peak eosinophilic count ≥ 15 eosinophils/high-power field in esophageal biopsy, in the setting of consistent symptoms and lack of other causes of eosinophilia. The controls from the Cincinnati Center for Eosinophilic Disorders consisted of clinically verified non-EoE subjects. Controls from the GCC consisted of non-allergic subjects as well as subjects with history of asthma, eczema, and allergic rhinitis but with no history of EoE or food allergy. The study was approved by the Institutional review boards at CCHMC and all participating sites. In total, 1847 (140 cases and 1447 controls) samples were selected for the study.
Genotyping
Genotyping was performed using Illumina’s Multi-Ethnic Global Array (MEGA) that contains SNP sets tailored towards admixed ancestry29. MEGA maximizes coverage and captures the genomic architecture of AA population. Genotypes were called using Genetrain2 algorithm in Illumina Genome Studio software. Genotype data were available for both cases and control individuals over 1.43 million variants.
Quality control
Participants with suboptimal call rate of < 95% were removed. SNPs were filtered for the suboptimal call rate of < 95%, MAF < 0.05 in case-control combine data, and significant deviation from HWE in control at P-value < 10−5. Possible duplicated and genetically related samples were determined using identity by descent (IBD) statistics from PLINK 2. IBD analysis was conducted using a set of LD-pruned SNPs and IBD score was used as a cutoff for filtering potential duplicate and cryptic related samples. Among the samples , samples with the highest call rate were selected and others were removed from the analysis. The quality control (QC) filtering resulted in 1,605 samples, with 138 EoE cases and 1,467 controls. All QC analysis were conducted using PLINK 1.938.
Principal Component Analysis
Principal component analysis (PCA) was performed using PLINK 2 and top 5 PCs were extracted. For the analysis, variants were pruned for linkage disequilibrium (LD) using PLINK 2 and only variants with LD < 0.1 were used.
Genotype Imputation
Imputation was carried out across the autosomal chromosomes using the Michigan Imputation Server which implemented the minimac4 algorithm39. Strand-aligned genotype data were loaded into the server. We performed the imputation using the Consortium on Asthma among African ancestry Populations in the Americas (CAAPA) reference panel40. All bi-allelic variants with imputation quality threshold of INFO score ≥ 0.3 were reported.
Local ancestry estimates
The chromosomes of admixed participants consisted of a mosaic of chromosomal blocks from the ancestral populations, which were called local ancestry blocks. Since the true ancestries were unknown, ancestry at each locus or block would be inferred computationally based on appropriate reference ancestral populations. Local ancestry for the participants were inferred by modeling African Americans as a two-way admixture between African and European populations that occurred approximately 8 generations prior41. Inference was carried out using the RFMix v2 which used supervised conditional Random Forest method to optimally infer the ancestries of the alleles at a marker locus42. The CEU and YRI panels from the 1000 Genome projects were used as the reference populations for the European and African ancestry. The sample genotype data was checked for strand alignment using coform-gt tool (https://faculty.washington.edu/browning/conform-gt.html). After removing the SNPs that did not conform the alignment to the reference panel, samples were phased using Beagle tool with the African and European as the reference panels43. The genetic map files were downloaded from the Beagle site (http://bochet.gcc.biostat.washington.edu/beagle/genetic_maps/). The number of generations since admixture was set to be 8 and the inference was carried out for each autosomal chromosome under Expectation-Maximization (EM) option with 5 iterations. The inference was carried out for each autosomal chromosome.
Global ancestry estimates
Global ancestry is the proportions of genomic contribution from the ancestral populations in the entire genome of an admixed sample. Using the RFMix v2 tool with CEU and YRI as the reference populations for European and African ancestry, the genomic proportions of European and African ancestries were estimated for each autosomal chromosome42. The global ancestry proportion for each sample was estimated using the weighted sum of the chromosomal ancestry estimates where weights were proportion for the size of the chromosomes. Samples with global African ancestry proportion < 0.1 were removed from further analysis.
Admixture mapping (AM)
To identify the association between EoE and African ancestry, AM was performed using case-only and case-control analyses25. Estimation of local and global ancestry were carried out as described before. In the case-only analysis, each sample’s local ancestry at a marker locus was compared to the respective global ancestry. In case-control analysis, deviations in local ancestries between the cases and controls were tested. The case-only and case-control AM approaches are described as below.
Let and be the proportion of African ancestry at a marker locus l and and be the global ancestry of i-th cases and j-th controls, respectively. Let n1and n2 be the number of cases and controls.
Case-only:
Define be average local ancestry at marker locus l and be average global ancestry among all cases. The test statistics for case-only is defined as , where is the standard error.
Case-control:
Define be average local ancestry at marker locus l and be average global ancestry among all controls. The test statistics for case-control analysis is defined as , where is the standard error. For large n1, n2, both T1 and T2 were approximated with standard normal distribution.
Post-hoc power analysis
The statistical power of the study was calculated using the Power Analysis in Multi-ancestry Admixture Mapping (PAMAM) web tool44. First, we estimated the testing burden using the R package CODA45. In particular, we fitted an autoregressive model (AR(1)) and evaluated the spectral density at frequency zero for each chromosome and each individual. The effective number of test were determined by adding the frequency across all chromosomes and averaging across all samples, which resulted in n = 1137 as the number of testing burden. Accordingly, the value 0.05/1137 = 4.4E-5 is used as the multiple testing adjusted level of significance for AM. Next, we used the PAMAM tool for the power estimation, with sample size of 137 cases and 1465 controls, a significance threshold of p < 4.4×10−5, and the known AA admixture of 0.78. Accordingly, this study achieved >80% power to detect an ancestral Odds Ratio (OR) > 2.7.
Fine mapping analysis and SNP prioritization
A functional-mapping study of the significant admixture mapping region was conducted. All the SNP variants mapped to the significant AM regions with MAF > 0.01 and imputation quality score (Rsq) > 0.3 were accessed from the genome wide imputed data. To identify the variants contributing towards admixture mapping signals, the allele frequency difference Δ = |fAFR − fEUR|, with allele frequency of the African (fAFR) and European (fEUR) were accessed from the 1000 Genomes project. Functional annotation of the variants with Δ ≥ 0.25 was performed with CADD32 and RegulomeDB scores33. The Variant Effect Predictor (VEP) tool from Ensembl (http://grch37.ensembl.org/info/docs/tools/vep/index.html) was used for the variant annotation including the CADD score and allele frequencies whereas RegulomeDB 2.0.3 web tool (https://regulomedb.org/regulome-search) was used for the RegulomeDB score. Overlap of variants with CADD score ≥ 10 and RegulomeDB score ≤ 3 are considered as the top prioritized variants associated to EoE. SNP association testing of the variants in AM regions was performed using PLINK 2, adjusted for the age, sex, principal components, and the global ancestry. Colocalization of the top association signals with eQTL signals on four tissue types from the GTEx Project v7 - Whole Blood, Esophagus_Mucosa, Esophagus_Muscularis, and Esophagus_Gastroesophageal_Junction, were tested using web-based tool LocusFocus (https://locusfocus.research.sickkids.ca)46. The tool implemented the Single Sum approach47 for testing the colocalization of GWAS signal with tissue-specific eQTL signal within ± 0.1 Mb region of the top GWAS signal. For LD matrix, we have used the African populations from the 1000 Genomes Project and performed the analysis under default setting.
Genome wide association analysis (GWAS)
Genome-wide association analysis was performed under the logistic regression framework, between the binary EoE phenotype (Y) and the genotype (X) adjusted for the covariates (W) which includes the global ancestry, sex, age, and principal components as
where β’s are the regression coefficients, ∈ is the normally distributed error terms. GWAS was performed using PLINK 238. By default, PLINK 2 performed the logistic regression analysis with ‘firth-fallout’ option which allows firth regression if the logistic regression fails to converge. To verify the robustness of the GWAS signals, we have also performed the firth regression using PLINK 2 ‘firth’ option. Variants were filtered for MAF < 0.05, missing rate > 0.05, HWE < 1e-5 in controls. Threshold for GWAS significance of 5×10−8 was used to access the significant association of genotype with EoE.
Gene association analysis
Gene based association analysis was performed with FUMA web server48 using the GWAS summary statistics. The FUMA pipeline uses MAGMA method to perform the gene association analysis49. Genes with p-value < 0.001 were selected as the prioritized genes for further downstream analysis.
Functional pathway and network analysis
Functional pathway and network analysis of the prioritized genes were performed using the web-based application Ingenuity Pathways Analysis (IPA, https://digitalinsights.qiagen.com). List of genes from the AM, GWAS, and gene analysis were used for the analysis. Pathways and networks were generated using the manually curated knowledge database. Networks were ranked based on the score with score ≥ 3 were considered significant. Significance of canonical pathways associated to the genes were assessed using the p-value.
Gene expression analyses
For gene expression analysis, RNAseq data set on the 10 EoE cases and 6 healthy controls of European ancestry were used35. Gene expression was determined from RNA sequence data from the esophageal biopsy specimen from the samples. Differential expression analysis was performed with NetworkAnalyst 3.0 web tool using limma algorithm50. Differential gene expression results for the targeted prioritized genes were accessed for significance at FDR adjusted p-value < 0.05 and a fold change ≥ 1.5.
Results
Cohort demographic characteristics
The cases included 137 individuals derived from three different cohorts (CCHMC (n = 74), CoFAR (n = 45), CEGIR (n = 18)) (Table 1A). Both external cohorts, CoFAR and CEGIR, consisted of EoE cases only. Participants of CoFAR cohort were significantly younger than the CEGIR cohort (average age = 9.95 vs. 17.32 years, p-value = 0.03). Proportion of African Ancestry was higher among the participants in CoFAR cohort than the CEGIR cohort but the difference was not significant (0.7339 vs. 0.6604 p-value = 0.22). Table 1B shows the demographic characteristics of cases and controls. Significant sex differences were observed among cases and controls (28.5% vs. 44.6% female, p-value < 0.0005). Global African ancestry proportion was 0.78 among overall samples, but significantly lower proportion of African ancestry was found among cases than controls (0.751 vs. 0.786, p-value = 0.011). Average age of EoE cases was 10.29 years and that of controls was 9.19 years, and the difference was not significant (p-value = 0.098). Social determinants and geographic location of cases were not available for this study.
Table 1.
Demographic information
| A. Cohort-wise demographic information | ||||
|---|---|---|---|---|
| Characteristics | CCHMC (n = 1539) | CoFAR (n = 45) | CEGIR (n = 18) | P-value (CoFAR vs CEGIR) |
| # of cases | 74 | 45 | 18 | |
| Female | 676 | 9 | 7 | 0.2166 |
| Mean age | 9.08 | 9.95 | 17.32 | 0.0301 |
| African Ancestry | 0.7857 | 0.7339 | 0.6604 | 0.2176 |
| B. Overall demographic information | ||||
| Characteristics | Total (n = 1602) | Cases (n = 137) | Control (n = 1465) | P-value |
| Female - count (%) | 692 (43.2%) | 39 (28.5%) | 653 (44.6%) | < 0.0005 |
| Mean Age in years (± SD) | 9.19 (5.6) | 10.29 (8.36) | 9.08 (5.26) | 0.098 |
| Mean African Ancestry (± SD) | 0.783 (0.12) | 0.750 (0.16) | 0.786 (0.115) | 0.0118 |
Figure 1A shows the distribution of global African ancestry the reference population and the sample data. The global African ancestry of AA individuals ranges from 10 – 99% with average of 78.3% in the combined dataset. The first principal component (PC1) is significantly and negatively correlated with the African ancestry, suggesting that the first PC explains the African ancestry variation (r = −0.997) (Fig 1C). The local ancestry of African American individuals alternates between blocks of African and European ancestry along the genome (Figure 1B). The genome of AA individuals consisted of 293 ancestral blocks on average, however there was high variation of numbers of blocks ranging from a minimum of 41 ancestral blocks to a maximum of 1124 blocks.
Figure 1. Ancestry Plots.
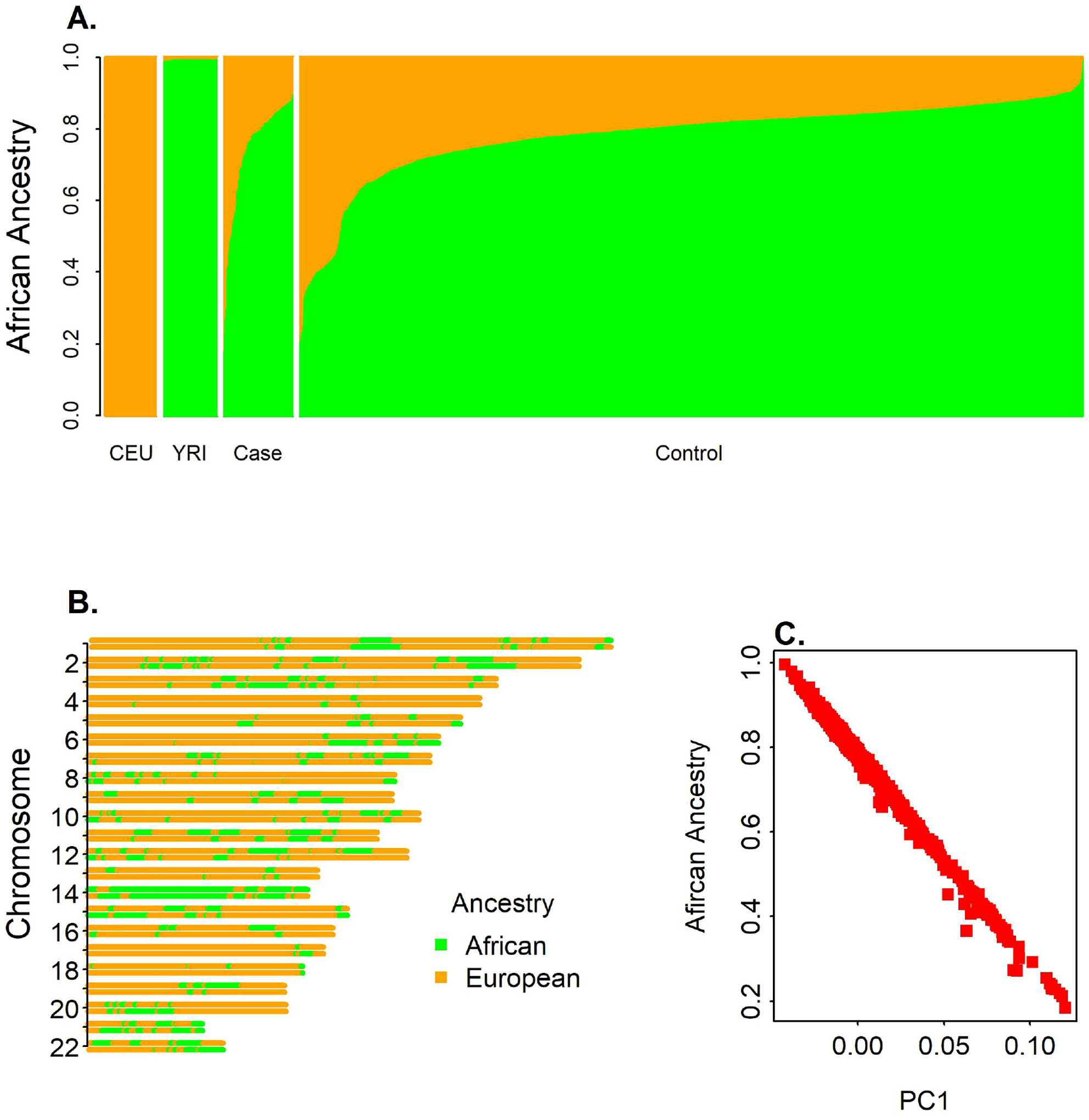
A. Distribution of Global ancestry proportion. B. Karyogram of a mosaic plot of Local ancestry across different chromosome for an African American individual. C. African ancestry proportion vs. PC1.
Admixture mapping
AM using the case-only and case-control approaches were performed to identify the loci associated to EoE cases and African ancestry. Figure 2 shows the Miami plot comparing the admixture mapping signals discovered in case-only and case-control analysis. The case-only analysis detected three signals on chromosomes 9p13, 12q24.22–23, and 15q11, at significance level of p-value < 4.4e-5 (Figure 2, Table 2A). The strongest signal was detected on chromosome 15q11, consisting of three admixture variants that reached the significance p-value < 1.4e-8 (Figure 1, Table 2A). The next strongest signal was observed at chromosome 12q24.22–23, with 9 admixture variants that reached the significance level. The locus 12q24.22–23 was also significantly associated in case-control analysis whereas the locus 9p13 showed near-significant association in case-control analysis (Table 2A). Additionally, a fourth signal on chromosome 12q24.33 (p-value ≤ 4.8e-5) was identified with p-value close to the significance level and strongly replicated in the case-control analysis.
Figure 2.
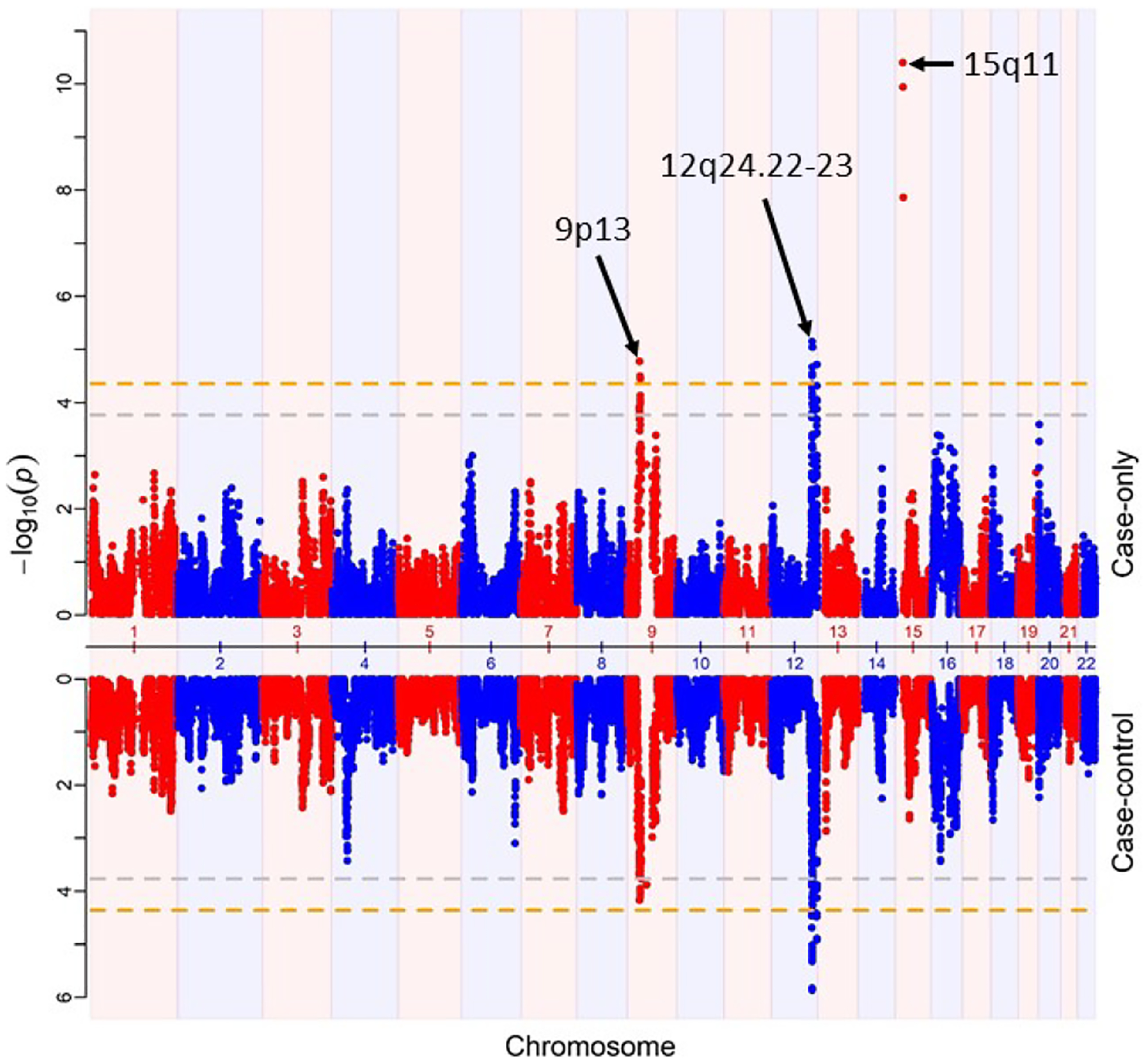
Miami Plot showing the AM results from case only (top) and case-control (bottom). Orange horizontal lines represent the genome-wide significance level p-value < 4.4e-5. Gray lines represent suggestive significance p-value < 1.17e-4.
Table 2.
Top signals and the prioritized variants from the admixture analysis of EoE in African American.
| A. Top admixture mapping signals. | ||||||
|---|---|---|---|---|---|---|
| CHR | START | END | ZCC | PCC | ZC | PC |
| 9p13.3 | 33524775 | 36319699 | 3.982 | 6.83E-05 | 4.304 | 1.67E-05 |
| 12q24.22-23 | 1.17E+08 | 1.19E+08 | 4.828 | 1.38E-06 | 4.493 | 7.02E-06 |
| 12q24.33 | 1.31E+08 | 1.31E+08 | 3.96 | 7.50E-05 | 4.063 | 4.84E-05 |
| CHR = Chromosome with cytogenetic location of the AM locus; START, END = Starting and Ending base pair position of loci; ZCC, ZC = Z-statistics for case-control and case-only analysis, respectively; PCC, PC = P-value for case-control and case-only analysis. | ||||||
| B. Functional prioritization of the AM loci. | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| SNP | CHR | POS | Consequence | Gene | AFR_AF | EUR_AF | CADD | Delta | RDB | eQTL gene |
| rs2297879 | 9 | 35662251 | missense | ARHGEF39 | 0.0628 | 0.325 | 14.72 | 0.2622 | 1f | ARHGEF39 |
| rs66898998 | 12 | 116398626 | 3_prime_UTR | MED13L | 0.0356 | 0.3628 | 12.45 | 0.3272 | 3a | |
| rs11068264 | 12 | 117396097 | intron | FBXW8 | 0.1793 | 0.8121 | 18.45 | 0.6328 | 1f | FBXWB,HRK, RP11-103B5.2 |
| rs7963451 | 12 | 117529889 | intron | TESC | 0.8003 | 0.502 | 11.21 | 0.2983 | 2b | FBXWB |
| rs10774904 | 12 | 117566689 | intron | RP11-103B5.2 | 0.1505 | 0.503 | 10.12 | 0.3525 | 3a | |
| rs7307331 | 12 | 118509191 | missense | VSIG10 | 0.7337 | 0.2813 | 14.88 | 0.4524 | 1f | VSIG10 |
| AFR_AF = Allele frequency in the African population; EUR_AF = Allele frequency in European population; CADD = CADD Score; RDB = RegulomeDB Score; Delta = |AFR_AF - EUR_AF|; eQTL gene = Gene with eQTL association to the SNP in one or more tissues from the GTEx portal. | ||||||||||
| C. Fine mapping of AM loci | ||||||||
|---|---|---|---|---|---|---|---|---|
| Locus | SNP | POS | OR | P_GWAS | P_CC | P_C | Gene | Consequence |
| 9p13.3 | rs7854218 | 35558136 | 2.36893 | 0.005773 | 6.83E-05 | 1.67E-05 | RUSC2 | Intron |
| 12q24.22-23 | rs115916534 | 118587866 | 3.70886 | 0.000183 | 1.38E-6 | 7.06E-6 | TAOK3 | 3 Prime UTR |
| 12q24.33 | rs7295352 | 130995467 | 1.64545 | 0.000349 | 7.50E-05 | 4.84E-05 | RIMBP2 | Intron |
| 15q11 | rs570427365 | 20161512 | 2.14371 | 0.016457 | 0.398029 | 1.37E-08 | ||
The directionality of the association showed ancestry-specific AM loci for EoE (Table 2A). The negative test statistics at the loci 15q11 showed that the higher European ancestry at the region was ancestry risk factor among the AA EoE cases. The positive z-statistics suggested the loci 9p13, 12q24.22–23, and 12q24.33 showed the higher African ancestry as the risk factor..
Functional prioritization of AM loci
To identify the putative genetic variants underlying the AM regions, we examined all variants mapped to the significant AM region using the 1000 Genomes Project. Variants were annotated and prioritized using CADD score, allele frequency difference (Δ), and RegulomeDB score (as detailed in the Method section). On locus 12q24.22–23, five variants were identified with the prioritization criteria CADD ≥ 10, Δ≥ 0.25, and RegulomeDB ≤ 3 (Table 2B). An intronic variant rs11068264 on gene FBXW8 has CADD score = 18.45 which is strongly suggestive of deleterious effect. The variant has high delta (Δ = 0.6328), implying that the variant contributes towards the ancestry association. The variant is scored 1f under RegulomeDB score categorization, which indicates the variant overlaps with a TF binding site or a DNAse peak and eQTL. The variant has a strong eQTL with genes FBXW8 and HRK in the esophagus and other tissues. A missense variant rs7307331 in VSIG10 has CADD score = 14.88, high delta (Δ = 0.4524), and 1f RegulomeDB score. The ancestral allele ‘A’ is the common variant in the African population but a minor allele in Europeans (0.7337 vs. 0.2813). The variant has an eQTL in whole-blood, cultured fibroblasts, and other tissues in the GTEx portal (https://gtexportal.org/home/). The intronic variant rs7963451 in TESC has RegulomeDB score 2b suggesting potential regulatory effects on nearby genes. The variant is found to have an eQTL with FBXW8 using skin tissue in the GTEx portal. The other two variants, rs66898998, a 3’ URT variant in MED13L and rs10774904, an intronic variant in lincRNA RP11-103B5.2, are scored 3a. No eQTL was found for these variants in the GTEx portal. The variant rs2297879 is a missense variant in ARHGEF39 (9p13) and has a CADD score = 14.72 and is predicted to be benign. The ancestral allele ‘C’ is more common in the European population (fEUR = 0.325) compared with the African population (fAFR = 0.0628). RegulomeDB scores the variant at 1f, suggesting evidence for an eQTL association and TF binding. The variant has an eQTL and sQTL with the gene in esophageal tissue. Thus, the loci 12q24.22 and 9q13 harbor potential variants associated with EoE in African Americans and the multi-omics functional annotations highlights loci encoding six genes FBXW8, TESC, HRK, MED13L, RP11-103B5.2, and VSIG10 on chromosome 12q24.22 and one gene ARHGEF39 on chromosome 9p13, respectively, as candidate loci.
Fine mapping of AM loci
Association of SNPs mapped to the four AM loci was performed using the logistic regression test, adjusted for the age, sex, global ancestry, and PC2 to PC5 (PC1 is highly correlated with global ancestry). All variants that passed the quality control criteria and imputation quality score Rsq > 0.3 were assessed for the association. The results showed some evidence of allelic association at two loci 12q24.22–23 and 12q24.33 with p-value < 0.05 (Table 2C). The strongest allelic association of variants on the AM locus 12q24.22–23 was observed at 3 prime UTR variant rs115916534 of gene TAOK3 with GWAS p-value = 0.00018. Similarly, the AM locus 12q24.33 showed some evidence of allelic association with EoE with GWAS p-value < 0.0003 at an intronic variant rs4759706 of RIMBP2. At 9p13.3, the strongest association was observed at rs7854218 (p-value = 0.005, intronic to gene RUSC2). At locus 5q11, the strongest association was observed at an intergenic variant rs373628495 (p-value = 0.027). The colocalization analysis of the top GWAS signal within each AM loci on the four EoE-relevant tissues was performed using the eQTL data from the GTEx project. Under default setting on LocusFocus tool, no colocalization test was performed on two loci 12q24.33 and 5q11. Two genes from the locus 9p13.3 and three genes from the locus 12q22–23 were identified within ± 0.1 Mb region of the top GWAS loci. Among the 20 gene-tissue (4 tissues, 5 genes) combinations, three combinations failed to meet the colocalization testing criteria under the default setting of LocusFocus tool. Based on the 17 tests performed, multiple testing adjustment p-value 0.0029 (= 0.05/17) was considered significant. The strongest colocalization was observed at gene RUSC2 (nominal p-value = 0.049) on whole blood (Supplemental Table T1) but failed to be significant under multiple testing adjustment (Supplemental Table T1). This could be due to the fact that the top GWAS association within each AM loci were not strong and this could have affected the colocalization test.
Genome-wide association analysis
GWAS was performed on 6 million variants with MAF > 0.01 using the logistics regression, adjusted for the age, sex, global ancestry, and PC2 to PC5 (PC1 is highly correlated with global ancestry) and resulted in one significant locus reaching the genome-wide significance at the p-value ≤ 5E-8 and multiple suggestive signals p-value < 1E-5 (Figure 3A). There was no genomic inflation with genomic control factor λ = 1.009 (Figure 3B). The strongest signal was observed in an intronic region on the gene DDAH1 (rs17131726, p-value = 2.39e-27). Based on the 1000 Genomes Project, the lead variant rs1713726 is low frequency variant among the African populations with MAF = 0.04 but the MAF is close to zero among other populations, suggesting that the variant is African ancestry specific. We identified additional 10 loci with significance p-value < 1e-6 (Table 3). The allele frequency distribution of the 11 GWAS loci with the p-value < 1e-6 showed that the variants were primarily low frequency variants with minor allele frequency (MAF) < 0.05 except for rs503078 which as MAF = 0.0914 (Supplemental Table T2). To investigate for the potential inflation due to low MAF and small number of cases, we have also performed firth regression analysis of the top variants. The results from the firth regression did not show any inflation in the results (Supplemental Table T2), which conformed the robustness of our analysis.
Figure 3.
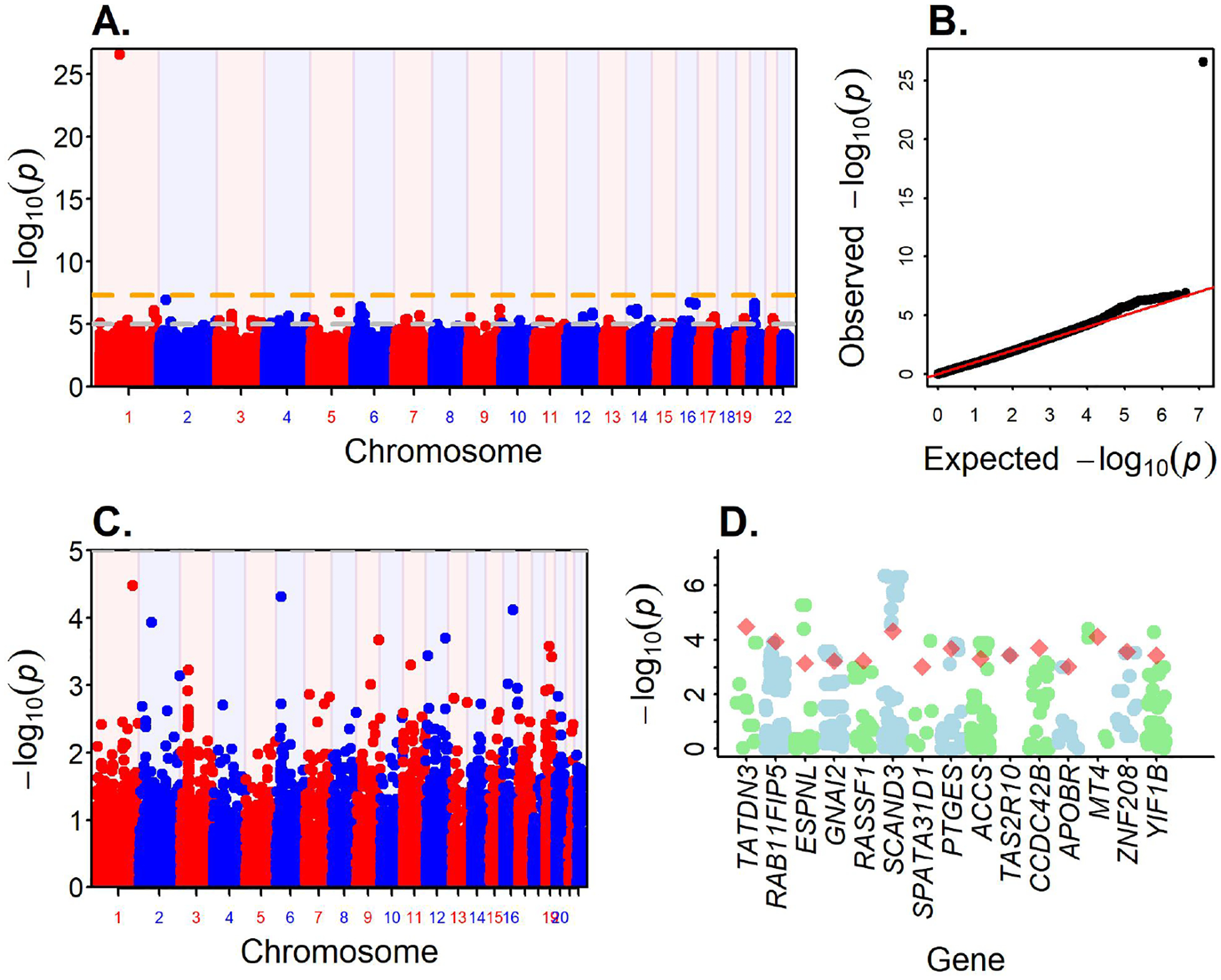
GWAS result of EoE in African Americans. A. Manhattan Plot shows the association of SNPs across genome. Red horizontal line marks the GWAS significance level p-value = 5e-8. B. QQ plot (genomic control lambda = 1.009). C. Gene association results from FUMA. D. Fifteen genes with p-value < 0.001 from the gene association test. For each gene, variants within ± 500 bp were identified from the GWAS data and the p-value are shown along with the p-value of the gene.
Table 3.
GWAS variants associated to EoE in African American at p-value < 1e-6.
| Lead SNP | CHR | POS | P | nSNPs | IndSignals | MAF_AFR | MAF_ EA | Variant consequence | Second SNP | Nearest Gene |
|---|---|---|---|---|---|---|---|---|---|---|
| rs17131726 | 1 | 85986140 | 2.39E-27 | 1 | 1 | 0.04 | 0 | intron_variant | DDAH1 | |
| rs503078 | 1 | 229111957 | 7.67E-07 | 2 | 1 | 0.04 | 0.33 | intergenic | ||
| rs145050353 | 2 | 29240681 | 1.16E-07 | 1 | 1 | 0.02 | 0 | missense | FAM179A | |
| rs75905640 | 5 | 120957676 | 9.92E-07 | 1 | 1 | 0 | 0.01 | |||
| rs56100858 | 6 | 28527321 | 3.81E-07 | 82 | 2 | 0.04 | 0.01 | intergenic | rs73740600 | SCAND3 |
| rs114834538 | 9 | 131570343 | 6.48E-07 | 1 | 1 | 0.04 | 0 | missense, 3_prime_UTR_variant | TBC1D13 | |
| rs142278943 | 14 | 20888602 | 8.39E-07 | 1 | 1 | 0.03 | 0 | Intergenic | TEP1, KLHL33 | |
| rs114643291 | 14 | 45071799 | 6.66E-07 | 9 | 1 | 0.02 | 0 | |||
| rs34800257 | 16 | 56641032 | 1.78E-07 | 1 | 1 | 0.04 | 0 | Promoter, Intergenic | MT2A | |
| rs56683615 | 16 | 77209792 | 2.34E-07 | 7 | 1 | 0.06 | 0 | intergenic | MON1B | |
| rs75293413 | 20 | 17381247 | 2.33E-07 | 13 | 2 | 0.02 | 0 | intron_variant | rs78011284 | PCSK2 |
Post-GWAS annotation using the summary statistics with FUMA tool identified two loci, 6p22.1 and 20p12.1, each consisted of two independent signals. Second signal on 6p22.1 was identified at rs73740600 (p-value = 1.2e-6) and on 20p12.1 at rs78011248 (p-value = 6.86e-7) (Table 3). Both signals on the locus 20p12.1 are intronic in the gene PCSK2 while signals on 6p22.1 span over 400Kb gene-rich region that encodes several genes from zinc finger and scan domain family including ZSCAN9, ZSCAN31, and ZBED9 (Supplementary Figure 1). At suggestive significance at p-value < 1e-5, FUMA analysis identified a total of 62 GWAS associations (Supplemental Table T3) across the 57 loci.
FUMA web tool was also used to perform gene association analysis. FUMA implemented the MAGMA algorithm for the analysis and identified several genes associated with EoE, however no gene reached the Bonferroni significant cutoff p-value of 0.05/18522 = 2.7e-6 for 18522 protein coding genes tested (Figure 3C). The top 3 genes were TATDN3, ZBED9, and MT4 with significance p-value < 0.0001. Additionally, 15 genes were identified with p-value < 0.001 (Figure 3D). GeneHancer showed TATDN3 gene is linked to asthma, eczema, and hay fever, phenotypes related to EoE and part of the atopic march51. The GeneHancer variants have eQTL in esophageal tissue, which suggest a possible biological link between EoE and TATDN3.
Functional pathway analysis
Functional prioritization of variants in the AM regions identified 7 genes from the two loci 12q24.22–23 and 9p13 as candidate genes for EoE. From the GWAS analysis, SNPs were mapped to within gene using the VEP annotation tool from Ensembl GRCH37 build (http://grch37.ensembl.org/Tools/VEP) and identified 40 genes were identified from the 57 risk loci with p-value < 1e-5 (Supplemental Table T3). Gene analysis using FUMA identified 15 genes with p-value < 0.001. There were two genes ZBED9 and ESPNL overlap between GWAS and gene analysis. In total, the three approaches identified 60 genes as candidate risk loci associated with EoE. To understand the potential functional role of the genes, we conducted network and pathway analysis using the Ingenuity Pathways Analysis (IPA) tool. The results from the IPA analysis are summarized in Supplemental Table T4. IPA identified 5 networks with score ≥ 3; networks related to respiratory diseases, gastrointestinal diseases, and cancers were among the top list (Supplemental Table T4). Seven canonical pathways related to the target genes were identified with significance ep-value < 0.05; Biotin-carboxyl Carrier Protein Assembly, Melatonin signaling, and Chemokine signaling pathways were the top three. Through the IPA analysis, five cellular and molecular functions were identified. Cellular growth and proliferation and cell-to-cell signaling and interaction were among the top functions with p-value (3.93E-02 – 4.49E-04) and overlapped with 10 and 9 genes, respectively.
Validation Analysis
Validation using GWAS catalog
The NHGRI-EBI GWAS Catalog was interrogated for known risk variants associated with EoE and identified 26 loci with p-value < 5e-8 as a risk variant associated to EoE. All GWAS discovery of EoE risk variants were based on EA population. Using LDlinkR tool52, variants in linkage disequilibrium (LD) were searched from the 1000 Genomes Project African population at r2 ≥ 0.6. Fourteen loci were identified with at least one SNP meeting the LD cutoff in our data (Table 4). At the significance level p-value < 0.05, three loci 15q22.2 (RORA), 9p24.1 (JAK2) and 15q13.3 (LINC02352 - KLF13) were replicated in the AA GWAS. The loci RORA and JAK2 were previously known risk factor for allergic disorder53, 54 and recently found to be associated to EoE23. In particular, JAK2 was found to be female-specific risk variant of EoE23 and JAK2 inhibitors have proven activity against Th2 cells in atopy.
Table 4.
Validation of EoE GWAS signals. Table shows the strongest p-value in the EoE GWAS of African American of SNPs in high LD (r2 ≥ 0.6) with the 14 EoE variants from GWAS Catalog. Out of the 26 loci in the GWAS Catalog, only 14 were found in the GWAS data.
| RSID | REGION | Pos37 | MAF | OR | P | Catalog SNP | Catalog MAF | R2 | GENE | Catalog_P |
|---|---|---|---|---|---|---|---|---|---|---|
| rs2279293 | 15q22.2 | 61057357 | 0.496 | 1.58 | 0.000673 | rs2279293 | 0.145 | 1 | RORA | 5.00E-11 |
| rs4593605 | 9p24.1 | 5107278 | 0.155 | 1.57 | 0.006845 | rs62541556 | 0.251 | 0.8189 | JAK2 | 4.00E-08 |
| rs17228227 | 15q13.3 | 31537646 | 0.044 | 0.392 | 0.032718 | rs8041227 | 0.28 | 0.968 | LINC02352 - KLF13 | 6.00E-10 |
| rs61894547 | 11q13.5 | 76248630 | 0.0129 | 0.191 | 0.10543 | rs61894547 | 0.043 | 1 | EMSY | 5.00E-15 |
| rs11124247 | 2p23.1 | 31411155 | 0.0234 | 0.423 | 0.150938 | rs143457388 | 0.047 | 0.8676 | CAPN14 | 3.00E-16 |
| rs2307472 | 2p22.2 | 37376247 | 0.0125 | 0.262 | 0.189446 | rs143457389 | 0.046 | 0.6662 | PRKD3 | 3.00E-16 |
| rs3806932 | 5q22.1 | 110405675 | 0.363 | 1.183 | 0.201364 | rs3806932 | 0.46 | 1 | TSLP | 3.00E-09 |
| rs371915 | 16q24.1 | 84578241 | 0.174 | 1.24 | 0.204934 | rs371915 | 0.13 | 1 | MEAK7 | 2.00E-08 |
| rs887992 | 2q12.1 | 103524931 | 0.324 | 0.862 | 0.276979 | rs887992 | 0.362 | 1 | TMEM182 | 4.00E-10 |
| rs2545357 | 19q13.11 | 33104610 | 0.448 | 0.891 | 0.384192 | rs3815700 | 0.14 | 0.7399 | ANKRD27 | 2.00E-09 |
| rs2753961 | 6p21.33 | 31754830 | 0.0535 | 1.23 | 0.421398 | rs599707 | 0.114 | 0.9838 | SNHG32 - NEU1 | 3.00E-09 |
| rs56062135 | 15q22.33 | 67455630 | 0.0947 | 1.114 | 0.597795 | rs56062135 | 0.228 | 1 | SMAD3 | 4.00E-10 |
| rs2706349 | 5q31.1 | 131906760 | 0.376 | 1.067 | 0.628782 | rs2106984 | 0.212 | 0.8568 | RAD50 | 4.00E-08 |
| rs34443974 | 16p13.13 | 11179305 | 0.0695 | 1.065 | 0.790302 | rs35099084 | 0.222 | 0.8598 | CLEC16A | 2.00E-12 |
Validation using RNA-seq data
We investigated the expression of 60 EoE associated genes using esophageal RNAseq data on 10 EoE and 6 controls. At the FDR < 0.05 and fold change ≥ 1.5, fourteen genes showed differential expression as a function of disease status (Figure 4). The genes DDAH1 (fold change = 6.62), PTGES (fold change = 5.85) and NRXN1 (fold change = 5.56) were the top genes with highest fold change observed (Supplemental Table T5). The genes DDAH1 showed increased expression among the healthy controls compared to EoE cases as indicated by the negative fold change. The other two genes PTGES and NRXN1 showed positive fold change and hence increased expression among cases than the controls.
Figure 4. Circle plot of EoE loci.
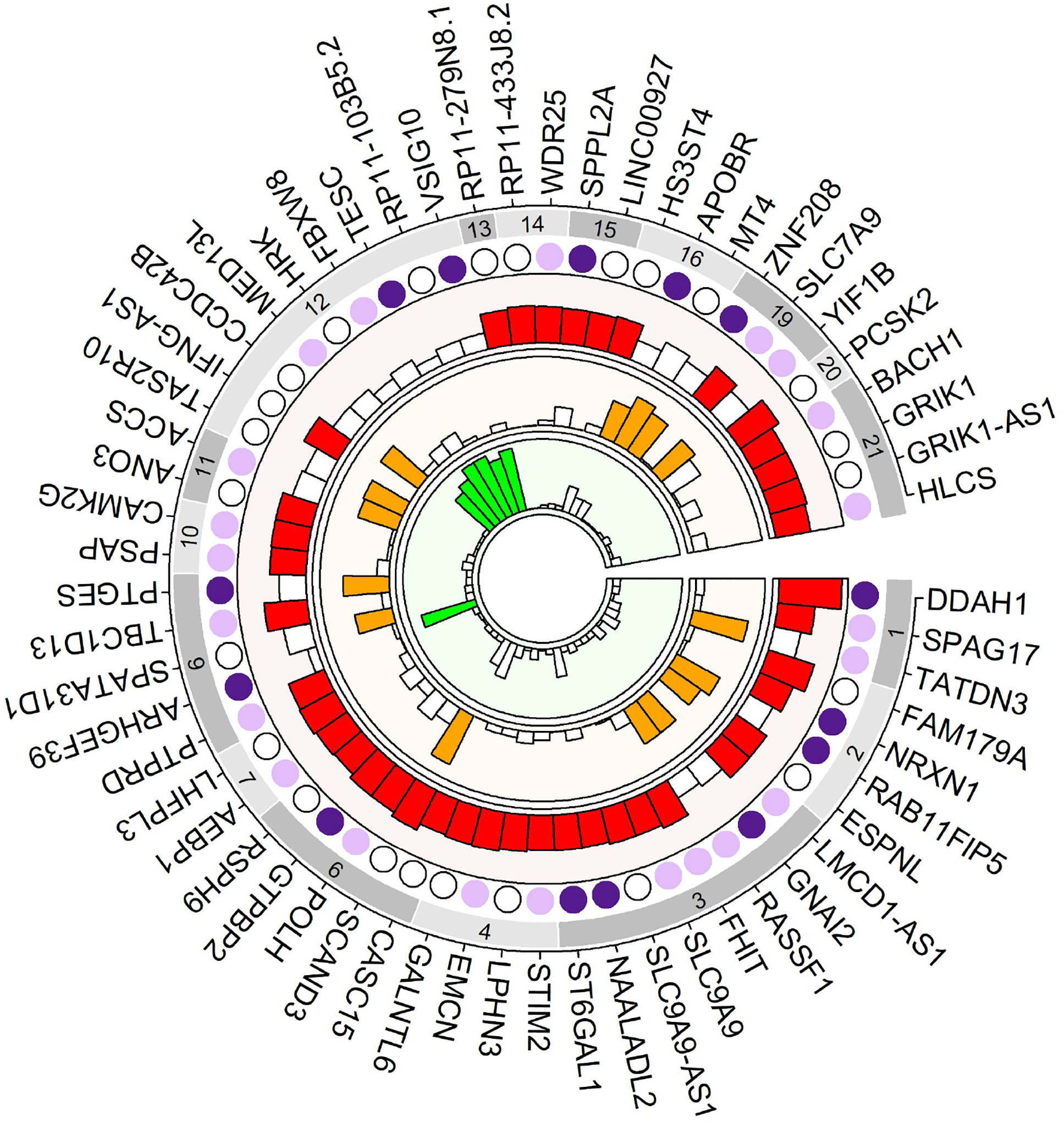
Outer track with gray rectangles shows the chromosomes. First inner shows the DEG replications. Dark filled circles show differentially expressed genes with fold change >= 1.5 and false discovery rate < 0.05; Light filled circle represent genes with fold change < 1.5; unfilled circle represent genes with expression result. Second inner track (light red) shows GWAS discovery results. The height of the rectangles represents the −log10(p-value) of the most significant SNP in the gene. Filled rectangles indicate −log10(p-value) < 1e-5. Value >9 are truncated to 9. Third track (light orange) shows −log10(p-value) from the gene analysis. Filled rectangles indicates −log10(p-value) < 1e-3. The innermost track (light green) shows AM results with the height of the rectangle indicating −log10(p-value) and filled rectangles indicating −log10(p-value) < 1e-3. EoE = Eosinophilic Esophagitis, DEG = Differentially expressed gene, GWAS = Genome wide association study, AM = Admixture mapping
Discussion and conclusion
EoE is a chronic inflammatory disease of the esophagus, clinically characterized by dysphagia, failure to thrive, vomiting and epigastric or chest pain. Discovery of genetic risk variants of EoE have previously been predominantly conducted on data with participants of European ancestry. Herein, we presented the first AM and GWAS of EoE on the African American population. We have genotyped 1602 samples (137 cases and 1465 controls) of AA individuals using the Illumina’s the Multi-Ethnic Genotyping Array (MEGA). To capture the African ancestry specific variants, genotype imputation was carried out using the CAAPA reference panel. Higher proportion of global European ancestry was observed among EoE cases than the controls. Significant sex difference was observed with more males were affected than the females which is consistent with the literature55, 56. Through GWAS, we have identified the strongest association in this study at the intronic variant rs17131726 (p-value = 2.39e-27) on gene DDAH1. GWAS analysis further identified 10 other suggestive loci including FAM179A (rs145050353), SCAND3 (rs56100858), TBC1D13 (rs114834583), MT2 (rs34800257) and PCSK2 (rs75293413) associated with EoE at p-value < 1×10−6. The variant rs17131726 and other GWAS variants were low frequency African-ancestry specific variants, which suggests the associations were ancestry-specific. The robustness of the results were confirmed with the firth regression analysis of the top signals. Functional annotations and gene association tests using the summary GWAS data were performed using the FUMA tool. Gene association analysis further identified 15 suggestive genes including, TATDN3, SCAND3, and MT4, associated with EoE in African Americans. The GWAS analysis replicated three loci RORA, JAK2, and LINC02352 - KLF13 at significance level p-value < 0.05. This finding suggests that the prior GWAS data may be ancestry specific to the European population, but the small sample size limits definitive conclusion. We have also identified four genome wide significant AM genomic regions (9p13.3, 12q24.22–23, 12q33, and 15q11) associated with EoE. Fine mapping and variant prioritization of the AM regions identified five SNPs (rs11068264, rs7307331, rs7963451, rs66898998, rs10774904) from chr12q24.22–23 and one SNP (rs2297879) from 9p13.3 with strong regulatory evidence and substantial differences in the ancestral allele frequency. Differential gene expression analysis using RNASeq data validated 14 genes including DDAH1, PTEGS, and VSIG10 with fold change > 1.5 and FDR ≤ 0.05. Even though GWAS and AM identified distinct set of risk loci associated to EoE, both results pointed towards similar ancestral sources to African ancestry. However, no GWAS locus including the most significant locus at 1q22.3 showed association in AM analysis. GWAS loci were low frequency variants and the allele frequency differences between the ancestral populations may not be large enough for AM approach to detect the signals.
Admixture mapping provide an opportunity for discovery of disease-susceptibility risk variants on the admixed populations by capturing the genetic architecture contributed from the different ancestral sources57. Our admixture mapping analysis of AA participants permitted not only a test of association in admixed populations but also the opportunity to identify more precisely the chromosomal region associated with EoE, in both African and European descent. For example, genomic region on 9q13.3 and 12q24.22–33 are associated with EoE specifically in participants of African ancestry whereas genomic region on 15q11 was specifically associated with European ancestry samples. This could attribute to differences in the underlying genomic architectures at these loci between persons of predominately African ancestry and those of predominately European ancestry. Further validation of such signals in African and European samples is required to identify the ancestry-specific risk variants of EoE.
Multi-omic annotation using CADD and RegulomeDB prioritized one SNP from the locus 9p13.3 and 5 SNPs from the locus 12q24.22–33 with evidence for potential regulatory functions and substantial difference in the allele frequency differences among the ancestral (African and European) populations. LD analysis of the five variants from the locus 12q24.22–33 showed no pairwise LD in the African population from the 1000 Genome project. The prioritized variant rs2297879 in locus 9p13.3 is missense variant in gene ARHGEF39 and more common in European population than the African (MAF = 0.325 vs. 0.0628). RegulomeDB scored the variant 1f which indicated strong evidence of regulatory function. The variant was eQTL for the gene in esophagus and other tissues in the GTEx portal. The RNASeq analysis validated the gene was differentially expressed among EoE cases (Figure 4). Among the five prioritized variants in the locus 12q24.22–23, the SNP rs11068264, intronic to gene FBXW8, was scored 1f in RegulomeDB and eQTL for genes FBXW8 and HRK on multiple tissues including esophagus on the GTEx portal. Large scale GWAS studies implicated the genes to be risk factors for several hematological traits including eosinophil counts58, 59, lung functions59 and brain volume measurements60. AM analysis also implicated TESC, MED13L, and VSIG10 genes, of which TESC and VSIG10 were further supported to be associated with EoE using RNASeq analysis (Figure 4, Supplementary Table T3). The prioritized variants intronic to gene MED13L was not related to expression level in the GTEx portal and the gene did not show differential expression in esophagus among EoE cases. On the other hand, gene TESC showed differential expression among EoE cases, but the prioritized variant rs7963451 was not eQTL for the gene on the GTEx portal. Interestingly, the variant was eQTL for the FBXW8 gene in skin tissue. The prioritized variant rs7307331 in gene VSIG10 was scored 1f in RegulomeDB, exhibited high allele frequency difference between African and European population with the ancestral allele being more common in the African than the European (freq = 0.73 vs. 0.28). Additionally, the variant was eQTL on multiple biologically relevant tissues including whole blood and cell culture fibroblast in the GTEx data.
The gene DDAH1 is involved in the metabolism of nitric oxide; dysregulation of the gene is linked to inflammatory effects on asthma61 and inflammatory bowel disease62 suggesting a biological role of this gene in inflammation. The gene is significantly downregulated in the esophagus among EoE cases (Supplementary Table T3). The lead SNP rs17131726 is a low frequency variant in the African population and near monomorphic in the European samples, indicating the association is African-specific. The GWAS locus at 6p22.1 consisted of two independent signals at rs56100858 and rs73740600. The locus spanned over 400KB region and implicated several zinc finger and scan domain genes (ZSCAN9, ZKSCAN4, ZSCAN26, and ZSCAN31), two glutathione peroxidase genes (GPX5 and GPX6) and ZBED9 (Supplementary Figure 1). ZBED9 was the closest gene to the most significant SNP (rs56100858) in the locus and also showed association with EoE on the gene-based association. Interrogation in the GWAS catalog showed that the genes in the locus were associated with psychological disorders (https://www.ebi.ac.uk/gwas/home, accessed on 12/09/2021). None of the genes were differentially expressed in the RNASeq analysis of the esophageal biopsies of EoE. However, ZSCAN31 was identified to be associated with biologically relevant phenotypes such as eosinophil counts63 and gastroesophageal reflux disease64.
The allele frequency distribution of the 11 GWAS loci with the significance association p-value < 1e-6 showed that the variants were primarily low frequency variants in the African population but rare variants in European population except for variants rs503078 and rs75905640 (Table 3). The variant rs503078 is a low frequency variant among Africans but a common variant among Europeans and rs75905640 is rare in both African and European populations but common in American and Asian populations in the 1000 Genomes Project III. The discovery of the African-specific variants could be due to the better tagging of African variants in the MEGA-chip accompanied with the denser coverage of the African genome by the CAAPA reference panel. These findings pointed towards the importance of the population-specific genotyping platform and reference panels to identify the ancestry-specific disease-susceptibility. Identifying the ancestry-specific variants are critical in unraveling the health disparity across different populations.
Gene analysis based on the GWAS summary statistics further complements the GWAS signals by accounting for the multiple weak association at the gene level. TATDN3 on the chromosome 1q32.3 is the top gene identified with FUMA gene association analysis. Previous study mapped the gene to type 1 diabetes65. An enhancer element GH01J212681 target to the gene TATDN3 is linked to asthma, eczema, and hay fever, the phenotypes related to EoE and part of the atopic march in EA (https://www.genecards.org/). GeneHancer variants are further identified as eQTL for TATDN3 in esophagus tissue in the GTEx portal. These evidences point to a biological link between EoE and TATDN3.
Validation of the genes using RNASeq analysis identified 15 genes with significant differential expression in esophagus biopsies of EoE cases. The top GWAS locus DDAH1, and three prioritized genes from the AM analysis, VSIG10, TESC, and ARHGEF39 were among the differentially expressed genes. Five other GWAS genes - NRXN1, GTPBP2, ST6GAL1, NAALADL2, and SPPL2A, were also differentially expressed. The gene ST6GAL1 was found to be associated with biologically relevant traits such as eosinophil count64 and esophageal carcinoma66. NAALADL2 was found to be associated with asthma among Latino67. Interrogating the genes in the GeneHancer database using the GeneCard Suite found several enhancer targets of the genes GTPBP2, ST6GAL1, NAALADL2, and SPPL2A, which were associated with blood-cell related traits such as monocyte count, basophil count, neutrophil counts, and white blood cell counts, and body mass index, and psychological disorders (https://www.genecards.org/). The lead SNPs in the loci were low frequency variants, and thus the eQTL association of the variants to the genes were not available in the GTEx portal. Additional analyses with African ancestry specific gene expression and eQTL analysis may be required to further confirm the associations at these the low-frequency ancestry-specific loci.
Five genes (PTGES, APOBR, ZNF208, RAB11FIP5, and GNAI2) identified from the Gene analysis using the FUMA tool were also validated with the RNASeq analysis (Figure 4, Supplementary Table T3). PTGES is found to be associated to asthma and the association is African American specific68. Deficiency of PTGES is linked to allergic inflammation of airways69, 70. RNASeq analysis of showed that the gene was upregulated in esophageal biopsies among EoE cases (Figure 4). The upregulation of the gene could be triggered due to pro-inflammation of the esophagus, further investigation may provide insight into the functional role of the gene in the EoE. APOBR is found to be associated with allergy71 and body mass index64, both of the traits are comorbid to EoE.
The present study has notable strengths. It is the first AM and GWAS of EoE in AA population. Together with the ancestry-specific genotyping array and imputation, we were able to identify ancestry-specific association of EoE in AA population. We have used multi-omic features including in-silico epigenomic annotations and transcriptomic to identify the potential variants associated with EoE. Our approach allowed prioritization of potential risk variants for further study of pathogenesis of EoE. There are also notable limitations in this study. First, the dataset only consisted of 137 EoE cases, which is small compared to typical AM and GWAS studies in the literature. This undoubtedly impacted the association analysis, in particular, replication of the known signals. Second, the DEGs were identified based on the samples of European ancestry. There is no publicly available transcriptome wide data set for EoE with AA participants. However, the genes were first identified through ancestry-specific analysis, so having replicated on the EA population could imply cross-ancestry risk variants of EoE albeit with smaller effects. Third, this study didn’t include environmental or geographic attributes that have in other studies been demonstrated to vary in prevalence based on ancestry72.Fourth, the TOPMed reference panel73 has higher representation of African ancestry than the CAAPA reference panel used in this study. TOPMed imputation could provide additional finding of ancestry-specific variants in the regions of interest and could be independently pursued in the future. Nevertheless, this study presents the first GWAS data in an AA cohort and complement discovery of genetic risk loci of EoE which are otherwise missed in GWAS of EA participants.
In summary, through a systematic and comprehensive screen of variants in individuals with EoE, we have identified multiple target variants and genes associated with EoE in the African American population. Both AM and GWAS results point towards novel genetic risk of EoE potentially attributed to African-ancestry genotyping. GWAS identified a strong African-specific EoE-risk locus at 1q22.3 (rs17131726, DDAH1). GWAS analysis primarily identified African-specific risk variants and suggests distinct genetic architecture of EoE in AA than EA. GWAS loci DDAH1, PTGES, and APOBR were previously known to be associated with allergic diseases, and were genetically and transcriptionally associated with EoE. AM loci 9p13.3 (ARHGEF39) and 12q24.22–23 (FBXW8, TESC, and VSIG10) were enriched for African ancestry, differentially expressed in EoE cases, and the prioritized variants in these loci showed genotype-dependent gene expression in the esophagus and other biologically relevant tissues. Our effort is a step forward from the current euro-centric genomics studies in human disease which impedes our ability to fully understand the genetic architecture of human diseases including EoE. Most importantly, our ability to translate genetic research into clinical practice may be less accurate due to the fact that an attempts to use incomplete estimates of genetic risk loci only from the European-based studies. Hence, there is an urgent need to increase representation of diverse ancestry in genomic research and to conduct similar population ancestry-specific assessment of pathogenic variants74. It has been shown that increasing diversity rather than studying additional individuals of European ancestry results in increased speed of fine-mapping functional variants and improved portability of polygenic prediction75. Our study highlights the need for population-specific genomic resources and creation of multi-ancestry cohorts for future studies.
Supplementary Material
{kind=link}
Clinical Implications.
There are approximately 26 GWAS risk loci, including CAPN14, TSLP, and EMSY, identified for EoE to date but this data is based primarily on European ancestry; this is the first EoE study in African American population.
Admixture mapping identified two genomic regions (9p13.3 and 12q24.22–23) with excess African ancestry and one genomic region on 15q11 with excess European ancestry associated with EoE in populations of African American.
Fine mapping and functional follow-up analysis using multi-omic annotations identified rs11068264 (FBXW8) and rs7307331 (VSIG10) on 12q24.22–23 and rs2297879 (ARHGEF39) on 9p13.3 as candidate causal variants at EoE-associated loci in African Americans.
Genome-wide association analysis identified a novel genomewide significant genetic locus DDAH1 (rs17131726, p-value = 2.39e-27) and several other suggestive loci including FAM179A, TBC1D13, MT2A, and PCSK2 associated with EoE.
Only three loci 15q22.2 (RORA), 9p24.1 (JAK2) and 15q13.3 (LINC02352 - KLF13) identified in European Ancestry population were replicated in the African American GWAS. This highlights the need of population-specific genomic resources in conducting genetic studies in EoE.
Acknowledgments
The authors would like to thank all patients who participated in the study. The authors are also grateful to their colleagues and clinical support staff for procuring biopsies, blood samples, and clinical data.
Funding
This work was supported by the National Institutes of Health (NIH) R01 HL132344 and R01 HG011411 grants support (T.B.M.) and by NIH R01 AI24355; the Campaign Urging Research for Eosinophilic Disease (CURED); and the Sunshine Charitable Foundation and its supporters, Denise and David Bunning, by CEGIR (U54 AI117804), which is part of the Rare Diseases Clinical Research Network (RDCRN), an initiative of the Office of Rare Diseases Research (ORDR), National Center for Advancing Translational Sciences (NCATS), and is co-funded by National Institute of Allergy and Infectious Diseases (NIAID), National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK), and NCATS, and in part by the Division of Intramural Research, NIAID. CEGIR is also supported by patient advocacy groups including the American Partnership for Eosinophilic Disorders (APFED), Campaign Urging Research for Eosinophilic Disease (CURED), and Eosinophilic Family Coalition (EFC). (MER, JMS).
Disclosures
MER is a consultant for Pulm One, Spoon Guru, ClostraBio, Serpin Pharm, Allakos, Celldex, Nextstone One, Bristol Myers Squibb, Astra Zeneca, Ellodi Pharma, GlaxoSmith Kline, Regeneron/Sanofi, Revolo Biotherapeutics, and Guidepoint and has an equity interest in the first seven listed, and royalties from reslizumab (Teva Pharmaceuticals), PEESSv2 (Mapi Research Trust) and UpToDate. MER is an inventor of patents owned by Cincinnati Children’s Hospital. JMS is consultant for Regeneron, Sanofi, Novaritis and royalities from Uptodate.
MC received consultant fees from Regeneron, Allakos, Adare/Ellodi, Shire/Takeda, AstraZeneca, Sanofi, Bristol Myers Squibb, Phathom; and research funding from Regeneron, Allakos, Shire/Takeda, AstraZeneca, Adare/Ellodi, Danone; none of which pose any conflict for this work.
Abbreviations:
- EoE
Eosinophilic esophagitis
- AM
Admixture mapping
- GWAS
Genome wide association study
- CAAPA
Consortium on Asthma among African-ancestry Populations in the Americas
- AA
African American
- EA
European American
- MAF
Minor Allele Frequency
- QC
Quality Control
- PCA
Principal Component Analysis
- IBD
Identity by Descent
- SNP
Single Nucleotide Polymorphism
- HWE
Hardy Weinberg Equilibrium
- LD
Linkage Disequilibrium
- TF
Transcription Factors
- eQTL
Expression quantitative trait loci
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflicts of interest: None
The list of CEGIR participants is provided in this article’s Supplementary Material
References
- 1.Papadopoulou A, Koletzko S, Heuschkel R, Dias JA, Allen KJ, Murch SH, et al. Management guidelines of eosinophilic esophagitis in childhood. J Pediatr Gastroenterol Nutr 2014; 58:107–18. [DOI] [PubMed] [Google Scholar]
- 2.Spergel JM, Dellon ES, Liacouras CA, Hirano I, Molina-Infante J, Bredenoord AJ, et al. Summary of the updated international consensus diagnostic criteria for eosinophilic esophagitis: AGREE conference. Ann Allergy Asthma Immunol 2018; 121:281–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Oliva S, Azouz NP, Stronati L, Rothenberg ME. Recent advances in potential targets for eosinophilic esophagitis treatments. Expert Rev Clin Immunol 2020; 16:421–8. [DOI] [PubMed] [Google Scholar]
- 4.Sleiman PM, Wang ML, Cianferoni A, Aceves S, Gonsalves N, Nadeau K, et al. GWAS identifies four novel eosinophilic esophagitis loci. Nat Commun 2014; 5:5593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mersha TB, Afanador Y, Johansson E, Proper SP, Bernstein JA, Rothenberg ME, et al. Resolving Clinical Phenotypes into Endotypes in Allergy: Molecular and Omics Approaches. Clin Rev Allergy Immunol 2021; 60:200–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Alexander ES, Martin LJ, Collins MH, Kottyan LC, Sucharew H, He H, et al. Twin and family studies reveal strong environmental and weaker genetic cues explaining heritability of eosinophilic esophagitis. J Allergy Clin Immunol 2014; 134:1084–92 e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Assa’ad AH, Putnam PE, Collins MH, Akers RM, Jameson SC, Kirby CL, et al. Pediatric patients with eosinophilic esophagitis: an 8-year follow-up. J Allergy Clin Immunol 2007; 119:731–8. [DOI] [PubMed] [Google Scholar]
- 8.Franciosi JP, Tam V, Liacouras CA, Spergel JM. A case-control study of sociodemographic and geographic characteristics of 335 children with eosinophilic esophagitis. Clin Gastroenterol Hepatol 2009; 7:415–9. [DOI] [PubMed] [Google Scholar]
- 9.Spergel JM, Brown-Whitehorn TF, Beausoleil JL, Franciosi J, Shuker M, Verma R, et al. 14 years of eosinophilic esophagitis: clinical features and prognosis. J Pediatr Gastroenterol Nutr 2009; 48:30–6. [DOI] [PubMed] [Google Scholar]
- 10.Sperry SL, Woosley JT, Shaheen NJ, Dellon ES. Influence of race and gender on the presentation of eosinophilic esophagitis. Am J Gastroenterol 2012; 107:215–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Veerappan GR, Perry JL, Duncan TJ, Baker TP, Maydonovitch C, Lake JM, et al. Prevalence of eosinophilic esophagitis in an adult population undergoing upper endoscopy: a prospective study. Clin Gastroenterol Hepatol 2009; 7:420–6, 6 e1–2. [DOI] [PubMed] [Google Scholar]
- 12.Weiler T, Mikhail I, Singal A, Sharma H. Racial differences in the clinical presentation of pediatric eosinophilic esophagitis. J Allergy Clin Immunol Pract 2014; 2:320–5. [DOI] [PubMed] [Google Scholar]
- 13.Chehade M, Jones SM, Pesek RD, Burks AW, Vickery BP, Wood RA, et al. Phenotypic Characterization of Eosinophilic Esophagitis in a Large Multicenter Patient Population from the Consortium for Food Allergy Research. J Allergy Clin Immunol Pract 2018; 6:1534–44 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Davis CM, Apter AJ, Casillas A, Foggs MB, Louisias M, Morris EC, et al. Health disparities in allergic and immunologic conditions in racial and ethnic underserved populations: A Work Group Report of the AAAAI Committee on the Underserved. J Allergy Clin Immunol 2021; 147:1579–93. [DOI] [PubMed] [Google Scholar]
- 15.Gupta J, Johansson E, Bernstein JA, Chakraborty R, Khurana Hershey GK, Rothenberg ME, et al. Resolving the etiology of atopic disorders by using genetic analysis of racial ancestry. J Allergy Clin Immunol 2016; 138:676–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Potaczek DP, Alashkar Alhamwe B, Miethe S, Garn H. Epigenetic Mechanisms in Allergy Development and Prevention. Handb Exp Pharmacol 2022; 268:331–57. [DOI] [PubMed] [Google Scholar]
- 17.Potaczek DP, Harb H, Michel S, Alhamwe BA, Renz H, Tost J. Epigenetics and allergy: from basic mechanisms to clinical applications. Epigenomics 2017; 9:539–71. [DOI] [PubMed] [Google Scholar]
- 18.Zhernov YV, Vysochanskaya SO, Sukhov VA, Zaostrovtseva OK, Gorshenin DS, Sidorova EA, et al. Molecular Mechanisms of Eosinophilic Esophagitis. Int J Mol Sci 2021; 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sherrill JD, Gao PS, Stucke EM, Blanchard C, Collins MH, Putnam PE, et al. Variants of thymic stromal lymphopoietin and its receptor associate with eosinophilic esophagitis. J Allergy Clin Immunol 2010; 126:160–5 e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kottyan LC, Davis BP, Sherrill JD, Liu K, Rochman M, Kaufman K, et al. Genome-wide association analysis of eosinophilic esophagitis provides insight into the tissue specificity of this allergic disease. Nat Genet 2014; 46:895–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kottyan LC, Maddox A, Braxton JR, Stucke EM, Mukkada V, Putnam PE, et al. Genetic variants at the 16p13 locus confer risk for eosinophilic esophagitis. Genes Immun 2019; 20:281–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rothenberg ME, Spergel JM, Sherrill JD, Annaiah K, Martin LJ, Cianferoni A, et al. Common variants at 5q22 associate with pediatric eosinophilic esophagitis. Nat Genet 2010; 42:289–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chang X, March M, Mentch F, Nguyen K, Glessner J, Qu H, et al. A genome-wide association meta-analysis identifies new eosinophilic esophagitis loci. J Allergy Clin Immunol 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kottyan LC, Trimarchi MP, Lu X, Caldwell JM, Maddox A, Parameswaran S, et al. Replication and meta-analyses nominate numerous eosinophilic esophagitis risk genes. J Allergy Clin Immunol 2021; 147:255–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gautam Y, Altaye M, Xie C, Mersha TB. AdmixPower: Statistical Power and Sample Size Estimation for Mapping Genetic Loci in Admixed Populations. Genetics 2017; 207:873–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Patterson N, Hattangadi N, Lane B, Lohmueller KE, Hafler DA, Oksenberg JR, et al. Methods for high-density admixture mapping of disease genes. Am J Hum Genet 2004; 74:979–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Grinde KE, Brown LA, Reiner AP, Thornton TA, Browning SR. Genome-wide Significance Thresholds for Admixture Mapping Studies. Am J Hum Genet 2019; 104:454–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mersha TB. Mapping asthma-associated variants in admixed populations. Front Genet 2015; 6:292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.DeWan AT, Egan KB, Hellenbrand K, Sorrentino K, Pizzoferrato N, Walsh KM, et al. Whole-exome sequencing of a pedigree segregating asthma. BMC Med Genet 2012; 13:95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jin W, Li R, Zhou Y, Xu S. Distribution of ancestral chromosomal segments in admixed genomes and its implications for inferring population history and admixture mapping. Eur J Hum Genet 2014; 22:930–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shriner D, Adeyemo A, Rotimi CN. Joint ancestry and association testing in admixed individuals. PLoS Comput Biol 2011; 7:e1002325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rentzsch P, Witten D, Cooper GM, Shendure J, Kircher M. CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res 2019; 47:D886–D94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Boyle AP, Hong EL, Hariharan M, Cheng Y, Schaub MA, Kasowski M, et al. Annotation of functional variation in personal genomes using RegulomeDB. Genome Res 2012; 22:1790–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.MacArthur J, Bowler E, Cerezo M, Gil L, Hall P, Hastings E, et al. The new NHGRI-EBI Catalog of published genome-wide association studies (GWAS Catalog). Nucleic Acids Res 2017; 45:D896–D901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sherrill JD, Kiran KC, Blanchard C, Stucke EM, Kemme KA, Collins MH, et al. Analysis and expansion of the eosinophilic esophagitis transcriptome by RNA sequencing. Genes Immun 2014; 15:361–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Prahalad S, Ryan MH, Shear ES, Thompson SD, Giannini EH, Glass DN. Juvenile rheumatoid arthritis: linkage to HLA demonstrated by allele sharing in affected sibpairs. Arthritis Rheum 2000; 43:2335–8. [DOI] [PubMed] [Google Scholar]
- 37.Cheng K, Gupta SK, Kantor S, Kuhl JT, Aceves SS, Bonis PA, et al. Creating a multi-center rare disease consortium - the Consortium of Eosinophilic Gastrointestinal Disease Researchers (CEGIR). Transl Sci Rare Dis 2017; 2:141–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chang CC, Chow CC, Tellier LC, Vattikuti S, Purcell SM, Lee JJ. Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience 2015; 4:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Das S, Forer L, Schonherr S, Sidore C, Locke AE, Kwong A, et al. Next-generation genotype imputation service and methods. Nat Genet 2016; 48:1284–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mathias RA, Taub MA, Gignoux CR, Fu W, Musharoff S, O’Connor TD, et al. A continuum of admixture in the Western Hemisphere revealed by the African Diaspora genome. Nat Commun 2016; 7:12522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen G, Shriner D, Zhou J, Doumatey A, Huang H, Gerry NP, et al. Development of admixture mapping panels for African Americans from commercial high-density SNP arrays. BMC Genomics 2010; 11:417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Maples BK, Gravel S, Kenny EE, Bustamante CD. RFMix: a discriminative modeling approach for rapid and robust local-ancestry inference. Am J Hum Genet 2013; 93:278–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Browning BL, Tian X, Zhou Y, Browning SR. Fast two-stage phasing of large-scale sequence data. Am J Hum Genet 2021; 108:1880–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gautam Y, Ghandikota S, Chen S, Mersha TB. PAMAM: Power analysis in multiancestry admixture mapping. Genet Epidemiol 2019; 43:831–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Plummer M, Best N, Cowles K, Vines K. CODA: convergence diagnosis and output analysis for MCMC. R news 2006; 6:7–11. [Google Scholar]
- 46.Panjwani N, Wang F, Mastromatteo S, Bao A, Wang C, He G, et al. LocusFocus: Web-based colocalization for the annotation and functional follow-up of GWAS. PLoS Comput Biol 2020; 16:e1008336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gong J, Wang F, Xiao B, Panjwani N, Lin F, Keenan K, et al. Genetic association and transcriptome integration identify contributing genes and tissues at cystic fibrosis modifier loci. PLoS Genet 2019; 15:e1008007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Watanabe K, Taskesen E, van Bochoven A, Posthuma D. Functional mapping and annotation of genetic associations with FUMA. Nat Commun 2017; 8:1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.de Leeuw CA, Mooij JM, Heskes T, Posthuma D. MAGMA: generalized gene-set analysis of GWAS data. PLoS Comput Biol 2015; 11:e1004219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhou G, Soufan O, Ewald J, Hancock REW, Basu N, Xia J. NetworkAnalyst 3.0: a visual analytics platform for comprehensive gene expression profiling and meta-analysis. Nucleic Acids Res 2019; 47:W234–W41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fishilevich S, Nudel R, Rappaport N, Hadar R, Plaschkes I, Iny Stein T, et al. GeneHancer: genome-wide integration of enhancers and target genes in GeneCards. Database (Oxford) 2017; 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Myers TA, Chanock SJ, Machiela MJ. LDlinkR: An R Package for Rapidly Calculating Linkage Disequilibrium Statistics in Diverse Populations. Front Genet 2020; 11:157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ferreira MA, Vonk JM, Baurecht H, Marenholz I, Tian C, Hoffman JD, et al. Shared genetic origin of asthma, hay fever and eczema elucidates allergic disease biology. Nat Genet 2017; 49:1752–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Johansson A, Rask-Andersen M, Karlsson T, Ek WE. Genome-wide association analysis of 350 000 Caucasians from the UK Biobank identifies novel loci for asthma, hay fever and eczema. Hum Mol Genet 2019; 28:4022–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Furuta GT, Liacouras CA, Collins MH, Gupta SK, Justinich C, Putnam PE, et al. Eosinophilic esophagitis in children and adults: a systematic review and consensus recommendations for diagnosis and treatment. Gastroenterology 2007; 133:1342–63. [DOI] [PubMed] [Google Scholar]
- 56.Moawad FJ, Dellon ES, Achem SR, Ljuldjuraj T, Green DJ, Maydonovitch CL, et al. Effects of Race and Sex on Features of Eosinophilic Esophagitis. Clin Gastroenterol Hepatol 2016; 14:23–30. [DOI] [PubMed] [Google Scholar]
- 57.Lin M, Park DS, Zaitlen NA, Henn BM, Gignoux CR. Admixed Populations Improve Power for Variant Discovery and Portability in Genome-Wide Association Studies. Front Genet 2021; 12:673167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chen MH, Raffield LM, Mousas A, Sakaue S, Huffman JE, Moscati A, et al. Trans-ethnic and Ancestry-Specific Blood-Cell Genetics in 746,667 Individuals from 5 Global Populations. Cell 2020; 182:1198–213 e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kichaev G, Bhatia G, Loh PR, Gazal S, Burch K, Freund MK, et al. Leveraging Polygenic Functional Enrichment to Improve GWAS Power. Am J Hum Genet 2019; 104:65–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhao B, Luo T, Li T, Li Y, Zhang J, Shan Y, et al. Genome-wide association analysis of 19,629 individuals identifies variants influencing regional brain volumes and refines their genetic co-architecture with cognitive and mental health traits. Nat Genet 2019; 51:1637–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kinker KG, Gibson AM, Bass SA, Day BP, Deng J, Medvedovic M, et al. Overexpression of dimethylarginine dimethylaminohydrolase 1 attenuates airway inflammation in a mouse model of asthma. PLoS One 2014; 9:e85148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Krzystek-Korpacka M, Fleszar MG, Bednarz-Misa I, Lewandowski L, Szczuka I, Kempinski R, et al. Transcriptional and Metabolomic Analysis of L-Arginine/Nitric Oxide Pathway in Inflammatory Bowel Disease and Its Association with Local Inflammatory and Angiogenic Response: Preliminary Findings. Int J Mol Sci 2020; 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sunadome H, Matsumoto H, Izuhara Y, Nagasaki T, Kanemitsu Y, Ishiyama Y, et al. Correlation between eosinophil count, its genetic background and body mass index: The Nagahama Study. Allergol Int 2020; 69:46–52. [DOI] [PubMed] [Google Scholar]
- 64.Sakaue S, Kanai M, Tanigawa Y, Karjalainen J, Kurki M, Koshiba S, et al. A cross-population atlas of genetic associations for 220 human phenotypes. Nat Genet 2021; 53:1415–24. [DOI] [PubMed] [Google Scholar]
- 65.Robertson CC, Inshaw JRJ, Onengut-Gumuscu S, Chen WM, Santa Cruz DF, Yang H, et al. Fine-mapping, trans-ancestral and genomic analyses identify causal variants, cells, genes and drug targets for type 1 diabetes. Nat Genet 2021; 53:962–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wu C, Kraft P, Zhai K, Chang J, Wang Z, Li Y, et al. Genome-wide association analyses of esophageal squamous cell carcinoma in Chinese identify multiple susceptibility loci and gene-environment interactions. Nat Genet 2012; 44:1090–7. [DOI] [PubMed] [Google Scholar]
- 67.Herrera-Luis E, Espuela-Ortiz A, Lorenzo-Diaz F, Keys KL, Mak ACY, Eng C, et al. Genome-wide association study reveals a novel locus for asthma with severe exacerbations in diverse populations. Pediatr Allergy Immunol 2021; 32:106–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Almoguera B, Vazquez L, Mentch F, Connolly J, Pacheco JA, Sundaresan AS, et al. Identification of Four Novel Loci in Asthma in European American and African American Populations. Am J Respir Crit Care Med 2017; 195:456–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Reeves SR, Kolstad T, Lien TY, Elliott M, Ziegler SF, Wight TN, et al. Asthmatic airway epithelial cells differentially regulate fibroblast expression of extracellular matrix components. J Allergy Clin Immunol 2014; 134:663–70 e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Liu T, Laidlaw TM, Feng C, Xing W, Shen S, Milne GL, et al. Prostaglandin E2 deficiency uncovers a dominant role for thromboxane A2 in house dust mite-induced allergic pulmonary inflammation. Proc Natl Acad Sci U S A 2012; 109:12692–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ferreira MAR, Vonk JM, Baurecht H, Marenholz I, Tian C, Hoffman JD, et al. Eleven loci with new reproducible genetic associations with allergic disease risk. J Allergy Clin Immunol 2019; 143:691–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Matsui EC, Adamson AS, Peng RD. Time’s up to adopt a biopsychosocial model to address racial and ethnic disparities in asthma outcomes. J Allergy Clin Immunol 2019; 143:2024–5. [DOI] [PubMed] [Google Scholar]
- 73.Taliun D, Harris DN, Kessler MD, Carlson J, Szpiech ZA, Torres R, et al. Sequencing of 53,831 diverse genomes from the NHLBI TOPMed Program. Nature 2021; 590:290–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Polygenic Risk Score Task Force of the International Common Disease A. Responsible use of polygenic risk scores in the clinic: potential benefits, risks and gaps. Nat Med 2021; 27:1876–84. [DOI] [PubMed] [Google Scholar]
- 75.Graham SE, Clarke SL, Wu KH, Kanoni S, Zajac GJM, Ramdas S, et al. The power of genetic diversity in genome-wide association studies of lipids. Nature 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.