

Abstract
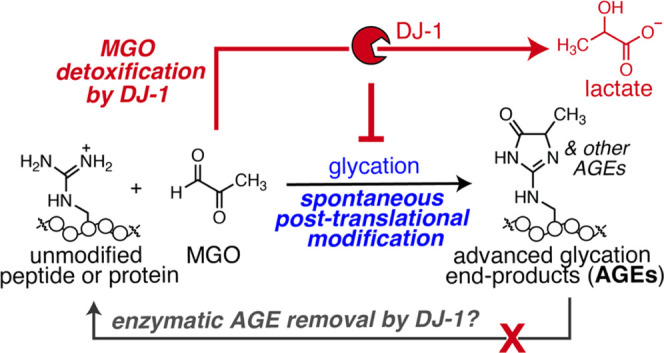
Advanced glycation end-products (AGEs) are irreversible protein modifications that are strongly associated with aging and disease. Recently, the Parkinsonism-associated protein DJ-1 has been reported to exhibit deglycase activity that erases early glycation intermediates and stable AGEs from proteins. In this work, we use mass spectrometry and western blot to demonstrate that DJ-1 is not a deglycase and cannot remove AGEs from protein or peptide substrates. Instead, our studies revealed that DJ-1 antagonizes glycation through glyoxalase activity that detoxifies the potent glycating agent methylglyoxal (MGO) to lactate. We further show that attenuated glycation in the presence of DJ-1 can be attributed solely to its ability to decrease the available concentration of MGO. Our studies also provide evidence that DJ-1 is allosterically activated by glutathione. Together, this work reveals that although DJ-1 is not a genuine deglycase, it still harbors the ability to prevent AGE formation and can be used as a valuable tool to investigate metabolic stress.
Introduction
Many protein post-translational modifications (PTMs) are part of signaling networks in which the modification itself enables information to be communicated within the cell.1 In general, these dynamic and responsive networks, like phosphorylation or glycosylation, are maintained by proteins that install (write), recognize (read), or remove (erase) the modification.2,3 However, this paradigm does not fit all PTMs, particularly those that occur without an enzyme and therefore have no “writer”. Glycation is one such PTM in which amino and guanidino groups are covalently modified by sugars and sugar-derived metabolites to generate advanced glycation end-products (AGEs) (Figure 1A).4,5 For instance, methylglyoxal (MGO) is a highly prevalent and reactive glycating agent that preferentially modifies arginine residues to form the MGO-derived hydroimidazolone isomers (MGH-1, -2, and -3), a dihydroxyimidazolidine (MGH-DH), carboxyethylarginine (CEA), tetrahydropyrimidine (THP), and argpyrimidine (APY), just to name a few (Figure 1B).4,6,7 AGE formation can lead to protein cross-linking, aggregation, functional impairment, and is correlated with aging and disease.8−13
Figure 1.

DJ-1 is reported to be a glycation eraser. (A) Glycation is a nonenzymatic post-translational modification (PTM) that occurs preferentially at Arg or Lys residues. Unlike most PTMs, glycation occurs spontaneously and has no writer. There is significant interest in enzymes, like the Parkinsonism-associated protein DJ-1, that have the potential to erase glycation events. (B) Reaction of Arg with methylglyoxal (MGO), one of the most potent and prevalent cellular glycating agents, can produce numerous advanced glycation end-products (AGEs). These include the methylglyoxal-derived hydroimidazolone isomers (MGH-1, -2, -3), the dihydroxyimidazolidine (MGH-DH), carboxyethylarginine (CEA), argpyrimidine (APY), and tetrahydropyrimidine (THP).
Because it occurs spontaneously, glycation has no writer and has historically been thought of as random cellular damage that occurs over time and/or through disrupted metabolism. Accordingly, most of the signaling that has been attributed to AGEs occurs through the only known glycation “reader”, the nonspecific cell surface receptor for AGEs (RAGE). In addition to AGE-modified proteins, RAGE broadly recognizes polyanionic species in the extracellular milieu, all of which activate a well-defined signaling cascade leading to immune cell activation, inflammation, and cell division.14−16 However, recent work points to the significance of glycation as a functional PTM that can impact gene expression and turn on the antioxidant response.17−19
The innate chemical diversity of AGEs presents a challenge for potential glycation “erasers”, or deglycase proteins. Thus, the discovery that the Parkinson-associated protein DJ-1 could remove glycation from a range of AGE-modified proteins and nucleotides has garnered major interest.17,18,20−22 DJ-1 was first discovered to be a novel glutathione-independent glyoxalase and has since been reported to remove a wide range of AGE-modified substrates.23,24 These proposed DJ-1 substrates include early glycation intermediates and, more recently, intact stable AGEs from chromatin and proteins like histone 3 and α-synuclein.18,20,23−25 Despite these prior reports, however, whether DJ-1 exhibits genuine deglycase activity remains an open question that has been hotly contested in the literature.26−29
Here, we provide a rigorous evaluation of DJ-1 activity and find that DJ-1 cannot be considered an AGE eraser. Instead, DJ-1 functions as a glycation antagonist. Using mass spectrometry and western-blotting-based deglycation assays, we demonstrate that DJ-1 is unable to remove stable AGEs from protein and peptide substrates glycated with MGO. We further demonstrate that DJ-1 cannot remove even early-stage AGEs, like the bis-hemiaminal dihydroxyimidazolidine (MGH-DH) from these substrates. However, concurrent incubation with DJ-1 and MGO did reduce levels of overall glycation. Time course studies revealed that this reduction could be attributed to DJ-1’s glyoxalase activity, which decreases the concentration of glycating agent and antagonizes AGE formation. Though DJ-1 does not possess true deglycase activity, it still plays an important role by minimizing the overall glycation burden and mitigating the impact of glycolytic and metabolic stress.
Materials and Methods
General
All chemical reagents were of analytical grade, obtained from commercial suppliers, and used without further purification unless otherwise noted. Methylglyoxal (40% w/v) was purchased from Sigma-Aldrich. Water used in biological procedures was distilled and deionized using an Atrium Pro purification system (Sartorius). All statistical analysis was conducted using Prism GraphPad.
DJ-1 Expression and Purification
The pET3a-His-DJ-1 plasmid was a gift from the Michael J Fox Foundation MJFF (Addgene plasmid #51488). The C106A mutant was generated using Agilent QuikChange Site-Directed Mutagenesis with the pET3a-His-DJ-1 plasmid as a template and the following reported primers, as previously reported:17
5′-GCCGCCATTGCCGCAGGCCCGACCGC-3′
5′-AATCAGGCCTTTGCGGTTCTCCTGCTCTTTCAGG-3′
Both DJ-1WT and DJ-1C106A proteins were expressed in BL21(DE3) E. coli (Agilent). Cultures were grown to an OD600 value of 0.6 at 37 °C and induced overnight at 18 °C using IPTG. Cells were then pelleted and frozen. Frozen pellets were thawed and then lysed with 50 mM Tris-HCl pH 7.0, 500 mM NaCl, 1 mM MgCl, 40 μM Benzonase (Millipore Sigma), and 0.4 mM PMSF via sonication on ice for 3.5 min. Lysates were loaded onto a Ni-NTA column (Cytiva) with 20 mM PBS pH 7.4, 20 mM imidazole, and 500 mM NaCl. His-tagged protein was eluted using 20 mM PBS pH 7.4, 500 mM imidazole, and 500 mM NaCl before being buffer-exchanged into 20 mM PBS pH 7.4 using a Nap25 column (Cytiva). Protein concentrations were determined using a Thermo Fisher Pierce BCA Protein Assay Kit and a Tecan 10M microplate reader.
Peptide Synthesis
Rink Amide MBHA resin (100–200 mesh, Novabiochem) was used for solid-phase peptide synthesis (0.5 mmol/g scale) in a 4 mL fritted syringe. Fmoc-deprotection was completed using 20% piperidine in DMF and washes were completed with DMF. Amino acid coupling was completed by adding 5 equivalences of amino acid to O-(benzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium hexafluorophosphate (HBTU) and N,N-diisopropylethyllamine (DIPEA) in DMF. All amino acids were purchased from ChemPep, Inc. or Advanced ChemTech, Inc. N-Terminal acetylation was accomplished using acetic anhydride and DIEA (both 3 equiv.) for 2 h in 3 mL of DMF. After completing synthesis and acetylation, resin was washed and dried using dichloromethane. Amidated peptides were then cleaved from resin using 95% trifluoroacetic acid (TFA), 2.5% triisopropylsilane (TIPS), and 2.5% water. The resulting crude peptide was dried under air flow and then dissolved in a mixture of water/acetonitrile based on solubility before purification.
Peptide Purification
After synthesis, peptide was purified using an Agilent 1260 LC system equipped with an Agilent ZORBAX SB-C18 column (9.4 × 250 mm2), 5 μm particle size employing water (A) and acetonitrile (B) mobile phase with 0.1% TFA. Crude peptide was eluted using the following method with a flow rate of 3.0 mL/min: gradient from 5% B to 45% B over 20 min. Absorbance at 215 and 280 nm was used to observe desired peptide peaks, and these peaks were eluted and collected using an automated fraction collector. The purity of the collected fractions was determined using matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) mass spectrometry (Bruker). Fractions deemed pure were combined and then lyophilized. Peptide stock solutions were prepared at 20 mM concentrations in DMF.
Protein Glycation Assays
Purchased purified HA-tagged ubiquitin (R&D Systems) or lyophilized RNase A or aldolase (Millipore Sigma) dissolved in water were used for protein glycation experiments. Reactions were conducted in Eppendorf tubes at a final volume of 100 μL containing 50 μM protein, 20 mM PBS pH 7.3, and 200 μM MGO. DJ-1WT or DJ-1C106A were added at a 10:1 ratio of protein:enzyme, as previously described.17 For “concurrent” conditions, a final concentration of 5 μM DJ-1WT or DJ-1C106A was used. Reactions were then incubated at 37 °C for 24 h and quenched with 3 μL of 500 mM Tris pH 7.3 (final concentration 15 mM Tris). For “subsequent” conditions, 5 μM DJ-1WT or DJ-1C106A was added after quenching, either with Tris (final concentration 15 mM Tris) or by buffer exchange into 20 mM PBS using a Nap5 column (Cytiva), and incubated again at 37 °C for an additional 24 h. Samples were diluted using ultrapure water before being analyzed by LC-MS or western blot.
Peptide Glycation Assays
For peptide glycation experiments, reactions were conducted in Eppendorf tubes at a final volume of 50 μL with final concentrations of 1 mM peptide, 20 mM PBS pH7.3, and 1 mM MGO. DJ-1WT or DJ-1C106A were added at a 10:1 ratio of peptide:enzyme. For concurrent conditions, a final concentration of 100 μM DJ-1WT or DJ-1C106A was used. Reactions were then incubated at 37 °C for 3 h and then quenched with 3 μL of 500 mM Tris pH 7.3 (final concentration, 28.3 mM Tris). For subsequent conditions, either 100 μM DJ-1WT or DJ-1C106A was added after quenching the 50 μL reaction initially with 3 μL of 500 mM Tris (final concentration 28.3 mM Tris) and then incubated again at 37 °C for 3 h. To evaluate the impact of free thiols on glycation reactions, the same reaction conditions were used in the presence of either 1 mM dithiothreitol (DTT) or glutathione (GSH). At the conclusion of the reaction, samples were diluted with ultrapure water and analyzed by LC-MS.
Liquid Chromatography Mass Spectrometry (LC-MS)
Reversed-phase liquid chromatography and mass spectrometry were conducted using an Agilent 1260 LC system connected in line to an Agilent 6530 Accurate Mass Q-TOF employing a mobile phase of water (A) and acetonitrile (B) with 0.1% formic acid.
Protein glycation reactions using intact proteins were injected onto a Zorbax RRHD 300 Å StableBond C8 (2.1 × 100 mm2, 1.8 μm, Agilent) column with the following method with a flow rate of 0.4 mL/min: isocratic at 5% B for 1.75 min, a gradient change from 5% B to 80% B over 24.25 min, gradient change from 80% B to 100% B over 0.5 min, isocratic column washing at 100% B for 7.50 min, and reequilibration at 5% B for 7 min. Intact protein deconvolutions were generated using Agilent MassHunter BioConfirm Qualitative Analysis software.
Peptide glycation reactions were injected onto an AdvanceBio Peptide 2.7 μm column (2.1 × 150 mm2, Agilent) with the following method with a flow rate of 0.4 mL/min: isocratic at 5% B for 1.75 min, gradient change from 5% B to 40% B over 14.25 min, gradient change from 40% B to 100% B over 4 min, isocratic column washing at 100% B for 3 min, and reequilibration at 5% B for 7 min. Peptide data were quantified using peak volumes determined by Agilent MassHunter Qualitative Analysis and the MassHunter Molecular Feature Extractor.
Percent glycation was determined by the formula below.
| 1 |
SDS-PAGE
Protein glycation reactions were treated with 6X SDS loading dye containing dithiothreitol (DTT) and boiled at 95 °C for 5 min. A precast protein gel (8–16%, mini-PROTEAN TGX, Bio-Rad) was used for SDS-PAGE, and the gel was run using a Tris/glycine/SDS running buffer at 226 mV for 18 to 24 min. Gels were stained using Coomassie dye (40% methanol, 40% acetic acid, 20% water, 0.05% Coomassie Brilliant Blue R-250 w/v) for 15 min after briefly heating. The gels were then destained 3× in 15 min intervals using a destain solution (25% methanol, 10% acetic acid, 65% water). The gels were imaged on a Bio-Rad ChemiDoc XRS+.
Western Blotting
Transfer onto PVDF membranes was accomplished with the iBlot 2 dry blotting system (Invitrogen). After transfer, the membrane was blocked in 1× TBST (20 mM Tris, 150 mM NaCl, and 0.1% Tween) with 5% (w/v) skim milk powder for an hour. Primary antibody mouse α-MGO (Cell BioLabs, STA-011) or mouse α-His tag (Cell Signaling Technology, 27E8) was added (1:1000) after blocking and incubated at 4 °C overnight while shaken. Membrane was washed three times with 1× TBST for 5 min each before adding HRP-linked secondary antibody (Cell Signaling Technology, 7076S) diluted into 5% milk and 1× TBST (1:2000). The membrane was incubated for 1 h with secondary antibody at room temperature before washing three times with 1× TBST for 5 min. Signal was developed using Clarity Western ECL Substrate (Bio-Rad) and imaged on a Bio-Rad ChemiDoc XRS+.
Lactate Detection Using a Luminescence Assay
To confirm DJ-1 glyoxalase activity, 1 μM DJ-1WT was incubated with 200 μM MGO for 0–30 min at 37 °C. The commercially available Lactate-Glo kit (Promega) was purchased, and manufacturer instructions were followed with the following modifications: 10 μM DJ-1C106A was added to the lactate standard reaction mix for more accurate determination of the standard curve. Reactions were initiated within a flat clear-bottom white 96-well plate (Corning) with the bottoms covered in white tape to minimize luminescence crosstalk between wells. Luminescence readings were recorded using a Tecan 10 M microplate reader with a 1 s integration time.
To detect both l- and d-lactate, lyophilized l- and d-lactate dehydrogenase (LDH) enzymes were purchased from Sigma-Aldrich and used in place of the provided enzyme from the Lactate-Glo kit. To confirm the specificity of purchased LDH enzymes, either l- or d-lactate (Sigma) was dissolved in water at concentrations from 0 to 200 μM and incubated with l- or d-LDH in the lactate standard reaction mix.
Evaluation of l- and d-lactate production by 10 μM DJ-1WT was conducted by incubating 1 mM MGO alone, or with reduced l-glutathione (GSH) or peptide 1 for 30 min at 37 °C. The latter two conditions can be used to model hemithioacetal or hemiaminal substrates. Following previously described methods,20,29 stock solutions of either 10 mM MGO and 10 mM GSH or 20 mM MGO and 20 mM peptide 1 were first incubated at room temperature for 1 h to allow for equilibration of the hemithioacetal or hemiaminal in solution before being treated with DJ-1 for a final substrate concentration of 1 mM. For the hemiaminal peptide 1 samples, 1 mM peptide stock solution was also added to lactate standard reaction mix to account for the presence of ∼5% DMF in experimental samples that would influence the luminescence measurement.
Methylglyoxal Quantification by HPLC
A standard curve was first generated where known MGO concentrations were determined by adding 100 μL of a 20 mM stock of 3,4-diaminobenzophenone (DABP) dissolved in DMF to 100 μL of MGO solutions (at 0, 0.05, 0.25, 1, 5, 7.5, or 10 mM) in 20 mM PBS and incubated for 30 min at 37 °C. Reversed-phase analytical high-performance liquid chromatography was performed using an Agilent Infinity LC system employing a mobile phase of water (A) and acetonitrile (B) with 0.1% TFA. Samples were injected (0.70 μL) onto an AdvanceBio Peptide 2.7 μm column (2.1 × 150 mm2, Agilent) with the following method using a flow rate of 0.400 mL/min: isocratic 5% B for 1 min, isocratic pumping at 30% B for 1 min, a gradient change from 30% B to 80% B over 1 min, a gradient change from 80% B to 100% B over 1 min, isocratic column washing at 100% B for 1.5 min, and ending with a gradient change back to 5% B for 2 min. Absorbance at a wavelength of 260 nm was used to detect both unreacted DABP and the addition product between MGO and DABP. The peak corresponding to the MGO-DABP addition product was integrated to generate a standard curve.
For samples with unknown MGO concentrations, 50 μL of the MGO-containing solution in 20 mM PBS was mixed with 50 μL of 20 mM DABP in DMF and incubated for 30 min at 37 °C. After analysis by HPLC, the product peaks were integrated and used to determine the concentration of MGO based on the standard curve.
Time Course Studies
For time course studies, 100 μM DJ-1WT was incubated with either 1 or 10 mM substrate concentrations in 20 mM PBS pH 7.3. Substrates include MGO alone, the modeled GSH hemithioacetal (equimolar GSH and MGO at 1 or 10 mM), and the modeled peptide hemiaminal (equimolar peptide 1 and MGO at 1 mM or 10 mM), as previously described. At predetermined timepoints, a 15 μL aliquot of the reaction mixture was removed and incubated with 15 μL of 20 mM DABP in DMF and incubated for 37 °C before being subjected to the previously described MGO quantification method.
Glutathione Titration
To determine if DJ-1WT is allosterically modulated by GSH, 100 μM DJ-1WT was incubated with 10 mM MGO and predetermined concentrations ranging from 0 to 10 mM GSH in 20 mM PBS at a final volume of 25 μL. Reactions were incubated at 37 °C for 30 min before being quenched with 25 μL of 20 mM DABP solution and incubated again at 37 °C for 30 min. The same steps were followed using BME in place of GSH with a concentration range of 0–2 mM BME. Samples were then subjected to the previously described MGO quantification method. To determine the extent of hemithioacetal formation, a range of glutathione (GSH) or β-mercaptoethanol (BME) concentrations (0–2 mM) were incubated with 10 mM MGO in 20 mM PBS pH 7.4 at 37 °C for 30 min in a 96-well plate. Absorbance measurements were then taken at 288 nm using a Tecan 10 M microplate reader.
Pretreatment Assays
To pretreat MGO with DJ-1, 10 mM MGO and 100 μM DJ-1WT or DJ-1C106A were co-incubated at a volume of 100 μL in 20 mM PBS pH 7.4 at 37 °C for 2.5 h. Separately, 25 μL of HisPur Ni-NTA Resin (Thermo Scientific) was spun down, and liquid from the slurry was pipetted out to reduce dilution upon addition to the pretreatment reaction. Next, the entirety of the 100 μL reaction was pipetted onto the resin, and the mixture was gently agitated and then spun on a rotisserie shaker for an additional 30 min at room temperature. Afterward, the slurry was transferred into a spin column and centrifuged at 13,000 rpm for 2 min; supernatant flowthrough was collected in microcentrifuge collection tubes (Pierce). Using the methods described above, the collected supernatant was used for peptide glycation reactions and LC-MS analysis. The supernatant was also quantified for MGO concentrations using the methods described above.
Results and Discussion
DJ-1 Is Unable to Remove Stable AGEs from Proteins
Although most of the available DJ-1 studies focus on its ability to reverse hemiaminal or hemithioacetal adducts, a few recent reports have suggested that DJ-1 is able to erase stable, late-stage AGEs like MGH-1 and carboxyethyllysine from proteins.17,18,21 Thus, our studies initiated with the goal of understanding which of the many possible AGEs could be removed by DJ-1. Our lab has previously evaluated protein glycation by MGO in vitro and found that the small protein ubiquitin (Ub) was among the most heavily glycated, making it an ideal substrate for these studies.30 To evaluate the ability of DJ-1 to remove AGEs, we subjected full-length ubiquitin (Ub) to our established glycation protocols. After glycation, the excess MGO was quenched using Tris (Figure S2). Next, the resulting AGE-modified Ub was treated with either wild-type DJ-1 (DJ-1WT) or a catalytically inactive variant (DJ-1C106A) (Figure 2A).17 Using western blot and mass spectrometry, we found that glycation levels remained unchanged even when DJ-1WT was incubated with AGE-modified Ub for 24 h (Figure 2B,C). We further confirmed that the DJ-1WT remained unable to remove AGEs from proteins when glycation reactions were quenched using buffer exchange into phosphate buffer, rather than sequestering by Tris (Figure S3).
Figure 2.

DJ-1 cannot remove stable AGEs from proteins. (A) To assess the potential deglycase activity of DJ-1, two sets of conditions were evaluated. In the concurrent treatment, DJ-1 was co-incubated with both ubiquitin (Ub) and MGO for 24 h at 37 °C in 20 mM PBS at pH 7.3. In the subsequent treatment, Ub was first glycated by MGO for 24 h at 37 °C in 20 mM PBS at pH 7.3, at which point the excess MGO was quenched with Tris buffer (see also Figure S2). Afterward, DJ-1 was incubated with the glycated protein for an additional 24 h at 37 °C in 20 mM PBS and 15 mM Tris at pH 7.3. (B) Using SDS-PAGE and western blotting against MGO, it was possible to observe a decrease in glycation from concurrent treatments with wild-type DJ-1 (DJ-1WT), but not a catalytically inactive DJ-1 variant (DJ-1C106A). (C) These same findings were obtained using intact protein mass spectrometry following subsequent or concurrent DJ-1 treatments, as observed in the representative deconvoluted mass spectra.
Although DJ-1WT was unable to remove previously generated AGEs, we found that DJ-1WT led to a major reduction in glycation when it was incubated concurrently with Ub during the 24 h MGO treatment (Figure 2A). As observed by both western blot and mass spectrometry, this reduction in glycation only occurred for DJ-1WT, not DJ-1C106A. This indicates that the decrease in glycation can be attributed to DJ-1WT activity (Figure 2B,C). This was also true for other glycated protein substrates including ribonuclease A and aldolase (Figures S4 and S5). In all cases, a reduction in AGE formation was only observed in conditions where DJ-1WT and MGO were concurrently incubated with the protein. While these results suggest that DJ-1 is unlikely to remove fully formed AGEs from proteins, they also confirm that DJ-1 possesses some activity that impedes glycation.
DJ-1 Does Not Erase Early AGEs from Peptides
Our past work has shown that many AGE adducts are isomeric and therefore difficult to discern on intact proteins. However, using peptide substrates that possess just a single Arg residue, it is straightforward to distinguish discrete AGEs. Thus, to further evaluate the ability of DJ-1 to remove specific AGEs, we used a glycated peptide substrate (peptide 1, Ac-LESRHYA) that our lab has studied extensively.7 Our past work has confirmed that the dihydroxyimidazolidine, MGH-DH, is the predominant adduct for peptide 1 after 3 h incubation with MGO.7 Based on the proposed mechanism for DJ-1 deglycase activity, this bis-hemiaminal adduct would be expected to be a substrate for DJ-1.20 After 3 h of MGO treatment, we subjected glycated peptide 1 to an additional 3 h incubation with DJ-1WT (Figure 3A). We found that this led to no change in overall glycation levels, as assessed by LC-MS (Figure 3B,C). Although we observed no change in the overall extent of glycation, we found that greater proportional levels of the [M + 54] adduct (MGH-1) were observed after DJ-1WT treatment. However, we confirmed that this was due to the spontaneous conversion of MGH-DH to MGH-1 that occurs during an additional 3 h of incubation in buffer, regardless of whether DJ-1WT was present (Figure 3C).7 We further established that the DJ-1WT does not affect the conversion of MGH-DH to MGH-1 using protocols previously developed in our lab (Figure S6).7
Figure 3.

DJ-1 cannot remove early AGEs from peptides. (A) Purified synthetic peptide Ac-LESRHYA, (peptide 1) was allowed to react with equimolar MGO (1 mM) in 20 mM PBS at pH 7.3 for 3 h at 37 °C, and was then analyzed by LC-MS after subsequent or concurrent treatment with DJ-1. (B) Representative combined mass spectra (8.5–10 min) show a reduction in glycation only for concurrent treatments with DJ-1WT. (C) Distribution of glycation products observed by LC-MS after DJ-1 treatment. Stacked bar graphs are plotted as mean ± standard deviation for each mass adduct. A nondirectional (two-tailed) one-way ANOVA using Dunnett’s multiple comparison test was used to determine statistically significant differences in total glycation compared to peptide 1 treated with MGO, p < 0.0001(****).
These data demonstrate that DJ-1WT is unable to remove previously generated AGEs, even early adducts like MGH-DH. However, when DJ-1WT and MGO were incubated concurrently with peptide 1, the extent of glycation was reduced to 10.7 ± 3.6%, compared to 28.3 ± 3.8% with no enzyme, or 28.8 ± 1.1% with DJ-1C106A (Figure 3B,C). Together, these results suggest that DJ-1’s protective effects against glycation cannot be attributed to genuine deglycase activity, but rather indicate that DJ-1 uses a different mechanism to compete with AGE formation.
Free Thiols Antagonize Glycation Independent of DJ-1 Activity
Most prior work has focused on DJ-1 activity in the presence of additional thiols, either as substrates (in the form of hemithioacetals) or to ensure that Cys 106, the key DJ-1 catalytic residue, remains reduced. Our initial studies were performed without any reducing agents present, and we confirmed that there was negligible DJ-1 oxidation using LC-MS and SDS-PAGE, even when stored in PBS (Figure S7). Nonetheless, we sought to determine if DJ-1 activity would be affected by the addition of thiols, like dithiothreitol (DTT). We first subjected peptide 1 to 3 h of MGO incubation and followed this treatment with either DJ-1WT, DTT, or DJ-1WT with DTT. Using LC-MS, we observed no change in glycation levels with any of these treatments, suggesting that additional thiol does not alter DJ-1 activity and is not sufficient for DJ-1 to remove preexisting AGEs (Figure S8). Next, we concurrently treated peptide 1 with MGO and DTT, with DJ-1WT, or with both DTT and DJ-1WT. Notably, we observed a significant reduction in glycation even when just DTT was co-incubated with peptide 1 and MGO (16.0 ± 2.6% glycation, compared to 34.7 ± 1.8% with MGO alone). Additionally, when peptide 1 was concurrently incubated with both DJ-1WT and DTT along with MGO, there were low levels of glycation (3.0 ± 2.0%).
Because DTT has two thiols that can each form hemithioacetals with MGO, we also evaluated peptide glycation in the presence of glutathione (GSH), which has only one thiol. We subjected peptide 1 to 3 h of MGO incubation and followed this treatment with either DJ-1WT, GSH, or DJ-1WT with GSH. Using LC-MS, we found that under these subsequent conditions, there were no differences in the extent of glycation after any of these treatments (Figure 4B). Next, we concurrently treated peptide 1 with MGO and GSH, with DJ-1WT, or with both GSH and DJ-1WT. We observed a significant reduction in glycation when just GSH was co-incubated with peptide 1 and MGO (23.3 ± 1.1% glycation, compared to 34.7 ± 1.8% with MGO alone). We also observed low levels of glycation when peptide 1 was concurrently incubated with both DJ-1WT and GSH along with MGO (4.1 ± 0.5% glycation) (Figure 4C). This behavior was similar to that what was observed when peptide 1 was co-incubated with MGO, DTT, and DJ-1WT. This suggests that there is an additive effect whereby thiols and DJ-1 minimize glycation through two different mechanisms. Further, these data are consistent with a model in which any compound that reacts with MGO will compete with AGE formation.
Figure 4.

Glutathione interferes with AGE formation even in the absence of DJ-1. (A) To determine the influence of free thiols on glycation reactions, 1 mM peptide 1 (Ac-LESRHYA) was allowed to react with 1 mM MGO and was then analyzed by LC-MS after subsequent or concurrent treatment with DJ-1WT and/or glutathione (GSH) in 20 mM phosphate buffer at pH 7.3 for 3 h at 37 °C. Distribution of glycation adducts observed by LC-MS after (B) subsequent or (C) concurrent DJ-1 and/or GSH treatment. Decreases in glycation were only observed in concurrent, not subsequent, conditions. While the addition of either GSH or DJ-1WT was able to reduce glycation during concurrent incubations, there was also an additive effect where even greater reductions in glycation were observed in the presence of both GSH and DJ-1WT. Stacked bar graphs are plotted as mean ± standard deviation for each mass adduct. A nondirectional (two-tailed) one-way ANOVA using Dunnett’s multiple comparison test was used to determine statistically significant differences in total glycation, p < 0.0001(****) and p < 0.001(***).
DJ-1 Has Glyoxalase Activity
DJ-1 has previously been reported to be a glyoxalase that detoxifies MGO to lactate.23 Because we observed a reduction in glycation only when DJ-1 co-incubated with MGO, we hypothesized that this glyoxalase activity could be the source of the protective effect against glycation, as has been previously proposed.27,29 Despite these two notable exceptions,27,29 most past efforts to evaluate DJ-1’s putative deglycase activity have largely ignored its glyoxalase function because of a handful of reports stating that it is inefficient.20,31 Nonetheless, using a commercially available enzyme-linked luminescence assay, we found that DJ-1WT generated considerable levels of l-lactate from MGO after short (30 min) exposures (Figure S9). We also adapted our luminescence assay for use with either l- or d-lactate dehydrogenase (LDH) (Figure S10). Using this approach, we found that both l- and d-lactate were generated, with 45% enantiomeric excess (ee) of l-lactate (Figure 5A,B). We also evaluated stereospecificity for DJ-1-mediated conversion of MGO to lactate in the presence of glutathione (GSH) or peptide 1. In this case, GSH or peptide 1 was co-incubated with MGO for 1 h at room temperature prior to DJ-1WT treatment, allowing hemithioacetals or hemiaminals with MGO to form. In both cases, we found that similar overall levels of lactate were produced (Figure 5B). However, the presence of GSH led to 80% ee, while the presence of peptide 1 produced 49% ee. The latter of these was similar to the 45% ee observed with MGO alone. These results reveal that DJ-1 glyoxalase activity is not stereospecific, as has been previously reported for MGO treated with DJ-1WT, even without peptide or GSH present.24
Figure 5.

DJ-1 is a glyoxalase that is allosterically activated by glutathione. (A) DJ-1 glyoxalase activity was measured in two ways: either by monitoring lactate production with a commercially available enzyme-linked luminescence assay or by monitoring the consumption of MGO in a quantitative HPLC chemical derivatization assay. (B) Using the luminescence assay, both d- and l-lactate were detected when 10 μM DJ-1WT was incubated for 30 min at 37 °C in 20 mM PBS at pH 7.3 with 1 mM MGO alone, 1 mM MGO and 1 mM GSH, or 1 mM MGO and 1 mM peptide 1. (C) Using the HPLC assay, time course studies were conducted using 100 μM DJ-1WT incubated with (top) 1 mM MGO, with or without equimolar GSH or peptide 1, or (bottom) 10 mM MGO, with or without equimolar GSH or peptide 1. Samples were incubated at 37 °C in 20 mM PBS at pH 7.3, and the resulting loss in MGO was monitored over 1 h. While DJ-1 exhibited modest differences in activity at low MGO (1 mM) concentrations, there was a major increase in activity when GSH was present at high (10 mM) MGO concentrations. (D) To evaluate if GSH could be an allosteric activator of DJ-1, we used the HPLC assay to monitor the reaction velocity (V0 = −d[MGO]/dt) over 30 min of treatment with 100 μM DJ-1WT and MGO (10 mM initial concentration), while titrating the GSH concentrations, up to 10 mM GSH (left). At low MGO concentrations (right), there was a sigmoidal relationship between GSH concentration and V0, suggesting that DJ-1 is allosterically activated by GSH. An unpaired Student’s t-test was used to determine statistically significant differences between V0 at 0 mM and either 1 mM or 2 mM GSH, p < 0.01(**), p < 0.05(*).
Because DJ-1WT produces varying amounts of l- and d- lactate in the presence of peptide 1 or GSH, we chose to monitor its glyoxalase activity by tracking the consumption of MGO with 3,4-diaminobenzophenone (DABP) in a quantitative HPLC-based assay (Figures 5A and S11). While several prior studies have determined kinetic parameters for DJ-1 with or without GSH, they have not all been directly comparable. For this reason, we chose instead to perform time course studies that would allow us to directly compare DJ-1 glyoxalase activity in the presence and absence of GSH or peptide at high (10 mM) and low (1 mM) MGO concentrations These concentrations were selected either to be well above (10 mM) or close to (1 mM) the reported Km values, which range from 0.3 to 0.9 mM.17,18,20,26
At low MGO concentrations, we found that DJ-1WT catalyzed the complete consumption of MGO after 1 h, regardless of whether GSH or peptide 1 was present. However, when equimolar (1 mM) GSH was present, there was noticeably more MGO consumed after 30 min (Figure 5C, top). At high MGO concentrations, this effect was substantially amplified, with greater than a 5-fold increase in the initial rate of MGO consumption in the presence of equimolar (10 mM) GSH (0.026 ± 0.003 mM/min for MGO only; 0.147 ± 0.020 mM/min with GSH) (Figure 5C, bottom). We also found that there was a modest increase in the rate of MGO consumption in the presence of peptide 1 (0.083 ± 0.013 mM/min). However, most of this difference could be attributed to the glycation of peptide 1, which we could account for by monitoring the loss of MGO over time in the presence of peptide 1 and DJ-1C106A.
Although the GSH-MGO hemithioacetal was originally proposed to be the ideal DJ-1 substrate, recent work has provided compelling data to the contrary, demonstrating that the hemithioacetal spontaneously decomposes due to the shifting equilibrium as DJ-1 decreases MGO concentrations.27 Nonetheless, our time course studies revealed a clear increase in DJ-1 glyoxalase activity in the presence of GSH. To reconcile these results, we postulated that glutathione could be an allosteric activator of DJ-1, rather than a co-substrate.
To evaluate this possibility, we monitored the extent of MGO consumption after 30 min of treatment with 100 μM DJ-1 across a range of GSH concentrations, using high (10 mM) initial concentrations of MGO. Using this approach, we confirmed that DJ-1WT activity increases, and then plateaus, with increasing concentrations of GSH, up to 10 mM GSH (Figure 5D, left). At lower GSH concentrations, between 0 and 1 mM, there was a sigmoidal relationship between the reaction velocity and GSH concentration (Figure 5D, right). This suggests that DJ-1 could be allosterically activated by GSH.
To further test this hypothesis, we compared the performance of GSH to another molecule containing a single thiol, β-mercaptoethanol (BME) (Figure S12A). First, we monitored the extent of hemithioacetal formation (λmax = 288 nm) when either GSH or BME (0–2 mM) was co-incubated with 10 mM MGO. We found that both thiols resulted in similar levels of hemithioacetal over this concentration range (Figure S12B). Next, we monitored the extent of MGO consumption after 30 min of treatment with 100 μM DJ-1WT across this same range of GSH or BME concentrations. For GSH, we once again observed a sigmoidal relationship between the V0 and GSH concentration (Figure S12C, left). Intriguingly, while V0 remains nearly constant between 1 and 2 mM GSH, the amount of hemithioacetal is doubled. Because there is not a concomitant increase in V0 as hemithioacetal levels rise, we can conclude that the GSH hemithioacetal is unlikely to be a significantly better substrate than MGO alone. Additionally, for BME, we observed virtually no change in V0 across the entire concentration range evaluated, even as BME hemithioacetal levels rise (Figure S12C, right). Thus, the observed increase in DJ-1 glyoxalase activity in the presence of GSH can be attributed to cooperativity between DJ-1 and GSH, rather than preferential deglycation of hemithioacetals.
DJ-1 Antagonizes Glycation through MGO Detoxification
Having established that DJ-1 functions as an effective glyoxalase, we next sought to determine if this activity alone could account for the observed protection against glycation. To test this hypothesis, we designed an experiment that would allow us to first treat MGO with DJ-1, next remove DJ-1, and finally, use the pretreated MGO to perform peptide glycation reactions (Figure 6A,B). Accordingly, 10 mM MGO was treated with 100 μM His-tagged DJ-1WT or DJ-1C106A for 3 h. At this point, the His-tagged DJ-1 was removed using Ni-NTA resin. We confirmed that this protocol resulted in undetectable levels of DJ-1 in the resulting supernatant (Figure S13), which could then be used to perform peptide glycation reactions. Using this approach, we found that similar levels of glycation were obtained for supernatants that had been previously exposed to DJ-1C106A or no enzyme at all (Figure 6C, left). There was an overall decrease in total glycation compared to our standard glycation protocols (Figure 3C), which we attribute to the nonspecific sequestering of MGO by the resin and spin columns used to collect the supernatant. We also observed a significant decrease in glycation for supernatants that were previously exposed to DJ-1WT, similar to the concurrent peptide glycation treatment condition that was observed previously.
Figure 6.

DJ-1 mitigates glycation by decreasing MGO concentration. (A) 10 mM MGO was pretreated with 100 μM His-tagged DJ-1 for 2.5 h at 37 °C in pH 7.3 20 mM PBS before addition of Ni-NTA resin for 30 min at room temperature to bind the His-tagged DJ-1. The resulting supernatant (PT-MGO stock) was then collected using spin columns and (B) used in peptide glycation reactions. These glycation reactions were conducted using 1 mM peptide 1 and a 1:10 dilution of the PT-MGO stock and were incubated for 3 h at 37 °C in 20 mM PBS at pH 7.3 before being analyzed by LC-MS. (C) Left: Distribution of glycation products observed by LC-MS using PT-MGO stocks obtained after incubation of MGO with no enzyme, DJ-1WT, or DJ-1C106A; middle: determined concentrations of MGO in the PT-MGO stocks, as measured by a quantitative HPLC assay; right: distribution of glycation products observed by LC-MS using new MGO stocks prepared at the previously determined concentrations. A nondirectional (two-tailed) one-way ANOVA using Dunnett’s multiple comparison test was used to determine statistically significant differences in total glycation concentrations, p < 0.0001(****).
To further confirm that DJ-1 decreases MGO levels, we quantified the amount of MGO remaining in the supernatant (PT-MGO stock) after the removal of DJ-1. Using the previously described quantitative HPLC-based assay, we found that the MGO concentration in supernatants in which no enzymes were added, but the 10 mM MGO solution was exposed to Ni-NTA resin led to supernatants with 8.29 ± 0.264 mM MGO, a 17.1% decrease from the original 10 mM stock. There was a somewhat larger loss of MGO (7.51 ± 0.240 mM, 24.9% decrease) after pretreatment with DJ-1C106A, likely due to the added influence of reversible nonspecific binding and/or glycation at some or all of the 25 possible glycation sites on DJ-1C106A itself. However, there was a dramatic reduction in MGO concentrations for supernatants that were previously treated with DJ-1WT (0.933 ± 0.256 mM, 90.7% decrease) (Figure 6C, middle).
Our next goal was to determine if the reduced MGO levels after DJ-1 treatment could be solely responsible for the observed decreases in glycation. To assess this, we prepared new stock solutions of MGO at the indicated concentrations, determined using the HPLC assay. We then performed peptide glycation reactions using these new MGO stocks. We found that these reactions produced identical levels of glycation compared to the ones performed using the pretreated supernatants (Figure 6C, right). This level of glycation was even less than what was observed during our original concurrent treatment condition (Figure 3C). This difference is likely because, during concurrent treatment with MGO and DJ-1, glycation competes with lactate formation during the reaction period. Taken together, these results demonstrate that DJ-1 glyoxalase, not deglycase, activity is solely responsible for decreasing glycation levels.
Conclusions
Here, we have demonstrated that DJ-1 is not a deglycase that erases AGEs from proteins. Instead, DJ-1 is a glyoxalase that prevents glycation by reducing MGO levels. While our conclusions differ from most studies that have evaluated DJ-1 as a potential deglycase, our findings are straightforward to reconcile with this prior work. Most prior studies have used conditions that match closely with our concurrent protocol.17,18,20−22,25,27−29,32−34 These include examples where the glycation reaction is started at an earlier time than the DJ-1 incubation, but free MGO remains unquenched when DJ-1 is added.17,20,21,27−29,34 Because MGO is a substrate for DJ-1, any attempt to measure “deglycase” activity without quenching free MGO will result in less glycation simply because there is less MGO available to react. In contrast, our work carefully included steps to remove excess, unbound MGO and quench glycation reactions prior to adding DJ-1. This allowed us to differentiate between a model in which DJ-1 could remove AGEs versus one in which it antagonizes AGE formation. Only one other report has described a subsequent DJ-1 treatment protocol similar to ours, which produced results consistent with our findings.32 Two other reports used buffer exchange via spin concentration to remove excess MGO, but subsequently evaluated only lactate formation33 or MGO consumption21—not protein or peptide glycation—in the presence of DJ-1. Based on the experimental details provided, which involved very high initial concentrations of MGO (5 mM33 or 100 mM21), it is likely that the observed lactate formation (measured by HPLC)33 or MGO consumption (measured by DNPH absorbance),21 was due to carryover free MGO and/or a spontaneous AGE reversal process that released free MGO (see Figure S6) and reproduced conditions more similar to our concurrent protocol.
Additionally, most prior studies of DJ-1 have been conducted in the presence of thiol-reducing agents that can amplify the apparent decreases in glycation by competing with AGE formation through an independent mechanism. In contrast, our work separately assessed the ability of thiols, like GSH or DTT, to interfere with glycation. As a result, our work clearly demonstrates that DJ-1 is not an AGE eraser and cannot reasonably be considered a deglycase. Despite this, it remains classified as such in most major databases. To avoid any confusion and aid in the correct interpretation of meaningful experimental results in the future, we echo previous calls20,27−29 to correct this misclassification.
Previous work has suggested that DJ-1 uses distinct mechanisms for its “deglycase” and glyoxalase activities. Many prior reports focus on a putative Cannizzaro-like glyoxalase mechanism involving a hydride shift that is postulated to occur stereospecifically and without any cofactors.20,26,34 However, there has been no experimental evidence obtained in support of this mechanism, and, to our knowledge, this type of transformation has not been observed in enzymes.35−39 While hydride transfers are frequently observed, these transformations required specialized cofactors like NADH.36−39 Instead, there is strong evidence that DJ-1 engages in a deprotonation/re-protonation mechanism that uses an enediol intermediate shared by all known glyoxalases.40,41 The observed stereoselectivity is easily reconciled by the presence of a planar enediol intermediate that is preferentially, but not exclusively, protonated on the re face.24
Furthermore, a conformational change associated with a more active form of the enzyme could enhance the observed stereoselectivity. Indeed, that is what we observed in the presence of glutathione, which we demonstrated to be an allosteric activator of DJ-1. Many prior studies have evaluated DJ-1 activity in the presence of glutathione;20,25,27−29,33 our results herein suggest that these may need to be reinterpreted in light of the finding that GSH augments DJ-1 activity. In particular, past kinetic studies may need to be revisited as allosteric enzymes do not obey traditional Michaelis–Menten kinetics and past work has typically co-varied equimolar concentrations of MGO and GSH.20,27,29 Additionally, recent work has suggested that DJ-1 has a distinct, but related, activity that protects metabolites and proteins from damage by 1,3-bisphoshoglycerate.42 It has also been reported to exhibit protease and esterase activities.43,44 Future studies that take these multiple activities into account will be helpful for understanding DJ-1’s biology.
Although DJ-1 is not a generalized AGE eraser, it still plays a valuable cellular role, helping mitigate glycolytic and metabolic stress in tandem with the glyoxalase system. Accordingly, DJ-1 activity can be up- or downregulated to protect functional proteins from glycation or to increase glycation-derived cellular damage in dysfunctional cells, similarly to Glo1.45 As a result, DJ-1 activity can be harnessed as a tool to study dysregulated metabolism and offer critical insight into how a nonenzymatic PTM, like glycation, can regulate cellular processes.
Acknowledgments
The authors thank C. Mace and J. Kritzer for helpful conversations regarding this work.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.biochem.3c00028.
Peptide and protein glycation under “concurrent” and “subsequent” treatments with DJ-1 or DTT, quenching and/or removal of MGO, lactate assay using d- or l-lactate dehydrogenase, quantitative HPLC assay used to measure MGO levels, and measurement of V0 and hemithioacetal levels using glutathione and β-mercaptoethanol (PDF)
This work was supported in part by NIH R01GM132422 and the Ashvin I. Patel, M.D., A84, M88 and Vidisha A. Patel, Ed.D. Summer Scholars fund.
The authors declare no competing financial interest.
Supplementary Material
References
- Duan G.; Walther D. The Roles of Post-Translational Modifications in the Context of Protein Interaction Networks. PLoS Comput Biol 2015, 11, e1004049 10.1371/journal.pcbi.1004049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dedola S.; Rugen M. D.; Young R. J.; Field R. A. Revisiting the Language of Glycoscience: Readers, Writers and Erasers in Carbohydrate Biochemistry. Chembiochem 2020, 21, 423–427. 10.1002/cbic.201900377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee M. J.; Yaffe M. B. Protein Regulation in Signal Transduction. Cold Spring Harb Perspect Biol 2016, 8, a005918 10.1101/cshperspect.a005918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabbani N.; Thornalley P. J. Methylglyoxal, Glyoxalase 1 and the Dicarbonyl Proteome. Amino Acids 2012, 42, 1133–1142. 10.1007/s00726-010-0783-0. [DOI] [PubMed] [Google Scholar]
- Harmel R.; Fiedler D. Features and Regulation of Non-Enzymatic Post-Translational Modifications. Nat. Chem. Biol. 2018, 14, 244–252. 10.1038/nchembio.2575. [DOI] [PubMed] [Google Scholar]
- Jaisson S.; Gillery P. Evaluation of Nonenzymatic Posttranslational Modification-Derived Products as Biomarkers of Molecular Aging of Proteins. Clin. Chem. 2010, 56, 1401–1412. 10.1373/clinchem.2010.145201. [DOI] [PubMed] [Google Scholar]
- McEwen J. M.; Fraser S.; Guir A. L. S.; Dave J.; Scheck R. A. Synergistic Sequence Contributions Bias Glycation Outcomes. Nat. Commun. 2021, 12, 3316 10.1038/s41467-021-23625-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stadtman E. R. Protein Oxidation and Aging. Free Radic. Res. 2006, 40, 1250–1258. 10.1080/10715760600918142. [DOI] [PubMed] [Google Scholar]
- Nandi S. K.; Nahomi R. B.; Rankenberg J.; Glomb M. A.; Nagaraj R. H. Glycation-Mediated Inter-Protein Cross-Linking Is Promoted by Chaperone-Client Complexes of Alpha-Crystallin: Implications for Lens Aging and Presbyopia. J. Biol. Chem. 2020, 295, 5701–5716. 10.1074/jbc.RA120.012604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iannuzzi C.; Irace G.; Sirangelo I. Differential Effects of Glycation on Protein Aggregation and Amyloid Formation. Front. Mol. Biosci. 2014, 1, 9. 10.3389/fmolb.2014.00009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes R. A.; Oliveira L. M.; Silva M.; Ascenso C.; Quintas A.; Costa G.; Coelho A. V.; Sousa Silva M.; Ferreira A. E.; Ponces Freire A.; Cordeiro C. Protein Glycation in Vivo: Functional and Structural Effects on Yeast Enolase. Biochem. J. 2008, 416, 317–326. 10.1042/BJ20080632. [DOI] [PubMed] [Google Scholar]
- Welsh K. J.; Kirkman M. S.; Sacks D. B. Role of Glycated Proteins in the Diagnosis and Management of Diabetes: Research Gaps and Future Directions. Diabetes Care 2016, 39, 1299–1306. 10.2337/dc15-2727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biemel K. M.; Friedl D. A.; Lederer M. O. Identification and Quantification of Major Maillard Cross-Links in Human Serum Albumin and Lens Protein. Evidence for Glucosepane as the Dominant Compound. J. Biol. Chem. 2002, 277, 24907–24915. 10.1074/jbc.M202681200. [DOI] [PubMed] [Google Scholar]
- Fritz G. RAGE: A Single Receptor Fits Multiple Ligands. Trends Biochem. Sci. 2011, 36, 625–632. 10.1016/j.tibs.2011.08.008. [DOI] [PubMed] [Google Scholar]
- Sparvero L. J.; Asafu-Adjei D.; Kang R.; Tang D.; Amin N.; Im J.; Rutledge R.; Lin B.; Amoscato A. A.; Zeh H. J.; Lotze M. T. RAGE (Receptor for Advanced Glycation Endproducts), RAGE Ligands, and Their Role in Cancer and Inflammation. J. Transl. Med. 2009, 7, 17. 10.1186/1479-5876-7-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie J.; Méndez J. D.; Méndez-Valenzuela V.; Aguilar-Hernández M. M. Cellular Signalling of the Receptor for Advanced Glycation End Products (RAGE). Cell. Signal. 2013, 25, 2185–2197. 10.1016/j.cellsig.2013.06.013. [DOI] [PubMed] [Google Scholar]
- Zheng Q.; Omans N. D.; Leicher R.; Osunsade A.; Agustinus A. S.; Finkin-Groner E.; D’Ambrosio H.; Liu B.; Chandarlapaty S.; Liu S.; David Y. Reversible Histone Glycation Is Associated with Disease-Related Changes in Chromatin Architecture. Nat. Commun. 2019, 10, 1289 10.1038/s41467-019-09192-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galligan J. J.; Wepy J. A.; Streeter M. D.; Kingsley P. J.; Mitchener M. M.; Wauchope O. R.; Beavers W. N.; Rose K. L.; Wang T.; Spiegel D. A.; Marnett L. J. Methylglyoxal-Derived Posttranslational Arginine Modifications Are Abundant Histone Marks. Proc. Natl. Acad. Sci. U.S.A. 2018, 115, 9228–9233. 10.1073/pnas.1802901115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bollong M. J.; Lee G.; Coukos J. S.; Yun H.; Zambaldo C.; Chang J. W.; Chin E. N.; Ahmad I.; Chatterjee A. K.; Lairson L. L.; Schultz P. G.; Moellering R. E. A Metabolite-Derived Protein Modification Integrates Glycolysis with KEAP1-NRF2 Signalling. Nature 2018, 562, 600–604. 10.1038/s41586-018-0622-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richarme G.; Mihoub M.; Dairou J.; Bui L. C.; Leger T.; Lamouri A. Parkinsonism-Associated Protein DJ-1/Park7 Is a Major Protein Deglycase That Repairs Methylglyoxal- and Glyoxal-Glycated Cysteine, Arginine, and Lysine Residues. J. Biol. Chem. 2015, 290, 1885–1897. 10.1074/jbc.M114.597815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma N.; Rao S. P.; Kalivendi S. V. The Deglycase Activity of DJ-1 Mitigates Alpha-Synuclein Glycation and Aggregation in Dopaminergic Cells: Role of Oxidative Stress Mediated Downregulation of DJ-1 in Parkinson’s Disease. Free Radic. Biol. Med. 2019, 135, 28–37. 10.1016/j.freeradbiomed.2019.02.014. [DOI] [PubMed] [Google Scholar]
- Richarme G.; Liu C.; Mihoub M.; Abdallah J.; Leger T.; Joly N.; Liebart J. C.; Jurkunas U. V.; Nadal M.; Bouloc P.; Dairou J.; Lamouri A. Guanine Glycation Repair by DJ-1/Park7 and Its Bacterial Homologs. Science 2017, 357, 208–211. 10.1126/science.aag1095. [DOI] [PubMed] [Google Scholar]
- Lee J. Y.; Song J.; Kwon K.; Jang S.; Kim C.; Baek K.; Kim J.; Park C. Human DJ-1 and Its Homologs Are Novel Glyoxalases. Hum. Mol. Genet. 2012, 21, 3215–3225. 10.1093/hmg/dds155. [DOI] [PubMed] [Google Scholar]
- Choi D.; Kim J.; Ha S.; Kwon K.; Kim E. H.; Lee H. Y.; Ryu K. S.; Park C. Stereospecific Mechanism of DJ-1 Glyoxalases Inferred from Their Hemithioacetal-Containing Crystal Structures. FEBS J. 2014, 281, 5447–5462. 10.1111/febs.13085. [DOI] [PubMed] [Google Scholar]
- Matsuda N.; Kimura M.; Queliconi B. B.; Kojima W.; Mishima M.; Takagi K.; Koyano F.; Yamano K.; Mizushima T.; Ito Y.; Tanaka K. Parkinson’s Disease-Related DJ-1 Functions in Thiol Quality Control against Aldehyde Attack in Vitro. Sci. Rep. 2017, 7, 12816 10.1038/s41598-017-13146-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jun Y. W.; Kool E. T. Small Substrate or Large? Debate Over the Mechanism of Glycation Adduct Repair by DJ-1. Cell Chem. Biol. 2020, 27, 1117–1123. 10.1016/j.chembiol.2020.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreeva A.; Bekkhozhin Z.; Omertassova N.; Baizhumanov T.; Yeltay G.; Akhmetali M.; Toibazar D.; Utepbergenov D. The Apparent Deglycase Activity of DJ-1 Results from the Conversion of Free Methylglyoxal Present in Fast Equilibrium with Hemithioacetals and Hemiaminals. J. Biol. Chem. 2019, 294, 18863–18872. 10.1074/jbc.RA119.011237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfaff D. H.; Fleming T.; Nawroth P.; Teleman A. A. Evidence Against a Role for the Parkinsonism-Associated Protein DJ-1 in Methylglyoxal Detoxification. J. Biol. Chem. 2017, 292, 685–690. 10.1074/jbc.M116.743823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazza M. C.; Shuck S. C.; Lin J.; Moxley M. A.; Termini J.; Cookson M. R.; Wilson M. A. DJ-1 Is Not a Deglycase and Makes a Modest Contribution to Cellular Defense against Methylglyoxal Damage in Neurons. J. Neurochem. 2022, 162, 245–261. 10.1111/jnc.15656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sjoblom N. M.; Kelsey M. M. G.; Scheck R. A. A Systematic Study of Selective Protein Glycation. Angew. Chem., Int. Ed. 2018, 57, 16077–16082. 10.1002/anie.201810037. [DOI] [PubMed] [Google Scholar]
- Smith N.; Wilson M. A.. Structural Biology of the DJ-1 Superfamily. In Advances in Experimental Medicine and Biology, 2017; Vol. 1037, pp 5–24. [DOI] [PubMed] [Google Scholar]
- Zheng Q.; Osunsade A.; David Y. Protein Arginine Deiminase 4 Antagonizes Methylglyoxal-Induced Histone Glycation. Nat. Commun. 2020, 11, 3241 10.1038/s41467-020-17066-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X.; Zhang N.; Hossain F.; Kandalai S.; Tian H.; Zheng Q. Biosynthesis of D/L-Lactate from Methylglyoxal. Tetrahedron 2022, 127, 133087 10.1016/j.tet.2022.133087. [DOI] [Google Scholar]
- Maksimovic I.; Finkin-Groner E.; Fukase Y.; Zheng Q.; Sun S.; Michino M.; Huggins D. J.; Myers R. W.; David Y. Deglycase-Activity Oriented Screening to Identify DJ-1 Inhibitors. RSC Med. Chem. 2021, 12, 1232–1238. 10.1039/D1MD00062D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin C.-I.; McCarty R. M.; Liu H. The Enzymology of Organic Transformations: A Survey of Name Reactions in Biological Systems. Angew. Chem., Int. Ed. 2017, 56, 3446–3489. 10.1002/anie.201603291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tassano E.; Hall M. Enzymatic Self-Sufficient Hydride Transfer Processes. Chem. Soc. Rev. 2019, 48, 5596–5615. 10.1039/C8CS00903A. [DOI] [PubMed] [Google Scholar]
- Whitlow M.; Howard A. J.; Finzel B. C.; Poulos T. L.; Winborne E.; Gilliland G. L. A Metal-Mediated Hydride Shift Mechanism for Xylose Isomerase Based on the 1.6 A Streptomyces Rubiginosus Structures with Xylitol and D-Xylose. Proteins 1991, 9, 153–173. 10.1002/prot.340090302. [DOI] [PubMed] [Google Scholar]
- Willistein M.; Bechtel D. F.; Muller C. S.; Demmer U.; Heimann L.; Kayastha K.; Schunemann V.; Pierik A. J.; Ullmann G. M.; Ermler U.; Boll M. Low Potential Enzymatic Hydride Transfer via Highly Cooperative and Inversely Functionalized Flavin Cofactors. Nat. Commun. 2019, 10, 2074 10.1038/s41467-019-10078-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji P.; Park J.; Gu Y.; Clark D. S.; Hartwig J. F. Abiotic Reduction of Ketones with Silanes Catalysed by Carbonic Anhydrase through an Enzymatic Zinc Hydride. Nat. Chem. 2021, 13, 312–318. 10.1038/s41557-020-00633-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chari R. V.; Kozarich J. W. Deuterium Isotope Effects on the Product Partitioning of Fluoromethylglyoxal by Glyoxalase I. Proof of a Proton Transfer Mechanism. J. Biol. Chem. 1981, 256, 9785–9788. 10.1016/S0021-9258(19)68690-4. [DOI] [PubMed] [Google Scholar]
- Hall S. S.; Doweyko A. M.; Jordan F. Glyoxalase I Enzyme Studies. 2. Nuclear Magnetic Resonance Evidence for an Enediol-Proton Transfer Mechanism. J. Am. Chem. Soc. 1976, 98, 7460–7461. 10.1021/ja00439a077. [DOI] [PubMed] [Google Scholar]
- Heremans I. P.; Caligiore F.; Gerin I.; Bury M.; Lutz M.; Graff J.; Stroobant V.; Vertommen D.; Teleman A. A.; Van Schaftingen E.; Bommer G. T. Parkinson’s Disease Protein PARK7 Prevents Metabolite and Protein Damage Caused by a Glycolytic Metabolite. Proc. Natl. Acad. Sci. U.S.A. 2022, 119, e2111338119 10.1073/pnas.2111338119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitsugi H.; Niki T.; Takahashi-Niki K.; Tanimura K.; Yoshizawa-Kumagaye K.; Tsunemi M.; Iguchi-Ariga S. M. M.; Ariga H. Identification of the Recognition Sequence and Target Proteins for DJ-1 Protease. FEBS Lett. 2013, 587, 2493–2499. 10.1016/j.febslet.2013.06.032. [DOI] [PubMed] [Google Scholar]
- Vázquez-Mayorga E.; Díaz-Sánchez Á. G.; Dagda R. K.; Domínguez-Solís C. A.; Dagda R. Y.; Coronado-Ramírez C. K.; Martínez-Martínez A. Novel Redox-Dependent Esterase Activity (EC 3.1.1.2) for DJ-1: Implications for Parkinson’s Disease. Int. J. Mol. Sci. 2016, 17, 1346. 10.3390/ijms17081346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabbani N.; Thornalley P. J. Emerging Glycation-Based Therapeutics-Glyoxalase 1 Inducers and Glyoxalase 1 Inhibitors. Int. J. Mol. Sci. 2022, 23, 2453. 10.3390/ijms23052453. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.