

Instructions for ClinVar submission spreadsheets
This page provide general information about filling in ClinVar's submission spreadsheet. The guidance here also applies to the fields that are available to submit in the online wizard and the submission API.
Not every column is described here; instructions are also included in each column in the spreadsheet itself.
- Checklist for faster processing
- Variants
- Condition
- Classification
- Evidence
- Citations
- Merge submissions
- Delete submissions
- Columns added by ClinVar
Checklist for faster processing
Help your ClinVar curator process your submission faster!
- Provide data in all required fields
- Validate your HGVS expressions with VariantValidator or Mutalyzer
- Do not modify the column headers
- Do not modify the cell validation; use allowed values when there is a list
- Check the instructions for each column on the spreadsheet for correct format and separators for a list
Variants
ClinVar welcomes submissions of variants classified for diseases and drug responses. We offer the following guidance:
- if each variant has been classified independently, please submit the variants separately to ClinVar. Distinct classifications for each variant are more useful to those using data in ClinVar.
- For example, if you observed two variants in compound heterozygosity and you determined that they are pathogenic for an autosomal recessive disease, submit each variant on a separate row as a pathogenic variant for that disease.
- The submission for each variant can note that it was observed with the other variant and the mode of inheritance.
- if multiple variants have been classified together because you cannot classify them independently, submit them on one row as the appropriate combination, either as a haplotype or as a compound heterozygote.
- if you are submitting a variant that was observed in a homozygote, do not submit the variant as an HGVS expression for the homozygote, e.g. c.[105C>A ]; [105C>A ]
- submit the single variant c.105C>A
- provide the classification based on the single variant's contribution to disease
- fill in the column "mode of inheritance" if possible
- if you provide aggregate evidence on the Variant tab, fill in the column "Number of homozygotes"
- if you provide individual evidence on the CaseData tab, enter "homozygote" in the column "Zygosity"
Sequence variants
HGVS expressions
- Check that your HGVS expressions are valid with VariantValidator or Mutalyzer.
- On the lite spreadsheet template, enter the HGVS expression in the 'HGVS' name column.
- On the full spreadsheet template, enter the accession.version number in the 'Reference sequence' column and the c./g. portion of the HGVS expression in the 'HGVS' column.
- We only accept NCBI RefSeq accession numbers as the reference sequence due to technical constraints (namely, that we do not have alignment datasets for GenBank accessions).
- Do not include the p. HGVS expression in these columns. It may be provided in the 'Alternate designations' column instead.
- If you have information on multiple nucleotide changes that result in the same protein change, submit each nucleotide change on a separate row.
- Spreadsheets with examples of valid HGVS expressions that ClinVar accepts and invalid HGVS expressions and corresponding error messages are available.
Chromosome coordinates
- Use the full spreadsheet template (SubmissionTemplate.xlsx).
- Note that you can delete any columns that are not required by ClinVar to make the spreadsheet simpler to use
- Provide the chromosome in the 'Chromosome' column.
- Provide the first and last positions of the variant in the 'Start' and 'Stop' columns.
- For variants of 15 or fewer nt, provide the reference and alternate alleles in the 'Reference allele' and 'Alternate allele' columns.
- Use VCF-style notation with an anchor nucleotide, e.g. ref AT alt T
- Do not fill in the 'Variant type' column; we will calculate this for you.
- For variants of more than 15 nt, fill in the 'Variant type' column.
- Do not fill in the 'Reference allele' and 'Alternate allele' columns.
- Spreadsheets with examples of valid cases with chromosome coordinates that ClinVar accepts and invalid cases and corresponding error messages are available.
CNVs/Structural variants
- Use the full spreadsheet template (SubmissionTemplate.xlsx).
- Note that you can delete any columns that are not required by ClinVar to make the spreadsheet simpler to use.
- Provide the chromosome in the 'Chromosome' column.
- Provide the type of variant in the 'Variant type' column.
- For deletions and duplications that are detected by array, use "copy number loss" and "copy number gain", rather than "deletion" and "duplication".
- If the exact coordinates (to basepair resolution) of the variant call are known, fill in the 'Start' and 'Stop' columns.
- If only the minimal region is known, use inner_start and inner_stop.
- If only the maximum region is known, use outer_start and outer_stop.
- Otherwise, use outer_start (lower value) and inner_start (upper value) to define the interval in which the call begins. Likewise, use inner_stop (lower value) and outer_stop (upper value) to define the interval in which the call ends.
- Provide the observed copy number in the 'Copy number' column
- Provide the expected copy number in the 'Reference copy number' column
Other considerations for CNVs/structural variants:
- a CNV or structural variant is often classified but not for a specific disease. You have a few options:
- You may enter "not provided" as the 'Preferred condition name'
- If you are also providing observed phenotypes in the 'Clinical features' column, you may enter "See cases" as the 'Preferred condition name' so that users know there is no condition for the classification but there is phentoype information about the case
- See the Condition section for more options.
- You may indicate the gene(s) affected by the variant, but it is not required.
- ClinVar will calculate the genes that are affected by the variant.
- ClinVar also displays the results of ClinGen's Dosage Sensitivity curation to flag genes that are known to cause a phenotype when there is a loss or gain of one copy of the gene.
Somatic variants
- Use the somatic spreadsheet template (SubmissionTemplateSomatic.xlsx)
- Define the variant
- Note: fusions are not in scope for submission to ClinVar at this time
- Enter the tumor type for the classification
- On each row, enter either a classification for somatic clinical impact or oncogenicity
- You can submit multiple classifications for the same variant, or same variant-tumor pair but they will be distinct submitted records (SCVs) in ClinVar so should be submitted on distinct rows in the spreadsheet
- For a classification of clinical impact
- Enter the appropriate tier for the evidence in the column "Somatic classification of clinical impact"
- Enter the type of assertion of clinical impact (e.g. therapeutic: resistance) for the variant in the column "Assertion type for clinical impact"
- If the assertion is for therapeutic impact, enter the drug in the column "Drug relevant for therapeutic assertion of clinical impact"
- For a classification of oncogenicity
- Enter the appropriate term in the column "Oncogenicity classification"
- For either clinical impact or oncogenicity, fill in columns for "Date last evaluated", "Classification citations", and "Comment on classification" if possible
- Enter the evidence for your classification
- If you submit classifications for both clinical impact and for oncogenicity and you provide assertion criteria, you likely need to submit the assertions of clinical impact in one spreadsheet and the assertions of oncogenicity in another
- Assertion criteria apply to all of the records with a spreadsheet, and it is likely that your classification rules are different for clinical impact vs oncogenicity, particularly if you are citing the PubMed IDs for the AMP/ASCO/CAP and ClinGen/CGC/VICC recommendations
Pharma variants
- Use the full spreadsheet template (SubmissionTemplate.xlsx)
- If the variant is a haplotype, provide the set of variants in the haplotype as an HGVS expression on a single row on the Variant tab
- Enter "drug response" for 'Germline classification'
- In 'Explanation if classification is other or drug response' provide a short description of the type of response, such as "poor metabolizer" or "likely responsive"
- Enter the condition for which the drug is relevant in the column “Drug response condition”
- if you want to provide your classification of a variant for a disease and also describe its effect on a drug response, provide that information as two rows on the Variant tab
- one row for the classification of pathogenicity for the disease
- a second row for the classification of a drug response
Haplotypes
Rarely, a classification may be submitted to ClinVar for a set of variants in cis, i.e. a haplotype.
- This should be done only if the combination of variants is important for the classification. For example, some star alleles for pharma variants are defined by several SNPs observed in cis.
- A classification for the haplotype is not necessary when the haplotype exists because a clinically important variant is always seen in combination with another variant, e.g. due to a founder effect. In that case, submit your classification of the clinically important variant.
Genotypes
Rarely, a classification for a genotype may be submitted to ClinVar. However, ClinVar is a variant-level database, not a case-level (or patient) database; thus a classification should be provided for each individual variant whenever possible.
- if you observed two variants in compound heterozygosity and you determined that they are pathogenic for an autosomal recessive disease, submit each variant separately as a pathogenic variant for that disease. Distinct classifications for each variant are more useful to those using data in ClinVar. The submission for each variant can note that it was observed with the other variant and the mode of inheritance should be indicated.
- if you observed a single variant in homozogosity, do not submit the variant as an HGVS expression for the homozygote, e.g. c.[105C>A ]; [105C>A ]. Instead, submit the single variant, e.g. c.105C>A, and provide the zygosity for the individual.
There are a few cases where a classification for a genotype is appropriate for submission to ClinVar:
- some practice guidelines are written based on the combination of variants seen in an individual. When the professional society who provides the practice guideline submits data to ClinVar, it may describe the genotype for each classification.
- in some cases, a combination of variants in trans causes a different phenotype than expected. For example, two pathogenic CFTR variants in trans may be expected to cause cystic fibrosis, but they may actually cause a less severe phenotype such as congenital bilateral absence of the vas deferens.
Autoclassified variants
If submitting a variant that was autoclassified as benign:
- in the “Comment on classification” field, note that the variant was autoclassified and filtered, not subjected to a comprehensive review
- do not provide assertion criteria for autoclassified variants; assertion criteria imply that the variant was subjected to a comprehensive review
Variation identifiers
Optionally, you can provide an identifier used for the variant in one of the following databases: OMIM (allelic variant ID), dbSNP (rs number), or dbVar (submitter or region identifier). Use the format database_name:database_identifier. For example:
- OMIM:611101.0001
- dbSNP:rs104894321
- dbVar:nsv491743 or dbVar:essv12345
Alternate designations
Optionally, you can provide other names for the variant. This is most important for variants that have legacy names that cannot be automatically calculated from current data, such as "deltaF508" or names that use old numbering systems. While you can provide other valid HGVS expressions in this column, such as the p. description, it is not necessary because ClinVar calculates other valid HGVS expressions for genomic DNA, cDNA, and protein automatically. Examples of useful alternate names include:
- deltaF508 - common name for NM_000492.3:c.1521_1523delCTT
- Z allele - common name for NM_001127701.1:c.1096G>A
- 3120G->A - legacy name for NM_000492.3:c.2988G>A
Condition
Variants in ClinVar are generally classified for a disease or a drug response, which ClinVar generically calls “condition”. Here are recommendations for how to provide the condition in various scenarios.
A general rule is that you can submit a condition as a database identifier (like an OMIM ID) or as a name, but not both.
Submission fields are named similarly in file, wizard, and API submissions, so these recommendations can guide all methods of submission.
The images below are from ClinVar’s full submission template, version 4.4. Some columns in the spreadsheet may not be shown in the images for display purposes.
For germline variants
- Classified for a known genetic disease
- Classified for a novel genetic disease
- Classified for several related diseases
- Classified for a combination of distinct diseases
- Classified for no specific disease because the variant is benign, likely benign or of unknown significance
- Classified but without a defined disease
- Interpreted for my patient’s phenotype
- Indication for testing
- Classified for its effect on a drug response
- Characterized by a functional assay
How to submit a germline variant classified for a known genetic disease
This scenario is common in ClinVar.
- Use a database identifier
- Enter the disease database as “Condition ID Type”, one of OMIM, MeSH, MedGen, Mondo, or Orphanet
- Enter the identifier in that database as “Condition ID value”
- OR if there is no database identifier for the disease, use a disease name
- Enter the disease name as “Preferred condition name”
- We strongly encourage the use of database identifiers whenever possible.

How to submit a germline variant classified for a novel genetic disease
A variant may be classified for a new disease that is not yet represented in a disease database like OMIM. This scenario is likely for a research laboratory that submits the variant and the novel disease to ClinVar while also publishing the data in a journal.
- If you have defined a name for the novel disease, enter the name as “Preferred condition name”
- If there is no name and the disease is defined by a set of clinical features
- Enter the set of clinical features as HPO IDs (“Condition ID Type” and “Condition ID value”) on a single row
- Enter “novel disease” as the “Explanation for multiple conditions”
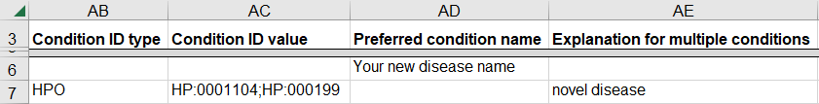
How to submit a germline variant classified for several related diseases
A variant may be classified for several related diseases, possibly a spectrum of disease, and you may be uncertain if the variant is associated with only one or more of the diseases. This scenario is common for clinical testing laboratories that consider all diseases that have an established relationship with the gene for the variant.
- Use a single row on the spreadsheet for the set of diseases
- Enter a single disease database as “Condition ID Type”, one of OMIM, MeSH, MedGen, Mondo, or Orphanet
- Enter the identifier in that database for each disease as “Condition ID value”
- Enter “uncertain” as the “Explanation for multiple conditions”
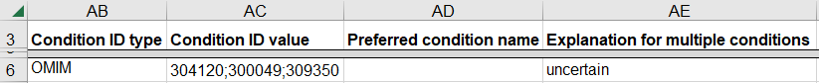
How to submit a germline variant classified for a combination of distinct diseases
This indicates that the variant causes a combination of distinct diseases in the same individual; this scenario is expected to be rare in ClinVar.
- Use a single row on the spreadsheet for the set of diseases
- Enter a single disease database as “Condition ID Type”, one of OMIM, MeSH, MedGen, Mondo, or Orphanet
- Enter the identifier in that database for each disease as “Condition ID value”
- Enter “co-occurring” as the “Explanation for multiple conditions”
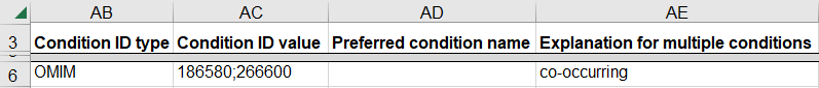
How to submit a germline variant classified for no specific disease because the variant is benign, likely benign or of unknown significance
Some clinical testing laboratories do not provide a disease for the classification to indicate that the variant is considered benign, likely benign or of unknown significance for Mendelian diseases in general. This is accepted in a ClinVar submission but it’s preferable to submit a disease name so that ClinVar users understand the context of the variant’s classification.
- Enter “not specified” as the “Preferred condition name”
How to submit a germline variant without a disease
Most submissions to ClinVar should indicate the disease for the classification. There are a few cases where this is not possible:
- A non-recurrent copy number variant (CNV) may be evaluated in an individual with a unique phenotype, as opposed to a recurrent CNV that is associated with a named syndrome like DiGeorge syndrome.
- Enter “See cases” as the “Preferred condition name”. Although not literally the name of a disease, this will direct ClinVar users to look at the clinical features provided as part of case-level data.
- Enter the phenotypes observed in the patient as “Clinical features”
- Some clinical laboratories do not store disease information at the level of the variant, making it difficult to provide it in ClinVar submissions.
- Enter “not provided” as the “Preferred condition name”
- For ClinVar users, it is preferable to provide a disease name. Contact us at [email protected] if we can help you develop an approach to include disease names in your submissions, rather than “not provided”.
- A variant in a gene of uncertain significance has no disease associated with the variant, so there is no condition for the classification to submit to ClinVar.
- If you can submit phenotype data for the patient
- Enter “See cases” as the “Preferred condition name”. Although not literally the name of a disease, this will direct ClinVar users to look at the clinical features provided as part of case-level data.
- Enter the phenotypes observed in the patient as “Clinical features”
- If you cannot submit phenotype data for the patient
- Enter “not provided” as the “Preferred condition name”
- If you can submit phenotype data for the patient
If you have another scenario where you are unable to provide a condition for the classification, contact us at [email protected] so that we can help develop a solution for your submission.
How to submit a germline variant interpreted for my patient’s phenotype
Submissions to ClinVar should be a variant-level classification for a disease, not the interpretation of a variant for a patient’s specific phenotype. This distinction is explained in more detail in the ACMG/AMP guidelines for the interpretation of sequence variants and the ACMG/ClinGen standards for interpretation of constitutional copy number variants.
However, phenotypic data is very valuable to ClinVar users so if you have it, we encourage you to submit it as follows. This scenario is common for clinical researchers, clinicians, and patient registries.
- Provide the disease for the classification
- Use a database identifier
- Enter the disease database as “Condition ID Type”, one of OMIM, MeSH, MedGen, Mondo, or Orphanet
- Enter the identifier in that database as “Condition ID value”
- OR if there is no database identifier for the disease, use a disease name
- Enter the disease name as “Preferred condition name”
- We strongly encourage the use of database identifiers whenever possible
- Use a database identifier
- Enter your patient’s phenotypes as “Clinical features”
- The phenotypes should be entered as a list of HPO identifiers or a list of features, not free text
- If you also have a free text comment to describe the patient’s phenotype, enter it as “Comment on clinical features"
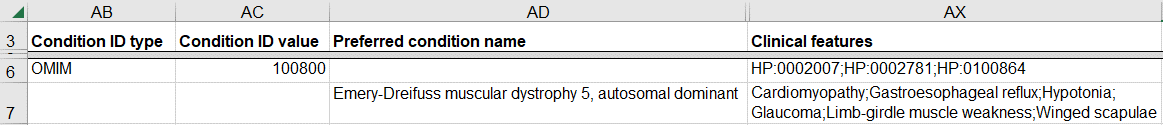
How to provide the indication for testing
Indication for testing is requested in submissions from clinical laboratories, which typically cannot provide any observed clinical features for the patient but can submit the indication for testing from the test requisition form.
If you do not know the patient’s actual phenotype but you do know the indication for testing:
- Provide the disease for the classification as described in How to submit a germline variant classified for a known genetic disease
- Enter the indication for testing as “Indication for testing”
If you do know the phenotype of the individual with the variant, then indication for testing is not requested. This scenario is common for clinical researchers, clinicians, and patient registries. In this case, it is preferable to:
- Indicate whether the individual with the variant was affected with the disease for the classification by entering “yes” for “Affected status”
- Enter observed clinical features as described in How to submit a germline variant interpreted for my patient’s phenotype
How to submit a germline variant classified for its effect on a drug response
For pharmacogenomic variants in ClinVar, the disease for the classification is the response to a drug instead of a disease.
- Provide the drug response for the classification using either a database identifier or a name for the drug response.
- Use a database identifier from MedGen
- Enter the database “MedGen” as “Condition ID type”
- Enter the MedGen identifier as “Condition ID value”
- OR if there is no MedGen identifier for the drug response, use a name for the drug response
- Enter the name as “Preferred condition name”
- Use the format “drug_name response”, e.g. warfarin response
- Use a database identifier from MedGen
- Enter “drug response” for “Clinical significance”
- Enter an “Explanation if clinical significance is other or drug response”, e.g. poor metabolizer. This is the term you would use on a lab report to describe the variant’s effect on drug response.
- Enter the condition that the drug is prescribed for as “Drug response condition”

How to submit a germline variant characterized by a functional assay
Functional data may be submitted to support a variant classification for a disease or drug response. For that case, submit the disease or drug response as described above.
If you are submitting only functional data with no classification for a disease, follow our instructions for submitting functional evidence.
For somatic variants
- Use a database identifier
- Enter the disease database as “Condition ID Type”, one of OMIM, MeSH, MedGen, Mondo, or Orphanet
- Enter the identifier in that database as “Condition ID value”
- OR if there is no database identifier for the tumor, use a name
- Enter the name of the tumor type as “Preferred condition name”
Classification
If you do not see an appropriate option for the germline classsification, e.g. for a pseudodeficiency allele
- Submit "other" as the classification
- Provide your value of classification as "Explanation if classification is other or drug response"
- You may also provide more information in the "Comment on classification" explaining how and why your laboratory reports the variant.
Assertion score
Submitters are encouraged to provide documentation describing the criteria they use to classify variants, referred to as assertion criteria. If your assertion criteria include a point-based scoring system, the final score, or point value, may be submitted as the Assertion score (Variant tab). The ACMG/ClinGen CNV Guidelines, 2019 is an example of a point-based scoring system for variant classification. If Assertion score is provided, assertion criteria must also be provided for the submission.
Assertion criteria and review status
We ask submitters to provide documentation describing the criteria they use to classify variants, which we call "assertion criteria".
- Read the requirements for assertion criteria.
- If you have questions about assertion criteria, contact us.
How to meet the requirements for the "criteria provider, single submitter" review status
Assertion criteria is provided through the ClinVar Submission Portal as either:
- a citation ID (PubMed ID, PubMedCentral ID, Bookshelf, or DOI) or
- an electronic document (a Word document or PDF)
See How to provide assertion criteria in your submission
A submission must reference one set of assertion criteria only, and these criteria must apply to every variant classification in the submission.
A review status of "criteria provided, single submitter" requires this documentation AND either supporting evidence for each classification OR a public contact for your organization. Supporting evidence in the submission includes:
- One or more of these columns on the Variant tab:
- comment on classification
- citations on classification
- citations or URLs that cannot be represented in classification citations column
- comment on evidence
- citations on evidence
- number of individuals with variant
- number of families with variant
- number of families with segregation observed
- number of homozygotes
- number of heterozygotes
- number of compound heterozygotes
- number of hemizygotes
- evidence citations
- OR data on the CaseData tab
Evidence
ClinVar requires that you provide some evidence for your variant classification.
The evidence is provided as observations, one of:
- individuals with the variant described in aggregate
- each case, or individual, with the variant described separately
- functional/experimental evidence
Aggregate observations
You can provide a single row on the Variant tab that represents your classification and all individuals in whom you observed the variant.
- only provide data in a column if it applies to everyone in the group, for example:
- provide an age range rather than one specific age
- "mixed gender group" if the group includes males and females
- only provide ethnicity if all individuals in the group are of the same ethnicity
- only provide zygosity if all individuals in the group have the same zygosity
Case observations
If you are able to submit information about each case in whom the variant was observed:
- Provide the variant classification on the Variant tab
- Fill in one row per variant/condition interpretation
- Provide a value for LinkingID; this represents your variant/condition classfication
- Do not fill in the columns in the sections for "Details of test and individuals tested", "Details of testing results", or "Methods"
- Provide your observations on the CaseData tab
- Each case that supports the variant/condition classification has its own row on CaseData, so there may be one or multiple CaseData rows for each Variant row
- The LinkingID links one row on the Variant tab to one or multiple rows on the CaseData tab
- One case may be described multiple times on CaseData. e.g. if you submit distinct classifications for both variants from a compound heterozygous individual:
- each variant is provided on a separate row on the Variant tab
- the case data is provided on two rows on the CaseData tab, once for each variant classification, represented by the LinkingID
Functional evidence
Research laboratories may generate functional data to support the functional impact, or consequence, of a variant. This type of evidence is submitted on the full spreadsheet template on the FunctionalEvidence tab.
This tab is to be used *only* to submit your own research results. If you are submitting classifications from another method such as clinical testing and you would like to note that there is functional evidence for the classification, please include that evidence on the Variant or CaseData tab, as appropriate, as one or more citations or as a comment.
- Use the full submission template (SubmissionTemplate.xlsx)
- On the Variant tab
- Define the variant , as described above.
- Provide a LinkingID to link this row on the Variant tab to one or more rows on the FunctionalEvidence tab
- If you are describing the functional consequence of the variant, but not a classification for a disease or a drug response:
- Enter "not provided" for 'Preferred condition name'
- Alternatively, you may provide a disease name if there is a clear gene-disease relationship that prompted the development of your functional assay
- Enter "not provided" for 'Classification'
- Enter a value for 'Functional consequence' based on your experimental results
- Optionally you may enter a 'Comment on functional consequence' to provide more details
- Enter "not provided" for 'Preferred condition name'
- On the FunctionalEvidence tab
- Enter each experimental observation on a separate row
- Multiple experiments may be submitted for the same variant classification
- Use the LinkingID to link multiple experiments on this tab to the corresponding row on the Variant tab
- Enter the appropriate 'Collection method'
- Enter the appropriate 'Allele origin'; "not applicable" may be the best option
- Enter the appropriate 'Affected status'; "not applicable" may be the best option
- Enter a summary description of the functional assay in the 'Method' column, including any scale or scoring system used, and the standard error of the assay if appropriate
- Enter the result of the assay for the specific variant in the 'Result' column
- This may be a numerical value or a short term like "non-functional", depending on the output of the assay
- A longer description of the effect should be provided in the 'Comment on functional consequence', described above
- Cite a publication that describes the assay in the 'Methods citations' column, when available
- Enter each experimental observation on a separate row
Collection method
Collection method describes the setting in which the variant classification is made.
It's important for users of ClinVar data to understand if it was collected:
- as part of clinical testing with very standardized classification
- as part of research where the classification may be standardized or more experimental
- from the literature which may be out of date
| Collection method | Use |
|---|---|
| clinical testing | For variants that were classified as part of clinical genetic testing, or as part of a large volume research study in which results compliant with CLIA, ISO, GLP, or an equivalent accreditation body are routinely returned to research subjects. Classification may be guided from the literature, but the number of individuals tested are reported only from the direct testing. |
| research | For variants that were classified as part of a research project but results are not routinely returned to research subjects and do not meet the requirements for clinical testing above. This is a general term to use when other more specific methods to not apply. |
| case-control | For variants gathered in a research setting but results are not routinely returned to research subjects and do not meet the requirements for clinical testing above. This term is for research projects specifically to compare alleles observed in cases and controls (without data about segregation). |
| in vitro |
For variants that were observed as part of an in vitro research project, such as experiments performed in cell culture, but results are not routinely returned to research subjects and do not meet the requirements for clinical testing above. This value is only used on the full spreadsheet template, on the FunctionalEvidence tab for experimental evidence. |
| in vivo |
For variants that were observed as part of an in vivo research project, such as a mouse model, but results are not routinely returned to research subjects and do not meet the requirements for clinical testing above. This value is only used on the full spreadsheet template, on the FunctionalEvidence tab for experimental evidence. |
| reference population | For variants gathered in a research setting but results are not routinely returned to research subjects and do not meet the requirements for clinical testing above. This term is used for baseline studies of a population group of apparently unaffected individuals to assess allele frequencies. |
| provider interpretation | For variants that were classified by a clinician, and for variants submitted by clinicians or researchers who are reinterpreting clinical test results. |
| phenotyping only | For variants that are submitted to ClinVar to provide individual observations with detailed phenotype data, such as submissions from clinicians or patient registries, without a variant classification from the submitter. The classification from the testing laboratory may be provided in a separate field. |
| literature only |
For variants extracted from published literature with or without a classification as reported in the citation. No additional curation has been performed by the submitter; the classification, if provided, is from the publication(s) only. This method is used by third parties, not the authors of the paper. To report results from your own paper, use one of the other collection methods, such as "research". |
| curation | For variants that were not directly observed by the submitter, but were classified by curation of multiple sources, including clinical testing laboratory reports, publications, private case data, and public databases. |
Allele Origin
Allele origin may generally describe whether the variant was germline or somatic. Alternatively, it may be a more specific description of germline, if you are providing case data or if it is part of your aggregate data.
- allowed values for allele origin are listed in the table below
- for some display and search purposes, submissions with allele origins other than "somatic" are grouped into "germline", but ClinVar retains the specific term you provide on your submission
|
Allele origin |
Use |
Grouping for search and display |
|
Germline |
Any germline variant |
germline |
|
Somatic |
Any somatic variant |
somatic |
|
De novo |
A germline variant that was not inherited |
germline |
|
Maternal |
A germline variant inherited from the mother |
germline |
|
Paternal |
A germline variant inherited from the father |
germline |
|
Inherited |
A germline variant that was inherited |
germline |
|
Unknown |
Allele origin is unknown |
germline |
|
Biparental |
A variant present on both chromosomes and inherited from both parents. |
germline |
Affected status
Affected status refers to the condition for the classification, i.e.
- whether all the individuals in that aggregate are affected or unaffected by the condition for the classification
- whether each case is affected or unaffected by the condition for the classification
- if you do not know the individuals are affected by that specific condition, provide "unknown" for affected status
Note that you can also provide information about the clinical features that were observed in individuals, whether or not they relate to the condition for the classification. See the Condition section for how to provide clinical features.
Control data
ClinVar does accept control data, for example, data from a specific ethnic group for variants that are reported elsewhere to be pathogenic.
- Provide "no" for 'Affected status'
- Provide "case-control" or "reference population" for 'Collection method'
- Provide descriptive information for the individuals in the control group, e.g. 'Population Group/Ethnicity'
Citations
Citations from PubMed, PubMed Central and Bookshelf, or a DOI, may be added in several fields. For example, you can add citations that you consulted while researching the classification of the variant in the column "Classification citations". Examples of each format, i.e. database source:identifier, include:
- PMID:123456
- PMCID:PMC3385229
- NBK:1535
- doi:10.21037/atm-21-2913
Merge submissions
Rarely you may find that you have more than one SCV record for the same variant and condition. For example, you may have submitted the same variant for a disease and for "not provided" as the condition.
To merge records, submit an update:
- Use the full submission template (SubmissionTemplate.xlsx) or the somatic template (SubmissionTemplateSomatic.xlsx)
- Include all the data that you would like to retain on the resulting record
- On the Variant tab
- enter the accession number that should be retained in the 'ClinVarAccession' column
- in the 'Novel or update' column, enter "update"
- in the 'Replaces ClinVarAccessions' column, enter the other accession number(s) that will not be retained. Note that this accession number will no longer be displayed but it will be searchable in ClinVar.
Delete submissions
Rarely you may need to delete an SCV record. For example, you may determine that the variant itself was miscalled. When possible, you should update your record (e.g. to change the classification, the condition, etc.) or merge it into a redundant record instead of deleting.
Note that deleting your record means that it is removed from the web display and from future ClinVar files (e.g. XML and VCF files). However, it remains archived in past files and it is retrievable on the web if a user queries with the appropriate VCV or RCV accession and version number.
To delete a submission:
- Use the full submission template (SubmissionTemplate.xlsx) or the somatic template (SubmissionTemplateSomatic.xlsx)
- Use the Deletes tab
- Indicate the SCV accession to be deleted in the ClinVarAccession column
- You have the option to provide a comment for public display indicating why the record is being deleted
Columns added by ClinVar
Some columns of data are added to the submission spreadsheet during pre-submission validation.
Error
The error column is added so that the submitter knows which rows have errors and cannot be processed by ClinVar. Contact us at [email protected] if you receive an error message that you don’t understand or know how to resolve.
ncbi_cluster_key
The ncbi_cluster_key column is added so that it can be used in ClinVar’s downstream processing. You may see this column in the spreadsheet that we return to you if you validate and stop the process to fix all errors before submission. You do not need to do anything with the data in this column.