Continuing Education Activity
Amyloidosis is a heterogeneous acquired or hereditary disease that results from the abnormal deposition of beta-sheet fibrillar protein aggregates in various tissues. This disease can be localized or systemic with amyloid accumulating in the spleen, liver, kidney, blood vessels and nerves. This activity illustrates the evaluation and management of amyloidosis and reviews the role of the interprofessional team in improving care for patients with this condition.
Objectives:
Describe the etiology of amyloidosis.
Review the pathophysiology of amyloidosis.
Summarize the treatment of amyloidosis using the Standard Mayo Clinic staging system.
Identify the importance of improving care coordination among the interprofessional team to improve outcomes for those affected by amyloidosis.
Access free multiple choice questions on this topic.
Introduction
Amyloidosis is a heterogeneous disease that results from the deposition of toxic insoluble beta-sheet fibrillar protein aggregates in different tissues. Amyloidosis can be acquired or hereditary. The disease can be localized or systemic. Amyloid can accumulate in the liver, spleen, kidney, heart, nerves, and blood vessels, causing different clinical syndromes, including cardiomyopathy, hepatomegaly, proteinuria, macroglossia, autonomic dysfunction, ecchymoses, neuropathy, renal failure, hypertension, and corneal and vitreous abnormalities.[1][2][3]
Previously multiple classification systems have been devised to classify different types of amyloidosis. In modern times it has been classified according to the chemical analysis of amyloid clinical entities. Amyloidosis can be classified according to systemic, hereditary, central nervous system, ocular, and localized etiology. However, the most common types are AL, AA, ATTR (amyloid transport protein transthyretin), and dialysis-related amyloidosis (beta2M type).
In AL amyloidosis, 'A' represents amyloid followed by the associated fibrillar protein, 'L' means light chain fragment or immunoglobulin light chain. In AA amyloidosis, the second A stands for the serum amyloid A protein.[4][5]
Etiology
The most common causes of amyloidosis are the immunoglobulin-light-chain relate amyloidosis (AL), ATTR amyloidosis, and reactive amyloidosis (AA) due to chronic inflammatory diseases like chronic infections and rheumatoid arthritis. AL amyloidosis is acquired and is caused by a small plasma cell clone that produces misfolded amyloidogenic light chains that deposit in different organs and tissues.[6][7]
AA amyloidosis is associated with various chronic inflammatory conditions, chronic or local microbial infections, and rarely with neoplasms. AA is the most common type of systemic amyloidosis. However, its incidence varies in different ethnic groups.[8]
Epidemiology
AL amyloidosis has an incidence of 1 case per 100,000 person-years in Western countries. In the United States, there are approximately 1275 to 3200 new cases per year. The annual proportion of new cases with AL is 78%.
Familial transthyretin-associated amyloidosis (ATTR) is a less common systemic type of amyloidosis with unknown incidence, but approximately 10% to 20% of diagnosed cases in tertiary centers are secondary to ATTR amyloidosis. Of these cases, seven percent are hereditary and result from mutated transthyretin (ATTRm) and approximately six percent represent the acquired age-related, wild-type ATTRwt amyloidosis. ATTRwt is seen more commonly in males and was formerly known as senile systemic amyloidosis. Secondary or AA amyloidosis represents 6% of all the amyloidosis cases diagnosed each year. AA amyloidosis is an acquired process and reactive due to chronic inflammation.[9][10]
Pathophysiology
Twenty-one different proteins have been identified as amyloidogenic agents. Polypeptides can adopt alternative misfolded states, making them prone to aggregation. There are multiple processes by which misfolding of protein precursors occur. The protein may be intrinsically likely to acquire a pathologic conformation with aging, as seen in patients with wild-type transthyretin senile systemic amyloidosis. This can also happen when there is a high serum concentration of protein precursors, as seen in long-term hemodialysis patients with increased levels of beta-2-microglobulin.
In hereditary amyloidosis, the replacement of single amino acids can lead to amyloidogenic misfolded proteins losing the biologic function of the native proteins and, in turn, aggregate. Proteolytic remodeling of beta-amyloid precursor proteins has been identified in Alzheimer's disease. In patients with AA amyloidosis, serum amyloid A, an acute-phase reactant protein deposits in different tissues.
The amyloidogenic variants of the various precursor proteins are thermodynamically less stable, resulting in tetramers dissociation into monomers in the case of transthyretin and destabilization of the tertiary structure leading to partially folded conformers in the case of lysozyme. Transthyretin monomers and lysozyme partially folded conformers have the propensity to aggregate and assemble into fibrils. Charged residues also play a role in modulating the aggregation using repulsive forces resulting in partial unfolding, rendering the protein susceptible to proteases attacks and the release of amyloidogenic polypeptides. This is the case of gelsolin, an amyloidogenic protein that causes systemic amyloidosis.
Low pH, increased temperatures, limited proteolysis, osmolytes, and metal ions can alter the tridimensional structure of proteins shifting the equilibrium towards the amyloidogenic state. The mechanism of tissue damage in amyloidosis involves alteration of tissue architecture, interaction with cell surface receptors, inflammation elicited by the amyloid protein deposition, oxidative stress, and apoptosis activation.[11][12][13]
Histopathology
One diagnostic and differentiating feature of amyloidosis is the apple-green birefringence of amyloid on congo red staining. Apart from this, amyloid has an amorphous eosinophilic appearance, when viewed with hematoxylin and eosin staining. On x-ray diffraction analysis, it has a beta-pleated sheet structure.[14]
History and Physical
The clinical features of amyloidosis vary depending on which type of amyloid fibrils are responsible. Systemic amyloidosis (AA) can lead to heart failure with left ventricular hypertrophy on echocardiogram with standard or low voltage electrocardiogram. Hepatomegaly, nephrotic syndrome, macroglossia, orthostatic hypotension, ecchymosis, and autonomic and peripheral neuropathy can be present. Carpal tunnel syndrome, jaw claudication, and articular deposits of amyloid can also be a manifestation of systemic amyloidosis. In secondary amyloidosis (AA), hepatosplenomegaly, proteinuria, renal failure, and orthostasis can be seen. ATTR amyloidosis onset is during midlife and presents with peripheral and autonomic neuropathy, cardiomyopathy, and vitreous opacities. Amyloid beta-amyloidosis is localized to the central nervous system and presents as sporadic Alzheimer's disease and aging.[15][16]
Other physical exam findings that can lead to suspicion of amyloidosis are hypertrophied shoulder pads from amyloid deposition, amyloid purpura, and raccoon eyes secondary to factor-X deficiency in the case of AL amyloidosis and prolonged PT, PTT, that correct with mixing studies pointing towards the involvement of factor deficiencies in the common pathway of coagulation.[17]
Evaluation
Clinical suspicion, family history, and tissue biopsy establish the diagnosis.[18][19]
Tissue biopsy
The subcutaneous abdominal fat aspirate is an easy and innocuous procedure that will stain Congo red positive with apple-green birefringence and has an 81% diagnostic sensitivity in AL amyloidosis.
Biopsy of minor salivary glands can establish systemic amyloidosis diagnosis 60% of the times when fat aspirates are negative.
Biopsy of the liver should be done by the transjugular route, since an amyloid-loaded liver may result in fatal bleeding.
[20]
Plasma cell clone identification
Serum and urine electrophoresis with immunofixation and free light chains (FLC) should be requested to rule out plasma cell dyscrasia. When monoclonal light chains are not present, bone marrow biopsy can help establish the diagnosis by immunohistochemical staining. Immunofluorescence in situ hybridization (FISH) should be ordered along with a skeletal survey.
When plasma cell dyscrasia is ruled out, other types of amyloidosis should be considered. Tissue typing can be done by mass spectrometry, immune electron microscopy or immunohistochemistry. Transthyretin can be detected by isoelectric focusing on the serum which separates wild-type (or formerly known senile cardiac amyloidosis) and variant transthyretin.[21]
Gene sequencing
Should be done when hereditary amyloidosis has to be ruled out based on clinical grounds.
When transthyretin has not been identified, and patients have macroglossia and other typical organ involvement, AL should still be suspected despite plasma cell dyscrasia absence. ATTRwt is diagnosed by cardiac biopsy showing positivity to antibodies against normal transthyretin. Cardiac Scintigraphy with bone tracers can help differentiate AL amyloidosis which shows mild or no uptake from transthyretin amyloidosis which has strong uptake. This could potentially spare cardiac biopsy.
AA amyloidosis is considered when transthyretin and AL have been ruled out, and there are kidney involvement and neuropathy. Immunohistochemistry aids in the diagnosis of AA.
Organ involvement and staging of the disease should be established to design the treatment plan. For cardiac function, evaluation should include an echocardiogram with an assessment of strain, NT-proBNP, troponins, ECG, Holter ECG, and cardiac MRI should be solicited. For kidney function, evaluation 24-hour urinary protein and eGFR are needed. Liver function tests and imaging (ultrasound (US), MRI, or CT scan) can help with hepatic function assessment.
Treatment / Management
AL amyloidosis: This disease is treated most likely in the framework of clinical trials and depending on the risk stratification according to the Standard Mayo Clinic staging system.[22][23][24]
Low-risk patients (NT-proBNP less than 5000 ng/L, troponins of less than 0.06 ng/ml, eGFR more than 50 ml/min per 1.73m, age less than 65 years, performance status of 0-2, NYHA class less than III, ejection fraction above 45%, systolic blood pressure above 90 mmHg standing and diffusion capacity of carbon monoxide (DLCO) above 90%). Low-risk patients receive autologous stem cell transplant (ASCT) with Melphalan 200mg/m2. Induction with cyclophosphamide, bortezomib, and dexamethasone should be considered if the bone marrow, plasma cell infiltration is more than 10% or if a patient refuses transplant. Post-transplant treatment with bortezomib increases the complete response (CR) rates, and if CR is not achieved, bortezomib and dexamethasone combination should be given.
Intermediate risk patients (ineligible for ASCT and stage I-IIIa): Melphalan and Dexamethasone are the preferred regimens especially in the case of neuropathy or t(11;14) translocation. Cyclophosphamide with Bortezomib and dexamethasone combination is a stem cell sparing regimen preferred in patients with renal failure and with 1q21 gain. If noninvolved Free Light Chains (FLC) is above 180 mg/l, the preferred regimen is bortezomib, melphalan, and dexamethasone combination.
High-risk patients (stage IIIb, NYHA class III or above): bortezomib can be preferred due to a rapid onset of action or low-dose combination regimens are preferred as well.
ATTR amyloidosis: Transthyretin is a protein predominantly synthesized in the liver. Upon liver transplant, mutant transthyretin disappeared from blood and neuropathy improvement was observed.
Supportive therapy: The treatment of systemic amyloidosis involves supportive therapy aiming to maintain the quality of life and prevent organ dysfunction. If patients are on a heart transplant waiting list, they should receive low-dose chemotherapy due to increased survival.
Splenectomy: An option with proper prior vaccination when there is severe factor-X deficiency leading to bleeding diathesis. Factor X deficiency is seen in 2.5% of patients with AL amyloidosis. Splenectomy is effective in patients who have splenomegaly, but not usually in patients with a normal size spleen.[25]
Differential Diagnosis
Chronic inflammatory conditions and systemic conditions are associated with amyloidosis. Systemic or secondary amyloidosis often coexists with immunoglobulin M associated disorders.[26]
The following is the differential diagnosis;
Familial Renal Amyloidosis
Immunoglobulin-Related Amyloidosis
Membranous Glomerulonephritis
Renal Vein Thrombosis due to amyloid
Cutis Verticis Gyrata
Mastocytosis
Nodular Localized Cutaneous Amyloidosis
Pseudoxanthoma Elasticum
Prognosis
The prognosis depends on the type of amyloidosis and the patient's response to treatment. Systemic amyloidosis if left untreated can be fatal. If the underlying chronic inflammatory condition is not picked up as a cause of amyloidosis or is misdiagnosed, amyloid aggregation usually accelerates. The survival rate depends on the organs affected. If the heart is involved, cardiovascular complications significantly increase morbidity and mortality.[27]
Complications
Amyloidosis affects multiple body organs and systems. Chronic deposition of amyloidosis leads to restrictive cardiomyopathy. Initially, cardiac involvement may cause angina, orthostatic hypotension or arrhythmias. Congestive heart failure accounts for the death of about 40% of patients that have primary systemic amyloidosis.[28]
Macroglossia can cause painful dysphagia. Infiltration of amyloid into blood vessels can cause leg or jaw claudication. Amyloid deposition in the skin can result in loss of scalp hair. Gastrointestinal tract infiltration with amyloid leads to hemorrhage that can cause malabsorption or can be fatal.
Consultations
Treatment of amyloidosis involves a multidisciplinary approach. Depending on the disease severity and the organs affected a clinical hematologist, cardiologist, nephrologist, pathologist, or other subspecialty specialists can be involved.
Enhancing Healthcare Team Outcomes
Amyloidosis is a heterogeneous disease that results from the deposition of toxic insoluble beta-sheet fibrillar protein aggregates in different tissues. Amyloidosis can be acquired or hereditary. The disease can be localized or systemic. Thus, it is best managed by an interprofessional team that includes an oncologist, hematologist, internist, nephrologist, and a neurologist. Amyloid can accumulate in the liver, spleen, kidney, heart, nerves, and blood vessels causing different clinical syndromes including cardiomyopathy, hepatomegaly, proteinuria, macroglossia, autonomic dysfunction, ecchymoses, neuropathy, renal failure, hypertension, and corneal and vitreous abnormalities. This disease is treated most likely in the framework of clinical trials and depending on the risk stratification according to the Standard Mayo Clinic staging system.[29]
Systemic AL Amyloidosis, Pathology, red-positive glomerular deposits in the mesangium, Capillary walls, Bowman's Capsule, Kidney Biopsy, Microscopy, Congo red stained Contributed by Desport et al.; BioMed Central (CC By 2.0) http://creativecommons.org/licenses/by/2.0 (more...)
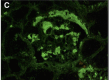
Systemic AL Amyloidosis, Pathology, Immunofluorescence, Glomerular amyloid deposits positively stained with anti-lambda conjugate Contributed by Desport et al.; BioMed Central (CC By 2.0) http://creativecommons.org/licenses/by/2.0
Systemic AL Amyloidosis, Pathology, Kidney Biopsy, Electron microscopy, Glomerular subepithelial amyloid deposits organized into randomly arranged fibrils 7 to 10 mm in external diameter Contributed by Desport et al.; BioMed Central (CC By 2.0) http://creativecommons.org/licenses/by/2.0 (more...)
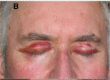
Systemic AL Amyloidosis, Pathology, Bilateral Periorbital purpura, Eyelids Contributed by Desport et al.; BioMed Central (CC By 2.0) http://creativecommons.org/licenses/by/2.0
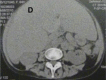
Systemic AL Amyloidosis, Pathology, Voluminous Hepatomegaly due to primary hepatic amyloidosis, Liver, CT scan Contributed by Desport et al.; BioMed Central (CC By 2.0) http://creativecommons.org/licenses/by/2.0
References
- 1.
Tini G, Vianello PF, Gemelli C, Grandis M, Canepa M. Amyloid Cardiomyopathy in the Rare Transthyretin Tyr78Phe Mutation.
J Cardiovasc Transl Res. 2019 Dec;12(6):514-516. [
PubMed: 30604309]
- 2.
Baiardi S, Rossi M, Capellari S, Parchi P. Recent advances in the histo-molecular pathology of human prion disease.
Brain Pathol. 2019 Mar;29(2):278-300. [
PMC free article: PMC8028685] [
PubMed: 30588685]
- 3.
Gavriatopoulou M, Fotiou D, Ntanasis-Stathopoulos I, Kastritis E, Terpos E, Dimopoulos MA. How I treat elderly patients with plasma cell dyscrasias.
Aging (Albany NY). 2018 Dec 18;10(12):4248-4268. [
PMC free article: PMC6326666] [
PubMed: 30568029]
- 4.
Thomas VE, Smith J, Benson MD, Dasgupta NR. Amyloidosis: diagnosis and new therapies for a misunderstood and misdiagnosed disease.
Neurodegener Dis Manag. 2019 Dec;9(6):289-299. [
PubMed: 31686587]
- 5.
Sipe JD, Benson MD, Buxbaum JN, Ikeda S, Merlini G, Saraiva MJ, Westermark P., Nomenclature Committee of the International Society of Amyloidosis. Amyloid fibril protein nomenclature: 2012 recommendations from the Nomenclature Committee of the International Society of Amyloidosis.
Amyloid. 2012 Dec;19(4):167-70. [
PubMed: 23113696]
- 6.
Yakupova EI, Bobyleva LG, Vikhlyantsev IM, Bobylev AG. Congo Red and amyloids: history and relationship.
Biosci Rep. 2019 Jan 31;39(1) [
PMC free article: PMC6331669] [
PubMed: 30567726]
- 7.
Sekijima Y, Nakamura K. Hereditary Transthyretin Amyloidosis. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors.
GeneReviews® [Internet]. University of Washington, Seattle; Seattle (WA): Nov 5, 2001. [
PubMed: 20301373]
- 8.
Buck FS, Koss MN, Sherrod AE, Wu A, Takahashi M. Ethnic distribution of amyloidosis: an autopsy study.
Mod Pathol. 1989 Jul;2(4):372-7. [
PubMed: 2668942]
- 9.
Alwahaibi NY, Al Issaei HK, Al Dhahli BS. Spectrum of glomerular diseases in Arab countries: A systematic review.
Saudi J Kidney Dis Transpl. 2018 Nov-Dec;29(6):1256-1266. [
PubMed: 30588955]
- 10.
Andreou S, Panayiotou E, Michailidou K, Pirpa P, Hadjisavvas A, El Salloukh A, Barnes D, Antoniou A, Agathangelou P, Papastavrou K, Christodoulou K, Tanteles GA, Kyriakides T. Epidemiology of ATTRV30M neuropathy in Cyprus and the modifier effect of complement C1q on the age of disease onset.
Amyloid. 2018 Dec;25(4):220-226. [
PubMed: 30572722]
- 11.
Buxbaum JN. The systemic amyloidoses.
Curr Opin Rheumatol. 2004 Jan;16(1):67-75. [
PubMed: 14673392]
- 12.
Mahmood S, Palladini G, Sanchorawala V, Wechalekar A. Update on treatment of light chain amyloidosis.
Haematologica. 2014 Feb;99(2):209-21. [
PMC free article: PMC3912950] [
PubMed: 24497558]
- 13.
Merlini G, Seldin DC, Gertz MA. Amyloidosis: pathogenesis and new therapeutic options.
J Clin Oncol. 2011 May 10;29(14):1924-33. [
PMC free article: PMC3138545] [
PubMed: 21483018]
- 14.
Chuang E, Hori AM, Hesketh CD, Shorter J. Amyloid assembly and disassembly.
J Cell Sci. 2018 Apr 13;131(8) [
PMC free article: PMC5963839] [
PubMed: 29654159]
- 15.
Maurer MS, Elliott P, Comenzo R, Semigran M, Rapezzi C. Addressing Common Questions Encountered in the Diagnosis and Management of Cardiac Amyloidosis.
Circulation. 2017 Apr 04;135(14):1357-1377. [
PMC free article: PMC5392416] [
PubMed: 28373528]
- 16.
Pinney JH, Whelan CJ, Petrie A, Dungu J, Banypersad SM, Sattianayagam P, Wechalekar A, Gibbs SD, Venner CP, Wassef N, McCarthy CA, Gilbertson JA, Rowczenio D, Hawkins PN, Gillmore JD, Lachmann HJ. Senile systemic amyloidosis: clinical features at presentation and outcome.
J Am Heart Assoc. 2013 Apr 22;2(2):e000098. [
PMC free article: PMC3647259] [
PubMed: 23608605]
- 17.
McCausland KL, White MK, Guthrie SD, Quock T, Finkel M, Lousada I, Bayliss MS. Light Chain (AL) Amyloidosis: The Journey to Diagnosis.
Patient. 2018 Apr;11(2):207-216. [
PMC free article: PMC5845075] [
PubMed: 28808991]
- 18.
Kapoor M, Rossor AM, Jaunmuktane Z, Lunn MPT, Reilly MM. Diagnosis of amyloid neuropathy.
Pract Neurol. 2019 Jun;19(3):250-258. [
PubMed: 30598431]
- 19.
Koop AH, Mousa OY, Wang MH. Clinical and endoscopic manifestations of gastrointestinal amyloidosis: a case series.
Clujul Med. 2018 Oct;91(4):469-473. [
PMC free article: PMC6296722] [
PubMed: 30564026]
- 20.
Vrana JA, Theis JD, Dasari S, Mereuta OM, Dispenzieri A, Zeldenrust SR, Gertz MA, Kurtin PJ, Grogg KL, Dogan A. Clinical diagnosis and typing of systemic amyloidosis in subcutaneous fat aspirates by mass spectrometry-based proteomics.
Haematologica. 2014 Jul;99(7):1239-47. [
PMC free article: PMC4077087] [
PubMed: 24747948]
- 21.
Lisenko K, Schönland SO, Jauch A, Andrulis M, Röcken C, Ho AD, Goldschmidt H, Hegenbart U, Hundemer M. Flow cytometry-based characterization of underlying clonal B and plasma cells in patients with light chain amyloidosis.
Cancer Med. 2016 Jul;5(7):1464-72. [
PMC free article: PMC4944872] [
PubMed: 27109862]
- 22.
Westerland OA, Pratt G, Kazmi M, El-Najjar I, Streetly M, Yong K, Morris M, Mehan R, Sambrook M, Hall-Craggs M, Silver D, Goh V. National survey of imaging practice for suspected or confirmed plasma cell malignancies.
Br J Radiol. 2018 Dec;91(1092):20180462. [
PMC free article: PMC6319860] [
PubMed: 30102561]
- 23.
Kalle A, Gudipati A, Raju SB, Kalidindi K, Guditi S, Taduri G, Uppin MS. Revisiting renal amyloidosis with clinicopathological characteristics, grading, and scoring: A single-institutional experience.
J Lab Physicians. 2018 Apr-Jun;10(2):226-231. [
PMC free article: PMC5896193] [
PubMed: 29692592]
- 24.
Eddou H, Zinebi A, Maaroufi HE, Moudden MK, Doghmi K, Mikdame M, Baaj ME. [Treatment of systemic AL amyloidosis: about 25 cases].
Pan Afr Med J. 2017;28:160. [
PMC free article: PMC5847059] [
PubMed: 29541306]
- 25.
Popkova T, Hajek R, Jelinek T. Monoclonal antibodies in the treatment of AL amyloidosis: co-targetting the plasma cell clone and amyloid deposits.
Br J Haematol. 2020 Apr;189(2):228-238. [
PubMed: 32072615]
- 26.
Terrier B, Jaccard A, Harousseau JL, Delarue R, Tournilhac O, Hunault-Berger M, Hamidou M, Dantal J, Bernard M, Grosbois B, Morel P, Coiteux V, Gisserot O, Rodon P, Hot A, Elie C, Leblond V, Fermand JP, Fakhouri F. The clinical spectrum of IgM-related amyloidosis: a French nationwide retrospective study of 72 patients.
Medicine (Baltimore). 2008 Mar;87(2):99-109. [
PubMed: 18344807]
- 27.
Gertz MA. Immunoglobulin light chain amyloidosis: 2018 Update on diagnosis, prognosis, and treatment.
Am J Hematol. 2018 Sep;93(9):1169-1180. [
PubMed: 30040145]
- 28.
Desai HV, Aronow WS, Peterson SJ, Frishman WH. Cardiac amyloidosis: approaches to diagnosis and management.
Cardiol Rev. 2010 Jan-Feb;18(1):1-11. [
PubMed: 20010333]
- 29.
Kumar S, Dispenzieri A, Lacy MQ, Hayman SR, Buadi FK, Colby C, Laumann K, Zeldenrust SR, Leung N, Dingli D, Greipp PR, Lust JA, Russell SJ, Kyle RA, Rajkumar SV, Gertz MA. Revised prognostic staging system for light chain amyloidosis incorporating cardiac biomarkers and serum free light chain measurements.
J Clin Oncol. 2012 Mar 20;30(9):989-95. [
PMC free article: PMC3675680] [
PubMed: 22331953]
Disclosure: Jean Bustamante declares no relevant financial relationships with ineligible companies.
Disclosure: Syed Rafay Zaidi declares no relevant financial relationships with ineligible companies.