

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
National Academies of Sciences, Engineering, and Medicine; Health and Medicine Division; Board on Health Sciences Policy; Committee on Processes to Evaluate the Safety and Efficacy of Drugs for Rare Diseases or Conditions in the United States and the European Union; Shore CK, Worku TL, Smith CW, et al., editors. Regulatory Processes for Rare Disease Drugs in the United States and European Union: Flexibilities and Collaborative Opportunities. Washington (DC): National Academies Press (US); 2024 Oct 30.

Regulatory Processes for Rare Disease Drugs in the United States and European Union: Flexibilities and Collaborative Opportunities.
Show detailsThere are an estimated 7,000 to 10,000 life-threatening and chronically debilitating rare diseases. While each disease affects only a small number of people, together rare diseases affect up to 30 million individuals in the United States, 36 million in the European Union, and at least 300 million across the globe. The impact of rare diseases extends well beyond the affected individual to include family members and caregivers, imposing a significant burden on an estimated 1 billion people globally, when accounting for both people living with a rare disease or condition and their caregivers.
The U.S. Food and Drug Administration (FDA) and the European Medicines Agency (EMA) play a critical role in protecting public health by ensuring that drugs to treat rare diseases and conditions are safe and effective. Additionally, the agencies help advance the public health by actively promoting scientific and technological innovation for advancing drug development. Before the Orphan Drug Act2 was passed in 1983 the development of drugs to treat rare diseases3 was largely neglected by the pharmaceutical industry. Following passage of the Orphan Drug Act and subsequent policy measures implemented around the world, including the European Union (EU) regulation on orphan medicinal products, which was adopted in 1999, there has been a marked increase in the investment and successful development of drugs to treat rare diseases and conditions. And yet today, less than 5 percent of rare diseases have approved products on the market.
For people living with rare disease and conditions, there is an urgent need to increase the pace and volume of drug development and of regulatory approval processes. Patient groups have expressed frustration with regulatory agencies, raising legitimate questions about how agencies analyze data gathered from small trials and consider patient and caregiver input in regulatory decision-making, noting seeming inconsistencies around drug products that are approved by one agency and not the other, and asking how and when FDA applies regulatory flexibilities across its centers and divisions.
The U.S. Congress called on FDA to contract with the National Academies of Sciences, Engineering, and Medicine (the National Academies) to conduct a study on processes for evaluating the safety and efficacy of drugs for rare diseases in the United States and the European Union. The statement of task includes: (1) flexibilities and mechanisms available to regulators, (2) the consideration and use of “supplemental data” submitted during the review process, and (3) an assessment of collaborative efforts between FDA and EMA.
The committee was specifically asked to focus on the regulatory processes for the review and approval of new molecular entities (NMEs) and biologics. As requested by the sponsor, drug repositioning or repurposing, new indications for drugs already approved, N-of-1 or single-participant clinical trials, devices, new modalities, and platform technologies were considered outside the scope of this report. While the committee looked at areas for collaboration between the United States and the European Union, recommendations are focused primarily on the United States.
The committee gathered information through open presentations from topic experts, public comments from interested parties, literature review, and semi-structured interviews. To supplement information gathered from the peer reviewed literature and during open sessions of committee meetings, the committee commissioned work to analyze: (1) success rates of orphan product authorization submissions and approvals by FDA and EMA; (2) distribution of products approved by FDA and EMA by therapeutic area; (3) use of expedited pathways by products approved by FDA and EMA; (4) use of “supplemental data” in applications for products approved by FDA and EMA.
Recommendations in this report seek to enhance strategic engagement of FDA with people living with a rare disease or condition, their caregivers, and patient representatives, especially patient groups that are small and under-resourced, throughout the full continuum of the drug development process; advance regulatory science, including the use of innovative study designs and methods and application of alternative and confirmatory data to inform regulatory decision-making for rare disease drug products; and improve collaboration between FDA and EMA.
It is important to note that at the time of this report’s writing, there were several activities underway that could affect the landscape for the approval of treatments for rare diseases, including the Consolidated Appropriations Act of 2023 (PL 117-328) and the Food and Drug Omnibus Reform Act of 2022, which contain multiple provisions intended to improve rare disease drug development.
REGULATORY FLEXIBILITIES, AUTHORITIES, AND MECHANISMS
In the United States, FDA has authority under the Federal Food, Drug, and Cosmetic Act to regulate medical products and devices to ensure that they are safe and effective for the intended use. EMA is a decentralized agency of the European Union that is responsible for evaluating the safety and efficacy of drugs in Europe. To gain access to the U.S. and European markets, drug sponsors must submit marketing applications to both the agencies, which have different organizational structures, applicable laws, risk management procedures, and regulations.
Both FDA and EMA have regulatory flexibilities and mechanisms designed to facilitate the development and approval of drug products to treat rare diseases. These include programs to expedite the review and approval of certain types of drug products; mechanisms for engaging patients, caregivers, and patient groups; incentives for orphan drug designated4 products (FDA) or orphan medicines5 (EMA); and guidance (FDA) and guidelines (EMA) on study design, methodologies, and the use of alternative and confirmatory data. Together, these efforts help the agencies execute their missions to protect and advance the public’s health with the goal of ensuring that patients have access to safe and effective treatments for rare diseases in a timely manner.
FDA and EMA Alignment on Evidence-Based Approaches and Programs
While FDA and EMA generally align on evidence-based approaches and have similar programs in place to expedite the review and approval of drugs to treat rare diseases and conditions, there is no required process for regulators to jointly discuss drug products under review. That said, the two agencies often reach the same regulatory decisions when it comes to submitted applications for marketing approval for drugs to treat rare diseases and conditions (see Figure S-1).
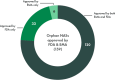
FIGURE S-1
Number of orphan drugs approved by FDA and EMA from 2018 to 2022. NOTE: NASs = new active substances. SOURCE: CIRS Data Analysis, 2024.
Inclusion of Pediatric Populations
The majority of rare diseases affect the pediatric population. Evidence has shown there can be substantial differences in the way that children respond to drug treatment compared to adults. Thus, the inclusion of pediatric populations in clinical trials should be a core component for rare disease drug development.
Over the past several decades, a combination of legislation, regulatory action and the accumulation of scientific evidence has enabled what some have considered to be a “revolutionary change” in pediatric drug development—a shift from considering pediatric populations as “therapeutic orphans” to a current state in which the number of drug products approved for use in children continues to increase. Two laws—the Pediatric Research Equity Act (PREA) of 2003, the Best Pharmaceuticals for Children Act (BPCA) of 2002—work together to address the need for pediatric drug development. In particular, PREA authorizes FDA to require pediatric studies for certain drugs and biological products. Despite these efforts, off-label drug use remains an issue for pediatric populations, particularly those living with rare diseases and conditions for whom there is no available treatment on the market. Notably, orphan designated drug products are generally exempted from PREA requirements.
In addition to measures taken by Congress to incentivize the inclusion of pediatric populations in rare disease drug development, FDA and the National Institutes of Health (NIH), in partnership with nongovernmental organizations—including patient and disease advocacy groups, academic clinical investigators, and biopharmaceutical companies—have an opportunity to better collaborate on approaches to include pediatric populations as early as possible in clinical trials and meet the needs of children living with rare diseases and conditions.
RECOMMENDATION 2-1: Congressional action is needed to encourage and incentivize more studies that provide information about the use of rare disease drug products in pediatric populations. To that end, Congress should remove the Pediatric Research Equity Act orphan exemption and require an assessment of additional incentives needed to spur the development of drugs to treat rare diseases or conditions.6
Additionally, the U.S. Food and Drug Administration (FDA) and the National Institutes of Health in partnership with nongovernmental organizations, including patient groups, clinical investigators, and biopharmaceutical companies, should work to provide clarity regarding the evolving regulatory policies and practices for the inclusion of pediatric populations as early as possible in rare disease clinical trials. Actions should include, but are not limited to the following:
Enhancing Mechanisms for Patient Input
FDA has made important strides through guidance and policy to engage people with lived experience—people who are living with a rare disease or condition and their caregivers—throughout the regulatory review process. FDA advisory committees offer independent expert advice and recommendations on scientific, technical, and policy matters related to FDA-regulated products. In addition to including a patient representative who provides lived experience with a particular disease, condition, or medical product, all advisory committee meetings include an open public hearing session during which patients and their caregivers have an opportunity to share relevant information about a given drug and disease or condition. In principle, open public hearing sessions should inform the advisory committee on the drug product under consideration.
Conclusion 2-2: FDA is using available mechanisms to gather patient input. However, there are opportunities to better ensure that patient input informs the development of treatments for rare diseases as well as the design and conduct of clinical trials for rare diseases. More clarity is needed on the part of patient groups and people with lived experience on how the agency is using patient input to inform regulatory decision-making and what types of patient input are most relevant.
Patients and caregivers are experts in their own experience of living with or caring for someone with a disease or condition and their perspectives and insights should be valued alongside those of regulators, sponsors, and researchers when it comes to informing the development of drugs to meet their needs.
RECOMMENDATION 2-2: The U.S. Food and Drug Administration (FDA) should strengthen mechanisms to integrate input from people living with a rare disease or condition, their caregivers, and patient representatives, especially patient groups that are small and under-resourced, throughout the full continuum of the drug development process. To that end, FDA should take the steps necessary to fully implement Section 1137 of the Food and Drug Administration Safety and Innovation Act (Public Law 112-144), which directs the Secretary of Health and Human Services to develop and implement strategies to solicit the views of rare disease patients during the full range of regulatory review discussions. This should include but not be limited to:
Enhancing Mechanisms for Sponsor Engagement
Sponsors developing new drugs must navigate a range of complex challenges when designing and conducting studies for regulatory submission. These challenges are heightened when it comes to rare diseases and conditions, particularly given that many companies developing rare disease drugs are small and medium-sized enterprises, which may have fewer resources and less in-house expertise than large pharmaceutical companies.
Conclusion 2-4: FDA engagement with rare disease drug development sponsors is of particular importance because compared to common diseases, rare diseases are less well understood, more often do not have regulatory precedent, more commonly lack validated endpoints and outcome measures, and involve small patient populations limit the size and number of clinical trials that can be conducted.
RECOMMENDATION 2-3: The U.S. Food and Drug Administration and the National Institutes of Health in collaboration with the European Medicines Agency, nongovernmental organizations, patient groups, and biopharmaceutical sponsors should implement a sponsor, investigator, and patient group navigation service to support the development of drugs to treat rare diseases and conditions (1) by advising on the range of available regulatory pathways and flexibilities and (2) by providing clarity on how to comply with regulatory policies, apply guidances, and meet requirements in rare disease drug development. Actions should include:
In addition to programs intended to facilitate and expedite development and review of new drugs, FDA has several newer programs relevant to rare diseases, including the Rare Disease Endpoint Advancement (RDEA) pilot program, the Model-Informed Drug Development (MIDD) Paired Meeting Program, Support for clinical Trials Advancing Rare disease Therapeutics (START) Pilot Program, the Complex Innovative Trial Design (CID) meeting program, the FDA-NIH Bespoke Gene Therapy Consortium, programs and pilots led by the Oncology Center of Excellence (e.g., real-time oncology review [RTOR], Project Orbis), and notably a newly announced Rare Disease Innovation Hub. These programs are welcome developments, but most are limited in scope and scale compared to the scale of unmet need in rare disease drug development. Several of these programs are early on in implementation, making it difficult to assess their impact on drug development for rare diseases and conditions.
RECOMMENDATION 2-4: The U.S. Food and Drug Administration (FDA) should assess the impact of new and ongoing programs and approaches that support drug development for rare diseases and conditions to improve the regulatory decision-making process; publicly share the results of these assessments in a timely manner; take steps to ensure that lessons learned across different programs are disseminated throughout FDA centers and divisions, including a summary of regulatory flexibilities and novel innovative approaches that were considered acceptable; scale-up and expand successful programs across therapeutic areas; and modify or sunset programs that are not improving the regulatory decision-making process. Programs and regulatory approaches should include, but not be limited to:
USE OF ALTERNATIVE AND CONFIRMATORY DATA
For the purposes of this report, the committee uses the term “alternative and confirmatory data” (ACD) to mean data that are collected outside the setting of a randomized controlled clinical trial and used as supplementary, alternative and/or confirmatory evidence in support of regulatory submission and review of a drug product. These data include natural history data, registry data, data from expanded access or compassionate use, data from open-label extension studies, data from studies using external control groups, patient reported outcomes, and real-world evidence.
The statutory requirements for drug review and approval for rare diseases and conditions are the same as for non-rare diseases or conditions. However, when a disease is life-threatening or severely debilitating with unmet need, both FDA and EMA have flexibility to consider the use of ACD along with a single adequate and well-controlled clinical trial. The agencies have published guidance on how these data sources can support a marketing application. However, in the United States, there is little publicly available information about whether and how these data are taken into account during regulatory decision-making.
Natural history data are particularly important to rare disease drug development. The use of biomarkers or a panel of biomarkers can help alleviate diagnostic challenges and facilitate clinical trials based on smaller sample sizes and shorter duration. However, limited populations and the heterogeneity in clinical presentation combined with a lack of information about disease emergence and progression makes it difficult to validate biomarkers for regulatory decision-making. Natural history studies can provide information about potential endpoints and the relationship between disease severity/progression and biomarker changes as well as clinical outcome assessments—measures that describe or reflect how a patient feels, functions, or survives. While natural history registries have been established for a growing number of rare diseases and conditions, they have not been established for the majority of rare diseases and conditions.
FDA supports programs aimed at developing alternative data sources, notably the Rare Disease Cures Accelerator-Data and Analytics Platform (RDCA-DAP®), which is funded by FDA and operated by the Critical Path Institute in collaboration with the National Organization of Rare Disorders (NORD). This platform is a centralized database and analytics hub that contains standardized data on a growing number of rare diseases and allows secure sharing of data collected across multiple sources, including natural history studies and patient registries, control arms of clinical trials, longitudinal observational studies, and real-world data. Drug developers and other data users can access the platform to better understand disease progression and heterogeneity, better target therapeutics, and inform trial design and other aspects of rare disease drug development. The European Reference Networks contribute work on registries.
RECOMMENDATION 4-1: The U.S. Food and Drug Administration (FDA) should enable the collection and curation of regulatory-grade natural history data to enhance the quality and accessibility of data for all rare diseases. This should include, but not be limited to:
Given proven examples of success, the evolution in regulatory thinking, and advances in new trial designs and methods for data analysis, there is a growing impetus to apply and expand available opportunities for collecting, analyzing, and using ACD to inform researchers, sponsors, regulators, and patient groups on when and how alternative and confirmatory data have informed regulatory decision-making to ensure the integration of lessons learned from past successes and failures.
EMA and FDA can facilitate the use of these types of data in marketing submission applications by standardizing, documenting, and publicly sharing information to enable stakeholders to track over time how alternative and confirmatory data have successfully and unsuccessfully informed regulatory decision-making for rare disease drug products. A publicly available and easily accessible (indexed and searchable) listing of products coupled with standardized information on the types and sources of alternative and confirmatory data that were considered as part of a marketing authorization application, would enable drug sponsors, patient and disease advocates, researchers, and regulators to improve the collection and use of these data for rare disease drug development going forward.
An understanding of the opportunities as well as the gaps and inadequacies in alternative and confirmatory data would help guide data collection strategies on the part of patients, caregivers, sponsors, and researchers, and ensure that the data gathered are both relevant and robust enough to support regulatory needs.
RECOMMENDATION 4-2: The U.S. Food and Drug Administration (FDA) should invite the European Medicines Agency (EMA) to jointly conduct systematic reviews of submitted and approved marketing authorization applications to treat rare diseases and conditions that document cases for which alternative and confirmatory data have contributed to regulatory decision-making. The systematic reviews should include relevant information on the context for whether these data were:
Findings from the systematic reviews should be made publicly available and accessible for sponsors, researchers, patients, and their caregivers through public reporting or publication of the results. EMA and FDA should establish a public database for these findings that is continuously updated to ensure that progress over time is captured, opportunities to clarify agency thinking over time are identified, and information on the use of alternative and confirmatory data to inform regulatory decision-making is publicly shared to inform the rare disease drug development community.
Several novel approaches for analyzing relevant data on drug safety and efficacy that can make it possible to generate useful information for regulatory decision-making based on limited data. Further acceptance of these methods on the part of regulatory agencies and sponsors would better enable the use of alternative and confirmatory data as well as data collected through traditional randomized clinical trials for rare diseases and conditions. While this report focuses on rare diseases and conditions, the committee notes that it is not uncommon for innovations in rare disease drug development to be a vanguard for applications across therapeutic areas, so lessons learned in the rare disease space could be considered by the agencies on a broader scale.
Conclusion 4-3: Given the variable and often longtime horizons for rare disease progression, gaps in the knowledge of disease etiology, ethical concerns, severity of disease, small sample sizes, and unmet medical need, rare diseases require additional methods of demonstrating substantial evidence of effectiveness. New approaches in study design and data analysis need not require lower regulatory standards, but rather they enable the consideration of alternative and confirmatory data and a nuanced interpretation of the benefit–risk assessments that take into account the limited availability of data, limited treatment availability and the risk acceptance threshold in these unique patient populations.
RECOMMENDATION 4-3: The U.S. Food and Drug Administration (FDA) should collect and disseminate information on how state-of-the art regulatory science; innovative study designs and methods; tools, including biomarkers and surrogate endpoints; and effective applications of alternative and confirmatory data inform regulatory decision-making for rare disease drug products by:
FDA AND EMA COLLABORATION
The complex regulatory landscape and the differences between regulatory agencies can have an outsized impact on patients with rare diseases and conditions. Due to the nature of rare disease drug development (e.g., small patient populations, high rates of morbidity and mortality), early collaboration and information exchange between the agencies to coordinate on study design and align on data requirements could help reduce duplication of clinical testing and streamline the regulatory process for sponsors submitting marketing authorization applications to both agencies.
Conclusion 5-1: Despite some key differences, FDA and EMA have similar approaches to the evaluation and approval of drugs for rare diseases. Given these parallel approaches, there are existing mechanisms for close collaboration between the two agencies, as well as opportunities for enhanced collaboration in the future, that would allow each agency to retain sovereign authority and accountability in regulatory decision-making.
Enhancing Information Sharing
Under EU regulations, transparency is an important feature of EMA’s operations. Starting in 2016, EMA has published clinical data submitted by sponsors in support of marketing applications for human medicines. EU law mandates that EMA make clinical trial data publicly available while also protecting personal data and commercially confidential information. In addition to the European public assessment report (EPAR) and clinical trial data, EMA makes other information available to improve transparency, including dates, agendas, minutes, and outcomes of its scientific committee meetings; information about staff and experts’ conflicts of interest; information about manufacturing inspections; pediatric investigation plans; and orphan designations.
Conclusion 5-2: To meet the needs of rare disease patients and their caregivers, there is an ethical obligation on the part of regulatory agencies to share relevant information on the review and approval of drugs to treat rare diseases and conditions. If researchers and sponsors working on rare disease drug development had a better understanding of the reasons for successes and failures of marketing authorization applications, they could better innovate new therapeutics that have a higher likelihood of reaching patients. Additionally, more transparency would enhance public understanding and confidence in the important work carried out by regulatory agencies.
RECOMMENDATION 5-1: The U.S. Food and Drug Administration (FDA) should take steps to make relevant information on marketing authorization submissions, review milestones, approval and negative review decisions (refusal to file, clinical hold, and complete response letters), and the use of regulatory flexibilities for rare disease drug products publicly available and easily accessible to inform sponsors, patients, researchers, and reviewers on decision-making rationales and when and how available policies are applied. While the committee acknowledges the legal challenges surrounding disclosure of information, actions should include, but not be limited to:
The committee recognizes there are multiple barriers to achieving greater transparency on the part of FDA, including laws that govern how the agency can or cannot share information. Some have argued that FDA has broad discretion on what is considered confidential. FDA has the ability to incentivize and facilitate pathways for enhancing information sharing, but there are practical and legal considerations, which may require modification of some of the laws that restrict the agency from sharing certain types of information. For these reasons, the committee recognizes the need for more transparency on the part of FDA, but acknowledges that additional consideration, assessment, and legal review are needed to determine how such measures should be implemented.
Clusters
One of the primary formal mechanisms for collaboration between EMA and FDA is holding so-called “clusters”—regular virtual meetings between EMA and FDA staff, that are focused on specific topics and therapeutic areas that would benefit from an “intensified exchange of information and collaboration.” Documents exchanged within clusters may include draft guidelines; assessment reports, review memos; and minutes from investigational new drug (IND), pre-IND, and pre-biologics-licenseapplication meetings. While clusters help inform drug development and approval processes and provide a valuable forum for collaboration between the regulatory agencies, there is substantial unfulfilled potential. The impact of the clusters on the drug development ecosystem may be limited by the fact that cluster discussions are largely focused on specific issues such as an existing development plan or safety concern. The existing cluster structure could instead more prospectively address common challenges for rare disease drug development, thereby harmonizing and streamlining the orphan designation and drug evaluation process.9
An expansion and shift in focus on the part of the clusters to include prospective issues facing rare disease drug development would align with current objectives and build on existing collaborative efforts and help inform regulatory decision-making. There may be concerns on the part of the agencies or sponsors that such an approach could constrain discussions if information were to be made publicly available. However, the agencies have in place mechanisms to share non-binding documents that lay out common thinking on how the agencies weigh urgency and pragmatic limitations against the need for data to support marketing authorization applications for rare diseases and conditions and considerations for how FDA and EMA might address areas of misalignment on clinical trial endpoints, the determination of non-inferiority (or similarity) margins, use and acceptance of statistical methodologies, and totality of evidence determinations.
RECOMMENDATION 5-2: To facilitate the efficient global development of orphan drugs, the U.S. Food and Drug Administration (FDA) and the European Medicines Agency (EMA) should build upon the existing clusters relevant for rare diseases by undertaking the following:
Increasing the Use of Parallel Scientific Advice
Another formal mechanism for collaboration between FDA and EMA is the Parallel Scientific Advice (PSA) program. PSA is a voluntary mechanism through which the two agencies can concurrently provide scientific advice to sponsors during the development of new drugs, biologics, vaccines, or advanced therapies. The program does not guarantee EMA and FDA alignment but can offer potential benefits for sponsors, including agency convergence on approaches for drug development, a better understanding of each agency’s concerns and requirements, and opportunity for sponsors and agencies to ask and answer questions. Despite the potential benefits of the PSA program, only a handful of sponsors apply each year. Reasons for the lack of uptake may include real and perceived concerns on the part of drug sponsors about the value of the program, practical limitations in participating in PSA, and a lack of incentives for using the program.
Conclusion 5-3: Despite the underuse of the PSA program and lack of available evidence related to its impact, the committee acknowledges and expects that, in principle, concurrent scientific discourse through PSA should better enable more streamlined clinical trials, regulatory review, and approval of drugs to treat rare diseases and conditions.
RECOMMENDATION 5-3: The U.S. Food and Drug Administration (FDA), along with the European Medicines Agency (EMA) and other key stakeholders, should assess the impact of the Parallel Scientific Advice (PSA) program over the past decade on drug development for rare diseases and conditions, publicly share the results of this assessment, seek sponsor input on approaches to improve and enhance the use and utility of the program, and take action to increase access, use, and impact of the PSA program going forward. This assessment and plan for improvement should include:
If the actions taken do not lead to an increased use and greater impact of the PSA program within a 5-year period after the assessment and improvement plan has taken place, FDA should implement other mechanisms for parallel advice between FDA and EMA on drug development programs for rare diseases and conditions.
This report evaluates and makes recommendations for one part of a multifaceted ecosystem that determines which diseases are studied, what types of drug research and development are prioritized, how the safety and efficacy of drug products are reviewed and approved, which products are brought to market, and how approved and marketed therapies are made available (or not) to patients. While the adoption of the recommendations in this report will serve to foster transparency, streamline regulatory processes, and facilitate more collaboration between FDA and EMA, regulators review what is submitted to them. The gap between the needs of patients living with rare diseases and conditions and the therapies available for treating them cannot be closed by focusing solely on regulatory processes. Increasing the number of available therapies for rare diseases and conditions will require additional attention upstream of regulatory decision-making—investment in basic research to understand the underlying biology of rare diseases and conditions, approaches to ensure patient input is incorporated early on and throughout the research process—as well as downstream from the regulatory process—policies, incentives, and business models to address issues with drug pricing and payer decisions that have outsized impacts on the accessibility and affordability of treatments for rare disease patients. The committee believes this framing is critical for understanding the report recommendations and considerations for implementation.
Footnotes
- 1
This summary does not include references. Citations for the discussion presented in the Summary appear in the subsequent report chapters.
- 2
P.L. 97-414
- 3
The Orphan Drug Act defines a rare disease as one that affects fewer than 200,000 people in the United States.
- 4
FDA has authority to grant orphan drug designation to a drug or biological product to prevent, diagnose or treat a rare disease or condition.
- 5
EMA may designate an orphan medicine for certain products intended to treat a rare disease or condition.
- 6
This sentence was edited after release of the prepublication version of the report to clarify the intent of the recommendation.
- 7
On May 19, 2010, the Transparency Task Force released a report containing 21 draft proposals about expanding the disclosure of information by FDA while maintaining confidentiality for trade secrets and individually identifiable patient information. FDA accepted public comment on the proposals, as well as on which draft proposals should be given priority, on this website from May 19, 2010, through July 20, 2010. https://wayback
.archiveit .org/7993/20171105152021 /https://www .fda.gov/AboutFDA/Transparency /PublicDisclosure /DraftProposalbyTopicArea /ucm211691.htm (accessed May 14, 2024). - 8
A national clinical trial number is an 8-digit unique identifier assigned to a clinical study when it is registered on ClinicalTrials
.gov. - 9
This section was edited after release of the prepublication version of the report to more precisely describe cluster discussions.
- Summary - Regulatory Processes for Rare Disease Drugs in the United States and E...Summary - Regulatory Processes for Rare Disease Drugs in the United States and European Union
Your browsing activity is empty.
Activity recording is turned off.
See more...