

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
National Academies of Sciences, Engineering, and Medicine; Health and Medicine Division; Board on Health Sciences Policy; Committee on Processes to Evaluate the Safety and Efficacy of Drugs for Rare Diseases or Conditions in the United States and the European Union; Shore CK, Worku TL, Smith CW, et al., editors. Regulatory Processes for Rare Disease Drugs in the United States and European Union: Flexibilities and Collaborative Opportunities. Washington (DC): National Academies Press (US); 2024 Oct 30.

Regulatory Processes for Rare Disease Drugs in the United States and European Union: Flexibilities and Collaborative Opportunities.
Show detailsA common understanding of how to develop a facilitating ecosystem would create a rising tide that would raise all boats in the pursuit of effective treatment and cures for rare diseases.
Robert Califf,
Commissioner of Food and Drugs, 2022 (FDA, 2022b)
Over the past several decades, drug development has grown increasingly complex and global. To gain access to the U.S. and European markets, drug sponsors must submit marketing applications to both the European Medicines Agency (EMA) and the U.S. Food and Drug Administration (FDA), which have different organizational structures, applicable laws, risk management procedures, and regulations. While both agencies generally align on evidence-based approaches and have similar programs in place to expedite the review and approval of drugs to treat rare disease and conditions, the differences between them can lead to variation in marketing authorization applications.
Once sponsors decide that they will be seeking regulatory approval and marketing of their product in both the United States and European Union (EU), they typically seek early regulatory advice from both jurisdictions. The results of these meetings may result in changes to sponsor decisions relating to the sequencing of submissions because one authority may present a more expedited pathway to approval. The types of studies and data required by each agency may not be the same. Without an established set of common requirements between FDA and EMA, sponsors may opt to submit a marketing authorization application to the agency perceived as offering the shortest time to approval. Additionally, market sizes and pricing considerations play a critical role in sponsor decisions about regulatory submission, given that the anticipated lifetime global revenues of a new drug are one of the primary factors impacting company decisions. The real and perceived concerns about EMA and FDA parallel scientific advice—a process that is often perceived as cumbersome and inefficient—is another barrier to use and may put smaller companies with fewer resources and capacity at a disadvantage. There is no required process for regulators to jointly discuss a potential product together. This can lead to an arbitrage-like process that is disconnected from considerations about patient needs and may result in sponsors choosing the path of least resistance rather than the path that is the most efficient way to demonstrate effectiveness and get the product to patients in both jurisdictions.
The complex regulatory landscape and the differences between regulatory agencies can have an outsized impact on patients with rare diseases and conditions. Due to the nature of rare disease drug development (e.g., small patient populations, high rates of morbidity and mortality), sponsors may only get “one bite at the apple” when it comes to designing and executing studies for regulatory decision-making (Lee et al., 2023). Early collaboration and information exchange between the agencies to coordinate on study design and align on data requirements could help reduce duplication of clinical testing, streamline the regulatory process for sponsors submitting marketing authorization applications to both agencies, and reduce incentives for regulatory arbitrage.
The agencies share a reliance on the best available data, including alternative and confirmatory data and novel approaches for data analysis, to reduce uncertainty regarding potential risks and benefits, and both agencies are committed to international collaborative efforts around best practices. However, both agencies have limited capacity to take advantage of these opportunities. Nevertheless, FDA, EMA, and other regulatory authorities have responded to pressure from patient groups, drug sponsors, and other key stakeholders by working collaboratively to better harmonize drug regulatory processes. This type of collaboration has the potential to have a significant impact on drug development and the health of patients; for example, FDA and EMA worked together closely during the COVID-19 pandemic to streamline the development of a vaccine and to quickly and efficiently grant approval through regulatory convergence (Marks, 2020). While the two authorities have the capacity to coordinate well under pressure, the usual practice is for them to work separately except under certain circumstances such as parallel reviews, which rarely occur.
This chapter is organized based on the following topics: similarities and differences between FDA and EMA, collaboration between regulatory agencies and opportunities for enhanced collaboration.
SIMILARITIES AND DIFFERENCES BETWEEN FDA AND EMA
While FDA and EMA are considered regulatory counterparts, there are a few key differences in their jurisdiction and authority as well as in how the organizations operate (see Appendix I for a comprehensive comparison between the agencies). FDA is a centralized regulatory body that oversees the evaluation of safety and efficacy of drugs approved in the United States and has a dedicated workforce and authority to issue guidance and make regulatory decisions on medications and medical devices. EMA is an independent agency of the European Union that is responsible for evaluating the safety and efficacy of drugs in Europe (EMA, n.d.-g). However, it does not have the authority to approve medications. Instead, EMA can issue guidance documents, review marketing authorization applications, and make authorization recommendations on medical products, which the European Commission then has the final decision as to whether they will be allowed to be marketed in the European Union (EMA, n.d.-g). Day-to-day operations at EMA are carried out by dedicated staff who rely on a network of experts from across Europe and collaboration with member states to pool resources and coordinate work to regulate medicines for use in humans (EMA, n.d.-q).
FDA and EMA each have regulatory policies in place to support and incentivize drug development for rare diseases and conditions.
Drug Review and Approval Process
The processes of drug evaluation and approval are fairly similar between FDA and EMA, though there are notable differences. FDA evaluates all drugs to be marketed in the United States1,2 and EMA evaluates almost all new drugs (orphan medicines, products with new active substances, products that are a significant innovation, and products that are in the interest of public health) (EMA, n.d.-b). Prior to the formal application for evaluation—called a new drug application (NDA) for FDA (FDA, 2022a) and a marketing authorization application (MAA) for EMA (EMA, n.d.-k)—a sponsor may request an expedited pathway or specific designation (e.g., orphan designation). Once the formal application is received, FDA assigns an internal review team to evaluate safety and efficacy (FDA, 2015a), while EMA assigns (co-)rapporteurs from member states to prepare an assessment report (EMA, n.d.-l). The FDA review team consists of staff experts in multiple disciplines (FDA, 2015a); they may also call on external advisory committees to provide opinions and recommendations (FDA, 2024d). EMA rapporteurs can establish multinational assessment teams in order to bring in experts from other member states (EMA, n.d.-j).
At EMA, the Committee for Medicinal Products for Human Use (CHMP), which is made up of experts nominated by EU member states, considers the assessment report that was prepared by the rapporteurs and attempts to reach a consensus opinion; if a consensus cannot be met, a vote is taken (EMA, n.d.-i). The CHMP’s opinion is sent to the European Commission, which reaches a final decision on marketing approval (EMA, n.d.-b).
Conclusion 5-1: Despite some key differences, FDA and EMA have similar approaches to the evaluation and approval of drugs for rare diseases. Given these parallel approaches, there are existing mechanisms for close collaboration between the two agencies, as well as opportunities for enhanced collaboration in the future that would allow each agency to retain sovereign authority and accountability in regulatory decision-making.
Standards of Evidence
As discussed in Chapters 2 and 3, FDA and EMA each define standards of evidence in different ways. By statute, FDA approval of a drug product requires a demonstration of substantial evidence of effectiveness, which has generally been interpreted as requiring at least two adequate and well-controlled studies. However, amendments have clarified that substantial effectiveness may be demonstrated with one adequate and well-controlled study along with confirmatory evidence. FDA and EMA approval processes require a risk-benefit assessment that requires consideration of many complex factors (e.g., therapeutic context, seriousness of condition). FDA has published a number of guidance documents that are relevant to rare disease drug development, including Demonstrating Substantial Evidence of Effectiveness for Human Drug and Biological Products (2019a), Rare Diseases: Considerations for the Development of Drugs and Biological Products (2023d), Benefit–Risk Assessment for New Drug and Biological Products (2023a), Demonstrating Substantial Evidence of Effectiveness with One Adequate and Well-Controlled Clinical Investigation and Confirmatory Evidence (2023b), and Rare Diseases: Natural History Studies for Drug Development (2019b). EMA has not issued general guidance on drug development for rare diseases and conditions, but there are a number of publications that apply to issues involved in rare disease drug development, including trials in small populations, real-world evidence, registry-based studies, single-arm trials, and use of one pivotal study in drug application (EMA, 2001, 2021a, 2021b, 2022b, 2023b).
Despite some of these differences, the two agencies often reach the same decisions regarding orphan products (see Figure 5-1). Of the 33 orphan new active substances (NASs) approved by FDA only, 7 were refused approval or withdrawn by the sponsor, while the rest were not submitted, approved outside the date range, or are still in review (see Figure 5-2). Of the six NASs approved by EMA only, one received a complete response letter, while the rest were not submitted or are still in review (see Appendix G for a full list of discordant decisions).
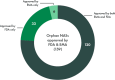
FIGURE 5-1
Number of orphan drugs approved by FDA and EMA from 2018 to 2022. NOTES: EMA = European Medicines Agency; FDA = U.S. Food and Drug Administration; NASs = new active substances. SOURCE: CIRS Data Analysis, 2024.
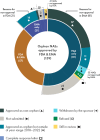
FIGURE 5-2
Discordance on the approval of orphan new active substances approved by FDA and EMA from 2018 to 2022a. a This figure was corrected after release of the prepublication version of the report to accurately reflect the orphan status of the drugs and decisions (more...)
Designation Programs
Both FDA and EMA offer an orphan designation for drugs that are targeted at rare diseases. The criteria are similar but not identical. FDA offers orphan designation to products that treat conditions affecting fewer than 200,000 individuals in the United States or that affect more than 200,000 individuals but there is no reasonable expectation that the cost of developing a drug for the condition would be recovered by sales of the drug.3 EMA offers orphan designation to products that treat conditions affecting not more than 5 in 10,000 individuals in the European Union or that affect more than 5 in 10,000 individuals but the market is unlikely to generate sufficient return on the investment. EMA further requires that the product targets a condition for which there is either no treatment available, or the product provides a “significant benefit” over available treatments. This significant benefit may be related to either improvements in clinical outcomes or improvements in patient care (e.g., ease of use).4 FDA also has a program for rare pediatric disease designation,5 while EMA does not have a designation specifically for rare pediatric conditions.
For the purposes of orphan designation, FDA and EMA define “condition” slightly differently. FDA requires that a product be targeted at a distinct condition, as determined by a variety of factors. A product targeted at a subset of a more common condition may be eligible if the drug itself has properties that make it inappropriate for patients with the more common version of the condition; for example, a drug that is only effective in patients with a specific biomarker may be eligible, given that those without the biomarker or drug-target would not be expected to respond (for example, mutationally-defined cancers).6 EMA also specifies that the targeted condition must be clearly distinct from other conditions, and states that differences in severity or stages do not make a condition distinct (European Commission, 2014). A treatment targeted at a subtype of the condition may be eligible if the characteristics of the subtype make the treatment ineffective for patients with a more common subtype of the condition, but biomarkers of a subtype are not currently accepted as evidence of a distinct condition (Thirstrup, 2023). For these reasons, it can be more difficult for a drug product to be granted and keep an orphan designation by EMA.
As stated in Chapter 1, orphan designation in the United States qualifies sponsors for incentives (see Box 1-1). Similarly, in the European Union, sponsors may also receive incentives including (EMA, n.d.-m):
- Reduced fees for regulatory activities, which may include reduced fees for protocol assistance, marketing-authorization applications, inspections before authorization, applications for changes to marketing authorizations made after approval, and reduced annual fees; and
- Potential 10 years of market exclusivity after approval.
Approval Rates for Orphan Designated Products
To understand the approval rates for orphan designated products, the committee commissioned work to examine FDA and EMA orphan designated products and marketing authorization applications and approvals of NASs to treat rare diseases (see Appendix D for full data analysis methodology). Between 2015 and 2020, more orphan NASs were submitted to FDA (184) than to EMA (123). Between 2015 and 2020, FDA approval rates for orphan NAS marketing applications were generally high (approaching 90 percent) (see Figure 5-3). EMA approval rates appeared slightly lower (closer to 80 percent). However, these differences could be accounted for differences in the number of granted orphan designations.
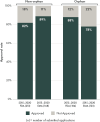
FIGURE 5-3
Approval rates for new active substance applications submitted to FDA and EMA between 2015 and 2020. NOTES: EMA = European Medicines Agency; FDA = U.S. Food and Drug Administration. SOURCE: CIRS Data Analysis, 2024.
When divided by therapeutic area (FDA office was used as a surrogate for therapeutic area), the number of applications was generally low, with the exception of oncology products, so the committee was unable to discern whether there are meaningful differences in approval rates across therapeutic areas by each agency (see Figures 5-4 and 5-5). These similar approval rates, despite the regulatory and other differences are consistent with the common FDA and EMA reliance on a range of data from randomized controlled trials (RCTs) to alternative and confirmatory data.
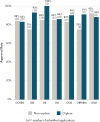
FIGURE 5-4
Novel approval rates for non-orphan and orphan new drug applications submitted to the Center for Drug Evaluation and Research from 2015 to 2020 by office. NOTES: OCHEN = Office of Cardiology, Hematology, Endocrinology and Nephrology; OID = Office of Infectious (more...)
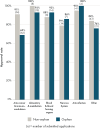
FIGURE 5-5
Approval rates for non-orphan and orphan new active substance applications submitted to European Medicines Agency from 2015 to 2020 per therapeutic area. NOTE: Other = other therapeutic areas not described in the top five therapeutic indications list. (more...)
Expedited Pathways
FDA and EMA both offer a number of expedited pathways that allow products to be approved on a shorter timeline or with preliminary or limited data or both. There are many similarities across the expedited pathways in both agencies (see Figure 5-6). The pathways fall into several categories: approval based on a shortened review timeline, approval based on preliminary data, and approval based on limited data.
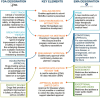
FIGURE 5-6
FDA and EMA expedited programs. NOTES: Drugs may qualify for more than one expedited program. For U.S. programs, drugs may be eligible for all of these programs, provided they meet the criteria. For EU programs, medicines may be eligible for most of these (more...)
Approval on a Shortened Review Timeline
FDA’s breakthrough therapy designation and EMA’s Priority Medicines (PRIME) scheme are similar programs; they are both designed to assist sponsors of products developed for conditions with an unmet need and offer the potential for shortened review. Breakthrough therapy designation is for products that are intended to treat a serious or life-threatening condition and for which preliminary clinical evidence indicates a substantial improvement on a clinically significant endpoint over available therapies (FDA, 2014). For PRIME designation, an applicant must provide data that demonstrate a meaningful improvement of clinical outcomes (EMA, n.d.o). Breakthrough therapy designation comes with intensive guidance on drug development, meetings and communication with FDA staff, and the potential for accelerated approval or priority review (FDA, 2014). A PRIME designation offers similar benefits including meetings with EMA experts, iterative and expedited scientific advice, and the potential for accelerated assessment (EMA, n.d.-o).
FDA has one program that shortens review time, called Priority Review. EMA has one program, called Accelerated Assessment. Priority Review, for products aimed at a serious condition that demonstrate a significant improvement in safety or effectiveness, which offers review of the application in 6 months (FDA, 2014). EMA’s accelerated assessment is for products that are of “major public health interest,” particularly ones that involve innovations or improvements for unmet needs. The program reduces the timeframe for application assessment from 210 days to 150 days (EMA, n.d.-a).
Approval Based on Preliminary Data
Both agencies have a mechanism that allows a product to be approved with preliminary data, with confirmatory data required to be provided after the approval. FDA’s accelerated approval pathway can be used when a product has a meaningful advantage over available therapies, and evidence demonstrates an effect on a surrogate or intermediate endpoint that is reasonably likely to predict clinical benefit. This pathway allows for a shorter development timeline and requires sponsors to collect data after approval to confirm the clinical benefit (FDA, 2014). EMA’s conditional marketing authorization is used when there is an unmet need and the benefits of making the product available to the public outweigh the risks; sponsors are required to collect additional data after approval to confirm the benefit-risk analysis (EMA, n.d.-e).
Approval Based on Limited Data
Only EMA has a mechanism for approving a product for which comprehensive data on safety and efficacy are not available. Under the exceptional circumstances pathway, EMA may grant approval to a product if it is not possible to collect comprehensive data because the condition is too rare, because of the current state of scientific knowledge, or because it would be unethical (EMA, n.d.-h; Prilla, 2018).
Both agencies have the legal authority to exercise a great degree of flexibility in the amount and type of data necessary for rare disease product approval.
Sponsor Engagement
Both agencies offer sponsors the opportunity to engage throughout the development and approval process. Sponsors may request a meeting with FDA at any time during drug development. In general, communication with the agency is through the regulatory project manager, and sponsors are discouraged from contacting reviewers directly (FDA, 2017). Sponsors may solicit advice on a variety of topics and may also request a formal meeting at critical junctures of development (FDA, 2023c). EMA offers preparatory meetings for sponsors early in the development process in order to avoid major issues, and sponsors are encouraged to reach out at any time for feedback. Scientific advice is available from EMA for a fee; the fee can be waived for orphan medicines, smaller sponsors, and in the case of public emergencies (EMA, n.d.-p).
While the committee found little evidence pointing to systemic differences between the agencies with regard to their capacity to engage with sponsors, in qualitative interviews conducted by National Academies staff, some interviewees noted that the structure of FDA allows for more accessible engagement between regulators and sponsors than EMA (see Appendix E for full methodology and results). Interviewees said that the timeline and structure of advice from FDA was helpful. In comparison, interviewees noted that the European process of engaging national regulatory bodies within the European Union before submitting a marketing authorization application to EMA was a barrier to agency engagement.
Expert Engagement
FDA uses advisory committees and special government employees to engage experts during drug development (FDA, 2016). EMA can also engage with experts, but engagement on the European side usually occurs through its Scientific Committees or Scientific Advisory Groups (EMA, n.d.-f). EMA also has a program in a pilot phase that intends to provide a mechanism for EMA and experts to collaborate (EMA, n.d.-d). Although there are differences in how FDA and EMA engage with scientific experts, there is no evidence that the differences are systematically impactful for rare disease drug sponsors.
Patient Engagement Programs
FDA and EMA both have several mechanisms for engaging with patients, caregivers, and patient groups. FDA uses the Patient-Focused Drug Development program as a systematic approach for incorporating patient perspectives into drug development (FDA, 2024b), while EMA uses the Patients’ and Consumers’ Working Party to provide a platform for the exchange of information between the agency and patients (EMA, n.d.-n). Individual patients can serve on FDA advisory committees and provide advice to FDA through the Patient Representative Program (FDA, 2024a), while at EMA patients and organizations can apply to be part of a database of patients who can be called on by EMA during the drug evaluation process (EMA, 2022a). Although there are differences in how each agency engages with patients, there is no evidence that one agency’s methods are superior.
Rare Disease Programs
The discrepancy between programs in the Unites States and the European Union also presents special challenges for developers. For example, using the Rare Disease Cures Accelerator-Data and Analytics Platform (RDCA-DAP®) in the United States could raise specific new review challenges in the European Union as there may be additional constraints in data usability and differences in data acceptance.
The data gathered by the committee indicate that there is significant overlap between FDA and EMA regulatory decision-making. Discrepancies between the agencies seem to be based on which location a marketing authorization for a rare disease product was first submitted. For the most part, the agencies seem to rely on the same data for marketing authorization applications. Programs, such as RDCA-DAP®, may benefit U.S.- and EU-based drug development programs, but only time will tell.
Receptiveness to Alternative and Confirmatory Data and Novel Methods of Data Analysis
In general, both agencies have demonstrated an openness to the use of alternative and confirmatory data (e.g., natural history studies) as well as novel approaches for data analysis (e.g., Bayesian statistical methods).
Enhancing Regulatory Transparency
EMA publicly shares information on marketing authorization submissions7 (European Union, 2004); FDA does not. FDA does not share information about unsuccessful sponsor applications; EMA does. These differences appear to stem from different legal frameworks for transparency in the United States and the European Union. Information about the reasons why sponsor applications are unsuccessful or successful would help guide companies in the rare disease space and avoid unnecessary duplication of effort and unnecessary exposure of the small number of patients with rare diseases to unproven drugs in clinical trials. In a worst-case scenario, the transparency of data relating to drug evaluation serves an important public benefit, but sponsors may see transparency as an impediment to their commercial competitiveness or, in the case of corporate sponsors, to their corporate reputation.
It is common that multiple sponsors investigate candidate drugs for the same indication, including some rare disease indications, simultaneously competing on speed to be the first to receive regulatory approval. Data that reveal agency views early on in a drug development program could be used by competitors to their advantage (e.g., modifying a scientific approach for a competitive program). Data may also be used by investigators for different indications, presumably increasing their chance of success. Arguably this improves the efficiency of drug development by allowing companies to avoid the mistakes of their peers, but it also may be seen as providing an unfair advantage to second-place sponsors in a competitive environment.
In both the United States and the European Union, data transparency on the part of regulators relies on complex policy and legal frameworks which are different in each jurisdiction. While the total amount of public data about marketing application review and submission has dramatically improved over time in both jurisdictions, important differences and gaps remain. As of 2024, only EMA posts public assessment reports, which describe the basis for regulatory opinions on how medicines should be used. As a general rule, EMA also publishes clinical reports submitted by sponsors in support of regulatory applications (EMA, n.d.-c). In contrast, FDA does not post complete response letters, which may include information on why a marketing authorization submission was considered by the agency to be inadequate or incomplete. The absence of such information may lead sponsors to repeat the methodological or regulatory missteps of other sponsors to the detriment of patients.
Conclusion 5-2: To meet the needs of rare disease patients and their caregivers, there is an ethical obligation on the part of regulatory agencies to share relevant information on the review and approval of drugs to treat rare diseases and conditions. If researchers and sponsors working on rare disease drug development had a better understanding of the reasons for successes and failures of marketing authorization applications, they could better innovate new therapeutics that have a higher likelihood of reaching patients. Additionally, more transparency would enhance public understanding and confidence in the important work carried out by regulatory agencies.
There have been calls for increased transparency at FDA. For example, the 2010 Transparency Initiative Task Force was launched in response to a memo issued by President Obama in 2009 to federal agencies and departments asking them to take steps to create more transparency, public participation, and collaboration (FDA, 2010). The task force published reports in 2010 and 2011 which contained draft proposals and action items, many of which sought to increase disclosure of the elements noted above (FDA, 2015b,c). While many of the task force’s recommendations were adopted, none of those covering product applications related to disclosure of applications and regulatory decisions were addressed. Information regarding the submission of drug marketing authorization applications and reasons for regulatory decision-making are generally not publicly disclosed by FDA. In the absence of such disclosure, the public must rely on sponsors to voluntarily share information on why a marketing authorization application was not approved, which is an unreliable source given that sponsors are not required to disclose this information and, as evidenced by the lack of press releases in response to complete response letters, may have strong disincentives for public sharing (Lurie et al., 2015). Third parties that aggregate disclosures by pharmaceutical companies may offer information for those who can pay for the cost of such services.
FDA should follow the early lead of EMA and adopt greater transparency with regard to regulatory rationale for approval or disapproval of marketing authorization submissions for rare disease drug products. Increased transparency at FDA would have multiple benefits:
- Sponsors would have a better understanding of the use of regulatory flexibilities for advancing the review and approval of drugs to treat rare diseases and conditions and of the reasons for the successes and failures of marketing authorization applications, which would enable them to better learn from past experience and develop new therapeutics that have a higher likelihood of reaching patients;
- Patients and patient groups would be better informed about progress in the development of much-needed treatments and could build on regulatory experiences across disease areas; and
- The public would have a better understanding of and increased confidence in the work of the agency.
To this end, the committee identified the following focus areas for which enhanced public disclosure would improve the likelihood of success for rare disease drug development:
- The impact of new and ongoing programs within FDA that support drug development for rare diseases and conditions to improve the regulatory/approval process.
- Lessons learned across therapeutic areas on the use of available regulatory flexibilities.
- Types of information relevant for public understanding of when and how available regulatory policies are applied for rare disease drug products.
Practical Considerations
The committee recognizes there are multiple barriers to achieving greater transparency on the part of FDA, including laws that govern how the agency can or cannot share information. Some have argued that FDA has broad discretion on what is considered confidential.8 Sharfstein et al. (2017) have suggested that FDA does not require an act of Congress to make progress on publicly sharing information about marketing authorization applications for drug products (see Box 5-1 for a case study on FDA using its discretion to share information with the public). FDA has the ability to incentivize and facilitate pathways for enhancing information sharing, but there are practical and legal considerations, which may require modification of some of the laws that restrict the agency from sharing certain types of information. For these reasons, the committee recognizes the need for more transparency on the part of FDA, but acknowledges that additional consideration, assessment, and legal review are needed to determine how such measures should be implemented.
BOX 5-1
FDA Demonstration of Discretion about Disclosure: COVID-19 Pandemic.
RECOMMENDATION 5-1: The U.S. Food and Drug Administration (FDA) should take steps to make relevant information on marketing authorization submissions, review milestones, approval and negative review decisions (refusal to file, clinical hold, and complete response letters), and the use of regulatory flexibilities for rare disease drug products publicly available and easily accessible to inform sponsors, patients, researchers, and reviewers on decision-making rationales and when and how available policies are applied. While the committee acknowledges the legal challenges surrounding disclosure of information, actions should include, but not be limited to:
COLLABORATION BETWEEN REGULATORY AGENCIES
Despite some key differences, FDA and EMA have similar approaches to the evaluation and approval of drugs for rare diseases. Given this overlap, there are several existing mechanisms for close collaboration between the two agencies as well as opportunities for enhanced collaboration in the future. Since the signing of a confidentiality agreement in 2003, which permits the agencies to share nonpublic information, including confidential commercial information, FDA and EMA have created multiple formalized mechanisms to facilitate communication and collaboration. These collaborations cover a wide range of topics and activities, including scientific advice, orphan designations, marketing authorizations, post-authorization requirements, inspections, pharmacovigilance, guidance documents, and other topics (EMA, 2024d) (see Figure 5-7). In addition to these formal mechanisms, which are further discussed below, there is ongoing informal communication between FDA and EMA, which enables staff to observe each other’s scientific meetings, participate in each other’s trainings, and share information on an ad hoc basis (Lee et al., 2023).
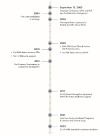
FIGURE 5-7
20 years of EU/U.S. collaboration on medicines regulation. SOURCE: Adapted from European Medicines Agency: 20 years EU/US collaboration on medicines regulation, https://www.ema.europa.eu/system/files/documents/other/2023-09_ema-fda-20-years-arrangement_en.pdf (more...)
Clusters
One of the primary formal mechanisms for collaboration between EMA and FDA is so-called “clusters”—regular virtual meetings between EMA and FDA staff focused on specific topics and therapeutic areas that would benefit from an “intensified exchange of information and collaboration” (EMA, 2024a). Documents exchanged within clusters may include draft guidances/guidelines, assessment reports, review memos, and meeting minutes. The agencies typically set the agenda for what is discussed at a cluster meeting. However, sponsors also have the option of asking that a drug program or topic be discussed. Topics discussed within clusters range from emerging scientific and ethical issues to challenges in product development to issues with the review of marketing authorization applications (see Figure 5-8).
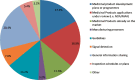
FIGURE 5-8
Top topic areas discussed in clusters. NOTES: Values shown are from aggregate results from a compiled list of the topic areas identified for all clusters. MAA = marketing authorization application; NDA = new drug application. SOURCE: Teixeira et al., (more...)
Since the first EMA–FDA cluster on oncology-hematology medicines was established in 2004, clusters have grown in number and size (EMA, 2024a). Clusters were initially designed as a forum for dialogue between FDA and EMA but have since expanded to include other regulators, including those from Canada, Australia, and Japan (EMA, 2024a; Lee et al., 2023). As of 2024, there were 31 clusters that cover a variety of topics and disease areas, including several that are relevant for rare diseases and conditions (see Table 5-1). In addition to regularly scheduled meetings, the relationships that are built through the work of the clusters have facilitated additional ad hoc conversations on time-sensitive issues (Tyner et al., 2023).
TABLE 5-1
Examples of Clusters Relevant for Rare Diseases.
Impact of Clusters
While the impact of the clusters on marketing authorization approval rates or regulatory harmonization cannot be easily quantified, discussions do help to disseminate information among regulatory staff and to facilitate better understanding of agency perspectives and approaches and can serve as a form of “peer review” among regulators (Teixeira et al., 2020). EMA and FDA staff have noted that clusters provide opportunities for regulators to discuss challenging issues for rare disease drug review and approval, such as novel endpoints, statistical methodologies, use of regulatory flexibilities, and post-marketing requirements (Tyner et al., 2023).
A 2017 survey conducted by EMA and FDA indicated that cluster participants from across regulatory agencies (EMA; FDA; Health Canada; Ministry of Health, Labour, and Welfare of Japan/Pharmaceuticals and Medical Devices Agency; Therapeutic Goods Administration; Swissmedic; and the European Directorate for the Quality of Medicines & HealthCare) found that this collaborative mechanism helps inform regulatory decision-making (Teixeira et al., 2020). Other reported impacts of the cluster program by the agencies include the establishment of new initiatives, such as the Patient Engagement Collaborative (FDA, 2024e), the FDA–EMA pediatric Common Commentaries (FDA, 2024c), a collaborative approach to facilitate pediatric drug development for Gaucher disease (EMA, 2017), joint publications and workshops, resolution of divergences, and professional growth among participants (Teixeira et al., 2020).
Patient Engagement Collaborative
In response to requests from patient communities, the Patient Engagement Collaborative (PEC) was established in 2018 as a joint project of FDA and the Clinical Trials Transformation Initiative (CTTI), a public–private partnership of Duke University and FDA (CTTI, 2023). PEC was modeled after EMA’s Patients’ and Consumers’ Working Party (FDA, 2024e), which facilitates EMA engagement with patients and consumer organizations and provides recommendations to EMA (EMA, n.d.-n). While PEC is not considered an FDA advisory committee and does not discuss specific medical products or treatments, meetings are an opportunity for patient communities to share views, ideas, and experiences with FDA and CTTI in order to inform communication, education, and engagement activities (CTTI, 2023).
Pediatric Common Commentary
In 2012, EMA and FDA began issuing common commentaries to share unofficial high-level summaries of discussions held during Pediatric Cluster meetings. Publicly and confidentially issued common commentaries may highlight scientific, ethical, or regulatory topics related to pediatric product development where EMA and FDA are working toward a harmonized view. Common commentaries might also identify discordant EMA and FDA agency views (FDA, 2024c). Common commentary for pediatric drug development has been critically important given the small patient populations for rare diseases, including childhood cancer.11
In 2021, EMA and FDA published a common commentary on pediatric oncology drug development, which lays out common issues requested for discussions by both agencies so that sponsors have an opportunity to address key issues early on and enable simultaneous submission of paediatric investigation plans (PIPs) to EMA and initial pediatric study plans (iPSPs) to FDA (FDA and EMA, 2021). Common commentary has also been issued on other topics relevant to rare disease, including discussions about trial design for Gaucher disease (see Box 5-2).
BOX 5-2
Gaucher Disease: A Strategic Collaborative Approach from EMA and FDA.
Opportunities for Clusters
While clusters help inform drug development and approval processes and provide a valuable forum for collaboration between the regulatory agencies, there is substantial unfulfilled potential. The impact of the clusters on the drug development ecosystem may be limited by the fact that cluster discussions are largely reactive, focused on specific issues, such as an existing development plan or safety concern. With the exception of a handful of targeted activities, such as the collaborative approach for Gaucher disease (see Box 5-2), clusters are not designed to more prospectively address challenges for rare disease drug development.
And yet, clusters have the potential to build and expand on current activities. In addition to strategic collaborative approaches for other rare diseases, clusters provide a mechanism for EMA and FDA to address early-stage barriers to successful marketing authorization approval that cut across therapeutic areas. The existing cluster structure could additionally be used to prospectively address common challenges for rare disease drug development, thereby harmonizing and streamlining the orphan designation and drug evaluation process. For example, increased use of EMA and FDA common commentary to include issues related to how drug development tools, extrapolation of efficacy, innovative trial design, new methodologies, and use of alternative and confirmatory data sources, such as patient registries and real-world data sources, could be used to strengthen rare disease development plans across therapeutic areas and would give sponsors more clarity on the regulatory considerations for rare disease drug programs.
An expansion and shift in focus on the part of the clusters to include prospective issues facing rare disease drug development would align with current objectives, build on existing collaborative efforts, and help inform regulatory decision-making. It is also important to note that this type of approach would further enhance scientific exchange and trust building between FDA and EMA staff and could have synergistic and cumulative impact—benefiting interagency cooperation, scientific review, and ultimately public health across therapeutic areas and drug types for disease both rare and common.
There may be concerns on the part of the agencies or sponsors that such an approach could constrain discussions if information were to be made publicly available. However, common commentary is intended to highlight issues where FDA and EMA are working toward a more harmonized view (or to provide clarity for sponsors on areas where the agency may be unable to reach alignment). These documents are not binding and could serve as a valuable source of information for sponsors, researchers, and patient groups. Agencies could use these documents to share common thinking on how they weigh urgency and pragmatic limitations against the need for data to support marketing authorization applications for rare diseases and conditions and considerations for how FDA and EMA might address areas of misalignment, such as on clinical trial endpoints, the determination of non-inferiority (or similarity) margins, use/acceptance of novel statistical methodologies, and totality-of-evidence determinations.
RECOMMENDATION 5-2: To facilitate the efficient global development of orphan drugs, the U.S. Food and Drug Administration (FDA) and the European Medicines Agency (EMA) should build on the existing clusters relevant for rare diseases by undertaking the following:
Parallel Scientific Advice
Established in 2005, the Parallel Scientific Advice (PSA) program is a voluntary mechanism through which FDA and EMA can concurrently provide scientific advice to sponsors during the development of new drugs, biological products, vaccines, or advanced therapies (EMA and FDA, 2021). The goals of the PSA program are to: (1) increase dialogue early on in the product lifecycle, (2) deepen understanding of regulatory decisions, (3) optimize product development, and (4) avoid unnecessary or duplicative testing (Thor et al., 2023). The program does not guarantee alignment between EMA and FDA, but it can offer a number of potential benefits for sponsors, including agency convergence on approaches to development, a better understanding of each agency’s concerns and requirements, and the opportunity for sponsors and agencies to ask and answer questions (Thor et al., 2023).
The process is typically requested by the drug sponsor but may also be initiated by EMA or FDA (EMA and FDA, 2021). Acceptance of the request by both agencies is required to proceed. The timelines for PSA are intended to align with what would be expected of similar processes to enable sponsors to predict when advice can be expected from EMA and FDA (see Table 5-2). A 5-year review of PSA indicated that the timelines were comparable to timelines for EMA’s Scientific Advice Working Party, which provides scientific advice and protocol assistance for sponsors, and to timelines for FDA Type B meetings (Thor et al., 2023).12
TABLE 5-2
Timeline for Parallel Scientific Advice.
Products designed for the treatment of rare diseases and conditions, as well as those targeted to address unmet medical need and treat special populations, may be good candidates for PSA due to their potential public health value and because they may be more likely to break new ground in terms of innovative science and treatment modalities (Kweder, 2022). Special consideration is given to products that may offer public health value, such as products that address an unmet medical need or are intended to treat rare diseases or special populations (Kweder, 2022). Products that are selected for PSA fall into a several categories (see Figure 5-9).
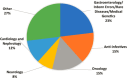
FIGURE 5-9
Accepted Parallel Scientific Advice requests (N=26) by product category from 2017 to 2021. SOURCE: Thor et al., 2023. CC BY 4.0 http://creativecommons.org/licenses/by/4.0/.
Impact of PSA Program
Regulatory staff who have overseen, coordinated, and participated in the PSA program describe it as “remarkably productive and positive for all parties” (Thor et al., 2023, 660). While this experience is difficult to quantify, regulators have said that the discussions between EMA and FDA have helped expand their thinking and given them opportunities to discuss ways to address common challenges, particularly for therapeutic areas for which there is limited experience or difficult scientific questions (Thor et al., 2023). For products that do not have a straightforward regulatory path, discussions between FDA and EMA allow for an exploration of alternative or innovative approaches and can add value to the advice given to the sponsor (Thor et al., 2023).
Despite the potential benefits of the PSA program, only a handful of sponsors apply each year. A review published by FDA and EMA staff showed that between 2017 and 2021, the agencies received 37 PSA requests, of which 26 (70 percent) were accepted (see Figure 5-9). Four of the sponsors withdrew their requests or chose not to proceed. Of the accepted PSA requests, 23 percent were for gastroenterological, inborn errors, rare diseases, and medical genetics13 (Thor et al., 2023). Compared to the number of marketing applications that are submitted and approved to EMA and FDA each year, few applications use the PSA program. Between 2017 and 2020, the PSA program accepted 19 submissions (see Figure 5-10). Similar to other therapeutic products, the majority of orphan-designated products submitted for marketing authorization do not receive PSA.
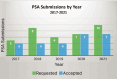
FIGURE 5-10
Parallel Scientific Advice submissions by year from 2017 to 2021. NOTE: PSA = Parallel Scientific Advice. SOURCE: Adapted from Thor et al., 2023. CC BY 4.0 http://creativecommons.org/licenses/by/4.0/.
When the PSA program was launched in 2005, uptake was so slow that the agencies considered ending the program (Gingery, 2021). Similar concerns have arisen for a related pilot project that offers PSA for complex generic drugs; FDA reported that some applicants were hesitant to join the program due to misperceptions about the consequences of conflicting advice, questions about the value of the advice because it is non-binding, and concerns about sharing confidential information (Ibrahim, 2024).
To better understand some of the real or perceived concerns that sponsors may have regarding the PSA program, the Committee considered results from a series of qualitative interviews carried out by National Academies staff, which showed that interviewees were aware of the PSA program but had varying opinions about its potential value (see Appendix E for full methodology and results). One interviewee stated, “PSA meetings can be protracted, discordant, and non-binding. Sponsors need them to be rapid, concordant, and binding.” Other interviewees listed issues with the PSA program that stem from the non-binding nature of PSA, potential for misaligned advice between EMA and FDA, issues with timing of the program, and concerns that in the event of divergent opinions, the agencies may follow the lead of the agency with the higher evidence threshold.
Discordant scientific advice between FDA and EMA can create confusion or uncertainty on the part of sponsors about how to proceed with a development program and could particularly impact smaller companies that are developing drugs to treat rare diseases and conditions. Smaller companies often have a limited product pipeline and contingent funding. As such, they are more likely to seek direction from a single regulatory agency that they consider most likely to view their development program favorably; and are more likely to disfavor use of PSA.
Another reason for the lack of uptake could be that the logistics of the PSA program—including timing, components of a request, and communicating with both agencies—may be difficult to navigate for companies, particularly those that are smaller or less experienced with regulatory submissions. Applying for PSA requires staff time, resources, and know-how. Additionally, it may be the case that for some companies, early access to the U.S. market is a key factor in decision making and the priority for development programs.
Participants at a public workshop convened by FDA and the DukeMargolis Center for Health Policy in 2017 suggested that more transparency on the requirements and timelines for the PSA program would help address some of the issues leading to lack of uptake (Richardson et al., 2018). FDA has made efforts to share information on the PSA program, but Thor et al., observed that by 2023, the agencies and pharmaceutical companies still had not widely promoted or publicized the program (Thor et al., 2023).
Despite potential issues with the PSA program, a few orphan drug products have used PSA and received marketing authorization (see Box 5-3).
BOX 5-3
Select Examples of Drug Products to Treat Rare Diseases and Conditions that Received Parallel Scientific Advice.
Opportunities for the PSA Program
There are limited data available on the approval rate of products in the PSA program, but seeking and complying with scientific advice from individual agencies has been positively correlated with the success of an application (Hofer et al., 2015; Regnstrom et al., 2009; Welch, 2015). One study found that two-thirds of applicants who received scientific advice were advised by EMA to alter their trial designs (Hofer et al., 2015). Of the applicants that changed their trial designs, 86 percent received marketing authorization; of those that did not comply with the scientific advice given, only 41 percent received authorization (Hofer et al., 2015).
As discussed above, under the current circumstances there may be pragmatic financial or business reasons that companies do not apply for PSA. In part, this may be due to business priorities within a company or a lack of resources. To overcome the practical hurdles and perceived drawbacks, it may be that sponsors need additional incentives for requesting PSA. An understanding of the resources required for sponsors to apply for PSA and publicly available reporting on the impact of PSA over time would better enable the agencies to identify appropriate mechanisms for increasing uptake of the use of PSA for companies seeking to develop new therapeutics to treat rare diseases and conditions.
Additionally, there may be ways to integrate the use of PSA more dynamically into the review process, which could help streamline procedures and timelines for rare disease programs. For example, offering PSA to sponsors as an option to consider at the point of orphan drug designation, after completion of Phase 1 and Phase 2 trials, or other key points throughout the drug development process. FDA and EMA discussions with the sponsor would be non-binding for any party but would provide a venue for the parties to prospectively discuss key issues and potential solutions, learn, and evolve in a collaborative and adaptive way.
When different experts discuss the available evidence for a given marketing authorization application, it is not unlikely that some may set the bar higher than others. From the perspective of drug developers, there may be a risk that the more demanding view will prevail given that evidence should be robust, and standards should not be lowered. This tension—whether real or perceived—could be alleviated by increased transparency on the part of FDA and EMA regarding the different agencies’ relative positions and the non-binding nature of scientific advice through PSA.
Conclusion 5-3: Despite underuse of the PSA program and lack of available evidence related to its impact, the committee acknowledges and expects that, in principle, concurrent scientific discourse through PSA should better enable more streamlined clinical trials, regulatory review, and approval of drugs to treat rare diseases and conditions.
The committee determined that EMA and FDA should undertake an assessment of the PSA program to include a review of how the types of scientific advice each agency has or would consider providing given the unmet medical need for rare disease patients and what can be accomplished by sponsors within a reasonable period of time. More generally speaking, increased transparency on the part of FDA and EMA on what is commonly held between the agencies regarding the types of questions asked and considerations for study design, data collection and analysis, and use of innovative tools would give sponsors better clarity on the regulatory pathway forward and help ensure that clinical trials are conducted in a scientifically rigorous and ethical manner and that rare disease patients are not exposed to unnecessary or duplicative studies.
RECOMMENDATION 5-3: The U.S. Food and Drug Administration (FDA), along with the European Medicines Agency (EMA) and other key stakeholders, should assess the impact of the Parallel Scientific Advice (PSA) program over the past decade on drug development for rare diseases and conditions, publicly share the results of this assessment, seek sponsor input on approaches to improve and enhance the use and utility of the program, and take action to increase access, use, and impact of the PSA program going forward. This assessment and plan for improvement should include:
If the actions taken do not lead to an increased use and greater impact of the PSA program within a 5-year period after the assessment and improvement plan has taken place, FDA should implement other mechanisms for parallel advice between FDA and EMA on drug development programs for rare diseases and conditions.
OPPORTUNITIES FOR ENHANCED COLLABORATION
FDA and EMA have the opportunity to serve as enablers of drug development for rare diseases and conditions by providing additional clarity on how alternative and confirmatory data (ACD) can be used to inform regulatory decision-making, reducing uncertainty about how to implement new trial designs and methodologies, and, when permissible, publicly sharing relevant information on marketing authorization submissions and approvals. Additionally, the agencies can work collaboratively with other organizations to proactively consider the use of new trial designs and methodologies in incorporating ACD into rare disease drug marketing applications.
Conclusion 5-4: FDA and EMA have mechanisms in place to coordinate and collaborate in ways that better enable drug review and approval, but more is needed to stimulate innovation and drug development for rare diseases and conditions. There is an opportunity for the agencies to take a more proactive approach when engaging with one another through existing collaborative mechanisms and to more effectively communicate this work with sponsors, patients, and other stakeholders. Collaboration between EMA and FDA can help maximize opportunities for each agency to leverage the other’s expertise and experience, share learnings, streamline the review process, and work through scientific and regulatory bottlenecks to help pave the way for new treatments for people living with rare diseases and conditions. 14
It is not uncommon for innovations in rare disease drug development to be a vanguard for applications across therapeutic areas, so lessons learned in the rare disease space could be considered by the agencies on a broader scale. For example, most rare diseases (roughly 80 percent) have well understood genetic precursors (Nature Genetics, 2022). Gene therapy—an intervention that modifies or manipulates the expression of a person’s gene to treat or cure disease—offers promise for the treatment of gene-based conditions. Although the phenotypic presentation varies, advancements in biomedical technologies (e.g., CRISPR), improved understanding of disease etiology and trajectory, and the high cumulative burden (health-related and economic) of rare diseases have led to growing interest in the applicability of gene therapies. Additionally, many rare diseases are not amenable to small molecule therapies although there are some exceptions (e.g., Imatinib for acute lymphoblastic leukemia) (Biondi et al., 2018; Papaioannou et al., 2023). The use of CRISPR (clustered regularly interspaced short palindromic repeats) and adeno-associated viruses in gene therapy is now being pursued by multiple drug sponsors for the treatment of rare and chronic diseases and conditions.
Over the past 20 years, there has been a shift in thinking on the part of FDA and EMA, which were founded to ensure that drug products are safe and effective. In addition to playing a critical role in protecting human health, the agencies serve as enablers by actively promoting scientific and technological innovation for advancing drug development. To meet the future needs of people living with rare diseases and conditions, it is important for the ecosystem that the regulatory agencies have the knowledge and capacity to review and assess the new innovative products they are charged with regulating. This will require thinking ahead to what the future will look like a decade from now—ensuring that there are experts with transversal knowledge across rare diseases and conditions and that people with lived experience are fully integrated throughout the regulatory process, to better enable patient-centered collaboration across and within the agencies.
Conclusion 5-5: Continued and intensified collaboration between FDA and EMA through the clusters, PSA program, and other shared initiatives has the potential to accelerate and scale up drug development for rare diseases and conditions by minimizing the number of trials conducted, improving the quality of trial design (i.e., analytical methods, study endpoints, use of biomarkers, safety mitigation approaches), and providing more clarity for sponsors on the evidence that is necessary for demonstrating that drugs are safe and effective.
EMA and FDA individually and collectively have an opportunity to strengthen their roles as facilitators of drug innovation, sharing insights of lessons learned within and across the agencies, as well as with researchers, drug sponsors, providers, patients, and their caregivers. The committee hopes that through the adoption of the recommendations in this report, the regulatory process for rare diseases will become more transparent, work more smoothly and collaboratively, and ultimately lead to more therapies for patients living with rare diseases and conditions.
SUMMARY OF CONCLUSIONS AND RECOMMENDATIONS
Conclusion 5-1: Despite some key differences, FDA and EMA have similar approaches to the evaluation and approval of drugs for rare diseases. Given these parallel approaches, there are existing mechanisms for close collaboration between the two agencies, as well as opportunities for enhanced collaboration in the future, that would allow each agency to retain sovereign authority and accountability in regulatory decision-making.
Conclusion 5-2: To meet the needs of rare disease patients and their caregivers, there is an ethical obligation on the part of regulatory agencies to share relevant information on the review and approval of drugs to treat rare diseases and conditions. If researchers and sponsors working on rare disease drug development had a better understanding of the reasons for successes and failures of marketing authorization applications, they could better innovate new therapeutics that have a higher likelihood of reaching patients. Additionally, more transparency would enhance public understanding and confidence in the important work carried out by regulatory agencies.
Conclusion 5-3: Despite the underuse of the PSA program and lack of available evidence related to its impact, the committee acknowledges and expects that, in principle, concurrent scientific discourse through PSA should better enable more streamlined clinical trials, regulatory review, and approval of drugs to treat rare diseases and conditions.
Conclusion 5-4: FDA and EMA have mechanisms in place to coordinate and collaborate in ways that better enable drug review and approval, but more is needed to stimulate innovation and drug development for rare diseases and conditions. There is an opportunity for the agencies to take a more proactive approach when engaging with one another through existing collaborative mechanisms and to more effectively communicate this work with sponsors, patients, and other stakeholders. Collaboration between EMA and FDA can help maximize opportunities for each agency to leverage the other’s expertise and experience, share learnings, streamline the review process, and work through scientific and regulatory bottlenecks to help pave the way for new treatments for people living with rare diseases and conditions. 15
Conclusion 5-5: Continued and intensified collaboration between FDA and EMA through the clusters, PSA program, and other shared initiatives has the potential to accelerate and scale up drug development for rare diseases and conditions by minimizing the number of trials conducted, improving the quality of trial design (i.e. analytical methods, study endpoints, use of biomarkers, safety mitigation approaches), and providing more clarity for sponsors on the evidence that is necessary for demonstrating that drugs are safe and effective.
RECOMMENDATION 5-1: The U.S. Food and Drug Administration (FDA) should take steps to make relevant information on marketing authorization submissions, review milestones, approval and negative review decisions (refusal to file, clinical hold, and complete response letters), and the use of regulatory flexibilities for rare disease drug products publicly available and easily accessible to inform sponsors, patients, researchers, and reviewers on decision-making rationales and when and how available policies are applied. While the committee acknowledges the legal challenges surrounding disclosure of information, actions should include, but not be limited to:
RECOMMENDATION 5-2: To facilitate the efficient global development of orphan drugs, the U.S. Food and Drug Administration (FDA) and the European Medicines Agency (EMA) should build on the existing clusters relevant for rare diseases by undertaking the following:
RECOMMENDATION 5-3: The U.S. Food and Drug Administration (FDA), along with the European Medicines Agency (EMA) and other key stakeholders, should assess the impact of the Parallel Scientific Advice (PSA) program over the past decade on drug development for rare diseases and conditions, publicly share the results of this assessment, seek sponsor input on approaches to improve and enhance the use and utility of the program, and take action to increase access, use, and impact of the PSA program going forward. This assessment and plan for improvement should include:
If the actions taken do not lead to an increased use and greater impact of the PSA program within a five-year period after the assessment and improvement plan has taken place, FDA should implement other mechanisms for parallel advice between FDA and EMA on drug development programs for rare diseases and conditions.
REFERENCES
- Biondi A, Gandemer V, Lorenzo PD, Cario G, Campbell M, Castor A, Pieters R, Baruchel A, Vora A, Leoni V, Stary J, Escherich G, Li C-K, Cazzaniga G, Cavé H, Bradtke J, Conter V, Saha V, Schrappe M, Valsecch MG. Imatinib treatment of paediatric Philadelphia chromosome-positive acute lymphoblastic leukaemia (EsPhALL2010): A prospective, intergroup, open-label, single-arm clinical trial. The Lancet. Haematology. 2018;5(12) [PubMed: 30501871]
- CTTI (Clinical Trials Transformation Initative). Framework of the FDA/CTTI Patient Engagement Collaborative. 2023. [August 15, 2024]. https:
//ctti-clinicaltrials .org/wp-content /uploads/2023/05/PEC-Framework _Compliant-Apr-10-2023 _FINAL.pdf . - Cox EM, Edmund AV, Kratz E, Lockwood SH, Shankar A. Regulatory affairs 101: Introduction to expedited regulatory pathways. Clinical and Translational Science. 2020;13(3):451–461. [PMC free article: PMC7214660] [PubMed: 31909876]
- EMA (European Medicines Agency). Points to consider on application with 1. Meta-analyses; 2. One pivotal study. 2001. [August 1, 2024]. https://www
.ema.europa .eu/en/documents/scientific-guideline /points-consider-application-1meta-analyses-2one-pivotal-study_en.pdf . - EMA. Recommendation for maintenance of orphan designation at the time of marketing authorisation. 2012. [August 1, 2024]. https://www
.ema.europa .eu/en/documents/orphan-review /recommendation-maintenance-orphan-designation-time-marketing-authorisation-kalydeco-ivacaftor-treatment-cystic-fibrosis_en.pdf . - EMA. Paediatric Gaucher disease: A strategic collaborative approach from EMA and FDA. 2017. [August 1, 2024]. https://www
.ema.europa .eu/en/documents/scientific-guideline /gaucher-diseasestrategic-collaborative-approach-european-medicines-agency-and-food-and-drug-administration_en.pdf . - EMA. Guideline on registry-based studies. 2021a. [August 1, 2024]. https://www
.ema.europa .eu/en/documents/scientific-guideline /guideline-registry-based-studies_en .pdf-0 . - EMA. A vision for use of real-world evidence in EU medicines regulation. 2021b. [August 15, 2024]. https://www
.ema.europa .eu/en/news/vision-use-real-world-evidence-eu-medicines-regulation . - EMA. Engagement framework: EMA and patients, consumers and their organizations. 2022a. [August 1, 2024]. https://www
.ema.europa .eu/system/files/documents /other/updated _engagement_framework _-_ema_and_patients _consumers_and_their _organisations_2022-en.pdf . - EMA. Good practice guide for the use of the metadata catalogue of real-world data sources. 2022b. [August 1, 2024]. https://www
.ema.europa .eu/en/documents/regulatory-procedural-guideline /goodpractice-guide-use-metadata-catalogue-real-world-data-sources_en .pdf . - EMA. 20 years EU/US collaboration on medicines regulation. 2023a. [August 15, 2024]. https://www
.ema.europa .eu/system/files/documents /other/2023-09 _ema-fda-20-years-arrangement_en.pdf . - EMA. Real-world evidence framework to support EU regulatory decision-making. 2023b. [August 1, 2024]. https://www
.ema.europa .eu/system/files/documents /report/real-world-evidenceframework-support-eu-regulatory-decision-making-report-experience-gained_en.pdf . - EMA. Soliris. 2023c. [August 15, 2024]. https://www
.ema.europa .eu/en/medicines/human/EPAR/soliris . - EMA. Cluster activities. 2024a. [August 15, 2024]. https://www
.ema.europa .eu/en/partners-networks /internationalactivities /cluster-activities . - EMA. Kalydeco. 2024b. [August 15, 2024]. https://www
.ema.europa .eu/en/medicines/human/EPAR/kalydeco . - EMA. Tegsedi. 2024c. [August 15, 2024]. https://www
.ema.europa .eu/en/medicines/human/EPAR/tegsedi . - EMA. United States. 2024d. [August 15, 2024]. https://www
.ema.europa .eu/en/partners-networks /internationalactivities /bilateral-interactions-non-eu-regulators /united-states . - EMA. Accelerated assessment. [August 15, 2024]. n.d.-a. https://www
.ema.europa .eu/en/human-regulatory-overview /marketing-authorisation /accelerated-assessment . - EMA. Authorisation of medicines. [March 18, 2024]. n.d.-b. https://www
.ema.europa .eu/en/about-us/what-wedo /authorisation-medicines . - EMA. Clinical data publication. [July 10, 2024]. n.d.-c. https://www
.ema.europa .eu/en/human-regulatoryoverview /marketing-authorisation /clinical-data-publication . - EMA. Collaborating experts. [August 15, 2024]. n.d.-d. https://careers
.ema.europa .eu/content/CollaboratingExpert /?locale=en_GB . - EMA. Conditional marketing authorization. [February 20, 2024]. n.d.-e. https://www
.ema.europa .eu/en/humanregulatory-overview /marketing-authorisation /conditional-marketing-authorisation . - EMA. European experts. [March 15, 2024]. n.d.-f. https://www
.ema.europa .eu/en/about-us/how-we-work /european-medicines-regulatory-network /european-experts . - EMA. European MedicinesAgency (EMA). [August 15, 2024]. n.d.-g. https:
//european-union .europa.eu/institutionslaw-budget /institutions-and-bodies /search-all-eu-institutions-and-bodies /europeanmedicines-agency-ema_en#:~:text =The %20European%20Medicines %20Agency%20(EMA,European %20Economic%20Area%20(EEA) - EMA. Exceptional circumstances. [March 10, 2024]. n.d.-h. https://www
.ema.europa .eu/en/glossary/exceptional-circumstances . - EMA. How EMA evaluates medicines for human use. [August 15, 2024]. n.d.-i. https://www
.ema.europa .eu/en/about-us/what-we-do /authorisation-medicines /how-ema-evaluates-medicines-human-use . - EMA. How the committees work. [March 15, 2024]. n.d.-j. https://www
.ema.europa .eu/en/committees/how-committees-work . - EMA. Marketing authorisation. [July 10, 2024]. n.d.-k. https://www
.ema.europa .eu/en/human-regulatoryoverview /marketing-authorisation . - EMA. Obtaining an EU marketing authorisation, step-by-step. [August 15, 2024]. n.d.-l. https://www
.ema.europa .eu/en/human-regulatory-overview /marketing-authorisation /obtaining-eu-marketingauthorisation-step-step . - EMA. Oprhan incentives. [February 19, 2024]. n.d.-m. https://www
.ema.europa .eu/en/human-regulatory-overview /research-and-development /orphan-designation-research-and-development/orphanincentives . - EMA. Patients’ and consumers’ working party. [August 6, 2024]. n.d.-n. https://www
.ema.europa .eu/en/committees/working-parties-other-groups /comp-working-parties-other-groups /patientsconsumers-working-party . - EMA. PRIME: Priority Medicines. [August 15, 2024]. n.d.-o. https://www
.ema.europa .eu/en/human-regulatoryoverview /research-development /prime-priority-medicines . - EMA. Scientific advice and protocol assistance. [March 10, 2024]. n.d.-p. https://www
.ema.europa .eu/en/humanregulatory-overview /research-development /scientific-advice-protocol-assistance . - EMA. Who we are. [July 9, 2024]. n.d.-q. https://www
.ema.europa .eu/en/about-us/who-we-are . - EMA and FDA (U.S. Food and Drug Administration). General principles: EMA-FDAParallel Scientific Advice. 2021. [August 1, 2024]. https://www
.ema.europa .eu/en/documents/other /general-principles-european-medicines-agency-food-and-drug-administration-parallel-scientific-advice_en.pdf . - European Commission. Guideline on the format and content of applications for designation as orphan medicinal products and on the transfer of designations from one sponsor to another. 2014. [August 1, 2024]. https://health
.ec.europa .eu/system/files /2016-11/2014-03_guideline_rev4_final_0 .pdf . - European Union. Regulation (EC) NO 726/2004 of the European Parliament and of the Council. 2004. [August 1, 2024]. https://eur-lex
.europa .eu/LexUriServ/LexUriServ .do?uri=OJ:L:2004 :136:0001:0033:en:PDF . - FDA. Soliris: Drug approval package. 2007. [August 15, 2024]. https://www
.accessdata .fda.gov/drugsatfda_docs /nda/2007/125166s0000TOC.cfm . - FDA. Food and Drug Administration Transparency Task Force: Request for comments. 2010. [August 1, 2024]. https://www
.federalregister .gov/documents /2010/03/12/2010-5377 /food-and-drug-administration-transparency-task-force-request-for-comments . - FDA. Drug approval package: Kalydeco (ivacaftor). 2012. [August 15, 2024]. https://www
.accessdata .fda.gov/drugsatfda_docs /nda/2012/203188s000TOC.cfm . - FDA. Expedited programs for serious conditions—Drugs and biologics: Guidance for industry. 2014. [August 1, 2024]. https://www
.fda.gov/media/86377/download . - FDA. Review team responsibilities. 2015a. [August 15, 2024]. https://www
.fda.gov/about-fda /center-drugevaluation-and-research-cder /review-team-responsibilities . - FDA. Transparency initiative: Phase II progress report. 2015b. [August 15, 2024]. https://wayback
.archive-it .org/7993/20171101192954 /https://www .fda.gov/AboutFDA/Transparency /TransparencyInitiative /ucm273854.htm . - FDA. Transparency initiative: Phase III progress report. 2015c. [August, 2024]. https://wayback
.archive-it .org/7993/20171101192958 /https://www .fda.gov/AboutFDA/Transparency /TransparencyInitiative /ucm273856.htm . - FDA. Advisory committees: Critical to the FDA’s product review process. 2016. [July 11, 2024]. https://www
.fda.gov/drugs /information-consumers-and-patients-drugs /advisory-committees-critical-fdas-product-review-process . [PubMed: 15032199] - FDA. Best practices for communication between IND sponsors and FDA during drug development guidance for industry and review staff: Good review practice. 2017. [August 1, 2024]. https://www
.fda.gov/media/94850/download . - FDA. Drug approval package: Tegsedi (inotersen). 2018. [August 1, 2024]. https://www
.accessdata .fda.gov/drugsatfda_docs /nda/2018/211172Orig1s000TOC.cfm . - FDA. Demonstrating substantial evidence of effectiveness for human drug and biological products: Guidance for industry. 2019a. [August 1, 2024]. https://www
.fda.gov/media /133660/download . - FDA. Rare diseases: Natural history studies for drug development: Guidance for industry. 2019b. [August 1, 2024]. https://www
.fda.gov/media /122425/download . - FDA. COVID-19 update: FDA’s ongoing commitment to transparency for COVID-19 EUAs. 2020. [August 15, 2024]. https://www
.fda.gov/news-events /press-announcements /covid-19-update-fdas-ongoing-commitment-transparency-covid-19-euas . - FDA. New drug application (NDA). 2022a. [June 25, 2024]. https://www
.fda.gov/drugs /types-applications /new-drug-application-nda . - FDA. Remarks by FDA Commissioner Robert M. Califf to the 2022 NORD Breakthrough Summit. 2022b. [July 9, 2024]. https://www
.fda.gov/news-events /speeches-fda-officials /remarks-fda-commissioner-robert-m-califf-2022-nord-breakthrough-summit-10172022 . - FDA. Benefit-risk assessment for new drug and biological products: Guidance for industry. 2023a. [August 1, 2024]. https://www
.fda.gov/media /152544/download . - FDA. Demonstrating substantial evidence of effectiveness with one adequate and well-controlled clinical investigation and confirmatory evidence: Guidance for industry. 2023b. [August 1, 2024]. https://www
.fda.gov/media /172166/download . - FDA. Formal meetings between the FDA and sponsors or applicants of PDUFA products. 2023c. [August 1, 2024]. https://www
.fda.gov/media /172311/download . - FDA. Rare diseases: Considerations for the development of drugs and biological products: Guidance for industry. 2023d. [August 1, 2024]. https://www
.fda.gov/media /119757/download . - FDA. About the FDA patient representative program. 2024a. [March 10, 2024]. https://www
.fda.gov/patients /learn-about-fda-patient-engagement /about-fda-patient-representative-program . - FDA. CDER patient-focused drug development. 2024b. [March 05, 2024]. https://www
.fda.gov/drugs /development-approval-process-drugs /cder-patient-focused-drug-development . - FDA. International collaboration/pediatric cluster. 2024c. [August 15, 2024]. https://www
.fda.gov/science-research /pediatrics /international-collaboration-pediatric-cluster . - FDA. Learn about FDA advisory committees. 2024d. [August 15, 2024]. https://www
.fda.gov/patients /learn-about-fda-advisory-committees . - FDA. Patient Engagement Collaborative. 2024e. [August 15, 2024]. https://www
.fda.gov/patients /learn-aboutfda-patient-engagement /patient-engagement-collaborative . - FDA, and EMA. Common commentary - EMA/FDA: Common issues requested for discussion by the respective agency (EMA/PDCO and FDA) concerning paediatric oncology development plans (Paediatric Investigation Plans [PIPs] and initial Pediatric Study Plans [iPSPs]). 2021. [August 1, 2024]. https://www
.fda.gov/media /147197/download?attachment . - Gingery D. FDA-EMA pilot could further push global generic harmonization, but will sponsors use it? 2021. [August 1, 2024]. https://generics
.citeline .com/GB151311/FDAEMA-Pilot-Could-Further-Push-Global-Generic-Harmonization-But-Will-Sponsors-Use-It . - Hofer MP, Jakobsson C, Zafiropoulos N, Vamvakas S, Vetter T, Regnstrom J, Hemmings RJ. Impact of scientific advice from the European Medicines Agency. Nature Reviews Drug Discovery. 2015;201514(5) [PubMed: 25881970]
- Ibrahim S. CDER’s OGD and EMA’s Parallel Scientific Advice pilot program for complex generics works to increase harmonization and bring generic drugs to patients. 2024. [August 1, 2024]. https://www
.fda.gov/drugs /our-perspective /cders-ogd-and-emas-parallel-scientific-advice-pilot-program-complex-generics-works-increase . - Kweder S. Considering a PSA request? Summary and best practices. 2022. [August 1, 2024]. https://www
.fda.gov/media /164718/download . - Lee KJ, Tyner K, Thirstrup S. Impact of FDA and EMA collaborative efforts: Processes to Evaluate the Safety and Efficacy of Drugs for Rare Diseases or Conditions in the United States and the European Union (Meeting 2 - Hybrid). 2023.
- Lurie P, Chahal SH, Sigelman WD, Stacy S, Sclar J, Ddamulira B. Comparison of content of FDA letters not approving applications for new drugs and associated public announcements from sponsors: Cross sectional study. BMJ: British Medical Journal. 2015;350:h2758. [PMC free article: PMC4462714] [PubMed: 26063327]
- Marks P. FDA and EMA collaborate to facilitate SARS-CoV-2 vaccine development. 2020. [August 1, 2024]. https://www
.fda.gov/news-events /fda-voices /fda-and-ema-collaborate-facilitate-sars-cov-2-vaccine-development . - Nature Genetics. Rare diseases, common challenges. Nature Genetics. 2022;54(3):215. [PubMed: 35288710]
- Papaioannou I, Owen JS, Yáñez-Muñoz RJ. Clinical applications of gene therapy for rare diseases: A review. International Journal of Experimental Pathology. 2023;104(4) [PMC free article: PMC10349259] [PubMed: 37177842]
- Prilla S. Legal basis & types of approvals. 2018. [August 1, 2024]. https://www
.ema.europa .eu/en/documents/presentation /presentation-legal-basis-and-types-approvals-s-prilla_en.pdf . - Reaman G, Karres D, Ligas F, Lesa G, Casey D, Ehrlich L, Norga K, Pazdur R. Accelerating the global development of pediatric cancer drugs: A call to coordinate the submissions of pediatric investigation plans and pediatric study plans to the European Medicines Agency and U.S. Food and Drug Administration. Journal of Clinical Oncology: Official Journal of the American Society of Clinical Oncology. 2020;38(36):4227–4230. [PubMed: 32946356]
- Regnstrom J, Koenig F, Aronsson B, Reimer T, Svendsen K, Tsigkos S, Flamion B, Eichler H-G, Vamvakas S, Regnstrom J, Koenig F, Aronsson B, Reimer T, Svendsen K, Tsigkos S, Flamion B, Eichler H-G, Vamvakas S. Factors associated with success of market authorisation applications for pharmaceutical drugs submitted to the European Medicines Agency. European Journal of Clinical Pharmacology. 2009;66(1) [PubMed: 19936724]
- Richardson E, Daniel G, Joy DR, Kweder SL, Maloney DM, Raggio MJ, Jarow JP. Regional approaches to expedited drug development and review. Therapeutic Innovation & Regulatory Science. 2018;52(6) [PubMed: 29714587]
- Sharfstein JM, Miller JD, Davis AL, Ross JS, McCarthy ME, Smith B, Chaudhry A, Alexander GC, Kesselheim AS. Blueprint for transparency at the US Food and Drug Administration: Recommendations to advance the development of safe and effective medical products. The Journal of Law, Medicine & Ethics. 2017;45(2_suppl):7–23.
- Teixeira T, Kweder SL, Saint-Raymond A. Are the European Medicines Agency, US Food and Drug Administration, and other international regulators talking to each other? Clinical Pharmacology & Therapeutics. 2020;107(3) [PMC free article: PMC7028217] [PubMed: 31449664]
- Thirstrup S. Orphan medicines in EU: Processes to Evaluate the Safety and Efficacy of Drugs for Rare Diseases or Conditions in the United States and the European Union (Meeting 2 - Hybrid). 2023.
- Thor S, Vetter T, Marcal A, Kweder S. EMA-FDA Parallel Scientific Advice: Optimizing development of medicines in the global age. Therapeutic Innovation & Regulatory Science. 2023;57(4) [PMC free article: PMC9985489] [PubMed: 36871110]
- Tyner K, Lee KJ, Arcidiacono J, Zaidi S. International clusters and collaboration: Processes to Evaluate the Safety and Efficacy of Drugs for Rare Diseases or Conditions in the United States and the European Union (Meeting 1 - Virtual). 2023.
- Ungstrup E, Vanags D. Aligning global drug development for pediatric populations. 2023. [August 15, 2024]. https://www
.pharmalex .com/thought-leadership /blogs/aligning-global-drug-developmentfor-pediatric-populations/ - Welch AR. FDA, EMA drug approval stats: Are we measuring success the wrong way. 2015. [August 1, 2024]. https://www
.bioprocessonline .com/doc/fda-ema-approval-stats-are-we-measuring-pharma-success-the-wrong-way-0001 .
Footnotes
- 1
P.L. 75–717. Federal Food, Drug, and Cosmetic Act (June 25, 1938).
- 2
P.L. 78–410. Public Health Service Act (July 1, 1944).
- 3
Federal Food, Drug, and Cosmetic Act, SEC. 526(a)(2).
- 4
Regulation (EC) 141/2000, 1999.
- 5
21 U.S.C. § 360ff.
- 6
78 FR 35117.
- 7
Regulation (EC) No 726/2004, Article 13 (3).
- 8
Relevant regulations include: 21 CFR. §312.130(a) (non-disclosure of Investigational New Drug applications for drugs); §601.50 (non-disclosure of Investigational New Drug applications for biological products); §314.430(b)(non-disclosure of New Drug Applications prior to approval); §601.51(b) (non-disclosure of Biologics Licenses Applications prior to approval); §814.9(b) & (c) (non-disclosure of Pre-Market Approval applications prior to approval).
- 9
On May 19, 2010, the Transparency Task Force released a report containing 21 draft proposals about expanding the disclosure of information by FDA while maintaining confidentiality for trade secrets and individually identifiable patient information. FDA accepted public comment on the proposals, as well as on which draft proposals should be given priority, on this website from May 19, 2010, through July 20, 2010. https://wayback
.archiveit .org/7993/20171105152021 /https://www .fda.gov/AboutFDA/Transparency /PublicDisclosure /DraftProposalbyTopicArea /ucm211691.htm (accessed May 14, 2024). - 10
A national clinical trial number is an 8-digit unique identifier assigned to a clinical study when it is registered on ClinicalTrials
.gov. - 11
This section was edited after release of the prepublication version of the report to more accurately reflect the content and approach to common commentaries.
- 12
This section was modified after release of the prepublication version of the report to more accurately reflect Type B meetings.
- 13
Authors combined therapeutic areas into this broad category because FDA reorganized during the period of the cohort analysis and its categorization of submissions changed.
- 14
This conclusion was modified after release of the prepublication version of the report to more accurately represent coordination and collaborative efforts of FDA and EMA.
- 15
This conclusion was modified after release of the prepublication version of the report to more accurately represent coordination and collaborative efforts of FDA and EMA.
- 16
On May 19, 2010, the Transparency Task Force released a report containing 21 draft proposals about expanding the disclosure of information by FDA while maintaining confidentiality for trade secrets and individually identifiable patient information. FDA accepted public comment on the proposals, as well as on which draft proposals should be given priority, on this website from May 19, 2010, through July 20, 2010. https://wayback
.archiveit .org/7993/20171105152021 /https://www .fda.gov/AboutFDA/Transparency /PublicDisclosure /DraftProposalbyTopicArea /ucm211691.htm (accessed May 14, 2024). - 17
A national clinical trial number is an 8-digit unique identifier assigned to a clinical study when it is registered on ClinicalTrials
.gov.
- FDA and EMA Collaboration - Regulatory Processes for Rare Disease Drugs in the U...FDA and EMA Collaboration - Regulatory Processes for Rare Disease Drugs in the United States and European Union
Your browsing activity is empty.
Activity recording is turned off.
See more...